
Abstract
Parkinson’s disease (PD) and atypical parkinsonian syndromes (APS) are symptomatically characterized by parkinsonism, with the latter presenting additionally a distinctive range of atypical features. Although the majority of patients with PD and APS appear to be sporadic, genetic causes of several rare monogenic disease variants were identified. The knowledge acquired from these genetic factors indicated that defects in vesicular transport pathways, endo-lysosomal dysfunction, impaired autophagy-lysosomal protein and organelle degradation pathways, α-synuclein aggregation and mitochondrial dysfunction play key roles in PD pathogenesis. Moreover, membrane dynamics are increasingly recognized as a key player in the disease pathogenesis due lipid homeostasis alterations, associated with lysosomal dysfunction, caused by mutations in several PD and APS genes. The importance of lysosomal dysfunction and lipid homeostasis is strengthened by both genetic discoveries and clinical epidemiology of the association between parkinsonism and lysosomal storage disorders (LSDs), caused by the disruption of lysosomal biogenesis or function. A synergistic coordination between vesicular trafficking, lysosomal and mitochondria defects exist whereby mutations in PD and APS genes encoding proteins primarily involved one PD pathway are frequently associated with defects in other PD pathways as a secondary effect. Moreover, accumulating clinical and genetic observations suggest more complex inheritance patters of familial PD exist, including oligogenic and polygenic inheritance of genes in the same or interconnected PD pathways, further strengthening their synergistic connection.
Here, we provide a comprehensive overview of PD and APS genes with functions in vesicular transport, lysosomal and mitochondrial pathways, and highlight functional and genetic evidence of the synergistic connection between these PD associated pathways.
Similar content being viewed by others

Introduction
Approximately 1–2% of the worldwide population aged over 65 years is affected with PD, and up to 4–5% people aged over 85 years [71]. The mean age at onset is 70 years, although 5–10% of patients develop PD before the age of 50, referred to as early-onset PD (EOPD) [298]. Clinically, PD patients present with bradykinesia, muscle rigidity, resting tremor and gait instability [270]. Various non-motor symptoms may occur as well, including cognitive impairment and dementia, depression and apathy, excessive daytime sleepiness and insomnia, impulse control disorder, and autonomic dysfunctions [267]. PD symptoms manifests when approximately 30% of dopaminergic neurons in the substantia nigra have been degenerated [96, 120]. The most effective symptomatic treatment of PD consists of replenishing dopamine levels by administering the dopamine precursor levodopa though prolonged use could result in the development of adverse effects such as dyskinesias or wearing off [14]. Parkinsonian syndromes, of which PD is the most common one, are symptomatically defined by parkinsonism, comprising the four core motor symptoms of PD, in addition to a distinctive range of atypical features. Under this category are numerous heterogeneous syndromes that are often misdiagnosed as PD due to considerable overlap in symptoms especially early in the disease course [219].
Although the majority of patients with PD and APS appear to be sporadic, genetic causes of several rare monogenic disease variants were identified. The knowledge acquired from the protein products of identified causal genes and risk factors of PD and APS indicates that defects in vesicular transport pathways, endo-lysosomal dysfunction, impaired autophagy-lysosomal protein and organelle degradation pathways, α-synuclein aggregation and mitochondrial dysfunction play key roles in PD pathogenesis [2, 29, 121, 236, 371]. More recent advances have revealed that several parkinsonism associated genes regulate membrane dynamics wherein mutations cause lipid pathway alterations associated with lysosomal dysfunction [82, 93, 201, 213]. Additionally, associations between parkinsonism and lysosomal storage disorders (LSDs), caused by disruption of lysosomal biogenesis or function, are emerging from genetic discoveries and clinical epidemiology [82, 169].
This review focusses on these molecular pathways affected in PD and their increasingly recognized synergistic relationship in PD pathogenesis, which emerged from the identification of causal genes and risk factors contributing to the development of PD and related APS. Emerging observation suggest complex inheritance patterns of PD, including oligogenic and polygenic inheritance of gene variants of interconnected PD pathways, suggesting crosstalk between PD associated pathways.
Vesicular transport pathways regulated by PD and APS genes
Intracellular vesicular transport pathways, which enables traffic of molecules between specific membrane-enclosed compartments, are especially vulnerable in neurons due to their highly complex organization of cell body and processes comprising axons, axon terminals and dendrites. Consequently, defect of vesicular transport pathways have been implicated in multiple neurodegenerative diseases [84, 354]. Several distinct pathways of complex, highly dynamic vesicular membrane structures with overlapping properties exist, including endocytosis, exocytosis, endosomal sorting and recycling, retrograde transport and autophagy. These membrane dynamics are important to maintain overall cellular homeostasis and organelle activities. During the regulation of membrane dynamics, the lipid and protein composition of the membranes changes. Several genes associated with PD and APS encode proteins that are involved in vesicular transport pathways. These genetic discoveries illuminate defects of the endosomal trafficking machinery and disrupted trafficking as pathological processes contributing to the development of PD.
LRRK2
Autosomal dominant mutations in LRRK2 are the most frequent cause of PD, accounting for 1–2% of all PD patients (Table 1) [251]. Although over 100 missense mutations in LRRK2 have been reported, only a few are considered to be pathogenic based on co-segregation with the disease [251]. LRRK2 encodes a large, multi-domain protein containing a kinase, GTPase and protein-interaction domains. Recent research revealed that LRRK2 plays a role in vesicular transport, autophagy and lysosomal function [283]. LRRK2 is able to phosphorylate a subgroup of Rab GTPases, including Rab8A and Rab10 at highly conserved positions in the center of the effector-binding motif (Fig. 1) [325, 326]. Vesicle formation, vesicle motility along cytoskeleton elements, and docking and fusion at target membranes in the endocytic pathway is controlled by a complex regulatory machinery, which includes Rab GTPases in which Rab GTPases play a major role [25]. The pathogenic LRRK2 mutations cluster within the GTPase and kinase domains, resulting in an increased kinase activity [10]. Phosphorylation of Rab8A and Rab10 by LRRK2 prevents these Rab proteins to bind to downstream effectors, causing perturbations in vesicular transport due to pathogenic LRRK2 mutations [325, 326]. Nevertheless, other regulatory mechanisms of vesicular transport may be affected as well by LRRK2 mutations. LRRK2 is localized in Rab5 positive early and Rab7 positive late endosomes, suggesting a role in endosomal trafficking as well as the autophagy lysosomal pathway [83, 117, 118, 140, 242].

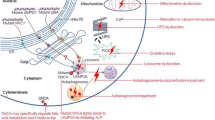
Schematic representation of vesicular transport and lysosomal pathways affected in Parkinson's disease. Mutations in α-synuclein (α-syn), LRRK2 and VPS35 are associated with autosomal dominant Parkinson's disease (PD), whereas mutations in VPS13C and ATP10B are associated with autosomal recessive PD. Mutations in ATP13A2, PLA2G6, DNAJC6, ATP6AP2 and SYNJ1 are associated with autosomal recessive atypical parkinsonian syndromes (APS). α-Synuclein interacts with membranes and functions in intracellular trafficking transport pathways. LRRK2 phosphorylates a subgroup of Rab GTPases which are important regulators of intracellular vesicle transport. VPS35, VPS26 and VPS29 form the retromer cargo-recognition complex involved in intracellular retrograde transport from endosomes to the trans-Golgi network, and associates with a dimer of sorting nexins. VPS13C tethers between the endoplasmic reticulum and late endosomes and lysosomes, and transports glycerolipids between membranes. ATP10B and ATP13A2 are both late endosomal/lysosomal P-ATPases, involved respectively in glucosylceramide export, and polyamine export/Mn2+ and Zn2+ import. ATP6AP2 is a subunit of the vacuolar H+ ATPase (V-ATPase) involved in maintaining a low lysosomal pH. PLA2G6 hydrolyzes the sn-2 ester bond of membrane glycerophospholipids to yield free fatty acids and lysophospholipids and interacts with the retromer subunits VPS35 and VPS26. DNAJC6 and SYNJ1 both play a crucial role in the detachment of the clathrin-coat after clathrin-mediated endocytosis
VPS35
The VPS35 p.Asp620Asn mutation is a rare cause of autosomal dominant inherited PD, with a prevalence of around 0.115% (Table 1) [275]. While other variants in VPS35 have been identified as well, the p.Asp620Asn mutation is the only recurrent mutation segregating with PD in different populations [275, 364]. VPS35 encodes a core component of the retromer cargo-recognition complex (CRC) involved in intracellular retrograde transport from endosomes to the trans-Golgi network [103]. The CRC trimer consists of VPS26, VPS29 and VPS35, and associates with a dimer of sorting nexins which is facilitated by Rab7 (Fig. 1) [177, 208, 301, 302]. In addition to carrying cargo from endosomes to the trans-Golgi network, the retromer carries cargo from endosomes to plasma membranes to recycle membrane bound receptors [343]. The cation-independent mannose 6-phosphate receptor (CI-MPR) is a carrier protein of the retromer system, involved in trafficking lysosomal proteases, such as the cathepsin D (CTSD), to lysosomes [40, 300]. In the trans-Golgi network, the mannose-6-phosphate residues of the CTSD signal peptide are recognized by CI-MPR and initiate trafficking towards the endosome [222]. Inside the endosome, CTSD is activated by proteolytic cleavage of the signal peptide and released for transport to the lysosome. Subsequently, CI-MPR is recycled from the endosome to the trans-Golgi network [187, 222]. The dominant negative VPS35 p.Asp620Asn mutation causes retromer dysfunction and a decreased delivery of CTSD to the lysosome, contributing to lysosomal dysfunction [98, 103]. Moreover, α-synuclein is known to be transported by the retromer complex, and knockdown of VPS35 in Drosophila leads to the accumulation of α-synuclein within the neurons [222].
DNAJC6
Missense and premature termination codon (PTC) mutations in DNAJC6, in line with autosomal recessive inheritance, have been reported in juvenile and early-onset APS called dystonia-parkinsonism (Table 1) [85, 87, 175, 240]. DNAJC6 encodes the neuron specific isoform of the co-chaperone auxilin-1, which plays a crucial role in the detachment of the clathrin-coat after clathrin-mediated endocytosis (Fig. 1). Auxilin-1 consists of a phosphoinositide phosphatase PTEN-like domain, which is required for the recruitment to a clathrin-coated pit, a clathrin-binding domain, and a J domain, which enables its interaction with Hsc70, a chaperone involved in diverse cellular processes [188]. The c.801-2A > G mutation in DNAJC6 generates two abnormal transcripts that lack either a significant part of the J domain or the PTEN-like domain, the p.Gln734* deletes 180 amino acid residues at the C-terminus of the protein, and the pathogenic p.Arg927Gly missense mutation in the J domain is predicted to reduce the positive charge on the protein surface, suggesting a loss-of-function mechanism for all pathogenic DNAJC6 mutations [85, 175, 240].
SYNJ1
The homozygous SYNJ1 p.Arg258Gln and p.Arg459Pro were identified in families affected with autosomal recessive juvenile APS (Table 1) [168, 180, 239, 273]. SYNJ1 encodes synaptojanin 1, a phosphoinositide phosphatase with a major role in endocytic recycling of synaptic vesicles [273]. SYNJ1 contains two different phosphatase domains, the 5-phosphatase and the Sac1 domain, which target different phosphoinositide phosphates (PIPs), and cooperates with Auxilin and Hsc70 to remove the clathrin-coat after clathrin-mediated endocytosis (Fig. 1) [180, 273]. The 5-phosphatase domain regulates synaptic vesicle endocytosis by dephosphorylating phosphatidylinositol 4,5-bisphosphate (PIP2) to facilitate uncoating of clathrin coated vesicles [60, 211]. Knock-in of the SYNJ1 p.Arg258Gln mutation in mice leads to accumulation of auxilin, clathrin and parkin, and impaired synaptic vesicle endocytosis in neurons [46].
PD and APS genes with endo-lysosomal functions
Lysosomes are the endpoint of various degradation pathways including endocytosis and autophagy and contain nearly 60 different hydrolytic enzymes including nucleases, proteases, phosphatases, lipases, sulfatases amongst others to degrade macromolecules and cellular components [290]. Many factors regulate lysosomal function including an acidic internal pH at which lysosomal hydrolases are active through the activity of vacuolar H+ ATPase (V-ATPase) [290]. Neurons are especially vulnerable to lysosomal dysfunction because, without the aid of cell division, they are largely dependent on autophagy to prevent the accumulation of cellular protein and damaged organelles. Insufficient degradation of neurotoxic proteins by lysosomes has been implicated in multiple neurodegenerative diseases [30]. Two novel PD genes, VPS13C and ATP10B, and several APS genes encode proteins involved in endo-lysosomal functions.
VPS13C
The vacuolar protein sorting 13 C (VPS13C) gene was first identified as a new risk gene for PD in a meta-analysis of genome-wide association studies (GWAS) [230], and later homozygous and compound heterozygous PTC mutations in VPS13C were associated with a distinct form of early-onset parkinsonism characterized by rapid and severe disease progression and early cognitive decline [193]. Two independent studies confirmed VPS13C loss-of-function mutations in autosomal recessive PD [69, 297]. To date, the mutation spectrum includes PTC mutations, and a large deletion comprising multiple exons (Table 1) [69, 193, 297]. The human VPS13 family consists of four proteins, VPS13A/Chorein, VPS13B, VPS13C and VPS13D, with all family members having a strong homology to yeast Vps13. Yeast studies have suggested that Vps13 may have a role in lipid exchange between organelles and showed that yeast mutants lacking Vps13 causes defects in mitochondrial membrane integrity [186, 259]. More recent research showed that human VPS13C is a lipid transport protein that functions as a tether between the endoplasmic reticulum (ER) and late endosomes and lysosomes, and between the ER and lipid droplets, enabling transport of glycerolipids between membranes (Fig. 1) [183]. Subsequently, loss of VPS13C implicates defects in membrane lipid homeostasis and lysosomal dysfunction. Interestingly, loss-of-function mutations in other human VPS13 genes are associated with different recessive neurological disorders [67, 174, 304].
ATP10B
We recently identified compound heterozygous loss-of-function mutations in the ATPase class V type 10B (ATP10B) gene increasing risk for PD (Table 1) [213]. ATP10B mRNA is mainly expressed in the gastrointestinal track and the brain [213]. Approximately 65% PD patients develop gastrointestinal disorders 4 years after diagnosis and Lewy body pathology is also observed in the enteric nervous system of PD patients [35, 210, 353]. Previously, ATP10B was identified as a P4-type transport ATPase present in the late endo−/lysosomal compartment [228]. P4 ATPases are lipid flippases that use ATP to drive the transport of lipids from the lumen to the cytosolic membrane leaflet, establishing the vitally important lipid asymmetry between two membrane leaflets [11, 254]. ATP10B forms a heteromeric complex with the Cell Cycle Control Protein 50A (CDC50A) to facilitate the trafficking from the ER to the late endosome and lysosome [11]. We established that ATP10B is involved in the translocation of glucosylceramide (GluCer) and phosphatidylcholine (PC) towards the cytosolic membrane leaflet of late endosomes/lysosomes (Fig. 1) [213]. Moreover, ATP10B might also transport glucosylsphingosine and sphingomyelin besides GluCer and PC [213]. ATP10B functionally belongs to the ATP10A/D sub-class of human lipid flippase isoforms that share highly conserved functional domains for GluCer and PC transport, pointing to a physiological role of the ATP10A, B and D transporters in GluCer/PC uptake or subcellular redistribution [281]. However, so far no neurological diseases are associated with ATP10A or ATP10D. In cellular overexpression models, the identified PD associated ATP10B mutants were shown to be catalytically inactive and failed to provide cellular protection against the environmental PD risk factors rotenone and manganese [213]. In isolated cortical neurons, knockdown of ATP10B led to a significant loss of lysosomal mass and a higher lysosomal pH resulting in a global reduction of lysosomal degradative capacity. Rotenone exposure in ATP10B knockdown cortical neurons also impaired lysosomal membrane integrity, which is a major driver of lysosome dependent cell death [5].
ATP13A2
Loss-of-function mutations in ATP13A2 cause Kufor-Rakeb syndrome, a rare form of juvenile onset autosomal recessive APS (Table 1) [277]. Pathogenic mutations in ATP13A2 are as well identified in patients with neuronal ceroid lipofuscinoses, a neurodegenerative LSD, and patients with hereditary spastic paraplegia [37, 90]. The considerable clinical heterogeneity of ATP13A2 mutation carriers could be partially explained by variable impact of different mutations on protein expression and functionality of ATP13A2 [257]. Interestingly, ATP13A2 has been identified in Lewy bodies in brains of sporadic PD patients [72, 226].
ATP13A2 encodes a P-type ATPase, mainly localized at endosomes and lysosomes, with a role in manganese (Mn2+) and zinc (Zn2+) homeostasis, mitochondrial bioenergetics, and the autophagy lysosomal pathway (Fig. 1) [23, 128, 258, 336, 345]. Recently, ATP13A2 was identified as a lysosomal polyamine exporter with a high affinity for spermine [348]. The protein is highly expressed in the brain, especially in the substantia nigra. Most of the pathogenic missense mutations occur in functional domains of ATP13A2, including the transmembrane domains and the E1-E2 ATPase domain, resulting in a loss of protein function [257]. Patient derived cells of ATP13A2 mutation carriers revealed an impaired Zn2+ homeostasis, with lysosomal and mitochondrial dysfunction as a consequence [258, 345]. Expression of wild-type but not mutant ATP13A2 protects mammalian cell lines and primary rat neuronal cultures against manganese induced cell death, also known as a PD environmental risk factor [336]. High concentrations of polyamines was shown to induce cell toxicity, which exacerbated by ATP13A2 loss due to lysosomal dysfunction [348]. Additionally, ATP13A2 has been shown to be involved in α-synuclein metabolism [205] and lipid homeostasis [212].
PLA2G6
Autosomal recessive mutations in the phospholipase A2 group 6 gene (PLA2G6) are causative for phospholipase A2-associated neurodegeneration (PLAN) syndromes, including classic infantile neuroaxonal dystrophy (INAD) and atypical neuroaxonal dystrophy with childhood-onset (atypical NAD), and adult onset APS called dystonia-parkinsonism, which is associated with Lewy bodies and neuroaxonal dystrophy (Table 1) [139, 157, 161, 249, 316, 370].
Mutations responsible for loss of PLA2G6 catalytic activity usually lead to INAD and atypical NAD whereas mutations altering substrate preference or regulatory mechanisms are usually causal for adult onset dystonia-parkinsonism [88]. However, patients carrying the same PLA2G6 mutation with different clinical phenotypes have been reported [225, 316]. The protein encoded by PLA2G6 is a calcium-independent group VI phospholipase A2 (iPLA-β), which hydrolyzes the sn-2 ester bond of membrane glycerophospholipids to yield free fatty acids and lysophospholipids (Fig. 1) [17]. iPLA-β expression is enriched in dendrites and axon terminals [241]. PLA2G6 is involved in repair of oxidative damage to membrane phosphopholipids, membrane remodeling and iron homeostasis [17, 309]. Recently, Lin and colleagues demonstrated that the fly homolog of iPLA-β binds the retromer subunits VPS35 and VPS26, and that loss of iPLA-β impairs retromer function, causes lysosomal ceramide accumulation, and leads to lysosomal dysfunction [200].
ATP6AP2
Two synonymous variants in ATP6AP2, p.Ser115Ser and p.Asp107Asp, increasing exon 4 skipping, were identified in patients with X-linked APS characterized by parkinsonism, spasticity, intellectual disability and epilepsy (Table 1) [176, 279]. ATP6AP2 encodes the lysosomal renin/prorenin receptor, an accessory unit of vacuolar H+ ATPase (V-ATPase) required for lysosomal degradative functions and autophagy (Fig. 1). Alterations in ATP6AP2 are involved in different human phenotypes, suggesting a critical function in various organ systems [44, 127, 135, 144, 176, 245, 279, 287, 358]. ATP6AP2 interacts with renin/prorenin at the cell membrane which enhances proteolytic activity toward Angiotensin II and causes activation of intracellular signaling pathways resulting in secretion of inflammatory and fibrotic factors [274]. Consistent with its role in renin signaling, ATP6AP2 polymorphisms have been linked to hypertension [135, 245, 358]. Moreover, as an accessory unit of the membrane transporter H+ ATPase, ATP6AP2 is involved in maintaining a low lysosomal pH and, thereby, degradation of cellular waste [255]. siRNA knockdown of ATP6AP2 in HEK293 cells results in perturbed autophagy, inhibited lysosomal clearance and in the accumulation of autophagosomes, suggesting that the impaired autophagy in ATP6AP2 mutation carriers is due to reduced vacuolar H+ ATPase activity [176]. Moreover, ATP6AP2 is a component of the WNT receptor complex involved in the canonical WNT signal transduction pathway in Drosophila and Xenopus [39, 63].
α-Synuclein aggregation connected to defects in vesicular transport and autophagy-lysosomal dysfunction
The autophagy-lysosomal pathway is one of the two major degradation pathways present in the cell for identifying and delivering cytosolic components to the lysosome for degradation and recycling [79]. Macro-autophagy is involved in the degradation of aggregated proteins and damaged organelles, via engulfment by a phagophore membrane, subsequent maturation into a vesicle called the autophagosome and afterwards fusion with a lysosome. Meanwhile, micro-autophagy is characterized by direct lysosomal engulfment of cytosolic material into the lysosomes via the formation of invaginations of the lysosomal membrane. Chaperone-mediated autophagy is involved in the degradation of soluble monomeric proteins containing the penta-peptide motif KFERQ, via transport to the lysosome by the chaperone HSC70.
PD is pathologically characterized by the presence of Lewy bodies and Lewy neurites, composed mainly of amyloid fibrils of α-synuclein, in neurons [78] of the central nervous system, e.g. basal ganglia, the dorsal motor nucleus of the vagus, the olfactory bulb, the locus coeruleus, and of the peripheral nervous system, e.g. the enteric nervous system [35, 353]. α-Synuclein, encoded by SNCA, was initially linked to PD as the main component of Lewy bodies [323] and subsequently, dominant mutations in SNCA were identified as the first genetic cause of familial PD (Table 1) [268]. Pathogenic SNCA mutations are present in approximately 1–2% familial and 0.2% sporadic PD patients [95, 192, 238]. SNCA mutations are also implicated in dementia with Lewy bodies (DLB) [124, 243], and single nucleotide polymorphisms (SNPs) in SNCA are associated with multiple system atrophy, an APS pathologically characterized by the presence of α-synuclein immunoreactive glial cytoplasmic inclusions [6, 263, 295].
α-Synuclein is abundantly expressed in the central nervous system, with a prominent presynaptic localization, as a lipid binding protein that interacts with membranes (Fig. 1) [70, 143]. The function of α-synuclein remains poorly understood but involves maturation of pre-synaptic vesicles, synaptic vesicle recycling, regulation of neurotransmitter release, and plasticity of dopaminergic neurons [202, 206, 233]. The protein contains an N-terminal domain, which includes an imperfect conserved repeat KTKGEV and acts as the membrane anchor region, and a central non-amyloid β component domain consisting of hydrophobic residues that renders α-synuclein susceptible to polymerization [115]. The latter domain also behaves as a lipid sensor and determines the membrane binding affinity of α-synuclein [102]. Under normal physiological conditions α-synuclein occurs as a monomeric disordered protein which could shift upon membrane binding to an amphipathic helical structure [347]. Nevertheless, α-synuclein can also convert from the disordered, monomeric form to polymerized β-sheets constructed of additional recruited α-synuclein monomers, which will eventually lead to the formation of protofilaments and amyloid fibrils (Fig. 1) [59]. α-Synuclein can be degraded by both chaperone-mediated autophagy and micro-autophagy, with both pathways reported to be impaired in PD pathogenesis, resulting in α-synuclein accumulation [64]. Meanwhile, studies in both animal models and human induced pluripotent stem cell (iPSC) derived dopaminergic neurons have shown that elevated levels of α-synuclein disrupt numerous intracellular trafficking transport pathways, including at the ER, early and late endosomes, and lysosomes [58, 216, 247]. α-Synuclein expression in yeast resulted in an early block in ER to Golgi vesicular trafficking [58, 247]. In human midbrain synucleinopathy models, generated through lentiviral overexpression of α-synuclein in control cultures, or through the generation of patient lines harboring PD-causing mutations, α-synuclein accumulation was found to reduce lysosomal degradation capacity by disrupting trafficking of lysosomal hydrolases [216]. Moreover, α-synuclein accumulation disrupted the ER-Golgi localization of Rab1a, a key mediator of vesicular transport [216].
PD and APS genes involved in mitochondrial pathways and mitophagy
Mitochondria are essential energy producing organelles that regulate cellular energy homeostasis and cell death. Mitochondria are highly dynamic and undergo fission and fusion to maintain a functional mitochondrial network [221]. Mitophagy, a process involved in the selective removal of damaged mitochondria through macro-autophagy, is therefore crucial for maintaining proper cellular functions. Mitophagy comprises three important steps: the recognition of impaired mitochondria and the formation of autophagic membranes, the engulfment by a phagophore membrane and subsequent maturation into a mitoautophagosome, and the fusion of the mitoautophagosome with a lysosome [51]. Defects in the autophagy-lysosomal pathway consequently lead to inappropriate removal of damaged mitochondria.
Mitochondrial dysfunction is known to contribute to several neurodegenerative diseases, including PD [121, 148]. A reduction in complex I mitochondrial respiratory chain activity was observed in in vivo and in vitro models of PD as well as in post-mortem brain tissue of idiopathic PD patients implicating a role for mitochondrial dysfunction in PD pathogenesis [256, 261, 294]. Later, loss-of-function homozygous and compound heterozygous mutations in PARK2, PINK1, PARK7/DJ-1 were found to be responsible for autosomal recessive EOPD. The proteins encoded by PARK2 (parkin), PINK1 and DJ-1 have various well described functions but appear to converge towards mitochondrial function, including mitophagy, mitochondrial dynamics and oxidative stress control. Moreover, FBXO7, in which autosomal recessive mutations cause juvenile Parkinsonian-pyramidal syndrome, is connected to the PARK2/PINK1 mitochondrial pathway (Fig. 2, Table 1).

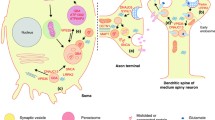
Schematic representation of the mitochondrial and oxidative stress pathways affected in Parkinson disease. Mutations in parkin, PINK1, DJ-1 and FBXO7 have been associated with autosomal recessive Parkinsonian syndromes. a In healthy, polarized mitochondria (∆Ψm) PINK1 translocates at the inner mitochondrial membrane via the mitochondrial import receptor TOMM20 machinery, which subsequently results in the degradation of PINK1. In damaged, depolarized mitochondria (∆Ψm↓), PINK1 accumulates at the outer mitochondrial membrane and recruits parkin upon phosphorylation. Moreover, parkin mediates the degradation of the parkin interacting substrate (PARIS), a repressor of the PGC1α transcriptional coactivator, leading to nuclear translocation of PGC1α and transcriptional activation of mitochondria associated genes. b In oxidative stress conditions or ∆Ψm↓, DJ-1 p.Cys106 will form a sulfonic acid, which will activate DJ-1 to regulate transcription of antioxidant genes and to promote mitophagy. c FBXO7 is a subunit of the SKP1-cullin-F-box (SCF) complex. PINK1 is involved in the recruitment of FBXO7 to damaged mitochondria which in turn leads to the recruitment of parkin
PARK2
Bi-allelic mutations in PARK2 are the most common cause of autosomal recessive PD (Table 1) [264]. Over 120 loss-of-function mutations have been identified so far, explaining approximately 15% of EOPD patients with an age at onset of 40–50 years [1]. Parkin, encoded by PARK2, plays a central role as a cytosolic E3 ubiquitin-ligating enzyme, and works in together with E1 ubiquitin-activating and E2 ubiquitin-conjugating enzymes of the ubiquitin proteasome system to ubiquitinate misfolded, damaged or unwanted proteins for degradation [307]. Later it was discovered that parkin is selectively recruited to impaired mitochondria during stress or upon membrane depolarization (Fig. 2a) [231]. Once localized at the outer mitochondrial membrane of depolarized mitochondria, parkin promotes the ubiquitination of outer mitochondrial membrane proteins involved in the upregulation of mitochondrial fusion, mitofusin 1 and mitofusin 2 (Fig. 2a) [112, 269]. The subsequent removal of these proteins would shift mitochondrial dynamics of fission and fusion and will eventually lead to mitochondrial fragmentation. Fragmented mitochondria are subsequently removed via mitophagy [48, 189]. Additionally, parkin regulates mitochondrial biogenesis by mediating the degradation of the parkin interacting substrate (PARIS), a repressor of the peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC1α) transcriptional coactivator, leading to nuclear translocation of PGC1α and transcriptional activation of mitochondria associated genes [308, 328]. Interestingly, loss of dopaminergic neurons have been demonstrated in animal models in which the PGC1α gene was silenced or knocked out [146, 235].
PINK1
Mutations in the PTEN-induced putative kinase 1 gene (PINK1) are the second leading causes of recessive PD (Table 1). The prevalence of homozygous and compound heterozygous PINK1 mutations varies from 0 to 4% [32, 54, 101, 170, 197, 238, 335, 361]. PINK1 is a highly conserved putative serine/threonine protein kinase localized to mitochondria via its mitochondrial targeting sequence at the N-terminus, and recognizes mitochondrial depolarisation, reactive oxygen species (ROS), or protein misfolding [107, 310, 355]. Most pathogenic mutations in PINK1 are found in the serine/threonine kinase domain, suggesting loss of kinase activity plays a crucial role in the pathogenesis of PINK1-associated PD. In healthy mitochondria, the electrical polarization of the inner mitochondrial membrane ensures PINK1 to translocate at the inner mitochondrial membrane via the mitochondrial import receptor TOMM20 machinery. Here, PINK1 is rapidly cleaved by mitochondrial proteases, including the rhomboid protease presenilin-associated rhomboid-like protein (PARL), and subsequently degraded by the ubiquitin-proteasome system in the cytosol (Fig. 2a) [130]. In damaged, depolarized mitochondria, translocation of PINK1 by the TOMM20 machinery is inhibited, resulting in the accumulation of PINK1 at the outer mitochondrial membrane. Uncleaved PINK1 recruits and activates parkin by phosphorylating both ubiquitin and the ubiquitin-like domain of parkin (Fig. 2a) [153, 158, 178]. Loss-of-function of either parkin or PINK1 results in the accumulation of dysfunctional mitochondria in the cytoplasm, resulting in oxidative stress and subsequently cell death [91, 110, 113, 119, 253].
DJ-1
Approximately 0.4–1% of EOPD is caused by homozygous or compound heterozygous loss-of-function mutations in DJ-1 (Table 1) [31, 163]. The protein encoded by DJ-1 is involved in transcriptional regulation, oxidative stress responses, anti-apoptotic signaling and protein quality control within the neuronal cells [13, 43, 52, 114, 246, 352]. In addition, DJ-1 is required for the degradation of dysfunctional mitochondria via mitophagy [357]. DJ-1 is predominantly located in the cytoplasm and to a lesser extent at mitochondria and in the nucleus [151, 376]. However, in oxidative stress conditions, the translocation of DJ-1 into the nucleus is enhanced. Simultaneously, the highly conserved cysteine residue (p.Cys106) of DJ-1 will form a sulfonic acid (SOH, SO2H), which will activate DJ-1 to regulate transcription of antioxidant genes (Fig. 2b) [13, 43, 166, 167]. Additionally, upon oxidative stress and a decrease in mitochondrial membrane potential, DJ-1 associates with mitochondria promoting mitophagy through mechanisms that are still unknown (Fig. 2b) [152, 357]. Knockdown of DJ-1 results in increased ROS, decreased mitochondrial membrane potential and changes in mitochondrial morphology, leading to cell death [12, 26, 179].
The parkin/PINK1/DJ-1 mitochondrial pathway
The functions of parkin, PINK1 and DJ-1 intersect at the mitochondria, but whether DJ-1 is directly or indirectly implicated in a common pathway involving parkin and PINK1 is still inconclusive. For example, DJ-1 is not necessary for the recruitment of parkin to depolarized mitochondria and is not able to rescue the mitochondrial defects caused by loss of parkin [131, 218, 341]. However, DJ-1 is able to reduce damage from the mitochondrial complex I inhibitor rotenone in the absence of PINK1, without altering PINK1 mitochondrial phenotypes [341]. Meanwhile, both PINK1 and parkin can rescue the mitochondrial fragmentation caused by the loss of DJ-1 in primary neurons and immortalized cells [49, 142, 341]. These data suggest that DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and mitophagy.
FBXO7
Loss-of-function mutations in the F-box protein 7 gene (FBXO7) are responsible for autosomal recessive APS with various heterogenic phenotypes (Table 1) [76, 311]. FBXO7 is a subunit of the SKP1-cullin-F-box (SCF) complex that acts as a multimeric E3 ubiquitin ligase, wherein cullin1 and SKP1 constitute the core of the E3 ligase and FBXO7 functions as substrate-recruiting subunit. To ubiquitinate a substrate in the ubiquitin proteasome pathway, the SCF complex brings the E2-ubiquitin-conjugate and the substrate in close proximity [149]. Interestingly, FBXO7 contains at its N-terminus an ubiquitin-related (UbR) domain, which mediates the interaction with parkin to regulate mitochondrial quality control [42]. Indeed, mediated by PINK1, FBXO7 is translocated to damaged mitochondria where it is required for a successful recruitment of parkin (Fig. 2) [42]. Drosophila models showed that overexpressing FBXO7 suppresses mitochondrial disruption and neurodegeneration in PARK2 mutants, confirming that they share a common role in mitochondrial biology [42, 378].
Lysosomal dysfunction and lipid homeostasis alterations in PD: insights from lysosomal storage disorders genes
LSDs are Mendelian-inherited metabolic disorders caused by dysfunction in lysosomal biogenesis or function resulting in the abnormal accumulation of non-degraded substrates. More than 50 LSDs exist with a broad spectrum of clinical manifestations depending on the specific substrate and the location of substrate accumulation caused by protein deficiencies associated with lysosomal function, including proteins involved in lipid metabolism (Fig. 3). Besides α-synuclein, Lewy body pathology consists of crowded membranes and lipids originating form vesicles and fragmented organelles, including mitochondria and lysosomes [306]. Compelling associations between parkinsonism and LSDs are emerging from clinical epidemiology and genetic discoveries (Table 2). Progressive cognitive and motor decline is present in more than two-thirds of LSDs, often including parkinsonism [305].

Lipid metabolism and lysosomal storage disorders associated with Parkinson's disease. Proteins indicated in orange are associated with Parkinson's disease (PD) and/or atypical Parkinsonian syndromes (APS). Proteins indicated in blue are causal for lysosomal storage disorders (LSDs) but are also linked to PD. The primary syndromes linked to the proteins are indicated in red. The lysosomal integral membrane protein 2 (LIMP-2) is involved in the transport of glucocerebosidase (GCase) from the endoplasmic reticulum to the lysosome. Once in the lysosome, GCase catalyzes the breakdown of glucosylceramide (GluCer) to ceramide and glucose. Ceramide is also obtained by acid sphingomyelinase (ASM) which catalyzes the hydrolysis of sphingomyelin to phosphocholine and ceramide, and GALC which hydrolyzes galactolipids, including galactosylceramide. Both α-synuclein (α-syn) and PLA2G6 have also been associated with ceramide levels, though the exact mechanisms are still unknown. Meanwhile, NPC1 has been associated with GluCer levels, even though NPC1 primarily mediates together with NPC2 intracellular cholesterol trafficking. Furthermore, ATP10B is involved in translocating GluCer towards the cytosolic membrane leaflet. Both ceramide and GluCer levels appear to play an important central role in PD pathogenesis. Meanwhile, NAG degrades heparan sulfate glycosaminoglycans by hydrolyzing terminal N-acetyl-D-glucosamine residues. Finally, VPS13C is a glycerolipid transporter between the endoplasmic reticulum and lysosomes
GBA
Homozygous and compound heterozygous loss-of-function mutations in GBA cause Gaucher disease (GD), the most common autosomal recessive LSD [344]. Clinical observations indicated parkinsonian features in a subset of GD patients and in heterozygous GBA relatives of GD patients, suggesting a role for GBA in the genetic etiology of PD. Indeed, a multicenter genetic analysis in 2009 confirmed an increased risk (odds ratio 5.43) of developing PD in heterozygous and homozygous GBA mutation carriers [313]. The missense mutations p.Asn370Ser and p.Leu444Pro are the most frequent observed pathogenic mutations in GD and PD patients, accounting for 17–31% in Ashkenazi Jewish PD patients and up to 4.5% in other PD patients [61, 234, 313, 349]. GBA patient carriers present an earlier age at onset and more frequent cognitive impairment compared to idiopathic PD patients [61, 234]. Widespread and abundant diffuse neocortical Lewy body pathology can be observed in brains of heterozygous GBA mutation carriers [234, 262]. Moreover, the activity of glucocerebosidase (GCase), the protein encoded by GBA, was found to be significantly reduced in postmortem brain tissue of PD patients with and without heterozygous GBA mutations, with the most profound reduction in the substantia nigra [111, 227]. The lysosomal enzyme GCase is involved in sphingolipid metabolism by catalyzing the breakdown of GluCer to ceramide and glucose (Fig. 3) [27, 82]. Loss of GCase and elevated GluCer levels were shown to increase α-synuclein aggregation, though the exact mechanism is unclear [165, 217]. The role of GCase in PD pathogenesis is extensively reviewed elsewhere [27, 82].
SMPD1
Recessive mutations in the sphingomyelin phosphodiesterase 1 (SMPD1) gene are responsible for neuropathic (Type A) and non-neuropathic (Type B) Niemann-Pick disease (NPD) [195, 299], while heterozygous mutations are associated with an increased risk of developing PD [106]. NPD is characterized by hepatosplenomegaly and progressive neurodegeneration, including ataxia, cognitive decline and seizures. In the Ashkenazi Jewish population, the pathogenic mutation p.Leu302Pro in SMPD1 was found to substantially increase risk for PD (odds ratio 9.4) [106]. More studies in different populations confirmed the association between several pathogenic SMPD1 mutations and PD [8, 55, 66, 74, 99, 105, 367]. SMPD1 encodes acid sphingomyelinase (ASM) which catalyzes the hydrolysis of sphingomyelin to phosphocholine and ceramide in late endosomes and lysosomes (Fig. 3). Subsequently, SMPD1 loss-of-function mutation carriers present an accumulation of sphingomyelin within lysosomes [53]. Strangely, Niemann-Pick type B patients with bi-allelic SMPD1 loss-of-function mutations, resulting in approximately 10% residual ASM activity [351], present rarely neurological symptoms, while a single heterozygous SMPD1 loss-of-function mutation, anticipating 50% residual ASM activity, increases the risk for PD. However, no differences in average ASM enzymatic activity in blood of sporadic PD patients compared to control individuals were observed [8, 9]. Meanwhile, an earlier age at onset in sporadic PD patients was found to be associated with reduced ASM activity levels [8]. Knockout and knockdown of SMPD1 in HeLa and BE (2)-M17 dopaminergic cells resulted in increased α-synuclein levels [8]. The same study provided evidence that the pathogenic p.Leu302Pro mutation impair the localization of ASM to the lysosome [8]. As the association between SMPD1 and PD is not fully understood, the functional effects of PD associated mutations need to be investigated.
SCARB2
Homozygous mutations in the scavenger receptor class B member 2 (SCARB2) gene cause action myoclonus-renal failure syndrome (AMRF), which is an autosomal recessive progressive myoclonic epilepsy, and are associated with significantly reduced GCase activity in patients [24, 68, 374]. Given the observation that homozygous SCARB2 mutation carriers have reduced GCase activity, heterozygous SCARB2 mutations might modify PD risk. Two common SNPs in SCARB2, rs6812193 and rs6825004, have been associated with PD and DLB in several genetic studies, including GWAS (odds ratio 0.84–0.91) [7, 36, 81, 136, 220, 230]. SCARB2 encodes the lysosomal integral membrane protein 2 (LIMP-2), a mannose-6-phosphate-independent trafficking receptor which transports GCase from the ER through the Golgi apparatus and endosomes to the lysosome (Fig. 3) [374]. However, the functional variants responsible for the association between SCARB2 and PD risk remain to be defined.
GALC
Recessive mutations in the galactosylceramidase (GALC) gene cause Krabbe disease (KD), also known as globoid cell leukodystrophy or galactosylceramide lipidosis, which is a rare, often fatal LSD resulting in progressive damage to the white matter of the peripheral and central nervous system [292]. More than 70 disease causing mutations have been identified, including missense mutations, and small deletions and insertions [362]. Recently, variants at the GALC locus were significantly associated with increased PD risk in a large GWAS meta-analysis [50]. The common SNP rs8005172, 12.6 kilobases proximal to the GALC promoter, was the most strongly associated variant, though the functional variant responsible for the association is not yet known. Interestingly, this SNP is significantly associated with GALC expression in multiple tissues, including the brain [19]. Moreover, brain tissue from the twitcher mouse model for KD and from patients affected with KD identified the presence of aggregated forms of α-synuclein and ubiquitin, which are both involved in Lewy bodies [318]. GALC is a lysosomal enzyme that hydrolyzes galactolipids, including galactosylceramide and galactosylsphingosine (Fig. 3). Mutations in GALC result in low enzymatic activity and a decreased ability to degrade galactolipids. Since galactosylceramide is an important glycosphingolipid in myelin, the pathological consequences of the GALC deficiency in KD are almost exclusively confined to the white matter of the central and peripheral nervous systems [45, 331]. The involvement of GALC in ceramide metabolism supports a role for GALC in PD risk, though further studies are needed to confirm the association with PD, and to unravel the underlying pathomechanism leading to the development of PD.
NPC1/NPC2
Niemann-Pick type C (NPC) disease is a rare autosomal recessive inherited disorder with a highly variable phenotype similar to Type A/B NPD and ranging from a fatal disorder within the first few months after birth to a late onset progressive disorder with predominantly neuropsychiatric symptoms of which the diagnosis is challenging. NPC is caused by mutations in the NPC intracellular cholesterol transporter 1 (NPC1) gene in 95% of cases or the NPC intracellular cholesterol transporter 2 (NPC2) gene in the remaining 5% of cases [47, 232, 288]. More than 260 mutations have been identified in NPC1, mostly missense mutations affecting the luminal domain of the protein [288]. Parkinsonism has been described in several NPC patients and their relatives [57, 150, 171]. Moreover, autopsy reports have described phosphorylated α-synuclein pathology in brain tissue of NPC patients [291], suggesting a possible link between NPC and PD. However, genetic studies investigating the association between NPC1/NPC2 variants and PD have shown conflicting results [171, 373].
NPC1 encodes a large protein that resides in the late endosomes and lysosomes which mediates cholesterol efflux [185]. When cholesterol is released from low-density lipoproteins in the lumen of the late endosomes/lysosomes, it is transferred by NPC2 to the N-terminal cholesterol-binding pocket of NPC1 [141, 359]. Loss-of-function of NPC1 leads to accumulation of cholesterol in the late endosome/lysosome [155, 375].
NAGLU
Recessive mutations in NAGLU are responsible for Sanfilippo syndrome B, also known as mucopolysaccharidosis III disease B (MPS-IIIB) [377]. Patients with MPS-IIIB present early-onset progressive neurological disturbances, including mental retardation, hyperactivity and seizures, in addition to mild somatic manifestations. Interestingly, immunoreactivity for phosphorylated α-synuclein was observed in brain tissue of MPS-IIIB patients [129]. The SNP rs2071046 tagging a common haplotype of NAGLU was shown to be associated with an increased risk for PD (odds ratio 1.23) [365]. Moreover, in a recent large meta-analysis of GWAS, NAGLU was nominated as a gene of interest in novel loci significantly associated with increased risk for PD [229].
NAGLU encodes α-N-acetylglucosminidase (NAG), a lysosomal hydrolase which degrades heparan sulfate glycosaminoglycans by hydrolyzing terminal N-acetyl-D-glucosamine residues (Fig. 3). Interestingly, heparan sulfate stimulates the formation of α-synuclein fibrils in vitro [56], and in neuroblastoma cells, cellular internalization of α-synuclein amyloid fibrils is dependent on heparan sulfate [138]. Glycosaminoglycans modulate the lysosome degradation pathway by regulating CTSD, the major lysosomal protease responsible of α-synuclein degradation. In a neuroblastoma cell model of PD, elevated glycosaminoglycans levels resulted in reduced CTSD activity and intracellular accumulation of α-synuclein [190].
Other lysosomal storage disorder genes genetically linked to PD
The LSD genes MCOLN1, ARSB, GUSB, GRN and NEU1 have been genetically connected with PD as well via gene based association studies, whole exome sequencing in unrelated PD patients or meta-analysis of GWAS, but await further replication [50, 145, 229]. Interestingly, a significant burden of rare LSD gene variants in PD patients was observed in a large whole exome sequencing dataset including > 1100 PD patients and > 1600 control individuals, taken into account 54 different LSD genes [280]. In this study, SLC17A5, CTSD, ASAH1 were identified as novel candidate susceptibility genes for PD risk [280].
Disrupted lipid homeostasis in PD pathogenesis
In recent years, lipid homeostasis has become a principal suspect in PD pathogenesis [368]. Binding of α-synuclein to membranes depends on its lipid composition, which has led to the hypothesis that PD pathology is induced by alterations in the binding properties of α-synuclein to membranes or lipid rafts. Indeed, α-synuclein is known to selectively interact phospholipids and sphingolipids, and disruption of lipid metabolism has been found to predispose α-synuclein toxicity [70, 77, 92, 93, 182, 214, 215, 276, 318, 329, 332, 333, 380].
Interestingly, both GCase and ATP10B play essential roles in the fate of lysosomal GluCer, and loss-of-function of both proteins results in the intra-lysosomal accumulation of GluCer combined with lysosomal dysfunction (Fig. 3) [97, 165, 213]. ATP10B may therefore synergize with GCase to maintain low levels of GluCer or GluSph in the lysosome. Accordingly, loss of ATP10B functionality might result in the same affected pathways associated with loss of GCase functionality. Besides lysosomal dysfunction, loss of GCase activity and elevated GluCer levels cause α-synuclein aggregation, mitochondrial impairment, inflammation, and ER stress [82, 165, 217, 314, 339], implying these pathways to be affected as a result of ATP10B dysfunctionality. Moreover, the LSD genes SMPD1, GALC and SCARB2, which have been associated with PD, are also involved in or linked to ceramide homeostasis (Fig. 3) [7, 50, 106]. Elevated levels of sphingolipids including GluCer, glucosylsphingosine, and sphingosine have been detected as well in NPC1 mutant cells, though the mechanism for accumulation of these lipids is still unclear (Fig. 3) [203, 340]. The brain is the organ with the highest proportion of lipid content, both in neurons and glial cells, in which ceramides are involved in a wide variety of functions to coordinate brain homeostasis, extensively reviewed elsewhere [62, 265]. Ceramide is enriched in lipid rafts, which are specialized membrane micro-domains, acting as assembling hubs for signaling complexes, to enable compartmentalization of various cellular processes [303]. Accumulation of ceramide in neuronal rafts was shown to be associated with impaired receptor trafficking and synapse loss [134].
Additionally, cholesterol homeostasis has been associated with PD pathogenesis, through the genetic association between NPC1 and PD (Fig. 3). Loss of NPC1 function has been shown to impede the clearance of α-synuclein [89, 172, 198], and accumulated oxidized metabolites of cholesterol, which have been identified in Lewy body brain pathology, can directly induce α-synuclein fibrilization [34]. Accumulated lysosomal cholesterol levels have been reported in fibroblasts of PD patient carriers with the GBA p.Asn370Ser pathogenic mutation [109]. Vice versa, increased intracellular cholesterol has been shown to initiate the breakdown of GCase via ER-associated degradation in proteasomes, which in turn resulted in reduced lysosomal GCase levels and increased glucosylceramide and α-synuclein levels [282].
Lysosomal PC export is also diminished by ATP10B dysfunction [213], which is in line with the observation of a disturbed PC lipid homeostasis in the substantia nigra of a PD rat model [94]. Loss of function mutations in PLA2G6, which is involved in oxidative damage repair to membrane phosphopholipids, membrane remodeling and iron homeostasis, also disturb neuronal lipid homeostasis, including ceramide levels (Fig. 3) [17, 200, 252, 309].
Finally, VPS13C enables the transport of glycerolipids between the ER and late endosomes/lysosomes, implicating defects in membrane lipid homeostasis and lysosomal dysfunction as a consequence of loss of VPS13C functionality (Fig. 3) [183].
Crosstalk between vesicular transport, lysosomes and mitochondria in Parkinson disease
Causal PD mutations in genes encoding proteins involved in one PD associated pathway are frequently associated secondary effects in other PD related pathways. The autophagy-lysosomal pathway is the only means by which damaged mitochondria are turned [15] and therefore, disruption in autophagy or lysosomal dysfunction could result in, if not exacerbate, mitochondrial dysfunction. Conversely, lysosomal function could be influenced by mitochondrial quality control, dynamics and/or respiration. Indeed, emerging evidence indicate that autophagy-lysosomal dysfunction impairs mitochondrial homeostasis, and in turn, mitochondrial defects also impact lysosomal functions, suggesting a complex relationship between these processes [260].
Mitochondrial dysfunction caused by alteration in genes primarily involved in transport pathways and lysosomal functioning
Increased levels of wild-type α-synuclein and α-synuclein with PD causing mutations have been associated with mitochondrial fragmentation and ROS accumulation. Moreover, α-synuclein has been found to localize at mitochondria-associated membranes (MAM), junctions that physically connect ER with mitochondria that are involved in Ca2+ signaling and apoptosis [122]. Pathogenic mutations in α-synuclein reduce the localization to MAM, and increase mitochondrial fragmentation, suggesting a direct role for α-synuclein in mitochondrial morphology [122]. Indeed, overexpressing wild-type or mutant α-synuclein was found to dissociate the ER and mitochondria at MAM, thereby impairing Ca2+ exchange and mitochondrial energy production [248]. Additionally, α-synuclein might regulate the PGC1α transcriptional network, involved in mitochondrial biogenesis and apoptosis. In a patient-derived stem cell model of PD carrying the SNCA p.Ala53Thr mutation, basal and mitochondrial toxin-induced nitrosative/oxidative stress resulted in S-nitrosylation of transcription factor myocyte-specific enhancer factor 2C (MEF2C), thereby inhibiting the transcription of PGC1α [289].
LRRK2 interacts with a number of key regulators of mitochondrial dynamics of fission and fusion, in the cytosol or at mitochondrial membranes. In mouse cortical neurons and human neuroblastoma cells, endogeneous LRRK2 interacts with the mitochondrial fission factor Dynamin like protein 1 (DLP1). DLP1 phosphorylates and transfers from the cytosol to mitochondria upon LRRK2 expression, resulting in mitochondrial fission. LRRK2 also interacts with the mitochondrial fusion regulators mitofusin 1 and mitofusin 2, and OPA1, a mitochondrial Dynamin like GTPase. Even though one study reported a direct interaction between LRRK2 and parkin in HEK293T cells [320], this finding could not be confirmed elsewhere [65]. However, LRRK2 is connected to the PINK1/parkin mediated mitophagy pathway via its substrate Rab10. Indeed, it was recently demonstrated that Rab10 binds the autophagy receptor optineurin (OPTN), promotes OPTN accumulation on depolarized mitochondria and thereby facilitating autophagosome formation around mitochondria, and consequently mitophagy [360]. Expression of LRRK2 p.Gly2019Ser in mouse cortical neurons causes defects in mitochondrial morphology and dynamics [237, 324]. Moreover, postmortem brain tissue of PD patients carrying the p.Gly2019Ser mutation demonstrates decreased levels of mature OPA1 [324]. In fibroblasts of PD patients with either the LRRK2 p.Gly2019Ser or p.Arg1441Cys mutation mitophagy of depolarized mitochondria is impaired [360].
VPS35 deficient mouse dopaminergic neurons and human fibroblasts also results in defects in mitochondrial fusion and mitochondrial fragmentation [338, 356]. VPS35-induced mitochondrial deficits can be prevented by inhibition of mitochondrial fission [356]. Moreover, VPS35 mutants show an increased interaction with the mitochondrial fission factor DLP1 [356].
VPS13C knockdown in COS-7 and in HEK293T cells resulted in mitochondrial fragmentation, decreased mitochondrial membrane potential, increased respiration rates and exacerbated PINK1/parkin-dependent mitophagy [193].
Knockdown of ATP13A2 in primary mouse cortical neurons and in SH-SY5Y cells shows an increase in mitochondrial fragmentation and an increase production of ROS [128, 278]. These effects could be mimicked by inhibiting autophagy induction using siRNA to Autophagy Related 7 (ATG7), a protein required for autophagy [128]. These results demonstrate that a decrease in autophagy influences mitochondrial quality control pathways, resulting in increased ROS production. Oppositely, overexpression of wild-type ATP13A2 in cultured midbrain dopaminergic neurons delays cadmium-induced mitochondrial fragmentation in neurons, consistent with a neuroprotective effect [278].
Knocking out PLA2G6 in mice results in degeneration of mitochondrial inner membranes and presynaptic membranes, triggering mitochondrial and synaptic dysfunction, and significant iron accumulation in brain [20,21,22].
Neuronal and glial cells of conditional GBA knockout mice present mitochondrial fragmentation, reduced respiratory chain complex activities, decreased mitochondrial membrane potential and lower oxygen consumption [244]. Analogous, overexpressing GBA p.Leu444Pro in SHSY-5Y neuroblastoma cells and knock-in of heterozygous GBA p.Leu444Pro in mice both trigger mitochondrial dysfunction by inhibiting autophagy and mitochondrial priming, a process by which autophagy receptor proteins are recruited to damaged mitochondria for degradation [196]. iPSC-derived dopaminergic neurons from GD and PD patients with GBA mutations showed increased glucosylceramide and α-synuclein levels, autophagic and lysosomal defects and a dysregulation of Ca2+ homeostasis [296]. Mitochondrial activities are driven in a Ca2+ dependent manner, and alterations in Ca2+ homeostasis may imply mitochondrial dysfunction [116, 286]. Induced mitophagy with carbonyl cyanide-m-chlorophenyl-hydrazine (CCCP) in 3D-neurosphere-models, consisting of neural stem cells with heterozygous and homozygous GBA p.Asn370Ser, resulted in a significant increase in lysosomal transcription factor EB (TFEB) mRNA levels, the master regulator of lysosomal and autophagy genes [224]. Interestingly, PGC1α mRNA levels were also significantly increased following CCCP-treatment in heterozygote, but not homozygote neurospheres, which might be explained by compensatory mechanism absent in homozygous lines [224]. In mouse cortical neurons, the chaperone ambroxol, which increases GCase mRNA levels and lysosomal GCase activity, was also shown to increase TFEB and PGC1α levels, block macro-autophagy flux and increased exocytosis [209]. These findings suggest that the GCase chaperone ambroxol might act on different pathways, including mitochondrial and lysosomal biogenesis, and the secretory pathway. Of note, most LSDs associated with PD or parkinsonism present some degree of mitochondrial dysfunction associated with primary lysosomal impairment [266].
Recently, proteome analysis of primary rat cortical neurons either overexpressing or silencing the lysosomal receptor LAMP2A, resulting in alterations in chaperone-mediated autophagy, identified a more than 2-fold difference in DJ-1 expression compared to control conditions [38]. Moreover, LAMP2A silencing, which results in DJ-1 depletion, sensitizes neurons to oxidative stress [38].
Endo-lysosomal dysfunction caused by alteration in genes primarily involved in mitophagy, mitochondrial dynamics and oxidative stress control
A mouse model of mitochondrial dysfunction, generated by deleting the mitochondrial transcription factor A (TFAM) in CD4+ T cells, demonstrated that mitochondrial respiratory defects impair lysosomal function, endo-lysosomal trafficking and autophagy, and increase sphingomyelin levels [16]. Similarly, large lysosomal vacuoles and lysosomal dysfunction were observed in brains from mice lacking the mitochondrial protein apoptosis inducing factor (AIF) and in embryonic fibroblasts from OPA1 knockout mice [73]. The lysosomal defects in the mouse embryonic fibroblasts were partially rescued by treatment with the antioxidants N-acetylcysteine or coenzyme Q10, suggesting that increased ROS from damaged mitochondria mediates lysosomal dysfunction [73]. Treatment of the mouse motor neuron NSC-34 cell line with the mitochondrial complex I inhibitor rotenone causes alterations in lysosomal biogenesis, function and morphology [123]. Interestingly, inducing TFEB via trehalose treatment in iPSC-derived dopaminergic neurons with compromised mitochondrial functioning, caused by long-term treatment with rotenone, restored the mitochondrial membrane potential and ATP production [312].
While parkin is well known to be involved in the regulation of mitophagy and mitochondrial biogenesis, the protective activity of parkin has been broadened to include roles in lipid metabolism and fat uptake [147, 164]. Moreover, parkin regulates the endo-lysosomal pathway by ubiquitinating the late-endosomal GTPase Rab7, which is a regulator of lysosomal dynamics [137, 321]. Loss of parkin function in primary fibroblasts of two PD patients with homozygous PARK2 mutations caused decreased endosomal tubulation and endosomal membrane association of VPS35 and sorting nexin 1, suggesting impairment of retromer pathway [321]. Substantia nigra tissue of PARK2 p.Q311* mutant mice displayed a late-stage block in autophagy, an increased PARIS expression and a PARIS-dependent reduced expression of both PGC1α and the lysosomal transcription factor TFEB [312]. Moreover, primary fibroblasts of a patient with juvenile PD with compound heterozygous deletions in PARK2 displayed abnormal abundance, acidification and morphology of the late endocytic compartment and lysosomal dysfunction [123].
PINK1 depletion in mouse embryonic fibroblasts from PINK1 knockout mice also impaired lysosomal activity and led to enlargement of lysosomal vacuoles [73] and silencing of DJ-1 in M17 neuroblastoma cells led to an accumulation of autophagy markers, in addition to mitochondrial membrane depolarization and mitochondrial fragmentation [341]. Moreover, a recent study in dopaminergic neurons derived from PD patients with homozygous DJ-1 p.E64D identified a time dependent pathological cascade starting with mitochondrial oxidative stress, followed by oxidized dopamine accumulation and finally resulting in reduced GCase activity, lysosomal dysfunction and α-synuclein accumulation [41].
Synergistic connection between the lysosomal and the mitochondrial compartment
Altogether, these data suggest synergistic effects of mitochondrial and lysosomal dysfunction in the pathogenesis of PD. The crosstalk between both pathways was also observed in a miRNA expression analysis to investigate the role of miRNAs in PD pathogenesis. In 6-hydroxydopamine induced stress conditions in SH-SY5Y cells, miR-5701 was shown to be significantly downregulated, and putative targets of miR-5701 are genes involved in lysosomal biogenesis and mitochondrial quality control [271]. The transfection of miR-5701 in SH-SY5Y cells induced both mitochondrial dysfunction and defects in autophagy flux [271]. The observed decrease in miR-5701 in 6-hydroxydopamine induced stress conditions might be a compensatory mechanism to simultaneously rescue lysosomal and mitochondrial function. Lastly, dynamic contact sites between lysosomes and mitochondria were recently identified using live-cell imaging, which was promoted by active GTP-bound lysosomal Rab7, a regulator of lysosomal dynamics [137, 366]. Moreover, these mitochondrial-lysosomal contact sites marked sites of mitochondrial fission, suggesting Rab7 may regulate mitochondrial dynamics [366]. Mitochondrial-lysosomal contact sites may be involved in a bidirectional regulation of mitochondrial and lysosomal dynamics, and might partially explain dysfunction of both organelles in PD.
Oligogenic and polygenic involvement of PD and APS genes
Because genes involved in mitochondrial and lysosomal function are associated with PD, and because of substantial evidence highlighting the crosstalk between these two pathways, it is reasonable to consider that oligogenic or polygenic inheritance of genes implicated in the mitochondrial-lysosomal pathway contribute to the genetic etiology of PD. Indeed, accumulating clinical and genetic observations suggest that besides monogenic inheritance, caused by dominant or recessive mutations in a single gene, more complex inheritance patters of familial PD exist.
Evidence pointing towards more complex inheritance patterns for PD
In 12 non-consanguine families affected with PD from Crete in which PD originated from both parental sides a high proportion (43%) of bilineal siblings were affected with PD, whereas only 5.7% of their offspring were affected [322]. This observation suggest recessive oligogenic inheritance in which disease predisposing alleles from two or more genes need to be present in the same individual for the expression of PD. Such alleles will be diluted again in the succeeding unilineal generation, resulting in a reduced proportion affected offspring. Moreover, single heterozygous mutations in PARK2, PINK1, DJ-1 and ATP13A2 are significantly more prevalent in PD or EOPD patients compared to control individuals [4, 75, 80, 133, 154, 170, 199, 238, 322, 330], suggesting these variants increase risk for PD, act as onset modifiers for PD, or contribute to the disease together with other mutations in an oligogenic fashion.
Oligogenic mutations in PD
In two Japanese families affected with autosomal recessive PD caused by homozygous or compound heterozygous PARK2 mutations, a heterozygous PINK1 mutation was identified as well [100]. The age at onset of patients with digenic mutations was lower than the age at onset of patients with the same recessive PARK2 mutations alone, indicating that heterozygous PINK1 might act as a modifier of age at onset in PARK2 mutation carriers. This study identified as well a sporadic PD patient with single heterozygous mutations in PARK2 and PINK1 [100]. Digenic heterozygous PARK2-PINK1 mutations have also been reported in two affected siblings with EOPD and one unrelated EOPD patient from Mexico [223]. These causative digenic PARK2-PINK1 mutations could be explained by the parkin/PINK1 pathway in which PINK1 acts directly upstream of parkin in regulating mitochondrial quality control and dynamics (Fig. 2). Similarly, digenic heterozygous PINK1 and DJ-1 missense mutations have been reported in a recessive family with two affected siblings [337]. This study also showed that PINK1 and DJ-1 physically associate and cooperate to protect cells against oxidative stress [337].
Most frequently reported to date are digenic PARK2-LRRK2 mutation carriers. So far, two carriers of LRRK2 p.Gly2019Ser and homozygous PARK2 mutations have been reported: a carrier of a homozygous deletion of exon 4–5-6 and a carrier of a homozygous triplication of exon 2 in PARK2 [108, 194]. The clinical manifestations of both patients are as typically seen in other PARK2 mutation carriers rather than in other LRRK2 mutation carriers, including dopa-responsive parkinsonism and an early age at onset. Additionally, several cases of digenic heterozygous LRRK2 and PARK2 mutations have been reported [65, 194, 238]. However, digenic PARK2-LRRK2 heterozygous mutations do not seem to cause a more severe disease progression or an earlier age at onset compared to single heterozygous PARK2 or LRRK2 mutations [65, 194, 238]. Noteworthy, one at risk member aged 52 of a family affected by the LRRK2 p.Gly2019Ser mutation carried as well as a heterozygous frameshift mutation in PARK2 and the pathogenic GBA p.Asn370Ser mutation [250]. The PARK2 and GBA mutations were absent from other members of the family and were likely inherited from the parent married into the family [250].
While the connection between PINK1 and SYNJ1 is yet unclear, digenic heterozygous mutations SYNJ1 p.Ser1422Arg and PINK1 p.W437* have been identified in a Brazilian EOPD patient [273].
The impact of oligogenic mutations on disease expression is currently not well understood due to the limited number oligogenic mutation carriers identified so far and the lack of large families to investigate segregation with PD. Disease expression in oligogenic mutation carriers is probably a joint effect of genetic background, gene-gene and gene-environment interactions.
Rare variant analysis suggests oligogenic and polygenic inheritance of PD
Rare variant analysis in 7900 PD patients with and without a known pathogenic mutation and 6166 control individuals revealed that more than 30% of PD patients with a recognized primary genetic cause of the disease had additional rare variants in Mendelian PD genes [207]. The carriers of additional rare variants in the PD genes had younger ages at onset of approximately 4 to 6 years, though this was not significant (p > 0.05) [207]. Exome sequencing analysis of postmortem human brains of 58 DLB and 39 PD patients identified a significant enrichment of rare oligogenic variants in neurodegenerative brain diseases (PD, DLB, Alzheimer’s disease, frontotemporal dementia and amyotrophic lateral sclerosis) genes in PD/DLB patients (23.71%) compared to control individuals (10.22%) [159]. Moreover, significant polygenic enrichment in PD patients of rare, non-synonymous variants of a gene-set of proteins involved in mitochondrial DNA maintenance was identified in both a Norwegian and a North American cohort consisting of PD patients and control individuals [104]. All of the above-mentioned observations suggest oligogenic and polygenic inheritance contribute to the expression of PD and might explain a part of the missing heritability in PD. Moreover, these observations may provide new insights for functional research to investigate how PD pathways are interconnected.
Concluding remarks
Numerous genes, either causing PD, APS or increasing risk, are directly and indirectly associated with defects in vesicular transport pathways, lysosomal dysfunction and mitochondrial dysfunction. Additionally, increasing evidence suggests a link between numerous LSDs and PD, though further clinical, genetic and biochemical studies are needed to clarify these associations. Nevertheless, these emerging associations highlight the involvement of lipid homeostasis and especially ceramide homeostasis in PD pathogenesis. However, why homozygous mutations in LSD genes cause LSDs, whether or not associated with neurological symptoms, and heterozygous mutation increase PD risk is unclear. We hypothesize that genetic modifying factors may influence the phenotype associated with these LSD gene mutations.
Our current understanding is that the lysosomal and the mitochondrial compartment is highly interconnected, given that a primary defect in either compartment leads to dysfunction of the other compartment. Moreover, while mitochondria-ER contact sites have been recognized for years [86], and recently implicated in PD pathogenesis via the discovery of VPS13C as a lipid transport tether between these two organelles [183], mitochondrial-lysosomal contact sites are only recognized more recently. However, dynamic contact sites and crosstalk may be important to transport metabolites and ions between the two organelles, including lipids, as seen for VPS13C [183]. Moreover, transcriptional regulation seems to play an important role in the crosstalk between the mitochondria and lysosomes, indicated by alterations in protein levels of the lysosomal and mitochondrial transcription factors, TFEB and PGC1α respectively, as a result of PD/APS gene defects and impairment of these organelles. Despite new evidence, significant gaps remain in our understanding of the functional and physical associations between these PD associated pathways [366].
Oligogenic mutations of the mitochondrial-lysosomal pathway have been identified in PD patients but have not been extensively studied yet. However, current observations suggest more complex inheritance patters of genes of interconnected PD pathways, further strengthening the crosstalk and synergistic connection between these pathways in PD pathogenesis. Nevertheless, segregation analysis, clinical and biochemical studies are needed provide insights in gene-gene interactions leading to the development of PD. The genetic architecture of PD, in terms of the number of variants needed to reach the threshold to express the disease and their respective effects size, probably ranges from monogenic highly penetrant variants, perhaps influenced by modifiers, to oligogenic rare variants with high to moderate effect sizes, to polygenic common risk factors which could also act as modifiers of more penetrant variants. Therefore, the presence of a mutation in one gene should not be an exclusion criterion for further genetic screening in PD patients. Mainly in PD patients with a single heterozygous mutation in a recessive gene, additional screening could reveal oligogenic variants in the same or connecting pathways. Possible oligogenic inheritance patterns may include both PD/APS genes and LSD genes associated with an increased risk for the development of PD.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated.
Abbreviations
- AIF:
-
Apoptosis inducing factor
- AMRF:
-
Action myoclonus-renal failure syndrome
- APS:
-
Atypical parkinsonian syndromes
- ASM:
-
Acid sphingomyelinase
- ATG7:
-
Autophagy Related 7
- ATP10B:
-
ATPase class V type 10B
- CCCP:
-
Cyanide-m-chlorophenyl-hydrazine
- CDC50A:
-
Cell Cycle Control Protein 50A
- CI-MPR:
-
Cation-independent mannose 6-phosphate receptor
- CRC:
-
Cargo-recognition complex
- CTSD:
-
Cathepsin D
- DLB:
-
Dementia with Lewy bodies
- DLP1:
-
Dynamin like protein 1
- EOPD:
-
Early-onset Parkinson’s disease
- ER:
-
Endoplasmic reticulum
- FBXO7:
-
F-box protein 7
- GALC:
-
Galactosylceramidase
- GCase:
-
Glucocerebosidase
- GD:
-
Gaucher disease
- GluCer:
-
Glucosylceramide
- GWAS:
-
Genome-wide association studies
- INAD:
-
Infantile neuroaxonal dystrophy
- iPLA-β:
-
Calcium-independent group VI phospholipase A2
- iPSC:
-
Induced pluripotent stem cell
- KD:
-
Krabbe disease
- LIMP-2:
-
Lysosomal integral membrane protein 2
- LSD:
-
Lysosomal storage disorders
- MAM:
-
Mitochondria-associated membrane
- MEF2C:
-
Myocyte-specific enhancer factor 2C
- MPS-IIIB:
-
Mucopolysaccharidosis III disease B
- NAD:
-
Neuroaxonal dystrophy
- NAG:
-
α-N-acetylglucosminidase
- NPD:
-
Niemann-Pick disease
- NPC1:
-
Niemann-Pick type C intracellular cholesterol transporter 1
- NPC2:
-
Niemann-Pick type C intracellular cholesterol transporter 2
- OPTN:
-
Optineurin
- PARIS:
-
Parkin interacting substrate
- PARL:
-
Presenilin-associated rhomboid-like protein
- PC:
-
Phosphatidylcholine
- PD:
-
Parkinson’s disease
- PGC1α:
-
Proliferator-activated receptor gamma coactivator 1-α
- PINK1:
-
PTEN-induced putative kinase 1
- PIP2 :
-
Phosphatidylinositol 4,5-bisphosphate
- PIPs:
-
Phosphoinositide phosphates
- PLA2G6:
-
Phospholipase A2 group 6
- PTC:
-
Premature termination codon
- ROS:
-
Reactive oxygen species
- SCARB2:
-
Scavenger receptor class B member 2
- SCF:
-
SKP1-cullin-F-box
- SMPD1:
-
Sphingomyelin phosphodiesterase 1
- SNPs:
-
Single nucleotide polymorphisms
- TFAM:
-
Mitochondrial transcription factor A
- TFEB:
-
Transcription factor EB
- UbR:
-
Ubiquitin-related
- VPS13C:
-
Vacuolar protein sorting 13 C
References
Abbas N, Lucking CB, Ricard S, Durr A, Bonifati V, De Michele G et al (1999) A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson's Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson's Disease. Hum Mol Genet 8:567–574. https://doi.org/10.1093/hmg/8.4.567
Abeliovich A, Gitler AD (2016) Defects in trafficking bridge Parkinson's disease pathology and genetics. Nature 539:207–216. https://doi.org/10.1038/nature20414
Abou-Sleiman PM, Healy DG, Quinn N, Lees AJ, Wood NW (2003) The role of pathogenic DJ-1 mutations in Parkinson's disease. Ann Neurol 54:283–286. https://doi.org/10.1002/ana.10675
Abou-Sleiman PM, Muqit MM, McDonald NQ, Yang YX, Gandhi S, Healy DG et al (2006) A heterozygous effect for PINK1 mutations in Parkinson's disease? Ann Neurol 60:414–419. https://doi.org/10.1002/ana.20960
Aits S, Jaattela M (2013) Lysosomal cell death at a glance. J Cell Sci 126:1905–1912. https://doi.org/10.1242/jcs.091181
Al-Chalabi A, Durr A, Wood NW, Parkinson MH, Camuzat A, Hulot JS et al (2009) Genetic variants of the alpha-synuclein gene SNCA are associated with multiple system atrophy. PLoS One 4:e7114. https://doi.org/10.1371/journal.pone.0007114
Alcalay RN, Levy OA, Wolf P, Oliva P, Zhang XK, Waters CH et al (2016) SCARB2 variants and glucocerebrosidase activity in Parkinson's disease. NPJ Parkinsons Dis 2. https://doi.org/10.1038/npjparkd.2016.4
Alcalay RN, Mallett V, Vanderperre B, Tavassoly O, Dauvilliers Y, Wu RYJ et al (2019) SMPD1 mutations, activity, and alpha-synuclein accumulation in Parkinson's disease. Mov Disord 34:526–535. https://doi.org/10.1002/mds.27642
Alcalay RN, Wolf P, Levy OA, Kang UJ, Waters C, Fahn S et al (2018) Alpha galactosidase A activity in Parkinson's disease. Neurobiol Dis 112:85–90. https://doi.org/10.1016/j.nbd.2018.01.012
Alessi DR, Sammler E (2018) LRRK2 kinase in Parkinson's disease. Science 360:36–37. https://doi.org/10.1126/science.aar5683
Andersen JP, Vestergaard AL, Mikkelsen SA, Mogensen LS, Chalat M, Molday RS (2016) P4-ATPases as Phospholipid Flippases-Structure, Function, and Enigmas. Front Physiol 7:275. https://doi.org/10.3389/fphys.2016.00275
Andres-Mateos E, Perier C, Zhang L, Blanchard-Fillion B, Greco TM, Thomas B et al (2007) DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci U S A 104:14807–14812. https://doi.org/10.1073/pnas.0703219104
Ariga H, Takahashi-Niki K, Kato I, Maita H, Niki T, Iguchi-Ariga SM (2013) Neuroprotective function of DJ-1 in Parkinson's disease. Oxid Med Cell Longev 2013:683920. https://doi.org/10.1155/2013/683920
Armstrong MJ, Okun MS (2020) Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 323:548–560. https://doi.org/10.1001/jama.2019.22360
Ashrafi G, Schwarz TL (2013) The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20:31–42. https://doi.org/10.1038/cdd.2012.81
Baixauli F, Acin-Perez R, Villarroya-Beltri C, Mazzeo C, Nunez-Andrade N, Gabande-Rodriguez E et al (2015) Mitochondrial Respiration Controls Lysosomal Function during Inflammatory T Cell Responses. Cell Metab 22:485–498. https://doi.org/10.1016/j.cmet.2015.07.020
Balsinde J, Balboa MA (2005) Cellular regulation and proposed biological functions of group VIA calcium-independent phospholipase A2 in activated cells. Cell Signal 17:1052–1062. https://doi.org/10.1016/j.cellsig.2005.03.002
Bargal R, Avidan N, Ben-Asher E, Olender Z, Zeigler M, Frumkin A et al (2000) Identification of the gene causing mucolipidosis type IV. Nat Genet 26:118–123. https://doi.org/10.1038/79095
Battle A, Brown CD, Engelhardt BE, Montgomery SB (2017) Genetic effects on gene expression across human tissues. Nature 550:204–213. https://doi.org/10.1038/nature24277
Beck G, Shinzawa K, Hayakawa H, Baba K, Sumi-Akamaru H, Tsujimoto Y et al (2016) Progressive Axonal Degeneration of Nigrostriatal Dopaminergic Neurons in Calcium-Independent Phospholipase A2beta Knockout Mice. PLoS One 11:e0153789. https://doi.org/10.1371/journal.pone.0153789
Beck G, Shinzawa K, Hayakawa H, Baba K, Yasuda T, Sumi-Akamaru H et al (2015) Deficiency of Calcium-Independent Phospholipase A2 Beta Induces Brain Iron Accumulation through Upregulation of Divalent Metal Transporter 1. PLoS One 10:e0141629. https://doi.org/10.1371/journal.pone.0141629
Beck G, Sugiura Y, Shinzawa K, Kato S, Setou M, Tsujimoto Y et al (2011) Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J Neurosci 31:11411–11420. https://doi.org/10.1523/JNEUROSCI.0345-11.2011
Bento CF, Ashkenazi A, Jimenez-Sanchez M, Rubinsztein DC (2016) The Parkinson's disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat Commun 7:11803. https://doi.org/10.1038/ncomms11803
Berkovic SF, Dibbens LM, Oshlack A, Silver JD, Katerelos M, Vears DF et al (2008) Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet 82:673–684. https://doi.org/10.1016/j.ajhg.2007.12.019
Bhuin T, Roy JK (2014) Rab proteins: the key regulators of intracellular vesicle transport. Exp Cell Res 328:1–19. https://doi.org/10.1016/j.yexcr.2014.07.027
Blackinton J, Lakshminarasimhan M, Thomas KJ, Ahmad R, Greggio E, Raza AS et al (2009) Formation of a stabilized cysteine sulfinic acid is critical for the mitochondrial function of the parkinsonism protein DJ-1. J Biol Chem 284:6476–6485. https://doi.org/10.1074/jbc.M806599200
Blandini F, Cilia R, Cerri S, Pezzoli G, Schapira AHV, Mullin S et al (2019) Glucocerebrosidase mutations and synucleinopathies: Toward a model of precision medicine. Mov Disord 34:9–21. https://doi.org/10.1002/mds.27583
Blauwendraat C, Kia DA, Pihlstrom L, Gan-Or Z, Lesage S, Gibbs JR et al (2018) Insufficient evidence for pathogenicity of SNCA His50Gln (H50Q) in Parkinson's disease. Neurobiol Aging 64:159 e155–159 e158. https://doi.org/10.1016/j.neurobiolaging.2017.12.012
Blesa J, Trigo-Damas I, Quiroga-Varela A, Jackson-Lewis VR (2015) Oxidative stress and Parkinson's disease. Front Neuroanat 9:91. https://doi.org/10.3389/fnana.2015.00091
Boland B, Yu WH, Corti O, Mollereau B, Henriques A, Bezard E et al (2018) Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat Rev Drug Discov 17:660–688. https://doi.org/10.1038/nrd.2018.109
Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E et al (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299:256–259. https://doi.org/10.1126/science.1077209
Bonifati V, Rohe CF, Breedveld GJ, Fabrizio E, De Mari M, Tassorelli C et al (2005) Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology 65:87–95. https://doi.org/10.1212/01.wnl.0000167546.39375.82
Bonten E, van der Spoel A, Fornerod M, Grosveld G, d'Azzo A (1996) Characterization of human lysosomal neuraminidase defines the molecular basis of the metabolic storage disorder sialidosis. Genes Dev 10:3156–3169. https://doi.org/10.1101/gad.10.24.3156
Bosco DA, Fowler DM, Zhang Q, Nieva J, Powers ET, Wentworth P Jr et al (2006) Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha-synuclein fibrilization. Nat Chem Biol 2:249–253. https://doi.org/10.1038/nchembio782
Braak H, de Vos RA, Bohl J, Del Tredici K (2006) Gastric alpha-synuclein immunoreactive inclusions in Meissner's and Auerbach's plexuses in cases staged for Parkinson's disease-related brain pathology. Neurosci Lett 396:67–72. https://doi.org/10.1016/j.neulet.2005.11.012
Bras J, Guerreiro R, Darwent L, Parkkinen L, Ansorge O, Escott-Price V et al (2014) Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum Mol Genet 23:6139–6146. https://doi.org/10.1093/hmg/ddu334
Bras J, Verloes A, Schneider SA, Mole SE, Guerreiro RJ (2012) Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum Mol Genet 21:2646–2650. https://doi.org/10.1093/hmg/dds089
Brekk OR, Makridakis M, Mavroeidi P, Vlahou A, Xilouri M, Stefanis L (2019) Impairment of chaperone-mediated autophagy affects neuronal homeostasis through altered expression of DJ-1 and CRMP-2 proteins. Mol Cell Neurosci 95:1–12. https://doi.org/10.1016/j.mcn.2018.12.006
Buechling T, Bartscherer K, Ohkawara B, Chaudhary V, Spirohn K, Niehrs C et al (2010) Wnt/Frizzled signaling requires dPRR, the Drosophila homolog of the prorenin receptor. Curr Biol 20:1263–1268. https://doi.org/10.1016/j.cub.2010.05.028
Bugarcic A, Zhe Y, Kerr MC, Griffin J, Collins BM, Teasdale RD (2011) Vps26A and Vps26B subunits define distinct retromer complexes. Traffic 12:1759–1773. https://doi.org/10.1111/j.1600-0854.2011.01284.x
Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S et al (2017) Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science 357:1255–1261. https://doi.org/10.1126/science.aam9080
Burchell VS, Nelson DE, Sanchez-Martinez A, Delgado-Camprubi M, Ivatt RM, Pogson JH et al (2013) The Parkinson's disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat Neurosci 16:1257–1265. https://doi.org/10.1038/nn.3489
Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S et al (2004) The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A 101:9103–9108. https://doi.org/10.1073/pnas.0402959101
Cannata Serio M, Rujano MA, Simons M (2018) Mutations in ATP6AP2 cause autophagic liver disease in humans. Autophagy 14:1088–1089. https://doi.org/10.1080/15548627.2018.1434370
Cantuti Castelvetri L, Givogri MI, Hebert A, Smith B, Song Y, Kaminska A et al (2013) The sphingolipid psychosine inhibits fast axonal transport in Krabbe disease by activation of GSK3beta and deregulation of molecular motors. J Neurosci 33:10048–10056. https://doi.org/10.1523/JNEUROSCI.0217-13.2013
Cao M, Wu Y, Ashrafi G, McCartney AJ, Wheeler H, Bushong EA et al (2017) Parkinson Sac Domain Mutation in Synaptojanin 1 Impairs Clathrin Uncoating at Synapses and Triggers Dystrophic Changes in Dopaminergic Axons. Neuron 93:882–896 e5. https://doi.org/10.1016/j.neuron.2017.01.019
Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C et al (1997) Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 277:228–231. https://doi.org/10.1126/science.277.5323.228
Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL et al (2011) Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20:1726–1737. https://doi.org/10.1093/hmg/ddr048
Chang C, Wu G, Gao P, Yang L, Liu W, Zuo J (2014) Upregulated Parkin expression protects mitochondrial homeostasis in DJ-1 konckdown cells and cells overexpressing the DJ-1 L166P mutation. Mol Cell Biochem 387:187–195. https://doi.org/10.1007/s11010-013-1884-3
Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F et al (2017) A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet 49:1511–1516. https://doi.org/10.1038/ng.3955
Chen C, Turnbull DM, Reeve AK (2019) Mitochondrial Dysfunction in Parkinson's Disease-Cause or Consequence? Biology (Basel) 8. https://doi.org/10.3390/biology8020038
Chen J, Li L, Chin LS (2010) Parkinson disease protein DJ-1 converts from a zymogen to a protease by carboxyl-terminal cleavage. Hum Mol Genet 19:2395–2408. https://doi.org/10.1093/hmg/ddq113
Chuang WL, Pacheco J, Cooper S, McGovern MM, Cox GF, Keutzer J et al (2014) Lyso-sphingomyelin is elevated in dried blood spots of Niemann-Pick B patients. Mol Genet Metab 111:209–211. https://doi.org/10.1016/j.ymgme.2013.11.012
Chung EJ, Ki CS, Lee WY, Kim IS, Kim JY (2006) Clinical features and gene analysis in Korean patients with early-onset Parkinson disease. Arch Neurol 63:1170–1174. https://doi.org/10.1001/archneur.63.8.1170
Clark LN, Chan R, Cheng R, Liu X, Park N, Parmalee N et al (2015) Gene-wise association of variants in four lysosomal storage disorder genes in neuropathologically confirmed Lewy body disease. PLoS One 10:e0125204. https://doi.org/10.1371/journal.pone.0125204
Cohlberg JA, Li J, Uversky VN, Fink AL (2002) Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha-synuclein in vitro. Biochemistry 41:1502–1511. https://doi.org/10.1021/bi011711s
Coleman RJ, Robb SA, Lake BD, Brett EM, Harding AE (1988) The diverse neurological features of Niemann-Pick disease type C: a report of two cases. Mov Disord 3:295–299. https://doi.org/10.1002/mds.870030403
Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B et al (2006) Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science 313:324–328. https://doi.org/10.1126/science.1129462
Cremades N, Dobson CM (2018) The contribution of biophysical and structural studies of protein self-assembly to the design of therapeutic strategies for amyloid diseases. Neurobiol Dis 109:178–190. https://doi.org/10.1016/j.nbd.2017.07.009
Cremona O, Di Paolo G, Wenk MR, Luthi A, Kim WT, Takei K et al (1999) Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99:179–188. https://doi.org/10.1016/s0092-8674(00)81649-9
Crosiers D, Verstraeten A, Wauters E, Engelborghs S, Peeters K, Mattheijssens M et al (2016) Mutations in glucocerebrosidase are a major genetic risk factor for Parkinson's disease and increase susceptibility to dementia in a Flanders-Belgian cohort. Neurosci Lett 629:160–164. https://doi.org/10.1016/j.neulet.2016.07.008
Cruciani-Guglielmacci C, Lopez M, Campana M, le Stunff H (2017) Brain Ceramide Metabolism in the Control of Energy Balance. Front Physiol 8:787. https://doi.org/10.3389/fphys.2017.00787
Cruciat CM, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, Ingelfinger D et al (2010) Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science 327:459–463. https://doi.org/10.1126/science.1179802
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305:1292–1295. https://doi.org/10.1126/science.1101738
Dachsel JC, Mata IF, Ross OA, Taylor JP, Lincoln SJ, Hinkle KM et al (2006) Digenic parkinsonism: investigation of the synergistic effects of PRKN and LRRK2. Neurosci Lett 410:80–84. https://doi.org/10.1016/j.neulet.2006.06.068
Dagan E, Schlesinger I, Ayoub M, Mory A, Nassar M, Kurolap A et al (2015) The contribution of Niemann-Pick SMPD1 mutations to Parkinson disease in Ashkenazi Jews. Parkinsonism Relat Disord 21:1067–1071. https://doi.org/10.1016/j.parkreldis.2015.06.016
Danek A, Jung HH, Melone MA, Rampoldi L, Broccoli V, Walker RH (2005) Neuroacanthocytosis: new developments in a neglected group of dementing disorders. J Neurol Sci 229–230:171–186. https://doi.org/10.1016/j.jns.2004.11.024
Dardis A, Filocamo M, Grossi S, Ciana G, Franceschetti S, Dominissini S et al (2009) Biochemical and molecular findings in a patient with myoclonic epilepsy due to a mistarget of the beta-glucosidase enzyme. Mol Genet Metab 97:309–311. https://doi.org/10.1016/j.ymgme.2009.04.011
Darvish H, Bravo P, Tafakhori A, Azcona LJ, Ranji-Burachaloo S, Johari AH et al (2018) Identification of a large homozygous VPS13C deletion in a patient with early-onset Parkinsonism. Mov Disord 33:1968–1970. https://doi.org/10.1002/mds.27516
Davidson WS, Jonas A, Clayton DF, George JM (1998) Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem 273:9443–9449. https://doi.org/10.1074/jbc.273.16.9443
de Lau LM, Breteler MM (2006) Epidemiology of Parkinson's disease. Lancet Neurol 5:525–535. https://doi.org/10.1016/S1474-4422(06)70471-9
Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E et al (2012) Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A 109:9611–9616. https://doi.org/10.1073/pnas.1112368109
Demers-Lamarche J, Guillebaud G, Tlili M, Todkar K, Belanger N, Grondin M et al (2016) Loss of Mitochondrial Function Impairs Lysosomes. J Biol Chem 291:10263–10276. https://doi.org/10.1074/jbc.M115.695825
Deng S, Deng X, Song Z, Xiu X, Guo Y, Xiao J et al (2016) Systematic Genetic Analysis of the SMPD1 Gene in Chinese Patients with Parkinson's Disease. Mol Neurobiol 53:5025–5029. https://doi.org/10.1007/s12035-015-9426-5
Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto G et al (2007) ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 68:1557–1562. https://doi.org/10.1212/01.wnl.0000260963.08711.08
Di Fonzo A, Dekker MC, Montagna P, Baruzzi A, Yonova EH, Correia Guedes L et al (2009) FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 72:240–245. https://doi.org/10.1212/01.wnl.0000338144.10967.2b
Di Pasquale E, Fantini J, Chahinian H, Maresca M, Taieb N, Yahi N (2010) Altered ion channel formation by the Parkinson's-disease-linked E46K mutant of alpha-synuclein is corrected by GM3 but not by GM1 gangliosides. J Mol Biol 397:202–218. https://doi.org/10.1016/j.jmb.2010.01.046
Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM et al (2009) Neuropathological assessment of Parkinson's disease: refining the diagnostic criteria. Lancet Neurol 8:1150–1157. https://doi.org/10.1016/S1474-4422(09)70238-8
Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19:349–364. https://doi.org/10.1038/s41580-018-0003-4
Djarmati A, Hedrich K, Svetel M, Lohnau T, Schwinger E, Romac S et al (2006) Heterozygous PINK1 mutations: a susceptibility factor for Parkinson disease? Mov Disord 21:1526–1530. https://doi.org/10.1002/mds.20977
Do CB, Tung JY, Dorfman E, Kiefer AK, Drabant EM, Francke U et al (2011) Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson's disease. PLoS Genet 7:e1002141. https://doi.org/10.1371/journal.pgen.1002141
Do J, McKinney C, Sharma P, Sidransky E (2019) Glucocerebrosidase and its relevance to Parkinson disease. Mol Neurodegener 14:36. https://doi.org/10.1186/s13024-019-0336-2
Dodson MW, Zhang T, Jiang C, Chen S, Guo M (2012) Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum Mol Genet 21:1350–1363. https://doi.org/10.1093/hmg/ddr573
Ebanks K, Lewis PA, Bandopadhyay R (2019) Vesicular Dysfunction and the Pathogenesis of Parkinson's Disease: Clues From Genetic Studies. Front Neurosci 13:1381. https://doi.org/10.3389/fnins.2019.01381
Edvardson S, Cinnamon Y, Ta-Shma A, Shaag A, Yim YI, Zenvirt S et al (2012) A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS One 7:e36458. https://doi.org/10.1371/journal.pone.0036458
Elbaz Y, Schuldiner M (2011) Staying in touch: the molecular era of organelle contact sites. Trends Biochem Sci 36:616–623. https://doi.org/10.1016/j.tibs.2011.08.004
Elsayed LE, Drouet V, Usenko T, Mohammed IN, Hamed AA, Elseed MA et al (2016) A Novel Nonsense Mutation in DNAJC6 Expands the Phenotype of Autosomal-Recessive Juvenile-Onset Parkinson's Disease. Ann Neurol 79:335–337. https://doi.org/10.1002/ana.24591
Engel LA, Jing Z, O'Brien DE, Sun M, Kotzbauer PT (2010) Catalytic function of PLA2G6 is impaired by mutations associated with infantile neuroaxonal dystrophy but not dystonia-parkinsonism. PLoS One 5:e12897. https://doi.org/10.1371/journal.pone.0012897
Eriksson I, Nath S, Bornefall P, Giraldo AM, Ollinger K (2017) Impact of high cholesterol in a Parkinson's disease model: Prevention of lysosomal leakage versus stimulation of alpha-synuclein aggregation. Eur J Cell Biol 96:99–109. https://doi.org/10.1016/j.ejcb.2017.01.002
Estrada-Cuzcano A, Martin S, Chamova T, Synofzik M, Timmann D, Holemans T et al (2017) Loss-of-function mutations in the ATP13A2/PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). Brain 140:287–305. https://doi.org/10.1093/brain/aww307
Exner N, Treske B, Paquet D, Holmstrom K, Schiesling C, Gispert S et al (2007) Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci 27:12413–12418. https://doi.org/10.1523/JNEUROSCI.0719-07.2007
Fantini J, Yahi N (2010) Molecular insights into amyloid regulation by membrane cholesterol and sphingolipids: common mechanisms in neurodegenerative diseases. Expert Rev Mol Med 12:e27. https://doi.org/10.1017/S1462399410001602
Fantini J, Yahi N (2011) Molecular basis for the glycosphingolipid-binding specificity of alpha-synuclein: key role of tyrosine 39 in membrane insertion. J Mol Biol 408:654–669. https://doi.org/10.1016/j.jmb.2011.03.009
Farmer K, Smith CA, Hayley S, Smith J (2015) Major Alterations of Phosphatidylcholine and Lysophosphotidylcholine Lipids in the Substantia Nigra Using an Early Stage Model of Parkinson's Disease. Int J Mol Sci 16:18865–18877. https://doi.org/10.3390/ijms160818865
Farrer M, Wavrant-De Vrieze F, Crook R, Boles L, Perez-Tur J, Hardy J et al (1998) Low frequency of alpha-synuclein mutations in familial Parkinson's disease. Ann Neurol 43:394–397. https://doi.org/10.1002/ana.410430320
Fearnley JM, Lees AJ (1991) Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain 114(Pt 5):2283–2301. https://doi.org/10.1093/brain/114.5.2283
Ferraz MJ, Marques AR, Appelman MD, Verhoek M, Strijland A, Mirzaian M et al (2016) Lysosomal glycosphingolipid catabolism by acid ceramidase: formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett 590:716–725. https://doi.org/10.1002/1873-3468.12104
Follett J, Norwood SJ, Hamilton NA, Mohan M, Kovtun O, Tay S et al (2014) The Vps35 D620N mutation linked to Parkinson's disease disrupts the cargo sorting function of retromer. Traffic 15:230–244. https://doi.org/10.1111/tra.12136
Foo JN, Liany H, Bei JX, Yu XQ, Liu J, Au WL et al (2013) Rare lysosomal enzyme gene SMPD1 variant (p.R591C) associates with Parkinson's disease. Neurobiol Aging 34(2890):e2813–e2895. https://doi.org/10.1016/j.neurobiolaging.2013.06.010
Funayama M, Li Y, Tsoi TH, Lam CW, Ohi T, Yazawa S et al (2008) Familial Parkinsonism with digenic parkin and PINK1 mutations. Mov Disord 23:1461–1465. https://doi.org/10.1002/mds.22143
Fung HC, Chen CM, Hardy J, Singleton AB, Lee-Chen GJ, Wu YR (2006) Analysis of the PINK1 gene in a cohort of patients with sporadic early-onset parkinsonism in Taiwan. Neurosci Lett 394:33–36. https://doi.org/10.1016/j.neulet.2005.10.005
Fusco G, De Simone A, Gopinath T, Vostrikov V, Vendruscolo M, Dobson CM et al (2014) Direct observation of the three regions in alpha-synuclein that determine its membrane-bound behaviour. Nat Commun 5:3827. https://doi.org/10.1038/ncomms4827
Fuse A, Furuya N, Kakuta S, Inose A, Sato M, Koike M et al (2015) VPS29-VPS35 intermediate of retromer is stable and may be involved in the retromer complex assembly process. FEBS Lett 589:1430–1436. https://doi.org/10.1016/j.febslet.2015.04.040
Gaare JJ, Nido GS, Sztromwasser P, Knappskog PM, Dahl O, Lund-Johansen M et al (2018) Rare genetic variation in mitochondrial pathways influences the risk for Parkinson's disease. Mov Disord 33:1591–1600. https://doi.org/10.1002/mds.64
Gan-Or Z, Orr-Urtreger A, Alcalay RN, Bressman S, Giladi N, Rouleau GA (2015) The emerging role of SMPD1 mutations in Parkinson's disease: Implications for future studies. Parkinsonism Relat Disord 21:1294–1295. https://doi.org/10.1016/j.parkreldis.2015.08.018
Gan-Or Z, Ozelius LJ, Bar-Shira A, Saunders-Pullman R, Mirelman A, Kornreich R et al (2013) The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 80:1606–1610. https://doi.org/10.1212/WNL.0b013e31828f180e
Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K et al (2009) PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell 33:627–638. https://doi.org/10.1016/j.molcel.2009.02.013
Gao L, Gomez-Garre P, Diaz-Corrales FJ, Carrillo F, Carballo M, Palomino A et al (2009) Prevalence and clinical features of LRRK2 mutations in patients with Parkinson's disease in southern Spain. Eur J Neurol 16:957–960. https://doi.org/10.1111/j.1468-1331.2009.02620.x
Garcia-Sanz P, Orgaz L, Bueno-Gil G, Espadas I, Rodriguez-Traver E, Kulisevsky J et al (2017) N370S-GBA1 mutation causes lysosomal cholesterol accumulation in Parkinson's disease. Mov Disord 32:1409–1422. https://doi.org/10.1002/mds.27119
Gautier CA, Kitada T, Shen J (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A 105:11364–11369. https://doi.org/10.1073/pnas.0802076105
Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW et al (2012) Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol 72:455–463. https://doi.org/10.1002/ana.23614
Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW (2010) Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 19:4861–4870. https://doi.org/10.1093/hmg/ddq419
Gegg ME, Cooper JM, Schapira AH, Taanman JW (2009) Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS One 4:e4756. https://doi.org/10.1371/journal.pone.0004756
Giaime E, Sunyach C, Druon C, Scarzello S, Robert G, Grosso S et al (2010) Loss of function of DJ-1 triggered by Parkinson's disease-associated mutation is due to proteolytic resistance to caspase-6. Cell Death Differ 17:158–169. https://doi.org/10.1038/cdd.2009.116
Giasson BI, Murray IV, Trojanowski JQ, Lee VM (2001) A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem 276:2380–2386. https://doi.org/10.1074/jbc.M008919200
Giorgi C, Agnoletto C, Bononi A, Bonora M, De Marchi E, Marchi S et al (2012) Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion 12:77–85. https://doi.org/10.1016/j.mito.2011.07.004
Gomez-Suaga P, Luzon-Toro B, Churamani D, Zhang L, Bloor-Young D, Patel S et al (2012) Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum Mol Genet 21:511–525. https://doi.org/10.1093/hmg/ddr481
Gomez-Suaga P, Rivero-Rios P, Fdez E, Blanca Ramirez M, Ferrer I, Aiastui A et al (2014) LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum Mol Genet 23:6779–6796. https://doi.org/10.1093/hmg/ddu395
Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ (2003) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A 100:4078–4083. https://doi.org/10.1073/pnas.0737556100
Greffard S, Verny M, Bonnet AM, Beinis JY, Gallinari C, Meaume S et al (2006) Motor score of the Unified Parkinson Disease Rating Scale as a good predictor of Lewy body-associated neuronal loss in the substantia nigra. Arch Neurol 63:584–588. https://doi.org/10.1001/archneur.63.4.584
Grunewald A, Kumar KR, Sue CM (2019) New insights into the complex role of mitochondria in Parkinson's disease. Prog Neurobiol 177:73–93. https://doi.org/10.1016/j.pneurobio.2018.09.003
Guardia-Laguarta C, Area-Gomez E, Rub C, Liu Y, Magrane J, Becker D et al (2014) alpha-Synuclein is localized to mitochondria-associated ER membranes. J Neurosci 34:249–259. https://doi.org/10.1523/JNEUROSCI.2507-13.2014
Guerra F, Girolimetti G, Beli R, Mitruccio M, Pacelli C, Ferretta A et al (2019) Synergistic Effect of Mitochondrial and Lysosomal Dysfunction in Parkinson's Disease. Cells 8. https://doi.org/10.3390/cells8050452
Guerreiro R, Ross OA, Kun-Rodrigues C, Hernandez DG, Orme T, Eicher JD et al (2018) Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet Neurol 17:64–74. https://doi.org/10.1016/S1474-4422(17)30400-3
Gunduz A, Eken AG, Bilgic B, Hanagasi HA, Bilguvar K, Gunel M et al (2014) FBXO7-R498X mutation: phenotypic variability from chorea to early onset parkinsonism within a family. Parkinsonism Relat Disord 20:1253–1256. https://doi.org/10.1016/j.parkreldis.2014.07.016
Guo YP, Tang BS, Guo JF (2018) PLA2G6-Associated Neurodegeneration (PLAN): Review of Clinical Phenotypes and Genotypes. Front Neurol 9:1100. https://doi.org/10.3389/fneur.2018.01100
Gupta HV, Vengoechea J, Sahaya K, Virmani T (2015) A splice site mutation in ATP6AP2 causes X-linked intellectual disability, epilepsy, and parkinsonism. Parkinsonism Relat Disord 21:1473–1475. https://doi.org/10.1016/j.parkreldis.2015.10.001
Gusdon AM, Zhu J, Van Houten B, Chu CT (2012) ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol Dis 45:962–972. https://doi.org/10.1016/j.nbd.2011.12.015
Hamano K, Hayashi M, Shioda K, Fukatsu R, Mizutani S (2008) Mechanisms of neurodegeneration in mucopolysaccharidoses II and IIIB: analysis of human brain tissue. Acta Neuropathol 115:547–559. https://doi.org/10.1007/s00401-007-0325-3
Harper JW, Ordureau A, Heo JM (2018) Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol 19:93–108. https://doi.org/10.1038/nrm.2017.129
Hasson SA, Kane LA, Yamano K, Huang CH, Sliter DA, Buehler E et al (2013) High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 504:291–295. https://doi.org/10.1038/nature12748
Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S et al (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol 7:583–590. https://doi.org/10.1016/S1474-4422(08)70117-0
Hedrich K, Hagenah J, Djarmati A, Hiller A, Lohnau T, Lasek K et al (2006) Clinical spectrum of homozygous and heterozygous PINK1 mutations in a large German family with Parkinson disease: role of a single hit? Arch Neurol 63:833–838. https://doi.org/10.1001/archneur.63.6.833
Hering H, Lin CC, Sheng M (2003) Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J Neurosci 23:3262–3271
Hirose T, Hashimoto M, Totsune K, Metoki H, Asayama K, Kikuya M et al (2009) Association of (pro) renin receptor gene polymorphism with blood pressure in Japanese men: the Ohasama study. Am J Hypertens 22:294–299. https://doi.org/10.1038/ajh.2008.357
Hopfner F, Schulte EC, Mollenhauer B, Bereznai B, Knauf F, Lichtner P et al (2013) The role of SCARB2 as susceptibility factor in Parkinson's disease. Mov Disord 28:538–540. https://doi.org/10.1002/mds.25349
Hutagalung AH, Novick PJ (2011) Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev 91:119–149. https://doi.org/10.1152/physrev.00059.2009
Ihse E, Yamakado H, van Wijk XM, Lawrence R, Esko JD, Masliah E (2017) Cellular internalization of alpha-synuclein aggregates by cell surface heparan sulfate depends on aggregate conformation and cell type. Sci Rep 7:9008. https://doi.org/10.1038/s41598-017-08720-5
Illingworth MA, Meyer E, Chong WK, Manzur AY, Carr LJ, Younis R et al (2014) PLA2G6-associated neurodegeneration (PLAN): further expansion of the clinical, radiological and mutation spectrum associated with infantile and atypical childhood-onset disease. Mol Genet Metab 112:183–189. https://doi.org/10.1016/j.ymgme.2014.03.008
Imai Y, Kobayashi Y, Inoshita T, Meng H, Arano T, Uemura K et al (2015) The Parkinson's Disease-Associated Protein Kinase LRRK2 Modulates Notch Signaling through the Endosomal Pathway. PLoS Genet 11:e1005503. https://doi.org/10.1371/journal.pgen.1005503
Infante RE, Wang ML, Radhakrishnan A, Kwon HJ, Brown MS, Goldstein JL (2008) NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc Natl Acad Sci U S A 105:15287–15292. https://doi.org/10.1073/pnas.0807328105
Irrcher I, Aleyasin H, Seifert EL, Hewitt SJ, Chhabra S, Phillips M et al (2010) Loss of the Parkinson's disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum Mol Genet 19:3734–3746. https://doi.org/10.1093/hmg/ddq288
Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA et al (1995) The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron 14:467–475. https://doi.org/10.1016/0896-6273(95)90302-x
Jansen EJ, Timal S, Ryan M, Ashikov A, van Scherpenzeel M, Graham LA et al (2016) ATP6AP1 deficiency causes an immunodeficiency with hepatopathy, cognitive impairment and abnormal protein glycosylation. Nat Commun 7:11600. https://doi.org/10.1038/ncomms11600
Jansen IE, Ye H, Heetveld S, Lechler MC, Michels H, Seinstra RI et al (2017) Discovery and functional prioritization of Parkinson's disease candidate genes from large-scale whole exome sequencing. Genome Biol 18:22. https://doi.org/10.1186/s13059-017-1147-9
Jiang H, Kang SU, Zhang S, Karuppagounder S, Xu J, Lee YK et al (2016) Adult Conditional Knockout of PGC-1alpha Leads to Loss of Dopamine Neurons. eNeuro 3. https://doi.org/10.1523/ENEURO.0183-16.2016
Johnson BN, Berger AK, Cortese GP, Lavoie MJ (2012) The ubiquitin E3 ligase parkin regulates the proapoptotic function of Bax. Proc Natl Acad Sci U S A 109:6283–6288. https://doi.org/10.1073/pnas.1113248109
Johri A, Beal MF (2012) Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther 342:619–630. https://doi.org/10.1124/jpet.112.192138
Joseph S, Schulz JB, Stegmuller J (2018) Mechanistic contributions of FBXO7 to Parkinson disease. J Neurochem 144:118–127. https://doi.org/10.1111/jnc.14253
Josephs KA, Matsumoto JY, Lindor NM (2004) Heterozygous Niemann-Pick disease type C presenting with tremor. Neurology 63:2189–2190. https://doi.org/10.1212/01.wnl.0000145710.25588.2f
Junn E, Jang WH, Zhao X, Jeong BS, Mouradian MM (2009) Mitochondrial localization of DJ-1 leads to enhanced neuroprotection. J Neurosci Res 87:123–129. https://doi.org/10.1002/jnr.21831
Kahle PJ, Waak J, Gasser T (2009) DJ-1 and prevention of oxidative stress in Parkinson's disease and other age-related disorders. Free Radic Biol Med 47:1354–1361. https://doi.org/10.1016/j.freeradbiomed.2009.08.003
Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA et al (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205:143–153. https://doi.org/10.1083/jcb.201402104
Kann M, Jacobs H, Mohrmann K, Schumacher K, Hedrich K, Garrels J et al (2002) Role of parkin mutations in 111 community-based patients with early-onset parkinsonism. Ann Neurol 51:621–625. https://doi.org/10.1002/ana.10179
Karten B, Peake KB, Vance JE (2009) Mechanisms and consequences of impaired lipid trafficking in Niemann-Pick type C1-deficient mammalian cells. Biochim Biophys Acta 1791:659–670. https://doi.org/10.1016/j.bbalip.2009.01.025
Kasten M, Hartmann C, Hampf J, Schaake S, Westenberger A, Vollstedt EJ et al (2018) Genotype-Phenotype Relations for the Parkinson's Disease Genes Parkin, PINK1, DJ1: MDSGene Systematic Review. Mov Disord 33:730–741. https://doi.org/10.1002/mds.27352
Kauther KM, Hoft C, Rissling I, Oertel WH, Moller JC (2011) The PLA2G6 gene in early-onset Parkinson's disease. Mov Disord 26:2415–2417. https://doi.org/10.1002/mds.23851
Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K et al (2014) Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J 460:127–139. https://doi.org/10.1042/BJ20140334
Keogh MJ, Wei W, Aryaman J, Wilson I, Talbot K, Turner MR et al (2018) Oligogenic genetic variation of neurodegenerative disease genes in 980 postmortem human brains. J Neurol Neurosurg Psychiatry 89:813–816. https://doi.org/10.1136/jnnp-2017-317234
Khan NL, Graham E, Critchley P, Schrag AE, Wood NW, Lees AJ et al (2003) Parkin disease: a phenotypic study of a large case series. Brain 126:1279–1292. https://doi.org/10.1093/brain/awg142
Khateeb S, Flusser H, Ofir R, Shelef I, Narkis G, Vardi G et al (2006) PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am J Hum Genet 79:942–948. https://doi.org/10.1086/508572
Kiely AP, Asi YT, Kara E, Limousin P, Ling H, Lewis P et al (2013) alpha-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson's disease and multiple system atrophy? Acta Neuropathol 125:753–769. https://doi.org/10.1007/s00401-013-1096-7
Kilarski LL, Pearson JP, Newsway V, Majounie E, Knipe MD, Misbahuddin A et al (2012) Systematic review and UK-based study of PARK2 (parkin), PINK1, PARK7 (DJ-1) and LRRK2 in early-onset Parkinson's disease. Mov Disord 27:1522–1529. https://doi.org/10.1002/mds.25132
Kim KY, Stevens MV, Akter MH, Rusk SE, Huang RJ, Cohen A et al (2011) Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J Clin Invest 121:3701–3712. https://doi.org/10.1172/JCI44736
Kim MJ, Jeon S, Burbulla LF, Krainc D (2018) Acid ceramidase inhibition ameliorates alpha-synuclein accumulation upon loss of GBA1 function. Hum Mol Genet 27:1972–1988. https://doi.org/10.1093/hmg/ddy105
Kim SJ, Park YJ, Hwang IY, Youdim MB, Park KS, Oh YJ (2012) Nuclear translocation of DJ-1 during oxidative stress-induced neuronal cell death. Free Radic Biol Med 53:936–950. https://doi.org/10.1016/j.freeradbiomed.2012.05.035
Kinumi T, Kimata J, Taira T, Ariga H, Niki E (2004) Cysteine-106 of DJ-1 is the most sensitive cysteine residue to hydrogen peroxide-mediated oxidation in vivo in human umbilical vein endothelial cells. Biochem Biophys Res Commun 317:722–728. https://doi.org/10.1016/j.bbrc.2004.03.110
Kirola L, Behari M, Shishir C, Thelma BK (2016) Identification of a novel homozygous mutation Arg459Pro in SYNJ1 gene of an Indian family with autosomal recessive juvenile Parkinsonism. Parkinsonism Relat Disord 31:124–128. https://doi.org/10.1016/j.parkreldis.2016.07.014
Klein AD, Mazzulli JR (2018) Is Parkinson's disease a lysosomal disorder? Brain 141:2255–2262. https://doi.org/10.1093/brain/awy147
Klein C, Djarmati A, Hedrich K, Schafer N, Scaglione C, Marchese R et al (2005) PINK1, Parkin, and DJ-1 mutations in Italian patients with early-onset parkinsonism. Eur J Hum Genet 13:1086–1093. https://doi.org/10.1038/sj.ejhg.5201455
Kluenemann HH, Nutt JG, Davis MY, Bird TD (2013) Parkinsonism syndrome in heterozygotes for Niemann-Pick C1. J Neurol Sci 335:219–220. https://doi.org/10.1016/j.jns.2013.08.033
Ko DC, Gordon MD, Jin JY, Scott MP (2001) Dynamic movements of organelles containing Niemann-Pick C1 protein: NPC1 involvement in late endocytic events. Mol Biol Cell 12:601–614. https://doi.org/10.1091/mbc.12.3.601
Koch J, Gartner S, Li CM, Quintern LE, Bernardo K, Levran O et al (1996) Molecular cloning and characterization of a full-length complementary DNA encoding human acid ceramidase. Identification Of the first molecular lesion causing Farber disease. J Biol Chem 271:33110–33115. https://doi.org/10.1074/jbc.271.51.33110
Kolehmainen J, Black GC, Saarinen A, Chandler K, Clayton-Smith J, Traskelin AL et al (2003) Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am J Hum Genet 72:1359–1369
Koroglu C, Baysal L, Cetinkaya M, Karasoy H, Tolun A (2013) DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism Relat Disord 19:320–324. https://doi.org/10.1016/j.parkreldis.2012.11.006
Korvatska O, Strand NS, Berndt JD, Strovas T, Chen DH, Leverenz JB et al (2013) Altered splicing of ATP6AP2 causes X-linked parkinsonism with spasticity (XPDS). Hum Mol Genet 22:3259–3268. https://doi.org/10.1093/hmg/ddt180
Kovtun O, Leneva N, Bykov YS, Ariotti N, Teasdale RD, Schaffer M et al (2018) Structure of the membrane-assembled retromer coat determined by cryo-electron tomography. Nature 561:561–564. https://doi.org/10.1038/s41586-018-0526-z
Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M et al (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510:162–166. https://doi.org/10.1038/nature13392
Krebiehl G, Ruckerbauer S, Burbulla LF, Kieper N, Maurer B, Waak J et al (2010) Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson's disease-associated protein DJ-1. PLoS One 5:e9367. https://doi.org/10.1371/journal.pone.0009367
Krebs CE, Karkheiran S, Powell JC, Cao M, Makarov V, Darvish H et al (2013) The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat 34:1200–1207. https://doi.org/10.1002/humu.22372
Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S et al (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet 18:106–108. https://doi.org/10.1038/ng0298-106
Kubo S, Nemani VM, Chalkley RJ, Anthony MD, Hattori N, Mizuno Y et al (2005) A combinatorial code for the interaction of alpha-synuclein with membranes. J Biol Chem 280:31664–31672. https://doi.org/10.1074/jbc.M504894200
Kumar N, Leonzino M, Hancock-Cerutti W, Horenkamp FA, Li P, Lees JA et al (2018) VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J Cell Biol 217:3625–3639. https://doi.org/10.1083/jcb.201807019
Kurian MA, Morgan NV, MacPherson L, Foster K, Peake D, Gupta R et al (2008) Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (PLAN). Neurology 70:1623–1629. https://doi.org/10.1212/01.wnl.0000310986.48286.8e
Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS et al (2009) Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 137:1213–1224. https://doi.org/10.1016/j.cell.2009.03.049
Lang AB, John Peter AT, Walter P, Kornmann B (2015) ER-mitochondrial junctions can be bypassed by dominant mutations in the endosomal protein Vps13. J Cell Biol 210:883–890. https://doi.org/10.1083/jcb.201502105
Laurent-Matha V, Derocq D, Prebois C, Katunuma N, Liaudet-Coopman E (2006) Processing of human cathepsin D is independent of its catalytic function and auto-activation: involvement of cathepsins L and B. J Biochem 139:363–371. https://doi.org/10.1093/jb/mvj037
Lee DW, Wu X, Eisenberg E, Greene LE (2006) Recruitment dynamics of GAK and auxilin to clathrin-coated pits during endocytosis. J Cell Sci 119:3502–3512. https://doi.org/10.1242/jcs.03092
Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP (2010) Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol 189:671–679. https://doi.org/10.1083/jcb.201001039
Lehri-Boufala S, Ouidja MO, Barbier-Chassefiere V, Henault E, Raisman-Vozari R, Garrigue-Antar L et al (2015) New roles of glycosaminoglycans in alpha-synuclein aggregation in a cellular model of Parkinson disease. PLoS One 10:e0116641. https://doi.org/10.1371/journal.pone.0116641
Lesage S, Anheim M, Letournel F, Bousset L, Honore A, Rozas N et al (2013) G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol 73:459–471. https://doi.org/10.1002/ana.23894
Lesage S, Brice A (2009) Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet 18:R48–R59. https://doi.org/10.1093/hmg/ddp012
Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A et al (2016) Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am J Hum Genet 98:500–513. https://doi.org/10.1016/j.ajhg.2016.01.014
Lesage S, Durr A, Tazir M, Lohmann E, Leutenegger AL, Janin S et al (2006) LRRK2 G2019S as a cause of Parkinson's disease in North African Arabs. N Engl J Med 354:422–423. https://doi.org/10.1056/NEJMc055540
Levran O, Desnick RJ, Schuchman EH (1991) Niemann-Pick disease: a frequent missense mutation in the acid sphingomyelinase gene of Ashkenazi Jewish type A and B patients. Proc Natl Acad Sci U S A 88:3748–3752. https://doi.org/10.1073/pnas.88.9.3748
Li H, Ham A, Ma TC, Kuo SH, Kanter E, Kim D et al (2019) Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 15:113–130. https://doi.org/10.1080/15548627.2018.1509818
Li Y, Tomiyama H, Sato K, Hatano Y, Yoshino H, Atsumi M et al (2005) Clinicogenetic study of PINK1 mutations in autosomal recessive early-onset parkinsonism. Neurology 64:1955–1957. https://doi.org/10.1212/01.WNL.0000164009.36740.4E
Liao G, Yao Y, Liu J, Yu Z, Cheung S, Xie A et al (2007) Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1 −/− mouse brain. Am J Pathol 171:962–975. https://doi.org/10.2353/ajpath.2007.070052
Lin CH, Tan EK, Chen ML, Tan LC, Lim HQ, Chen GS et al (2008) Novel ATP13A2 variant associated with Parkinson disease in Taiwan and Singapore. Neurology 71:1727–1732. https://doi.org/10.1212/01.wnl.0000335167.72412.68
Lin G, Lee PT, Chen K, Mao D, Tan KL, Zuo Z et al (2018) Phospholipase PLA2G6, a Parkinsonism-Associated Gene, Affects Vps26 and Vps35, Retromer Function, and Ceramide Levels. Similar to alpha-Synuclein Gain. Cell Metab 28:605–618.e6. https://doi.org/10.1016/j.cmet.2018.05.019
Lin G, Wang L, Marcogliese PC, Bellen HJ (2019) Sphingolipids in the Pathogenesis of Parkinson's Disease and Parkinsonism. Trends Endocrinol Metab 30:106–117. https://doi.org/10.1016/j.tem.2018.11.003
Liu S, Ninan I, Antonova I, Battaglia F, Trinchese F, Narasanna A et al (2004) alpha-Synuclein produces a long-lasting increase in neurotransmitter release. EMBO J 23:4506–4516. https://doi.org/10.1038/sj.emboj.7600451
Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ et al (2008) Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med 14:1247–1255. https://doi.org/10.1038/nm.1876
Lohmann E, Coquel AS, Honore A, Gurvit H, Hanagasi H, Emre M et al (2015) A new F-box protein 7 gene mutation causing typical Parkinson's disease. Mov Disord 30:1130–1133. https://doi.org/10.1002/mds.26266
Lopes da Fonseca T, Pinho R, Outeiro TF (2016) A familial ATP13A2 mutation enhances alpha-synuclein aggregation and promotes cell death. Hum Mol Genet 25:2959–2971. https://doi.org/10.1093/hmg/ddw147
Lotharius J, Brundin P (2002) Impaired dopamine storage resulting from alpha-synuclein mutations may contribute to the pathogenesis of Parkinson's disease. Hum Mol Genet 11:2395–2407. https://doi.org/10.1093/hmg/11.20.2395
Lubbe SJ, Escott-Price V, Gibbs JR, Nalls MA, Bras J, Price TR et al (2016) Additional rare variant analysis in Parkinson's disease cases with and without known pathogenic mutations: evidence for oligogenic inheritance. Hum Mol Genet 25:5483–5489. https://doi.org/10.1093/hmg/ddw348
Lucas M, Gershlick DC, Vidaurrazaga A, Rojas AL, Bonifacino JS, Hierro A (2016) Structural Mechanism for Cargo Recognition by the Retromer Complex. Cell 167:1623–1635 e1614. https://doi.org/10.1016/j.cell.2016.10.056
Magalhaes J, Gegg ME, Migdalska-Richards A, Schapira AH (2018) Effects of ambroxol on the autophagy-lysosome pathway and mitochondria in primary cortical neurons. Sci Rep 8:1385. https://doi.org/10.1038/s41598-018-19479-8
Makaroff L, Gunn A, Gervasoni C, Richy F (2011) Gastrointestinal disorders in Parkinson's disease: prevalence and health outcomes in a US claims database. J Parkinsons Dis 1:65–74. https://doi.org/10.3233/JPD-2011-001
Mani M, Lee SY, Lucast L, Cremona O, Di Paolo G, De Camilli P et al (2007) The dual phosphatase activity of synaptojanin1 is required for both efficient synaptic vesicle endocytosis and reavailability at nerve terminals. Neuron 56:1004–1018. https://doi.org/10.1016/j.neuron.2007.10.032
Marcos AL, Corradi GR, Mazzitelli LR, Casali CI, Fernandez Tome MDC, Adamo HP et al (2019) The Parkinson-associated human P5B-ATPase ATP13A2 modifies lipid homeostasis. Biochim Biophys Acta Biomembr 1861:182993. https://doi.org/10.1016/j.bbamem.2019.05.015
Martin S, Smolders S, Van den Haute C, Heeman B, van Veen S, Crosiers D et al (2020) Mutated ATP10B increases Parkinson's disease risk by compromising lysosomal glucosylceramide export. Acta Neuropathol. https://doi.org/10.1007/s00401-020-02145-7
Martinez Z, Zhu M, Han S, Fink AL (2007) GM1 specifically interacts with alpha-synuclein and inhibits fibrillation. Biochemistry 46:1868–1877. https://doi.org/10.1021/bi061749a
Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA et al (2011) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146:37–52. https://doi.org/10.1016/j.cell.2011.06.001
Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D (2016) alpha-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci U S A 113:1931–1936. https://doi.org/10.1073/pnas.1520335113
Mazzulli JR, Zunke F, Tsunemi T, Toker NJ, Jeon S, Burbulla LF et al (2016) Activation of beta-Glucocerebrosidase Reduces Pathological alpha-Synuclein and Restores Lysosomal Function in Parkinson's Patient Midbrain Neurons. J Neurosci 36:7693–7706. https://doi.org/10.1523/JNEUROSCI.0628-16.2016
McCoy MK, Kaganovich A, Rudenko IN, Ding J, Cookson MR (2014) Hexokinase activity is required for recruitment of parkin to depolarized mitochondria. Hum Mol Genet 23:145–156. https://doi.org/10.1093/hmg/ddt407
McFarland NR, Hess CW (2017) Recognizing Atypical Parkinsonisms: "Red Flags" and Therapeutic Approaches. Semin Neurol 37:215–227. https://doi.org/10.1055/s-0037-1602422
Michelakakis H, Xiromerisiou G, Dardiotis E, Bozi M, Vassilatis D, Kountra PM et al (2012) Evidence of an association between the scavenger receptor class B member 2 gene and Parkinson's disease. Mov Disord 27:400–405. https://doi.org/10.1002/mds.24886
Mishra P, Chan DC (2016) Metabolic regulation of mitochondrial dynamics. J Cell Biol 212:379–387. https://doi.org/10.1083/jcb.201511036
Miura E, Hasegawa T, Konno M, Suzuki M, Sugeno N, Fujikake N et al (2014) VPS35 dysfunction impairs lysosomal degradation of alpha-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson's disease. Neurobiol Dis 71:1–13. https://doi.org/10.1016/j.nbd.2014.07.014
Monroy-Jaramillo N, Guerrero-Camacho JL, Rodriguez-Violante M, Boll-Woehrlen MC, Yescas-Gomez P, Alonso-Vilatela ME et al (2014) Genetic mutations in early-onset Parkinson's disease Mexican patients: molecular testing implications. Am J Med Genet B Neuropsychiatr Genet 165B:235–244. https://doi.org/10.1002/ajmg.b.32228
Moren C, Juarez-Flores DL, Chau KY, Gegg M, Garrabou G, Gonzalez-Casacuberta I et al (2019) GBA mutation promotes early mitochondrial dysfunction in 3D neurosphere models. Aging (Albany NY) 11:10338–10355. https://doi.org/10.18632/aging.102460
Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S et al (2006) PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet 38:752–754. https://doi.org/10.1038/ng1826
Murphy KE, Cottle L, Gysbers AM, Cooper AA, Halliday GM (2013) ATP13A2 (PARK9) protein levels are reduced in brain tissue of cases with Lewy bodies. Acta Neuropathol Commun 1:11. https://doi.org/10.1186/2051-5960-1-11
Murphy KE, Gysbers AM, Abbott SK, Tayebi N, Kim WS, Sidransky E et al (2014) Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson's disease. Brain 137:834–848. https://doi.org/10.1093/brain/awt367
Naito T, Takatsu H, Miyano R, Takada N, Nakayama K, Shin HW (2015) Phospholipid flippase ATP10A translocates phosphatidylcholine and is involved in plasma membrane dynamics. J Biol Chem. https://doi.org/10.1074/jbc.M115.655191
Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D et al (2019) Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet Neurol 18:1091–1102. https://doi.org/10.1016/S1474-4422(19)30320-5
Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M et al (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet 46:989–993. https://doi.org/10.1038/ng.3043
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803. https://doi.org/10.1083/jcb.200809125
Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R et al (2000) Identification of HE1 as the second gene of Niemann-Pick C disease. Science 290:2298–2301. https://doi.org/10.1126/science.290.5500.2298
Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK et al (2010) Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65:66–79. https://doi.org/10.1016/j.neuron.2009.12.023
Neumann J, Bras J, Deas E, O'Sullivan SS, Parkkinen L, Lachmann RH et al (2009) Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain 132:1783–1794. https://doi.org/10.1093/brain/awp044
Ng CH, Basil AH, Hang L, Tan R, Goh KL, O'Neill S et al (2017) Genetic or pharmacological activation of the Drosophila PGC-1alpha ortholog spargel rescues the disease phenotypes of genetic models of Parkinson's disease. Neurobiol Aging 55:33–37. https://doi.org/10.1016/j.neurobiolaging.2017.03.017
Nguyen M, Wong YC, Ysselstein D, Severino A, Krainc D (2019) Synaptic, Mitochondrial, and Lysosomal Dysfunction in Parkinson's Disease. Trends Neurosci 42:140–149. https://doi.org/10.1016/j.tins.2018.11.001
Niu J, Yu M, Wang C, Xu Z (2012) Leucine-rich repeat kinase 2 disturbs mitochondrial dynamics via Dynamin-like protein. J Neurochem 122:650–658. https://doi.org/10.1111/j.1471-4159.2012.07809.x
Nuytemans K, Meeus B, Crosiers D, Brouwers N, Goossens D, Engelborghs S et al (2009) Relative contribution of simple mutations vs. copy number variations in five Parkinson disease genes in the Belgian population. Hum Mutat 30:1054–1061. https://doi.org/10.1002/humu.21007
Olgiati S, De Rosa A, Quadri M, Criscuolo C, Breedveld GJ, Picillo M et al (2014) PARK20 caused by SYNJ1 homozygous Arg258Gln mutation in a new Italian family. Neurogenetics 15:183–188. https://doi.org/10.1007/s10048-014-0406-0
Olgiati S, Quadri M, Fang M, Rood JP, Saute JA, Chien HF et al (2016) DNAJC6 Mutations Associated With Early-Onset Parkinson's Disease. Ann Neurol 79:244–256. https://doi.org/10.1002/ana.24553
Ong WY, Yeo JF, Ling SF, Farooqui AA (2005) Distribution of calcium-independent phospholipase A2 (iPLA 2) in monkey brain. J Neurocytol 34:447–458. https://doi.org/10.1007/s11068-006-8730-4
Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I et al (2013) Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 16:394–406. https://doi.org/10.1038/nn.3350
Orme T, Guerreiro R, Bras J (2018) The Genetics of Dementia with Lewy Bodies: Current Understanding and Future Directions. Curr Neurol Neurosci Rep 18:67. https://doi.org/10.1007/s11910-018-0874-y
Osellame LD, Rahim AA, Hargreaves IP, Gegg ME, Richard-Londt A, Brandner S et al (2013) Mitochondria and quality control defects in a mouse model of Gaucher disease--links to Parkinson's disease. Cell Metab 17:941–953. https://doi.org/10.1016/j.cmet.2013.04.014
Ott C, Schneider MP, Delles C, Schlaich MP, Hilgers KF, Schmieder RE (2011) Association of (pro) renin receptor gene polymorphism with blood pressure in Caucasian men. Pharmacogenet Genomics 21:347–349. https://doi.org/10.1097/FPC.0b013e328344cdd2
Ottolini D, Cali T, Negro A, Brini M (2013) The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum Mol Genet 22:2152–2168. https://doi.org/10.1093/hmg/ddt068
Outeiro TF, Lindquist S (2003) Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302:1772–1775. https://doi.org/10.1126/science.1090439
Paillusson S, Gomez-Suaga P, Stoica R, Little D, Gissen P, Devine MJ et al (2017) alpha-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca (2+) homeostasis and mitochondrial ATP production. Acta Neuropathol 134:129–149. https://doi.org/10.1007/s00401-017-1704-z
Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW et al (2009) Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol 65:19–23. https://doi.org/10.1002/ana.21415
Paisan-Ruiz C, Lang AE, Kawarai T, Sato C, Salehi-Rad S, Fisman GK et al (2005) LRRK2 gene in Parkinson disease: mutation analysis and case control association study. Neurology 65:696–700. https://doi.org/10.1212/01.wnl.0000167552.79769.b3
Paisan-Ruiz C, Lewis PA, Singleton AB (2013) LRRK2: cause, risk, and mechanism. J Parkinsons Dis 3:85–103. https://doi.org/10.3233/JPD-130192
Paisan-Ruiz C, Li A, Schneider SA, Holton JL, Johnson R, Kidd D et al (2012) Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging 33:814–823. https://doi.org/10.1016/j.neurobiolaging.2010.05.009
Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M et al (2004) Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem 279:18614–18622. https://doi.org/10.1074/jbc.M401135200
Palmgren MG, Nissen P (2011) P-Type ATPases. Ann Rev Biophys 40(40):243–266. https://doi.org/10.1146/annurev.biophys.093008.131331
Pamarthy S, Kulshrestha A, Katara GK, Beaman KD (2018) The curious case of vacuolar ATPase: regulation of signaling pathways. Mol Cancer 17:41. https://doi.org/10.1186/s12943-018-0811-3
Papa S, De Rasmo D (2013) Complex I deficiencies in neurological disorders. Trends Mol Med 19:61–69. https://doi.org/10.1016/j.molmed.2012.11.005
Park JS, Blair NF, Sue CM (2015) The role of ATP13A2 in Parkinson's disease: Clinical phenotypes and molecular mechanisms. Mov Disord 30:770–779. https://doi.org/10.1002/mds.26243
Park JS, Koentjoro B, Veivers D, Mackay-Sim A, Sue CM (2014) Parkinson's disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum Mol Genet 23:2802–2815. https://doi.org/10.1093/hmg/ddt623
Park JS, Thorsness MK, Policastro R, McGoldrick LL, Hollingsworth NM, Thorsness PE et al (2016) Yeast Vps13 promotes mitochondrial function and is localized at membrane contact sites. Mol Biol Cell 27:2435–2449. https://doi.org/10.1091/mbc.E16-02-0112
Park JT, Lee YS, Cho KA, Park SC (2018) Adjustment of the lysosomal-mitochondrial axis for control of cellular senescence. Ageing Res Rev 47:176–182. https://doi.org/10.1016/j.arr.2018.08.003
Parker WD Jr, Boyson SJ, Parks JK (1989) Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann Neurol 26:719–723. https://doi.org/10.1002/ana.410260606
Parkkinen L, Neumann J, O'Sullivan SS, Holton JL, Revesz T, Hardy J et al (2011) Glucocerebrosidase mutations do not cause increased Lewy body pathology in Parkinson's disease. Mol Genet Metab 103:410–412. https://doi.org/10.1016/j.ymgme.2011.04.015
Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J et al (2014) Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson's disease-type pathology. Neurobiol Aging 35(2180):e2181–e2185. https://doi.org/10.1016/j.neurobiolaging.2014.03.024
Periquet M, Latouche M, Lohmann E, Rawal N, De Michele G, Ricard S et al (2003) Parkin mutations are frequent in patients with isolated early-onset parkinsonism. Brain 126:1271–1278. https://doi.org/10.1093/brain/awg136
Plotegher N, Bubacco L, Greggio E, Civiero L (2019) Ceramides in Parkinson's Disease: From Recent Evidence to New Hypotheses. Front Neurosci 13:330. https://doi.org/10.3389/fnins.2019.00330
Plotegher N, Duchen MR (2017) Mitochondrial Dysfunction and Neurodegeneration in Lysosomal Storage Disorders. Trends Mol Med 23:116–134. https://doi.org/10.1016/j.molmed.2016.12.003
Poewe W (2008) Non-motor symptoms in Parkinson's disease. Eur J Neurol 15(Suppl 1):14–20. https://doi.org/10.1111/j.1468-1331.2008.02056.x
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A et al (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276:2045–2047. https://doi.org/10.1126/science.276.5321.2045
Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L (2010) The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One 5:e10054. https://doi.org/10.1371/journal.pone.0010054
Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W et al (2015) MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 30:1591–1601. https://doi.org/10.1002/mds.26424
Prajapati P, Sripada L, Singh K, Roy M, Bhatelia K, Dalwadi P et al (2018) Systemic Analysis of miRNAs in PD Stress Condition: miR-5701 Modulates Mitochondrial-Lysosomal Cross Talk to Regulate Neuronal Death. Mol Neurobiol 55:4689–4701. https://doi.org/10.1007/s12035-017-0664-6
Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL et al (2013) A novel alpha-synuclein missense mutation in Parkinson disease. Neurology 80:1062–1064. https://doi.org/10.1212/WNL.0b013e31828727ba
Quadri M, Fang M, Picillo M, Olgiati S, Breedveld GJ, Graafland J et al (2013) Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat 34:1208–1215. https://doi.org/10.1002/humu.22373
Rafiq K, Mori H, Masaki T, Nishiyama A (2013) (Pro) renin receptor and insulin resistance: possible roles of angiotensin II-dependent and -independent pathways. Mol Cell Endocrinol 378:41–45. https://doi.org/10.1016/j.mce.2012.05.016
Rahman AA, Morrison BE (2019) Contributions of VPS35 Mutations to Parkinson's Disease. Neuroscience 401:1–10. https://doi.org/10.1016/j.neuroscience.2019.01.006
Ramakrishnan M, Jensen PH, Marsh D (2003) Alpha-synuclein association with phosphatidylglycerol probed by lipid spin labels. Biochemistry 42:12919–12926. https://doi.org/10.1021/bi035048e
Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP et al (2006) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 38:1184–1191. https://doi.org/10.1038/ng1884
Ramonet D, Podhajska A, Stafa K, Sonnay S, Trancikova A, Tsika E et al (2012) PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum Mol Genet 21:1725–1743. https://doi.org/10.1093/hmg/ddr606
Ramser J, Abidi FE, Burckle CA, Lenski C, Toriello H, Wen G et al (2005) A unique exonic splice enhancer mutation in a family with X-linked mental retardation and epilepsy points to a novel role of the renin receptor. Hum Mol Genet 14:1019–1027. https://doi.org/10.1093/hmg/ddi094
Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J et al (2017) Excessive burden of lysosomal storage disorder gene variants in Parkinson's disease. Brain 140:3191–3203. https://doi.org/10.1093/brain/awx285
Roland BP, Naito T, Best JT, Arnaiz-Yepez C, Takatsu H, Yu RJ et al (2019) Yeast and human P4-ATPases transport glycosphingolipids using conserved structural motifs. J Biol Chem 294:1794–1806. https://doi.org/10.1074/jbc.RA118.005876
Ron I, Horowitz M (2008) Intracellular cholesterol modifies the ERAD of glucocerebrosidase in Gaucher disease patients. Mol Genet Metab 93:426–436. https://doi.org/10.1016/j.ymgme.2007.10.132
Roosen DA, Cookson MR (2016) LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol Neurodegener 11:73. https://doi.org/10.1186/s13024-016-0140-1
Rosborough K, Patel N, Kalia LV (2017) alpha-Synuclein and Parkinsonism: Updates and Future Perspectives. Curr Neurol Neurosci Rep 17:31. https://doi.org/10.1007/s11910-017-0737-y
Ross OA, Toft M, Whittle AJ, Johnson JL, Papapetropoulos S, Mash DC et al (2006) Lrrk2 and Lewy body disease. Ann Neurol 59:388–393. https://doi.org/10.1002/ana.20731
Rostovtseva TK, Gurnev PA, Protchenko O, Hoogerheide DP, Yap TL, Philpott CC et al (2015) alpha-Synuclein Shows High Affinity Interaction with Voltage-dependent Anion Channel, Suggesting Mechanisms of Mitochondrial Regulation and Toxicity in Parkinson Disease. J Biol Chem 290:18467–18477. https://doi.org/10.1074/jbc.M115.641746
Rujano MA, Cannata Serio M, Panasyuk G, Peanne R, Reunert J, Rymen D et al (2017) Mutations in the X-linked ATP6AP2 cause a glycosylation disorder with autophagic defects. J Exp Med 214:3707–3729. https://doi.org/10.1084/jem.20170453
Runz H, Dolle D, Schlitter AM, Zschocke J (2008) NPC-db, a Niemann-Pick type C disease gene variation database. Hum Mutat 29:345–350. https://doi.org/10.1002/humu.20636
Ryan SD, Dolatabadi N, Chan SF, Zhang X, Akhtar MW, Parker J et al (2013) Isogenic human iPSC Parkinson's model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell 155:1351–1364. https://doi.org/10.1016/j.cell.2013.11.009
Saftig P, Klumperman J (2009) Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 10:623–635. https://doi.org/10.1038/nrm2745
Saito Y, Suzuki K, Hulette CM, Murayama S (2004) Aberrant phosphorylation of alpha-synuclein in human Niemann-Pick type C1 disease. J Neuropathol Exp Neurol 63:323–328. https://doi.org/10.1093/jnen/63.4.323
Sakai N, Inui K, Fujii N, Fukushima H, Nishimoto J, Yanagihara I et al (1994) Krabbe disease: isolation and characterization of a full-length cDNA for human galactocerebrosidase. Biochem Biophys Res Commun 198:485–491. https://doi.org/10.1006/bbrc.1994.1071
Samaranch L, Lorenzo-Betancor O, Arbelo JM, Ferrer I, Lorenzo E, Irigoyen J et al (2010) PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 133:1128–1142. https://doi.org/10.1093/brain/awq051
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem 54:823–827. https://doi.org/10.1111/j.1471-4159.1990.tb02325.x
Scholz SW, Houlden H, Schulte C, Sharma M, Li A, Berg D et al (2009) SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol 65:610–614. https://doi.org/10.1002/ana.21685
Schondorf DC, Aureli M, McAllister FE, Hindley CJ, Mayer F, Schmid B et al (2014) iPSC-derived neurons from GBA1-associated Parkinson's disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun 5:4028. https://doi.org/10.1038/ncomms5028
Schormair B, Kemlink D, Mollenhauer B, Fiala O, Machetanz G, Roth J et al (2018) Diagnostic exome sequencing in early-onset Parkinson's disease confirms VPS13C as a rare cause of autosomal-recessive Parkinson's disease. Clin Genet 93:603–612. https://doi.org/10.1111/cge.13124
Schrag A, Schott JM (2006) Epidemiological, clinical, and genetic characteristics of early-onset parkinsonism. Lancet Neurol 5:355–363. https://doi.org/10.1016/S1474-4422(06)70411-2
Schuchman EH (2007) The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. J Inherit Metab Dis 30:654–663. https://doi.org/10.1007/s10545-007-0632-9
Seaman MN (2007) Identification of a novel conserved sorting motif required for retromer-mediated endosome-to-TGN retrieval. J Cell Sci 120:2378–2389. https://doi.org/10.1242/jcs.009654
Seaman MN (2012) The retromer complex - endosomal protein recycling and beyond. J Cell Sci 125:4693–4702. https://doi.org/10.1242/jcs.103440
Seaman MN, Harbour ME, Tattersall D, Read E, Bright N (2009) Membrane recruitment of the cargo-selective retromer subcomplex is catalysed by the small GTPase Rab7 and inhibited by the Rab-GAP TBC1D5. J Cell Sci 122:2371–2382. https://doi.org/10.1242/jcs.048686
Sebastiao AM, Colino-Oliveira M, Assaife-Lopes N, Dias RB, Ribeiro JA (2013) Lipid rafts, synaptic transmission and plasticity: impact in age-related neurodegenerative diseases. Neuropharmacology 64:97–107. https://doi.org/10.1016/j.neuropharm.2012.06.053
Seong E, Insolera R, Dulovic M, Kamsteeg EJ, Trinh J, Bruggemann N et al (2018) Mutations in VPS13D lead to a new recessive ataxia with spasticity and mitochondrial defects. Ann Neurol 83:1075–1088. https://doi.org/10.1002/ana.25220
Shachar T, Lo Bianco C, Recchia A, Wiessner C, Raas-Rothschild A, Futerman AH (2011) Lysosomal storage disorders and Parkinson's disease: Gaucher disease and beyond. Mov Disord 26:1593–1604. https://doi.org/10.1002/mds.23774
Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP et al (2019) Lewy pathology in Parkinson's disease consists of crowded organelles and lipid membranes. Nat Neurosci 22:1099–1109. https://doi.org/10.1038/s41593-019-0423-2
Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S et al (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25:302–305. https://doi.org/10.1038/77060
Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O et al (2011) PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell 144:689–702. https://doi.org/10.1016/j.cell.2011.02.010
Shinzawa K, Sumi H, Ikawa M, Matsuoka Y, Okabe M, Sakoda S et al (2008) Neuroaxonal dystrophy caused by group VIA phospholipase A2 deficiency in mice: a model of human neurodegenerative disease. J Neurosci 28:2212–2220. https://doi.org/10.1523/JNEUROSCI.4354-07.2008
Shirihai OS, Song M, Dorn GW 2nd (2015) How mitochondrial dynamism orchestrates mitophagy. Circ Res 116:1835–1849. https://doi.org/10.1161/CIRCRESAHA.116.306374
Shojaee S, Sina F, Banihosseini SS, Kazemi MH, Kalhor R, Shahidi GA et al (2008) Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am J Hum Genet 82:1375–1384. https://doi.org/10.1016/j.ajhg.2008.05.005
Siddiqui A, Bhaumik D, Chinta SJ, Rane A, Rajagopalan S, Lieu CA et al (2015) Mitochondrial Quality Control via the PGC1alpha-TFEB Signaling Pathway Is Compromised by Parkin Q311X Mutation But Independently Restored by Rapamycin. J Neurosci 35:12833–12844. https://doi.org/10.1523/JNEUROSCI.0109-15.2015
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER et al (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 361:1651–1661. https://doi.org/10.1056/NEJMoa0901281
Sidransky E, Samaddar T, Tayebi N (2009) Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 73:1424–1425, author reply 1425–1426. https://doi.org/10.1212/WNL.0b013e3181b28601
Siintola E, Partanen S, Stromme P, Haapanen A, Haltia M, Maehlen J et al (2006) Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain 129:1438–1445. https://doi.org/10.1093/brain/awl107
Sina F, Shojaee S, Elahi E, Paisan-Ruiz C (2009) R632W mutation in PLA2G6 segregates with dystonia-parkinsonism in a consanguineous Iranian family. Eur J Neurol 16:101–104. https://doi.org/10.1111/j.1468-1331.2008.02356.x
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J et al (2003) alpha-Synuclein locus triplication causes Parkinson's disease. Science 302:841. https://doi.org/10.1126/science.1090278
Smith BR, Santos MB, Marshall MS, Cantuti-Castelvetri L, Lopez-Rosas A, Li G et al (2014) Neuronal inclusions of alpha-synuclein contribute to the pathogenesis of Krabbe disease. J Pathol 232:509–521. https://doi.org/10.1002/path.4328
Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M et al (2012) Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet 90:1102–1107. https://doi.org/10.1016/j.ajhg.2012.04.021
Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB et al (2005) Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci U S A 102:18676–18681. https://doi.org/10.1073/pnas.0508052102
Song P, Trajkovic K, Tsunemi T, Krainc D (2016) Parkin Modulates Endosomal Organization and Function of the Endo-Lysosomal Pathway. J Neurosci 36:2425–2437. https://doi.org/10.1523/JNEUROSCI.2569-15.2016
Spanaki C, Plaitakis A (2004) Bilineal transmission of Parkinson disease on Crete suggests a complex inheritance. Neurology 62:815–817. https://doi.org/10.1212/01.wnl.0000113720.71387.88
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840. https://doi.org/10.1038/42166
Stafa K, Tsika E, Moser R, Musso A, Glauser L, Jones A et al (2014) Functional interaction of Parkinson's disease-associated LRRK2 with members of the dynamin GTPase superfamily. Hum Mol Genet 23:2055–2077. https://doi.org/10.1093/hmg/ddt600
Steger M, Diez F, Dhekne HS, Lis P, Nirujogi RS, Karayel O et al (2017) Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife 6. https://doi.org/10.7554/eLife.31012
Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M et al (2016) Phosphoproteomics reveals that Parkinson's disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5. https://doi.org/10.7554/eLife.12813
Steinfeld R, Reinhardt K, Schreiber K, Hillebrand M, Kraetzner R, Bruck W et al (2006) Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am J Hum Genet 78:988–998. https://doi.org/10.1086/504159
Stevens DA, Lee Y, Kang HC, Lee BD, Lee YI, Bower A et al (2015) Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc Natl Acad Sci U S A 112:11696–11701. https://doi.org/10.1073/pnas.1500624112
Stockl M, Fischer P, Wanker E, Herrmann A (2008) Alpha-synuclein selectively binds to anionic phospholipids embedded in liquid-disordered domains. J Mol Biol 375:1394–1404. https://doi.org/10.1016/j.jmb.2007.11.051
Sun M, Latourelle JC, Wooten GF, Lew MF, Klein C, Shill HA et al (2006) Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: the GenePD study. Arch Neurol 63:826–832. https://doi.org/10.1001/archneur.63.6.826
Suzuki K (1990) Myelin pathology in the twitcher mouse. Ann N Y Acad Sci 605:313–324. https://doi.org/10.1111/j.1749-6632.1990.tb42405.x
Suzuki M, Fujikake N, Takeuchi T, Kohyama-Koganeya A, Nakajima K, Hirabayashi Y et al (2015) Glucocerebrosidase deficiency accelerates the accumulation of proteinase K-resistant alpha-synuclein and aggravates neurodegeneration in a Drosophila model of Parkinson's disease. Hum Mol Genet 24:6675–6686. https://doi.org/10.1093/hmg/ddv372
Taguchi YV, Liu J, Ruan J, Pacheco J, Zhang X, Abbasi J et al (2017) Glucosylsphingosine Promotes alpha-Synuclein Pathology in Mutant GBA-Associated Parkinson's Disease. J Neurosci 37:9617–9631. https://doi.org/10.1523/JNEUROSCI.1525-17.2017
Taipa R, Pereira C, Reis I, Alonso I, Bastos-Lima A, Melo-Pires M et al (2016) DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain 139:1680–1687. https://doi.org/10.1093/brain/aww080
Tan EK, Yew K, Chua E, Puvan K, Shen H, Lee E et al (2006) PINK1 mutations in sporadic early-onset Parkinson's disease. Mov Disord 21:789–793. https://doi.org/10.1002/mds.20810
Tan J, Zhang T, Jiang L, Chi J, Hu D, Pan Q et al (2011) Regulation of intracellular manganese homeostasis by Kufor-Rakeb syndrome-associated ATP13A2 protein. J Biol Chem 286:29654–29662. https://doi.org/10.1074/jbc.M111.233874
Tang B, Xiong H, Sun P, Zhang Y, Wang D, Hu Z et al (2006) Association of PINK1 and DJ-1 confers digenic inheritance of early-onset Parkinson's disease. Hum Mol Genet 15:1816–1825. https://doi.org/10.1093/hmg/ddl104
Tang FL, Liu W, Hu JX, Erion JR, Ye J, Mei L et al (2015) VPS35 Deficiency or Mutation Causes Dopaminergic Neuronal Loss by Impairing Mitochondrial Fusion and Function. Cell Rep 12:1631–1643. https://doi.org/10.1016/j.celrep.2015.08.001
Tayebi N, Walker J, Stubblefield B, Orvisky E, LaMarca ME, Wong K et al (2003) Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol Genet Metab 79:104–109. https://doi.org/10.1016/S1096-7192(03)00071-4
te Vruchte D, Lloyd-Evans E, Veldman RJ, Neville DC, Dwek RA, Platt FM et al. (2004) Accumulation of glycosphingolipids in Niemann-Pick C disease disrupts endosomal transport. J Biol Chem 279:26167–26175. doi:https://doi.org/10.1074/jbc. M311591200
Thomas KJ, McCoy MK, Blackinton J, Beilina A, van der Brug M, Sandebring A et al (2011) DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet 20:40–50. https://doi.org/10.1093/hmg/ddq430
Tomatsu S, Fukuda S, Sukegawa K, Ikedo Y, Yamada S, Yamada Y et al (1991) Mucopolysaccharidosis type VII: characterization of mutations and molecular heterogeneity. Am J Hum Genet 48:89–96
Trousdale C, Kim K (2015) Retromer: Structure, function, and roles in mammalian disease. Eur J Cell Biol 94:513–521. https://doi.org/10.1016/j.ejcb.2015.07.002
Tsuji S, Choudary PV, Martin BM, Stubblefield BK, Mayor JA, Barranger JA et al (1987) A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher's disease. N Engl J Med 316:570–575. https://doi.org/10.1056/NEJM198703053161002
Tsunemi T, Krainc D (2014) Zn (2)(+) dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum Mol Genet 23:2791–2801. https://doi.org/10.1093/hmg/ddt572
Ujiie S, Hatano T, Kubo S, Imai S, Sato S, Uchihara T et al (2012) LRRK2 I2020T mutation is associated with tau pathology. Parkinsonism Relat Disord 18:819–823. https://doi.org/10.1016/j.parkreldis.2012.03.024
Uversky VN, Eliezer D (2009) Biophysics of Parkinson's disease: structure and aggregation of alpha-synuclein. Curr Protein Pept Sci 10:483–499. https://doi.org/10.2174/138920309789351921
van Veen S, Martin S, Van den Haute C, Benoy V, Lyons J, Vanhoutte R et al (2020) ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 578:419–424. https://doi.org/10.1038/s41586-020-1968-7
Velayati A, Yu WH, Sidransky E (2010) The role of glucocerebrosidase mutations in Parkinson disease and Lewy body disorders. Curr Neurol Neurosci Rep 10:190–198. https://doi.org/10.1007/s11910-010-0102-x
Verheijen FW, Verbeek E, Aula N, Beerens CE, Havelaar AC, Joosse M et al (1999) A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases. Nat Genet 23:462–465. https://doi.org/10.1038/70585
Viana MB, Giugliani R, Leite VH, Barth ML, Lekhwani C, Slade CM et al (1990) Very low levels of high density lipoprotein cholesterol in four sibs of a family with non-neuropathic Niemann-Pick disease and sea-blue histiocytosis. J Med Genet 27:499–504. https://doi.org/10.1136/jmg.27.8.499
Waak J, Weber SS, Waldenmaier A, Gorner K, Alunni-Fabbroni M, Schell H et al (2009) Regulation of astrocyte inflammatory responses by the Parkinson's disease-associated gene DJ-1. FASEB J 23:2478–2489. https://doi.org/10.1096/fj.08-125153
Wakabayashi K, Takahashi H, Takeda S, Ohama E, Ikuta F (1989) Lewy bodies in the enteric nervous system in Parkinson's disease. Arch Histol Cytol 52(Suppl):191–194. https://doi.org/10.1679/aohc.52.suppl_191
Wang C, Telpoukhovskaia MA, Bahr BA, Chen X, Gan L (2018) Endo-lysosomal dysfunction: a converging mechanism in neurodegenerative diseases. Curr Opin Neurobiol 48:52–58. https://doi.org/10.1016/j.conb.2017.09.005
Wang D, Qian L, Xiong H, Liu J, Neckameyer WS, Oldham S et al (2006) Antioxidants protect PINK1-dependent dopaminergic neurons in Drosophila. Proc Natl Acad Sci U S A 103:13520–13525. https://doi.org/10.1073/pnas.0604661103
Wang W, Wang X, Fujioka H, Hoppel C, Whone AL, Caldwell MA et al (2016) Parkinson's disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat Med 22:54–63. https://doi.org/10.1038/nm.3983
Wang X, Petrie TG, Liu Y, Liu J, Fujioka H, Zhu X (2012) Parkinson's disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J Neurochem 121:830–839. https://doi.org/10.1111/j.1471-4159.2012.07734.x
Wang Y, Bao MH, Zhang QS, Li JM, Tang L (2015) Association of ATP6AP2 Gene Polymorphisms with Essential Hypertension in a South Chinese Han Population. Asian Pac J Cancer Prev 16:8017–8018. https://doi.org/10.7314/apjcp.2015.16.17.8017
Watari H, Blanchette-Mackie EJ, Dwyer NK, Glick JM, Patel S, Neufeld EB et al (1999) Niemann-Pick C1 protein: obligatory roles for N-terminal domains and lysosomal targeting in cholesterol mobilization. Proc Natl Acad Sci U S A 96:805–810. https://doi.org/10.1073/pnas.96.3.805
Wauters F, Cornelissen T, Imberechts D, Martin S, Koentjoro B, Sue C et al (2019) LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy:1–20. https://doi.org/10.1080/15548627.2019.1603548
Weng YH, Chou YH, Wu WS, Lin KJ, Chang HC, Yen TC et al (2007) PINK1 mutation in Taiwanese early-onset parkinsonism : clinical, genetic, and dopamine transporter studies. J Neurol 254:1347–1355. https://doi.org/10.1007/s00415-007-0534-7
Wenger DA, Rafi MA, Luzi P, Datto J, Costantino-Ceccarini E (2000) Krabbe disease: genetic aspects and progress toward therapy. Mol Genet Metab 70:1–9. https://doi.org/10.1006/mgme.2000.2990
Wicker G, Prill V, Brooks D, Gibson G, Hopwood J, von Figura K et al (1991) Mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). An intermediate clinical phenotype caused by substitution of valine for glycine at position 137 of arylsulfatase B. J Biol Chem 266:21386–21391
Williams ET, Chen X, Moore DJ (2017) VPS35, the Retromer Complex and Parkinson's Disease. J Parkinsons Dis 7:219–233. https://doi.org/10.3233/JPD-161020
Winder-Rhodes SE, Garcia-Reitbock P, Ban M, Evans JR, Jacques TS, Kemppinen A et al (2012) Genetic and pathological links between Parkinson's disease and the lysosomal disorder Sanfilippo syndrome. Mov Disord 27:312–315. https://doi.org/10.1002/mds.24029
Wong YC, Ysselstein D, Krainc D (2018) Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554:382–386. https://doi.org/10.1038/nature25486
Wu RM, Lin CH, Lin HI (2014) The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 82:283. https://doi.org/10.1212/WNL.0000000000000004
Xicoy H, Wieringa B, Martens GJM (2019) The Role of Lipids in Parkinson's Disease. Cells 8. https://doi.org/10.3390/cells8010027
Yalcin-Cakmakli G, Olgiati S, Quadri M, Breedveld GJ, Cortelli P, Bonifati V et al (2014) A new Turkish family with homozygous FBXO7 truncating mutation and juvenile atypical parkinsonism. Parkinsonism Relat Disord 20:1248–1252. https://doi.org/10.1016/j.parkreldis.2014.06.024
Yoshino H, Tomiyama H, Tachibana N, Ogaki K, Li Y, Funayama M et al (2010) Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology 75:1356–1361. https://doi.org/10.1212/WNL.0b013e3181f73649
Zaltieri M, Longhena F, Pizzi M, Missale C, Spano P, Bellucci A (2015) Mitochondrial Dysfunction and alpha-Synuclein Synaptic Pathology in Parkinson's Disease: Who's on First? Parkinsons Dis 2015:108029. https://doi.org/10.1155/2015/108029
Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I et al (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55:164–173. https://doi.org/10.1002/ana.10795
Zech M, Nubling G, Castrop F, Jochim A, Schulte EC, Mollenhauer B et al (2013) Niemann-Pick C disease gene mutations and age-related neurodegenerative disorders. PLoS One 8:e82879. https://doi.org/10.1371/journal.pone.0082879
Zeigler M, Meiner V, Newman JP, Steiner-Birmanns B, Bargal R, Sury V et al (2014) A novel SCARB2 mutation in progressive myoclonus epilepsy indicated by reduced beta-glucocerebrosidase activity. J Neurol Sci 339:210–213. https://doi.org/10.1016/j.jns.2014.01.022
Zervas M, Dobrenis K, Walkley SU (2001) Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J Neuropathol Exp Neurol 60:49–64. https://doi.org/10.1093/jnen/60.1.49
Zhang L, Shimoji M, Thomas B, Moore DJ, Yu SW, Marupudi NI et al (2005) Mitochondrial localization of the Parkinson's disease related protein DJ-1: implications for pathogenesis. Hum Mol Genet 14:2063–2073. https://doi.org/10.1093/hmg/ddi211
Zhao HG, Li HH, Bach G, Schmidtchen A, Neufeld EF (1996) The molecular basis of Sanfilippo syndrome type B. Proc Natl Acad Sci U S A 93:6101–6105. https://doi.org/10.1073/pnas.93.12.6101
Zhou ZD, Sathiyamoorthy S, Angeles DC, Tan EK (2016) Linking F-box protein 7 and parkin to neuronal degeneration in Parkinson's disease (PD). Mol Brain 9:41. https://doi.org/10.1186/s13041-016-0218-2
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S et al (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44:601–607. https://doi.org/10.1016/j.neuron.2004.11.005
Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H et al (2018) Reversible Conformational Conversion of alpha-Synuclein into Toxic Assemblies by Glucosylceramide. Neuron 97:92–107 e110. https://doi.org/10.1016/j.neuron.2017.12.012
Acknowledgements
We thank many of our colleagues for their helpful suggestions.
Funding
Research in the authors’ group is funded in part by the Flemish government initiated Methusalem excellence program; the Flanders Impulse Program on Networks for Dementia Research (VIND); and the Research Foundation Flanders (FWO); Belgium.
Author information
Authors and Affiliations
Contributions
SS reviewed the literature, prepared the manuscript, and designed and developed the figures. CVB reviewed the manuscript and gave valuable feedback. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Smolders, S., Van Broeckhoven, C. Genetic perspective on the synergistic connection between vesicular transport, lysosomal and mitochondrial pathways associated with Parkinson’s disease pathogenesis. acta neuropathol commun 8, 63 (2020). https://doi.org/10.1186/s40478-020-00935-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-020-00935-4