
Abstract
Background
Human adenoviruses (HAdVs) cause a wide range of diseases worldwide, including respiratory infections. Studies on HAdV molecular epidemiology are limited in Cameroon. The purpose of this study is to document the different types HAdV circulating in Cameroon in children with acute respiratory infections.
Methods
Nasopharyngeal swabs were collected from 811 children under 15 years from 2011 to 2014. The HAdV detection was assessed by semi-quantitative generic PCR r-gene®. The HAdV-positive samples were typed by amplification and sequencing of partial hexon gene and a real-time PCR. Demographic data were collected and analyzed. The infection and hospitalization risk factors were assessed thought the Chi-square test.
Results
A total of 137/220 HAdV-positive samples were amplified successfully. Six species of HAdV (Mastadenovirus A to F) were detected with B (108/220) and C (47/220) being the predominant strains. Hospitalization and age were significantly associated to HAdV-B and HAdV-C respectively. Phylogenetic analysis of HAdV-B3 virus (18) and B7 (5) shows a conserved and a significant temporal stability in relation to the reference sequence (99.1 to 100% of similarity).
Conclusion
This study reported HAdV species and types detected in children with acute respiratory infections in Cameroon between September 2011 and July 2014. These results support further evaluation of the spatio-temporal circulation pattern of HAdV species and types in Cameroon.
Similar content being viewed by others


Background
Human adenoviruses (HAdVs) are one of the common pathogens found in acute respiratory infections worldwide. About 6–50% of respiratory infections are caused by HAdV in children [1,2,3,4,5]. This virus is also one of the most common causes of morbidity and mortality among children, particularly in developing countries. A study by McMorrow and colleagues shows that HAdV was detected in 6% of 834 deaths in patients with severe respiratory infections recorded across eight African countries [6]. HAdV is consisting of unenveloped capsid with icosahedral symmetry and diameter between 70 and 90 nm. This box surrounds the genome that is associated to 4 polypeptides (V, VII, X, and μ) and consisting of linear double-stranded DNA, non-segmented and ranging in size from 26 kb to 48 according to the gender. HAdVs are members of the family Adenoviridae and gender Mastadenovirus. Traditionally these viruses were classified by neutralization reactions, hemagglutination and hydrolysis with restriction enzymes in 51 serotypes and multiple types. To date, they are classified into 7 groups named species Mastadenovirus A to G with a total of 54 types officially recognized by the International Committee on Taxonomy of Viruses (ICTV). This classification is based on the partial (Hexon, penton base and fiber) or complete viral genome [7, 8]. New types of HAdV (HAdV-55 to 69) have recently been identified based on whole genome analysis. According to their tissue tropism, the various HAdV types are responsible of a wide range of human diseases. HAdVs species B, C, and, E are involved in upper and lower acute respiratory tract infections. Effective vaccine against HAdV strains is available only for types E (4) and B (7) [9]. To date, there is no antiviral treatment approved against HAdV infections. However, significant progress has been reported on cidofovir, a nucleotide analog that inhibits viral DNA polymerase [10]. The epidemiology, clinical presentation, pathogenesis, vaccination and treatment of HAdV infections depend on the viral type implicated. Thus, typing HAdV is therefore crucial for patient care and limiting the spread of infection [11]. Although HAdV has been studied in great detail for more than half century, data on circulating types in Cameroon remain completely unknown. HAdVs could have a particular epidemiology in African countries. High prevalence of HAdV have also been reported in African countries such as South Africa (31.4%) [12], Senegal (50%) [5], and Kenya (29.6%) [3] among patients of all ages with severe or mild acute respiratory infections. Similarly, a previously study on HAdV among Cameroonian hospitalized children showed that HAdV was the predominantly detected respiratory viruses (27.3%) [13]. In this study we perform molecular typing of HAdV strains detected in Cameroon between 2011 and 2014 and we also find hospitalization risk factors related to the different HAdV types isolated in Cameroon.
Methods
Study design
This cross-sectional study was performed between September 2011 and July 2014. The samples were collected from outpatient or inpatient children at the pediatric service of the National Social Insurance hospital in Yaounde, Cameroon. Pediatric patients recruited met the case definition of WHO Influenza-Like illnesses (fever> 38 °C accompanied by cough or sore throat with symptoms dating the previous 5 days of inclusion). Overall 231 (28.4%) out of 811 tested samples were positive for HAdV (Fig. 1).

Flow chart of 811 patients with influenza-like illness enrolled in Cameroun, 2011–2014. *PCR-1: AdV/HBoV Respiratory Multi Well System r-geneTM (bioMérieux, Lyon, France), *PCR-2: PCR end point developed by Sriwanna et al. [15], *PCR3: In-house real-time PCR developed Leruez-Ville et al. and Watzinger et al. [16, 17]
Ethical approval
Ethical approval was obtained from the National Research Ethics Committee and the Ministry of Health of Cameroon for the main study (IMMI Project) associated with this work. This protocol was carried out in accordance with all the regulations establishing the protection of study participants. Informed written consent was acquired from the parents or guardians of all children involved in the study. A coding system was used for all participants to protect their confidential data. The caregivers of children were properly informed about the purpose of the study, the benefits and risks of participation in the study.
Sampling, extraction of nucleic acids, detection, and typing of HAdV
Nasopharyngeal swabs were screened to detect respiratory viruses using r-gene commercial duplex AdV/HBoV r-gene™ (Respiratory Multi Well System r-gene™, BioMerieux, Lyon, France) as described elsewhere [14]. A total of 220 HAdV positive samples were used for further characterization by PCR carried out on hexon gene. To this end the test was carried out using primers published by Sriwanna et al. in 2013 [15]. Briefly, 5 μL of extracted DNA were added to a PCR mixture containing: 5 μL of 10X PCR buffer, 1.5 μL of MgCl2 at 50 mM, 1 μL of dNTP at 10 mM, 2.5 μL of W1 solution at 1%, 0.5 μL of Taq DNA polymerase (Invitrogen, USA), 29.5 μL of distilled water, 2.5 μL of the forward primer ADV_F2 (5’-TTY GCN ACC ATG CCC AAC AC-3 ‘), and 2.5 μL of the reverse primer AdV_R2 (GYY TCR AYG GCC ATG CGG TG) to a final volume of 50 μL. The thermal profile used consisted of initial denaturation at 94 °C for 3 min; followed by 40 cycles of 94 °C for 45 s, 50 °C for 30 s and 72 °C for one minute 30 s; and a final extension at 72 °C for 10 min. The primers amplify a partial hexon gene of 956 bp. Amplified hexon sequences were sequenced bidirectionally using the same PCR primers. The species of not amplified samples (121 cases) after research by end point PCR of the Hexon gene have been identified as HAdV-A, B, C, D, E, and F at the virology laboratory of Caen University Hospital, France. Samples analyzed were those with threshold cycle values of r-gene detection technique lower than 34. This additional characterization was performed by real-time PCR techniques using primers described in 2004 by Leruez-Ville et al. and Watzinger et al. [16, 17]. Amplifications were carried out using Hot Star Taq® DNA Polymerase kit (Qiagen, USA). A volume of 5 μL of DNA extracts were added to 20 μL of PCR mixture containing 11 μL of water, 2.5 μL of 10X buffer, 2 μL of MgCl2 at 25 mM, 1 μl of dNTPs at 10 μM, and 1 μl of enzyme. The PCR program used was 50 °C for 2 min, 95 °C for 15 min, 40 cycles at 95 °C for 15 s, and 60 °C for 1 min.
Sequence analysis
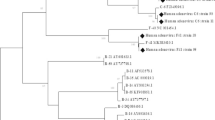
Our sequences were cleaned and assembled using the program EDITSEQ of the software Seqman™ II Lasergene (DNA, Madison, WI, USA). Alignments were performed using the BLAST program (http://www.ncbi.nlm.nih.gov/BLAST/) for the research on Genbank of nucleotide sequences similar to the raw sequences of this study. Phylogenetic analyzes were performed using the MEGA software version 6 [18]. The gene sequences obtained in this work were aligned with reference sequences of the 54 recognized types of HAdV by the Clustal W method. We generated the phylogenetic tree using the maximum-likelihood method and Tamura Nei with the site heterogeneity gamma and invariant sites, which was the best model for our sequences. The stability of the phylogenetic tree was assessed by a 1000 bootstrap repetitions. Evolutionary distances were calculated by the Kimura-2-parameter method. The Genbank accession numbers for all sequences used in this work are included in the phylogenetic tree (Fig. 2). The nucleotide sequences of the hexon obtained in this work have been deposited in the Genbank database with accession numbers KX452120 to KX452142.

Phylogenetic analysis of partial sequences of the hexon gene (560 nucleotides) of adenovirus type 3 and type 7 collected in Cameroon from 2011 to 2014. The nucleotide sequences were aligned using the Clustal W method. The tree topology was built according to the maximum-likelihood method under the Tamura-Nei substitution model with the site heterogeneity gamma and invariant sites using the MEGA software 6. The scale bars represent the frequency of nucleotide substitutions. The accuracy of the tree was assessed by 1000 bootstrap replicates. Only Bootstrap values> 70% are presented. Cameroonian sequences are indicated by black triangles (▲) and the other sequences are the prototypes of 54 HAdV formally recognized by the ICTV. Cameroonian samples are identified in the following format: CMR (Cameroon), the year of detection and the laboratory number. The number of identical Cameroonian sequences is indicated in parenthesis following the name of the strain. Genbank accession numbers of all sequences used are shown in the tree. The scale bar represents the number of nucleotide substitutions per site between close relatives
Data analysis
Descriptive statistics such as numbers, proportions and medians were calculated to summarize the sociodemographic data. The data were also presented in tables and figures. The Chi-square test was used to assess the significance of associations between categorical variables. Associations were considered significant for p-value less than 5%. The data were analyzed using the R software version 3.1.0.
Results
The gender and outpatient/inpatient ratio of recruited participants were well distributed with ratio of 1.1: 1 and 1.2: 1 respectively. All children included were under 15 years, with 63.4% having less than 2 years. The median age of these children was 19 months, IQR, (9–36) months.
HAdV genotyping
For this study, we considered 220 HAdV positive samples. After analysis by endpoint PCR of a partial region of the hexon gene and sequencing, only 23/220 (10.5%) were amplified. The samples amplified successfully were those with low Ct (15.4 ± 4.5 vs 29.9 ± 5.9, p < 0.001). All 23 amplified samples were positive for HAdV-B species. The phylogenetic analysis of the 23 sequences obtained reveals that 18 of these samples are HAdV-B3 type while the remaining 5 samples are HAdV-B7 type (Fig. 2). Cameroonian HAdV-B strains clustered on the phylogenetic tree with sequences from China, Taiwan and South Korea. Of the 197 samples non-amplified by endpoint PCR, 121 were analyzed by real time PCR for screening of HAdV-A to F species. HAdV was found in 114 of the 121 analyzed samples. Six HAdV species were identified and HAdV-B (85 cases) was the predominant strain followed by 47 HAdV-C, 6 HAdV-F, 5 HAdV-E, 1 HAdV-A and 1 HAdV-D.
HAdV-associated factors
HAdV-B was significantly more present in hospitalized patients (Table 1). The detection rate of HAdV-C was inversely proportional to the age of the children recruited in the study (p = 0.04). HAdV and the remaining HAdV species prevalence were not associated to age, gender, and hospitalization.
Temporal distribution HAdV-positive samples
The HAdV-B and C species were the most common and were found throughout the study period (Table 2). HAdV-E and F have noticed sporadically between 2012 and 2014. The only cases HAdV-D and A appeared in February and June 2014 respectively. Of the 23 strains that have been sequenced and analyzed, the HAdV-B3 was the predominant type and was found continuously throughout the investigation. In contrast, 5 strains of HAdV-B7 were found sporadically in 2011 and 2012.
Distribution of HAdV species mixed detections
HAdV species of 137/220 positive samples were obtained. Mixed HAdV interspecies detections were obtained in 30 cases (29 double infections and 1 triple infection). It is noteworthy that 22 double infections were the couple HAdV-B/HAdV-C and all HAdV-A, HAdV-D, and HAdV-F infections occur in co-detection (Table 3).
Phylogenetic analysis
A similarity rate of 99.9 to 100% was obtained when comparing the 861 nucleotides and deduced amino acid of the Cameroonian hexon gene sequences to reference strains B3 (NC_011203.1) and B7 (AC_000018.1). In comparison with the B7 reference strain, B7 sequences of this study have only two variable residues in all sequences from Cameroon. Eight variable positions were observed with respect to the Cameroonian B3 sequences and B3 prototype strain.
Discussion
This study reports the different HAdV species involved in respiratory infections in Yaounde, Cameroon between September 2011 and July 2014. To the best of the information available to date, this is the first study that performed a molecular characterization of HAdV strains isolated from patients with acute respiratory infections in Cameroon. Only 23 samples positives in generic quantitative PCR could be amplified by typing PCR on the hexon gene during this work. This low rate of amplification may mainly be due to the significantly low viral load of unamplified samples (mean Ct unamplified samples = 28.9 +/− 5.9 vs. 15.4 +/− 4, 5 for amplified samples, p < 0.001). As nested PCR is known to be more sensitive than PCR, it would have been possible to amplify and sequencing a larger number of positive samples if nested PCR had been used. Out of the 137 HAdV positive samples that were characterized, 78.5% (108) were virus HAdV-B species and 34.8% (47) HAdV-C. In fact, these species represent the most common species identified worldwide in acute respiratory infections [15, 19,20,21,22,23,24,25,26,27]. In contrast to this study, where HAdV-B was the predominantly species, two other African studies conducted in Egypt and Senegal have showed the predominance of the HAdV-C species [22, 26]. This could be partially explained by the inclusion of only patients with ILI in these studies, contrary to this work which also recruited patients with severe acute respiratory infections. In fact, the HAdV-B species was significantly predominant in hospitalized patients during this study (p < 0.001). Another study in Chile from 1988 to 1996 had also shown that HAdV-B infections were more severe than HAdV-C infections [27]. Previous studies have shown that HAdV species A (31) and F (40 and 41) were commonly found in the gastrointestinal tract [28, 29]. Similarly, HAdV-D (8, 19 and 37) are found in ocular system [29,30,31,32]. Similar to other authors [19, 33], we detected these species HAdV in respiratory samples, showing that their tropism can include respiratory tract. Unlike previous studies [22, 24], during this work 21.9% (30/137) of interspecies HAdV mixed infections were recorded. Nevertheless, this interspecies coinfection frequency remains similar to that (19%) recorded by Wang et al., in 2013 in Taiwan [25]. The comparison of the partial sequence of the hexon obtained for the Cameroonian HAdV-7 and HAdV-3 types and the reference strains reveals a very low rate of divergence. This result confirms the high stability of the HAdV genome also reported in previous studies [34]. The principal limitation of the present study is the low rate of amplification of HAdV-positive samples by end point PCR.
Conclusion
The various HAdV species involved in acute respiratory infections in Cameroon are showed in this study. HAdV-B and C were the predominant species and HAdV-B was associated to hospitalization. These results will form the basis for the implementation of the work to determine the burden of HAdV infection in Cameron.
Abbreviations
- HAdV:
-
Human adenovirus
References
El Kholy AA, Mostafa NA, Ali AA, El-Sherbini SA, Ismail RI, Magdy RI, et al. Risk factors of prolonged hospital stay in children with viral severe acute respiratory infections. J Infect Dev Ctries. 2014;8:1285–93.
Lekana-Douki SE, Nkoghe D, Drosten C, Ngoungou EB, Drexler JF, Leroy EM. Viral etiology and seasonality of influenza-like illness in Gabon, march 2010 to June 2011. BMC Infect Dis. 2014;14:373.
Breiman RF, Cosmas L, Njenga M, Williamson J, Mott JA, Katz MA, et al. Severe acute respiratory infection in children in a densely populated urban slum in Kenya, 2007-2011. BMC Infect Dis. 2015;25:15–95.
Hindiyeh MY, Keller N, Mandelboim M, Ram D, Rubinov J, Regev L, et al. High rate of human Bocavirus and adenovirus coinfection in hospitalized Israeli children. J Clin Microbiol. 2008;46:334–7.
Assane D, Makhtar C, Abdoulaye D, Amary F, Djibril B, Amadou D, et al. Viral and bacterial etiologies of acute respiratory infections among children under 5 years in Senegal. Microbiol Insights. 2018;1178636118758651:11.
McMorrow ML, Wemakoy EO, Tshilobo JK, Emukule GO, Mott JA, Njuguna H, et al. Severe acute respiratory illness deaths in sub-Saharan Africa and the role of influenza: a case series from 8 countries. J Infect Dis. 2015;212:853–60.
Walsh MP, Seto J, Jones MS, Chodosh J, Xu W, Seto D. Computational analysis identifies human adenovirus type 55 as a re-emergent acute respiratory disease pathogen. J Clin Microbiol. 2010;48:991–3.
Singh G, Robinson CM, Dehghan S, Jones MS, Dyer DW, Seto D, et al. Homologous recombination in E3 genes of human adenovirus species D. J Virol. 2013;87:12481–8.
Radin JM, Hawksworth AW, Blair PJ, Faix DJ, Raman R, Russell KL, et al. Dramatic decline of respiratory illness among US military recruits after the renewed use of adenovirus vaccines. Clin Infect Dis. 2014;59:962–8.
Wold WS, Toth K. New drug on the horizon for treating adenovirus. Expert Opin Pharmacother. 2015;16:2095–9.
Ghanaiem H, Averbuch D, Koplewitz BZ, Yatsiv I, Braun J, Dehtyar N, et al. An outbreak of adenovirus type 7 in a residential Facility for Severely Disabled Children. Pediatr Infect Dis J. 2011;30:948–52.
Annamalay AA, Abbott S, Sikazwe C, Khoo S-K, Bizzintino J, Zhang G, et al. Respiratory viruses in young south African children with acute lower respiratory infections and interactions with HIV. J Clin Virol. 2016;81:58–63.
Kenmoe S, Tchendjou P, Vernet M, Moyo-Tetang S, Mossus T, Njankouo-Ripa M, et al. Viral etiology of severe acute respiratory infections in hospitalized children in Cameroon, 2011–2013. Influenza Other Respir Viruses. 2016;10:386–93.
Kenmoe S, Vernet M-A, Miszczak F, Dina J, Schoenhals M, Beng VP, et al. Genetic diversity of human respiratory syncytial virus isolated among children with acute respiratory infections in southern Cameroon during three consecutive epidemic seasons, 2011–2013. Trop Med Health. 2018;46:7.
Sriwanna P, Chieochansin T, Vuthitanachot C, Vuthitanachot V, Theamboonlers A, Poovorawan Y. Molecular characterization of human adenovirus infection in Thailand, 2009–2012. Virol J. 2013;10:193.
Leruez-Ville M, Minard V, Lacaille F, Buzyn A, Abachin E, Blanche S, et al. Real-time blood plasma polymerase chain reaction for Management of Disseminated Adenovirus Infection. Clin Infect Dis. 2004;38:45–52.
Watzinger F, Suda M, Preuner S, Baumgartinger R, Ebner K, Baskova L, et al. Real-time quantitative PCR assays for detection and monitoring of pathogenic human viruses in immunosuppressed pediatric patients. J Clin Microbiol. 2004;42:5189–98.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Abbas KZ, Lombos E, Duvvuri VR, Olsha R, Higgins RR, Gubbay JB. Temporal changes in respiratory adenovirus serotypes circulating in the greater Toronto area, Ontario, during December 2008 to April 2010. Virol J. 2013;10:15.
Ampuero JS, Ocaña V, Gómez J, Gamero ME, Garcia J, Halsey ES, et al. Adenovirus respiratory tract infections in Peru. PLoS One. 2012;7(10):e46898.
Chen M, Zhu Z, Huang F, Liu D, Zhang T, Ying D, et al. Adenoviruses associated with acute respiratory diseases reported in Beijing from 2011 to 2013. PLoS One. 2015;e0121375:10.
Demian PN, Horton KC, Kajon A, Siam R, Hasanin AMN, Elgohary Sheta A, et al. Molecular identification of adenoviruses associated with respiratory infection in Egypt from 2003 to 2010. BMC Infect Dis. 2014;14:50.
Li Y, Zhou W, Zhao Y, Wang Y, Xie Z, Lou Y, et al. Molecular typing and epidemiology profiles of human adenovirus infection among Paediatric patients with severe acute respiratory infection in China. PLoS One. 2015;e0123234:10.
Qurei L, Seto D, Salah Z, Azzeh M. A molecular epidemiology survey of respiratory adenoviruses circulating in children residing in southern Palestine. PLoS One. 2012;7(8):e42732.
Wang S-L, Chi C-Y, Kuo P-H, Tsai H-P, Wang S-M, Liu C-C, et al. High-incidence of human adenoviral co-infections in Taiwan. PLoS One. 2013;8(9):e75208.
Niang MN, Diop NS, Fall A, Kiori DE, Sarr FD, Sy S, et al. Respiratory viruses in patients with influenza-like illness in Senegal: focus on human respiratory adenoviruses. PLoS One. 2017;12(3):e0174287.
Larrañaga C, Kajon A, Villagra E, Avendaño LF. Adenovirus surveillance on children hospitalized for acute lower respiratory infections in Chile (1988-1996). J Med Virol. 2000;60:342–6.
Jones MS. Harrach B, Ganac RD, Gozum MMA, dela Cruz WP, Riedel B, et al. new adenovirus species found in a patient presenting with gastroenteritis. J Virol. 2007;81:5978–84.
Lynch JP, Fishbein M, Echavarria M. Adenovirus. Semin Respir Crit Care Med. 2011;32:494–511.
Ishiko H, Aoki K. Spread of epidemic Keratoconjunctivitis due to a novel serotype of human adenovirus in Japan. J Clin Microbiol. 2009;47:2678–9.
Robinson CM, Singh G, Henquell C, Walsh MP, Peigue-Lafeuille H, Seto D, et al. Computational analysis and identification of an emergent human adenovirus pathogen implicated in a respiratory fatality. Virology. 2011;409:141–7.
Walsh MP, Chintakuntlawar A, Robinson CM, Madisch I, Harrach B, Hudson NR, et al. Evidence of molecular evolution driven by recombination events influencing tropism in a novel human adenovirus that causes epidemic Keratoconjunctivitis. PLoS One. 2009;4(6):e5635.
Mandelboim M, Dror P, Azar R, Bromberg M, Mendelson E. Adenovirus infections in hospitalized patients in Israel: epidemiology and molecular characterization. J Clin Microbiol. 2011;49:597–601.
Mahadevan P, Seto J, Tibbetts C, Seto D. Natural variants of human adenovirus type 3 provide evidence for relative genome stability across time and geographic space. Virology. 2010;397:113–8.
Funding
This work was funded by the U.S. Department of Health and Human Services (DHHS) grant number 6 DESP060001–01-01 via the International Network of Pasteur Institutes and the ‘Institut de Microbiologie et de Maladies Infectieuses’ (IMMI) in France.
Availability of data and materials
The nucleotide sequences of this study were submitted to Genbank and registered with the accession numbers KX452120 to KX452142.
Author information
Authors and Affiliations
Contributions
N.J. designed the study; K.S. and V.M-A. carried out and interpreted experiments; K.S. conducted phylogenetic analyses. K.S. wrote the first draft of the paper; All authors reviewed the manuscript critically and approved of the final manuscript as submitted.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Samples were submitted to the “Centre Pasteur du Cameroon” as part of influenza surveillance in Cameroon and the IMMI (Institut de Microbiologie et de Maladies Infectieuses) project. Written informed consent was obtained from parents or guardians of all children enrolled in the study. The procedures of the original study were evaluated and approved by the National Ethics Committee and the Ministry of Public Health of Cameroon.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kenmoe, S., Vernet, MA., Le Goff, J. et al. Molecular characterization of human adenovirus associated with acute respiratory infections in Cameroon from 2011 to 2014. Virol J 15, 153 (2018). https://doi.org/10.1186/s12985-018-1064-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-018-1064-x