
Abstract
Background
Human adenoviruse (HAdV) is a major pathogen of paediatric respiratory tract infections (RTIs). Mutation or recombination of HAdV genes may cause changes in its pathogenicity and transmission. We described the epidemiology and genotypic diversity of HAdV in hospitalized children with RTIs in Beijing, China.
Methods
Nasopharyngeal aspirates were collected from hospitalized children with RTIs from April 2018 to March 2019. HAdVs were detected by a quantitative real-time PCR, and the hexon gene was used for phylogenetic analysis.
Results
Among 1572 samples, 90 (5.72%) were HAdV-positive. The HAdV detection rate was highest in November and July. Among HAdV-positive children, 61.11% (55/90) were co-infected with other respiratory viruses, the most common of which were human respiratory syncytial virus and human rhinovirus. The main diagnosis was bronchopneumonia, most patient have cough and fever. Children with a high viral load were more likely to have a high fever (P = 0.041) and elevated WBC count (P = 0.000). Of 55 HAdV-positive specimens, HAdV-B (63.64%), HAdV-C (27.27%), and HAdV-E (9.09%) were main epidemic species. Phylogenetic analysis indicated that hexon sequences of three samples were on the same branch with the recombinant HAdV strain (CBJ113), which was circulating in Beijing since 2016.
Conclusion
The HAdV-B3 and HAdV-B7 are the main epidemic strains in Beijing, and the recombinant HAdV-C strain CBJ113 has formed an epidemic trend.
Similar content being viewed by others


Background
Both well-known and emerging viruses affect human health by causing various diseases, and sometimes they even have a devastating impact on the entire society, such as the newly emerged human coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Adenoviruses (AdVs), members of the family Adenoviridae, are non-enveloped double-stranded DNA viruses, found widely in the biosphere. Since they were first discovered by Rowe et al. in 1953 [1], AdVs have been the focus of intense research. AdVs can infect various tissues and organs, sometimes with serious consequences, especially in children. The infectivity and cell entry mechanism of AdVs make them suitable for drug delivery, vaccination and gene therapy for many diseases including cancer [2]. Research on adenoviruses has greatly contributed to the fields of life sciences and medicine over the past decades.
Currently, about 110 human adenovirus (HAdV) genotypes are recognized; these are classified into seven species (A–G) in the genus Mastadenovirus based on their physical, chemical and biological properties [3]. HAdV direct or indirect transmission of occurs through throat, faeces, eyes or urine, depending on the virus type. Certain HAdV types are predominantly associated with specific pathologies, such as acute respiratory outbreaks (HAdV-B/C/E) [4, 5], epidemic keratoconjunctivitis (HAdV-D) [6, 7], gastroenteritis, and/or acute hemorrhagic cystitis (HAdV-F/G) [8, 9]. The major disease-associated HAdV genotypes detected in various countries and regions differ and change over time [10]. Although most HAdVs cause only mild symptoms, some can cause severe infections, most of which occur in children, the elderly, and people with severely compromised immune systems [11,12,13]. However, there have also been reported cases of severe pneumonia and death in adults with normal immunity caused by HAdV-55 [14]. HAdVs can cause outbreaks, which usually occur in crowded places, such as hospitals, nursing homes, military bases, schools, and swimming pools, and these have been reported in several countries [15,16,17,18,19]. HAdV-B (HADV-3, -7, -14, and -55) cause outbreaks of respiratory-related diseases [4, 16, 20, 21], whereas HAdV-D (HAdV-8, -19, -37, -53, and -54) cause outbreaks of epidemic keratoconjunctivitis [18, 22, 23].
HAdV-G is a novel species that has been typed and named by using whole genome sequencing and phylogenetics rather than by applying traditional serology. At present, the specific and frequently mutated hexon gene of HAdV has been widely used in the molecular diagnosis and genotyping of this virus. Owing to the frequent recombination of HAdVs, as exemplified by HAdV-85/89 in Japan, HAdV-D56 in France, and the HAdV-55 and CBJ113 strains in China [15, 24,25,26,27,28], whole-genome sequencing remains the gold standard for proper classification of HAdVs [28]. Among them, HAdV-55, reconstituted from HAdV-B11 and HAdV-B14, has repeatedly caused outbreaks in densely populated areas such as schools and the military in China [15, 16]. The purpose of this study was to evaluate the epidemiological, clinical, and molecular characteristics of HAdV infections occurring among hospitalized children with respiratory tract infections (RTIs) in Beijing Friendship Hospital in China from April 2018–March 2019. In addition, this work explored the relationship between HAdV infection and RTI symptoms to provide information for the control and prevention of HAdV infection in China.
Materials and methods
Patient specimens
The 1572 nasopharyngeal aspirate (NPA) samples used in this study were collected from hospitalized children (aged < 14 years) with RTIs at Beijing Friendship Hospital during the period from April 2018–March 2019. Informed consent was received from the parents or guardians of the children enrolled in the study. An RTI was defined as an illness that presented during the previous week with at least two of the following clinical manifestations: fever, cough, nasal obstruction, expectoration, sneeze and dyspnoea. Patients who were diagnosed with pneumonia by chest radiography were also included in the study, even if they did not show the clinical features described above [29]. The collected samples were stored in virus preservation solution (1640 medium with 2.5 mg/mL Bovine Serum Albumin, 25 µg/mL amphotericin B and 1% Penicillin–Streptomycin Solution), transported to the laboratory on ice, and stored at − 80 °C until further processing. The clinical data were collected and sorted out from the hospital database.
Detection of HAdVs and other common respiratory viruses
For molecular detection, total viral nucleic acid was extracted from 200 µL of each clinical NPA specimen by using the QIAamp MinElute Kit (Qiagen, Germany) in accordance with the manufacturer’s instructions. HAdV detection was performed by using a quantitative real-time polymerase chain reaction (qPCR) assay targeting the highly conserved 132-bp region of the HAdV hexon gene, as previously decribed [30]. TaqMan Universal PCR Master Mix (Applied Biosystems, USA) was used to amplify HAdV hexon DNA with specific primers (Forward:5′-GCCCCAGTGGTCTTACATGCACATC-3′; Reverse: 5′-GCCACGGTGGGGTTTCTAAACTT-3′) and probe (5′-FAM-TGCACCAGACCCGGGCTCAGGTACTCCGA-3′-TAMRA); qPCR was performed using the Mx3005P qPCR System (Agilent Stratagene, USA). Samples with a cycle threshold (CT) of < 38 were retested with qPCR to confirm their classification as positive samples. The positive samples were quantified by applying a standard curve, as described previously [31].
Detection of viral co-infection in HAdV-positive specimens
The HAdV-positive specimens were screened for simultaneous co-infection with 15 common respiratory viruses: influenza virus types A, B, and C (IFV A/B/C), parainfluenza virus types 1–4 (HPIV 1–4), human coronavirus HKU1/229E/OC43/NL63, human respiratory syncytial virus (HRSV), human rhinoviruses (HRV), human bocavirus (HBoV) and human metapneumovirus (HMPV). All viruses were detected by qPCR (Additional file 1: Table S1) [30, 32,33,34,35,36,37,38,39,40,41,42], RNA viruses were tested using AgPath-ID™ One-Step RT-PCR Kit (Ambion, USA), and DNA viruses were tested using TaqMan™ Gene Expression Master Mix (Thermo Fisher, USA) in accordance the corresponding manufacture’s protocols.
HAdV genotyping
Nested PCR targeting the hypervariable region of the HAdV hexon gene was employed for genotyping as previously described [31]. The outer primers used were forward 5′-GCCACCTTCTTCCCCATGGC-3′ and reverse 5′-GTAGCGTTGCCGGCCGAGAA-3′, and the internal primers were forward 5′-TTCCCCATGGCCCACAACAC-3′ and reverse 5′-GCCTCGATGACGCCGCGGTG-3′. Specimens that failed to be amplified were classified as untyped. Nested-PCR products were confirmed by sequencing, and a phylogenetic tree was constructed by applying the Maximum Likelihood (ML) method with MEGA 7.0 using 1000 bootstrap replicates. Reference HAdV strains (Additional file 1: Table S2) were selected based on the HAdV reference strain recommended by International Committee on Taxonomy of Viruses (ICTV) and also included CBJ113 strain (KR699642). Homology between sequences on the same evolutionary branch with CBJ113 was analyzed using BioEdit.
Statistical analysis
Data analysis was performed using SAS 9.4 software, and the significance of the difference in rates among categorical data was tested by chi-squared and Fisher’s exact tests. Wilcoxon’s test and independent-samples t-test were used to analyze continuous variables. Two-sided P-values < 0.05 were considered indicative of statistical significance.
Results
HAdV epidemiology
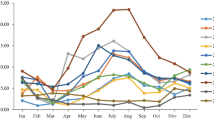
A total of 1572 hospitalized children were enrolled during the period from April 2018–March 2019, among which 900 (57.25%) were male and 672 (42.75%) were female (sex ratio: 1.34:1). The age range was from 1 day old to 14 years old with a median age of 3 years old (IQR: 1–5 years old). Among the 1572 samples, 90 (5.72%) tested positive for HAdV. As shown in Table 1, among the 90 HAdV-positive children, 60 (66.67%) were male and 30 (33.33%) were female (sex ratio: 2:1). No significant difference in HAdV-positivity was observed between the male and female patients (P = 0.063). Infected children were aged range from 1 month to 13 years old (Fig. 1). The detection rate of HAdV infection differed significantly among age groups (P = 0.008); the HAdV detection rate was highest in the group aged > 4 but ≤ 5 years old (12.22%, 11/90). Cases of HAdV infection were detected throughout the whole year, but the month with the highest detection rate was November 2018 (17.07%, 28/164), followed by July 2018 (10%, 13/130). The HAdV detection rates differed significantly between spring, summer, autumn and winter (3.31%, 5.72%, 9.38% and 4.23%, respectively, P = 0.001).

Age distribution (a) and seasonal distribution (b) of hospitalized children with RTIs who were infected with HAdV
Detection of viral co-infection in HAdV-positive specimens
Among the 90 HAdV-positive specimens, single infection samples accounted for 38.89% (35/90), among these patients (23 males and 12 females, male–female ratio of 1.92:1), the difference between the numbers of male and female patients was not statistically different (P = 0.878). Of the 55 HAdV-positive children who were co-infected with other respiratory viruses, the most common co-infecting viruses were HRSV and HRV (Table 2). The viral load of the 90 HAdV positive samples ranged from 17 to 12.8 × 106 copies/mL NPA. The log numbers of HAdV genome copies were 2.85 ± 1.45 and 2.63 ± 1.32 in the NPAs of children infected with HAdV only and those co-infected with HAdV and other respiratory virus, respectively; however there is no statistical difference in the viral load between HAdV mono- and co-infections (P = 0.061).
Clinical characteristics of HAdV infections
Among the 90 HAdV-positive children, the main diagnosis was bronchopneumonia (68.89%, 62/90), followed by mycoplasma pneumoniae pneumonia (8.89%, 8/90); only 3 cases were diagnosed with adenovirus pneumonia. The average duration hospitalization among these patients was 5.85 days. Eighty (88.89%) of the 90 children had an abnormal chest radiograph, and 48 (53.33%) of the 90 exhibited an elevated WBC count (> 10 × 109 cells/L). The main clinical features of the HAdV infections included cough (83.33%, 75/90) and fever (temperature ≥ 38 °C; 90%, 81/90). Five cases experienced convulsion as a symptom. A small number of children presented with gastrointestinal symptoms, such as vomiting (14.44%, 13/90) and diarrhoea (4.44%, 4/90). There were no significant differences in the clinical characteristics of HAdV infection between HAdV-positive patients with or without a viral co-infection (Table 3). Among children with an exclusive HAdV infection (no viral co-infection), the relationships between viral load and patient age, sex, disease duration, and body temperature were not statistically significant (Table 4), but children with a high viral load were more likely to have a high fever (P = 0.041) and an elevated WBC count(P = 0.000).
Phylogenetic analysis of HAdVs
The hexon gene (758-bp) was used for nested PCR amplification, and 55 HAdV-positive samples were amplified. Phylogenetic analysis revealed that 63.64% (35/55) of strains belonged to HAdV-B, including HAdV-B3 (68.57%, 24/35) and HAdV-B7 (31.43%, 11/35). Additionally, 27.27% (15/55) of the strains belonged to HAdV-C, including HAdV-C1 (40%, 6/15), HAdV-C2 (20%, 3/15), HAdV-C6 (13.33%, 2/15), HAdV-C5 (6.67%, 1/15) and CBJ113 (20%, 3/15). Furthermore, 5 cases were revealed to be HAdV-E, all of which were HAdV-E4 (Fig. 2). The genotyping of ADV1623/1630/2521 was consistent with that of CBJ113, and the homology between their hexon genes was 99.7%–99.8%.

Phylogenetic analysis of HAdV hexon gene (758-bp) compared with reference strains. The reference strain is marked with “●”
Discussion
The respiratory tract-related clinical symptoms caused by HAdV infections are similar to those caused by infection with IFA, HRSV and other respiratory pathogens. Consequently, correct diagnosis of HAdV infection is often difficult. In this study, qPCR and Sanger sequencing were used to analyse the phylogenetic sequence of the hexon gene, and the epidemiological characteristics and genotypic diversity of HAdVs in children hospitalized during the period from April 2018–March 2019 in Beijing, China were investigated. Of the 1572 collected specimens, 90 (5.73%) were positive for HAdV; this HAdV detection rate is very similar to that of the previous year in this hospital (5.64%) [31] and is also consistent with those reported from China and other countries (3.71%–35.5%) [31, 38,39,40,41,42]. The HAdV detection rate was 3.71% in Hebei Province, China. However, it was slightly higher among hospitalized children with RTIs in southern China; the HAdV detection rate in hospitalized children with RTIs during the period from 2009 to 2012 in Chongqing was 8.55%, and that in Hunan Province was 9.4%. It should be noted that different HAdV detection rates may be the result of differences in detection method, sample collection site, collection time and other factors. Thus, it is necessary to establish a unified and continuous epidemiological surveillance over a wider area.
The HAdV detection rate was not significantly affected by patient sex, but it was significantly affected by patients age (P = 0.008). The main age group affected by HAdV was children aged ≤ 5 years (73.3%, 66/90), specifically, the group of patients aged 3–5 years had the highest HAdV detection rate (10.11%, 27/267), whereas the group aged 2–3 years had the lowest (3.35%, 11/328). The reason for this difference remains to be determined.
Previous studies have shown that the HAdV detection rate is positively correlated with monthly average temperature, sunshine hours, and air temperature [39]. The number of HAdV infections in southern China reaches its peak during summer. In this study, the HAdV detection rate in Beijing had an obvious seasonal distribution difference (P = 0.001), and peaking in autumn (9.33%), which is consistent with a previous report by Duan et al. [43].
The clinical symptoms caused by HAdV infection in our patients were similar to those commonly caused by infections with other respiratory viruses, such as HRSV and IFV; their most common clinical symptoms and signs were fever and cough, and a few cases of HAdV also experienced gastrointestinal symptoms, such as vomit and nasal obstruction. The most common diagnosis among our HAdV-positive subjects was bronchopneumonia (68.89%, 62/90). The duration of hospital stay was generally less than 7 days, which is consistent with the results of previous studies. The co-infection of HAdVs and other respiratory viruses has been reported many times [39, 40]. In this study, the co-infection rate was 61.11% (55/90), and the viruses with the highest frequency of mixed infection were HRSV and HRV. No significant difference was observed in clinical symptoms and duration of hospitalization between the mono- and co-infections. The severity of HAdV infection is affected by many factors, including the patient age, immune status, and socioeconomic status. Although, some studies have shown that HAdV-7 may cause a more severe infection [44]; others have found that the HAdV type has no obvious influence on the severity of respiratory tract infection in children. Additionally, HAdV-infected patients with a long-lasting fever often experience more serious disease [40]. In agreement with previous work, this study found that there was no significant association between the HAdV genotype and disease severity (Additional file 1: Table S3), and only one child who was co-infected with HRSV had dyspnea. In the analysis of HAdV simple infection, the viral load in NPAs had no significant statistical association with patient age, sex or hospital stay duration, but children with a high viral load were more likely to have a high fever and elevated WBC count.
The most common species of HAdV worldwide, affecting the respiratory tracts of both adults and children, are HAdV-B, HAdV-C and HAdV-E, among which HAdV-B3 and HAdV-B7 are the main epidemic strains [44,45,46]. HAdV-55, which often causes outbreaks, is also detected at high rates in cases of adult respiratory tract infection [4, 6]. The prevalent HAdVs in China are mainly genotypes HAdV-2, -3 and -7. The dominant genotypes in northern China are HAdV-3 and -7, while those in southern China were HAdV-2 and 3 [43]. In this study, hexon gene sequencing and phylogenetic analysis were performed on 55 samples. The results show that HAdV species B and C were the most common species, accounting for 63.64% (35/55) and 29.10% (16/55) of HAdV cases, respectively. HAdV-B3 was the most common genotype (43.64%, 24/55), followed by HAdV-B7 (20.00%, 11/55), HAdV-C1 (10.91%, 6/55), and HAdV-E4 (9.09%, 9/55), which is consistent with other reports. HAdVs are prone to gene mutation and recombination [7, 25, 45, 47], and the CBJ113 strain isolated in Beijing in 2016 contained HAdV-C2, HAdV-C6, HAdV-C1, HAdV-C5 and HAdV-C57 sequences, which were recombined in several genes, including the hexon and fiber genes. Notably, three of the hexon sequences detected here were on the same branch as strain CBJ113, and they showed maximum homology with strain CBJ113. This study demonstrates that there are at least eight different HAdV genotypes circulating in Beijing; and that the HAdV species C strain CBJ113 has been prevalent in China for a long time. The hexon gene is commonly used for typing and is common in many molecular epidemiological studies of HAdV [48, 49]. However, because adenoviruses are prone to mutating and recombining, it is more accurate to use the whole genotype, which is a limitation of this study.
Our data allows CDC and health officials to understand the importance of adenovirus infection more deeply, thereby the government will give more attention and financial support to adenovirus research. In addition, our study can also provide clinicians more information of adenovirus infection, and patients can get the accurate diagnosis and better treatment for viral infection.
Conclusion
This study described the epidemiological, clinical, and molecular characteristics of HAdV infections occurring among children with RTIs in a Chinese tertiary hospital during the period April 2017–March 2018. Our results show the latest trends of HAdV epidemic genotypes in Beijing, China. Notably, the HAdV-C strain CBJ113 has formed an epidemic trend in Beijing; therefore it is necessary to establish a nationwide epidemiological surveillance program for adenovirus infection because the epidemic data from a single region are not necessarily representative. The detection of HAdV should be carried out across multiple regions in China. Additionally, it is not sufficient to use only a single gene fragment for HAdV typing; the whole HAdV genome should be sequenced to provide a more comprehensive understanding of the evolutionary characteristics of HAdVs and to provide theoretical support for HAdV prevention and control strategies.
Availability of data and materials
Condensed anonymized data are available from the corresponding author on reasonable request.
Abbreviations
- CT:
-
Cycle threshold
- HAdV:
-
Human adenoviruse
- HBoV:
-
Human bocavirus
- HCoV:
-
Human coronavirus
- HMPV:
-
Human metapneumovirus
- HPIVs:
-
Human parainfluenza viruses
- HRV:
-
Human rhinovirus
- ICTV:
-
International Committee on Taxonomy of Viruses
- IFV-A:
-
Influenza A
- IQR:
-
Interquartile Range
- MEGA:
-
Molecular evolutionary genetics analysis
- ML:
-
Maximum Likelihood method
- NPAs:
-
Nasopharyngeal aspirates
- qPCR:
-
Quantitative real-time polymerase chain reaction
- RSV:
-
Respiratory syncytial virus
- RTIs:
-
Respiratory tract infections
- SAS:
-
Statistics Analysis System
- WBC:
-
White Blood Count
References
Fukumi H, Nishikawa F, Kurimoto U, et al. Epidemiological studies of an outbreak of epidemic keratoconjunctivitis in Ogaki City and its vicinity, Gifu Prefecture in 1957. Jpn J Med Sci Biol. 1958;11(6):467–81.
Crenshaw BJ, Jones LB, Bell CR, Kumar S, Matthews QL. Perspective on adenoviruses: epidemiology, pathogenicity, and gene therapy. Biomedicines. 2019;7(3):61.
Greber UF. Adenoviruses—infection, pathogenesis and therapy. Febs Lett. 2020;594(12):1818–27.
Yi L, Zou L, Lu J, et al. A cluster of adenovirus type B55 infection in a neurosurgical inpatient department of a general hospital in Guangdong, China. Influenza Other Respir Viruses. 2017;11(4):328–36.
Tan D, Zhu H, Fu Y, et al. Severe community-acquired pneumonia caused by human adenovirus in immunocompetent adults: a multicenter case series. PLoS ONE. 2016;11(3):e0151199.
Akello JO, Kamgang R, Barbani MT, et al. Epidemiology of human adenoviruses: a 20-year retrospective observational study in hospitalized patients in Bern, Switzerland. Clin Epidemiol. 2020;12:353–66.
Hiroi S, Morikawa S, Takahashi K, Komano J, Kase T. Molecular epidemiology of human adenoviruses d associated with epidemic keratoconjunctivitis in Osaka, Japan, 2001–2010. Jpn J Infect Dis. 2013;66(5):436–8.
Kattareeya K, Pattara K, Hiroshi U, Niwat M. Enteric and non-enteric adenoviruses associated with acute gastroenteritis in pediatric patients in Thailand, 2011 to 2017. Plos One. 2019;14(8):e0220263.
Li P, Yang L, Guo J, et al. Circulation of HAdV-41 with diverse genome types and recombination in acute gastroenteritis among children in Shanghai. Sci Rep. 2017;7(1):3548.
Lynch JR, Kajon AE. Adenovirus: epidemiology, global spread of novel serotypes, and advances in treatment and prevention. Semin Respir Crit Care Med. 2016;37(4):586–602.
Westerberg S, Hagbom M, Rajan A, et al. Interaction of human enterochromaffin cells with human enteric adenovirus 41 leads to serotonin release and subsequent activation of enteric glia cells. J Virol. 2018;92(7):e00026-e118.
Kajon AE, Lamson DM, Bair CR, et al. Adenovirus type 4 respiratory infections among civilian adults, Northeastern United States, 2011–2015(1). Emerg Infect Dis. 2018;24(2):201–9.
Fang X, Xu M, Fang Q, et al. Real-time utilization of metagenomic sequencing in the diagnosis and treatment monitoring of an invasive adenovirus B55 infection and subsequent herpes simplex virus encephalitis in an immunocompetent young adult. Open Forum Infect Dis. 2018;5(6):ofy114.
Shu-Yan Z, Yan-Ping L, Dou-Dou H, et al. Fatal pneumonia cases caused by human adenovirus 55 in immunocompetent adults. Infect Dis. 2016;48(1):40–7.
Cheng Z, Yan Y, Jing S, et al. Comparative genomic analysis of re-emergent human adenovirus type 55 pathogens associated with adult severe community-acquired pneumonia reveals conserved genomes and capsid proteins. Front Microbiol. 2018;9:1180.
Wang W, Liu Y, Zhou Y, et al. Whole-genome analyses of human adenovirus type 55 emerged in Tibet, Sichuan and Yunnan in China, in 2016. PLoS ONE. 2017;12(12):e0189625.
Li D, Zhou JN, Li H, et al. An outbreak of epidemic keratoconjunctivitis caused by human adenovirus type 8 in primary school, southwest China. BMC Infect Dis. 2019;19(1):624.
Lamson BD, Kajon AE, Shudt M, et al. Molecular typing and whole genome next generation sequencing of human adenovirus 8 strains recovered from four 2012 outbreaks of keratoconjunctivitis in New York State. J Med Virol. 2018;90(9):1471–7.
Mei YF, Skog J, Lindman K, Wadell G. Comparative analysis of the genome organization of human adenovirus 11, a member of the human adenovirus species B, and the commonly used human adenovirus 5 vector, a member of species C. J Gen Virol. 2003;84(8):2061–71.
Zuiyuan G, Libo T, Shuang X, et al. Epidemiological analysis of an outbreak of an adenovirus type 7 infection in a boot camp in China. PLoS ONE. 2020;15(6):e0232948.
Tang L, Wang L, Tan X, Xu W. Adenovirus serotype 7 associated with a severe lower respiratory tract disease outbreak in infants in Shaanxi Province. China Virol J. 2011;8:23.
Peigert SM, Jagat R, Archit K, et al. Molecular epidemiology of circulating human adenovirus types in acute conjunctivitis cases in Chandigarh, North India. Indian J Med Microb. 2018;36(1):113–5.
Li J, Lu X, Jiang B, et al. Adenovirus-associated acute conjunctivitis in Beijing, China, 2011–2013. BMC Infect Dis. 2018;18(1):135.
Hashimoto S, Gonzalez G, Harada S, et al. Recombinant type Human mastadenovirus D85 associated with epidemic keratoconjunctivitis since 2015 in Japan. J Med Virol. 2018;90(5):881–9.
Takahashi K, Gonzalez G, Kobayashi M, et al. Pediatric infections by human mastadenovirus C types 2, 89, and a recombinant type detected in Japan between 2011 and 2018. Viruses. 2019;11(12):1131.
Robinson CM, Singh G, Henquell C, et al. Computational analysis and identification of an emergent human adenovirus pathogen implicated in a respiratory fatality. Virology. 2011;409(2):141–7.
Wang Y, Li Y, Lu R, et al. Phylogenetic evidence for intratypic recombinant events in a novel human adenovirus C that causes severe acute respiratory infection in children. Sci Rep. 2016;6:23014.
Singh G, Robinson CM, Dehghan S, et al. Overreliance on the hexon gene, leading to misclassification of human adenoviruses. J Virol. 2012;86(8):4693–5.
Zhang L, Liu W, Liu D, et al. Epidemiological and clinical features of human metapneumovirus in hospitalised paediatric patients with acute respiratory illness: a cross-sectional study in Southern China, from 2013 to 2016. BMJ Open. 2018;8(2):e019308.
Weinberg GA, Schnabel KC, Erdman DD, et al. Field evaluation of TaqMan Array Card (TAC) for the simultaneous detection of multiple respiratory viruses in children with acute respiratory infection. J Clin Virol. 2013;57(3):254–60.
Yao LH, Wang C, Wei TL, et al. Human adenovirus among hospitalized children with respiratory tract infections in Beijing, China, 2017–2018. Virol J. 2019;16(1):78.
Hammitt LL, Kazungu S, Welch S, et al. Added value of an oropharyngeal swab in detection of viruses in children hospitalized with lower respiratory tract infection. J Clin Microbiol. 2011;49(6):2318–20.
Esposito S, Bosis S, Niesters HG, et al. Impact of human coronavirus infections in otherwise healthy children who attended an emergency department. J Med Virol. 2006;78(12):1609–15.
Kodani M, Yang G, Conklin LM, et al. Application of TaqMan low-density arrays for simultaneous detection of multiple respiratory pathogens. J Clin Microbiol. 2011;49(6):2175–82.
Mcleish NJ, Witteveldt J, Clasper L, et al. Development and assay of RNA transcripts of enterovirus species A to D, rhinovirus species a to C, and human parechovirus: assessment of assay sensitivity and specificity of real-time screening and typing methods. J Clin Microbiol. 2012;50(9):2910–7.
Ligozzi M, Diani E, Lissandrini F, et al. Assessment of NS1 gene-specific real time quantitative TaqMan PCR for the detection of human bocavirus in respiratory samples. Mol Cell Probes. 2017;34:53–5.
Maertzdorf J, Wang CK, Brown JB, et al. Real-time reverse transcriptase PCR assay for detection of human metapneumoviruses from all known genetic lineages. J Clin Microbiol. 2004;42(3):981–6.
Lu QB, Tong YG, Wo Y, et al. Epidemiology of human adenovirus and molecular characterization of human adenovirus 55 in China, 2009–2012. Influenza Other Respir Viruses. 2014;8(3):302–8.
Xie L, Zhang B, Xiao N, et al. Epidemiology of human adenovirus infection in children hospitalized with lower respiratory tract infections in Hunan. China J Med Virol. 2019;91(3):392–400.
Zhao M, Guo Y, Qiu F, et al. Molecular and clinical characterization of human adenovirus associated with acute respiratory tract infection in hospitalized children. J Clin Virol. 2020;123:104254.
De Conto F, Conversano F, Medici MC, et al. Epidemiology of human respiratory viruses in children with acute respiratory tract infection in a 3-year hospital-based survey in Northern Italy. Diagn Microbiol Infect Dis. 2019;94(3):260–7.
Shafiei-Jandaghi N, Yavarian J, Malekshahi SS, et al. Identification of adenovirus species in Iranian pediatric population with severe acute respiratory infections. Future Virol. 2019;14(9):577–83.
Duan YL, Zhu Y, Xu BP, et al. Multicenter study of human adenovirus infection in pediatric community-acquired pneumonia in China. Zhonghua Er Ke Za Zhi 2019;57(1):27–32 (in Chinese).
Cai R, Mao N, Dai J, et al. Genetic variability of human adenovirus type 7 circulating in mainland China. PLoS ONE. 2020;15(4):e0232092.
Ezazul H, Urmila B, Tahmina M, Leela A, Kumar AA. Worldwide increased prevalence of human adenovirus type 3 (HAdV-3) respiratory infections is well correlated with heterogeneous hypervariable regions (HVRs) of hexon. PLoS ONE. 2018;13(3):e0194516.
Siew JX, Seah XFV, Chew YR, et al. Epidemiology of adenovirus infections and outcomes of cidofovir treatment in severely Ill children. Pediatr Infect Dis J. 2020;39(10):907–13.
Torres S, Chodosh J, Seto D, Jones MS. The revolution in viral genomics as exemplified by the bioinformatic analysis of human adenoviruses. Viruses. 2010;2(7):1367–81.
Hui D, Hongli X, Li H, et al. Molecular epidemiology and clinical features analysis of respiratory adenovirus infections reveals correlations between genotype, inflammatory biomarkers, and disease severity. Biomed Res Int. 2020;66:4357910.
Gu-Lung L, Chun-Yi L, Jong-Min C, et al. Molecular epidemiology and clinical features of adenovirus infection in Taiwanese children, 2014. J Microbiol Immunol Infect. 2019;52(2):215–24.
Acknowledgements
We thank the Department of Pediatrics, Beijing Friendship Hospital (Capital Medical University) for providing the NPA specimens. We also thank Katie Oakley, PhD, from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Funding
This work was supported by the Key Technologies R&D Program of the National Ministry of Science, China (2018ZX10305-410-003-002, 2018ZX10713-002). The funding body played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
YMH, CW and LSZ conceived and designed the experiments. YMH, CW, FLM and AJC performed the experiments. YMH, LHY and XYL analyzed the data; YMH, CW and QG contributed reagents/materials/analysis tools; YMH and LSZ wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The project was approved by the Ethical Committee of National Institute for Viral Disease Control and Prevention, China CDC, and the committee's reference number is IVDC2018-012.
Consent for publication
Written informed consent for specimen collection, testing and publication was obtained from all individuals (or their parents) in the study population.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
The tables of primers and probes used to detect co-infection respiratory viruses, reference strain of HAdV strains, clinical characteristics of children infected with different HAdV genotypes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Huang, Y., Wang, C., Ma, F. et al. Human adenoviruses in paediatric patients with respiratory tract infections in Beijing, China. Virol J 18, 191 (2021). https://doi.org/10.1186/s12985-021-01661-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-021-01661-6