
Abstract
Multiple system atrophy (MSA) is a unique and fatal α-synucleinopathy associated with oligodendroglial inclusions and secondary neurodegeneration affecting striatum, substantia nigra, pons, and cerebellum. The pathogenesis remains elusive; however, there is emerging evidence suggesting a prominent role of neuroinflammation. Here, we critically review the relationship between αS and microglial activation depending on its aggregation state and its role in neuroinflammation to explore the potential of TNFα inhibitors as a treatment strategy for MSA and other neurodegenerative diseases.
Similar content being viewed by others

Background
Multiple system atrophy (MSA) is a rare, rapidly progressing and fatal neurodegenerative disease of unknown etiology that is clinically characterized by a variable combination of parkinsonism, cerebellar impairment, and autonomic and motor dysfunction [1, 2]. Together with Parkinson disease (PD) and dementia with Lewy bodies (DLB), MSA belongs to the neurodegenerative group of α-synucleinopathies which are characterized by the deposition of aberrant α-synuclein (αS) in both glial cells and neurons. MSA is considered a synucleinopathy with specific glioneural degeneration involving striatonigral, olivopontocerebellar, autonomic, and peripheral nervous systems [3, 4]. The neuropathological hallmark of this unique proteinopathy is the deposition of aberrant fibrillary αS in glial cells, mainly oligodendroglia, forming glial cytoplasmic inclusions (GCI) [5], which may even represent a primary pathologic event [3, 6, 7]. Less frequent are neuronal cytoplasmic inclusions (NCI) and other cellular deposits. Inclusion pathology is accompanied by neuronal loss, widespread demyelination, and gliosis. Degeneration of multiple neuronal pathways over the course of the disease causes a multifaceted clinical picture of this multisystem disorder [2]. The etiology and pathogenesis of MSA are not fully understood, but converging evidence suggests the propagation of misfolded αS from diseased neurons to oligodendroglia and its spreading from cell to cell in a “prion-like” manner [8, 9], inducing oxidative stress (OS), proteosomal and mitochondrial dysfunction, dysregulation of myelin lipids, decreased neurotrophic factor activity, neuroinflammation, and energy failure that result in a multisystem involvement [3, 4, 10,11,12]. Recent experimental and human studies demonstrated that deposition of αS and other pathologic proteins induces neuroinflammation not only in MSA but also in other neurodegenerative diseases, e.g., PD and Alzheimer disease (AD) [13,14,15,16,17,18,19,20,21,22,23,24]. In MSA, αS has been shown to mediate formation of abnormal inclusion bodies and to induce neuroinflammation, which, interestingly, may also favor the formation of intracellular αS aggregates as a consequence of cytokine release and the shift to a pro-inflammatory environment [23]. αS may directly activate microglia, and recent studies have shown that only fibrillary αS is an important inducer of pro-inflammatory immune responses [25], associated with increased production of key pro-inflammatory cytokines, like tumor necrosis factor (TNF)-α and interleukin-1β (IL-1β) [26]. The association of activated microglial cells and GCI burden [27] suggests that pathologic αS triggers inflammatory response in α-synucleinopathies by affecting αS aggregation and provoking cell death [28]. This was corroborated by a number of experimental studies in vitro and in vivo [29,30,31]. These and other studies supported the notion that microglial activation may contribute to the progression of the neurodegenerative process in MSA and in other synucleinopathies via increased levels of reactive oxygen species (ROS) [20, 32, 33], like in other neurodegenerative diseases [31]. Although this mechanism is non-specific, it may be exploited for therapeutic and neuroprotective interventions.
TNFα in the central nervous system
TNFα, one of the key regulators in inflammation, belongs to the TNF ligand superfamily and is synthesized as a type II integral membrane protein occurring in a vast number of cell types. Within the central nervous system (CNS), microglia, astrocytes, and neurons are capable of synthesizing TNFα; however, activated microglia represent the main production site during neuroinflammatory processes [34, 35]. Following translation, it is synthesized as a transmembrane protein (tmTNFα) and cleavage by TNFα-converting enzyme (TACE) releases soluble TNFα (sTNFα). Both forms exert their functions on two receptors, TNFα receptor (TNFR) type I and II, with sTNFα preferentially binding to TNFR I, whereas tmTNFα has higher affinity towards TNFR II [36, 37]. The downstream signal-transduction cascades of TNFR I and TNFR II differ and imply the activation of numerous transcription factors including nuclear factor-kappa light chain enhancer of activated B cells (NF-κB) resulting in the regulation of various homeostatic and pathologic functions [38, 39]. In neurons, depending on the eventually activated transcription factor down the signaling pathway, TNFα drives either pro-apoptotic or pro-survival cell fate via TNFR I or TNFR II, respectively. Excessive release of TNFα, especially in a chronic manner as can be seen in many neurodegenerative diseases, leads to a shift towards receptor-independent neuronal cell death, directly through the activation of caspase 8 and 10 and indirectly by mediating glutamate excitotoxicity independent of receptor subtype. However, TNFR II also seems to exhibit neuroprotective properties as TNFR II has been shown to be critical for maintaining the oligodendrocyte progenitor pool in a cuprizone model of demyelination [40]. Furthermore, there is upcoming evidence on the role of TNFR II in oligodendroglial differentiation and thus remyelination capacity [41]. In addition, after knock-out of TNFR II, hippocampal neurons reveal increased sensitivity towards TNFα toxicity, whereas loss of TNFR I did not have this effect [42]. This raises the question whether inhibition of TACE and thereby largely depleting TNFR I activation while preserving tmTNFα triggered TNFR II signaling is to be considered in MSA, which will be discussed later on.
TNFα in neurodegenerative disorders
Neuroinflammation characterized by microglial activation with secretion of many pro-inflammatory cytokines, in particular IL-1β and TNFα, has been implicated as main effector of the functional consequence of neurotoxicity, resulting in mitochondrial dysfunction [43], thereby contributing to the progress of neurodegeneration [13, 17, 44,45,46,47]. The two cytokines are potent mediators of microglial functions and modulate the complex networks of interactions of microglial-secreted molecules. The role of neuroinflammation has been demonstrated repeatedly in animal models of PD [21, 29, 48, 49] and of MSA [49,50,51]. The major pro-inflammatory cytokine released by activated microglia is TNFα secreted by the brain resident microglia/macrophages in response to various stimuli. Microglial TNFα plays a major role in angiotensin-induced dopaminergic cell death. Microglial release of TNFα is mediated by activation of angiotensin type 1 receptors, NADPH (nicotinamide adenine dinucleotide phosphate)-oxidase, Rho-kinase, and NFK-β [52]. An early increase in TNF, which leads to protein thiol oxidation resulting in activation of ASK1 (apoptosis signal-regulating kinase 1)-p38 signaling, may be critical for neuroprotection in PD [53]. Increased levels of TNFα and IL-1β have been detected in the cerebrospinal fluid of PD patients [54]; in serum of patients with MSA [55]; in p t-mortem tissue of DLB, PD, and MSA [56, 57]; and in animal models of PD [45, 58,59,60,61,62]. It has been demonstrated to play a major role in neuroinflammation-related cell death in PD, AD, and other CNS disorders. Experimental studies indicated that TNFα is toxic for dopaminergic neurons in vivo [63] and in vitro [64]. Long-term expression of TNFα seems to be necessary to exert univocal toxic effects in the substantia nigra (SN) [65]. The increased frequency of TNF1031, a high producer allele of TNF, in Japanese MSA patients compared with controls [66] and increase of TNFα rs1799964 and IL-1β rs16944 polymorphisms in Chinese patients with MSA [67] suggest that they may represent genetic risk factors for MSA and that TNF may have a toxic effect in MSA. Interleukin-8, intercellular adhesion molecule-1, and TNFα polymorphisms significantly increased the risk of MSA [68].
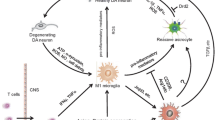
The importance of TNFα in the processes of development of PD is strengthened by animal models of PD, where elevated levels of TNFα are seen in a manner consistent with that of clinical PD [59, 61, 62]. Agents that interfere with TNFα synthesis and release seem to be protective in experimental models of PD [69, 70]. In other mouse models, conflicting findings regarding the protective effects of TNFα receptor deletion in toxin-induced PD have been reported [45, 64]. However, if lower levels of TNFα were expressed in the SN, a transient neuroprotective effect against 6-hydroxydopamine toxicity was observed [71]. On the other hand, when the synthesis of TNFα is unregulatory overproduced, this results in inappropriate cell death. TNFα and NF-κB expression and microglial activation in MSA [72] indicate an important role of TNFα in oligodendroglial cell death in this disease. High expression of TNFα in degenerating regions suggests that this potent pro-inflammatory cytokine is a mediator of neuronal injury. Furthermore, markers of inflammation such as serum T-lymphocyte-associated cytokine concentrations give evidence of immune mechanisms contributing to PD and MSA disease progression [73]. The pathobiological effects of TNFα in neurodegeneration and, in particular, in PD have been critically reviewed recently [17]. Figure 1 illustrates the effects of chronic TNFα-mediated neuroinflammation on oligodendroglia and neurons: TNFα drives oligodendroglial and neuronal cell death via activation of pro-apoptotic pathways and by increasing αS misfolding and aggregation.

TNFα in MSA pathogenesis. Misfolded αS leads to microglial activation and subsequent microglial release of pro-inflammatory cytokines. High levels of TNFα in vulnerable brain areas in MSA contribute to αS aggregation and establish a pro-apoptotic environment in chronic disease
Due to the possible cyclic nature of αS aggregation/release and gliosis in MSA, interventions that target neuroinflammation may have the potency to slow the progression of the disease and increase the quality of life [23]. Recent studies have approached α-synucleinopathies including MSA, by use of immunotherapy [74,75,76]. They have been shown to reduce αS co-localization in oligodendrocytes and astrocytes but increased microglial uptake, with decreasing demyelination, neuronal death, and motor deficit [76]. Other studies aimed at microglia as therapeutic target, e.g., treatment of the PLP-αS mouse model with minocycline reduced the density of activated microglia and inducible nitric oxidase synthase and toll-like receptor 4 immunoreactivity [51]. Recently, an inducer of heat shock proteins (HSP), carbenoxalone, was shown to decrease pro-inflammatory cytokines and OS in a rotenone model of PD [77]. Other treatments targeting neuroinflammation in glia and immune cells may be promising therapeutic strategies for MSA [23].
TNFα inhibition as treatment strategy
Therapeutic regimes that interfere with either the synthesis of TNFα or the downregulation of TNFα/receptor interactions may have a considerable benefit to patients with such conditions. The use of agents similar to thalidomide and its analogs will inhibit TNFα protein biosynthesis, which will circumvent classical receptor subtype agonistic effects on signaling pathways. Furthermore, thalidomide will display biological activity in the CNS as it is a relatively small molecule and can cross the blood-brain barrier (BBB). Thus, it offers a better strategy for treating TNFα overproduction-induced disorders than that of currently available large proteins that bind and lower soluble TNFα [22]. Agents that interfere with pro-inflammatory events connected with TNFα synthesis and release have been shown to be protective in animal models of PD. Thalidomide has been shown to afford partial protection to striatal dopaminergic neurons in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model [69, 70]. In transgenic mice, in which the gene for TNFα was functionally deleted, dopaminergic neurons in the striatum exhibited a greater degree of protection than wild-type littermates when challenged with MPTP [70]. However, in other mouse models employing gene targeting events on either one or both of the TNFα receptors (p55TNFR or p75TNFR), conflicting results regarding the protective effects of TNFα receptor deletion were reported [45, 64].
Thalidomide is widely known for its deleterious side effects since the late 50s/early 60s when birth defects were observed after pregnant women were prescribed thalidomide to treat morning sickness [78]. In addition, neuropathies may occur [79]. Meanwhile, however, thalidomide has been re-evaluated for immunomodulatory purposes as it enhances TNFα mRNA destabilization and degradation and, thereby, lowers its rate of synthesis and secretion [80, 81]. Furthermore, it is a co-stimulator of both CD8+ and CD4+ T cells [82], an inhibitor of angiogenesis [83] via its inhibitory actions on basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF), and an inhibitor of the transcription factor NF-κB, whereas TNFα-induced leucine-rich α-2-glycoprotein-1 (LRG1) promotes angiogenesis and mesenchymal stem cell migration, this inhibition being a potential therapeutic approach [84].
Inhibition of the biological effects of sTNFα by etanercept and infliximab displayed beneficial properties against rheumatoid arthritis and other peripheral inflammatory diseases. Unfortunately, these agents are large macromolecules that minimally pass the BBB and thus will preclude their utility in CNS neurodegenerative disorders. BMS-561392 reduces the amount of sTNFα by inhibiting TACE; however, clinical phase II trials for rheumatoid arthritis have been halted due to mild hepatotoxicity [85], which might be due to the accumulation of tmTNFα and TNFRII increase. This effect has been described in available anti-TNF treatment, but in this case, hepatic injury usually takes a self-limiting course. In addition, BMS-561392 has shown poor ability to penetrate the BBB and can therefore not be considered a suitable candidate substrate. In contrast, thalidomide analogs can readily and rapidly pass through the BBB and, if well tolerated in animal studies, may be of potential in a wide spectrum of CNS diseases. The mechanisms underlying thalidomide’s actions have been summarized by [22]. In brief, the agent comprises two conjoined heterocyclic moieties—a phthalimide and a glutarimide ring showing structural modifications [86,87,88,89]. Recent studies confirmed that the actions of thiothalidomide agents were identical to that of thalidomide. They caused a concentration-dependent reduction in luciferase activity—consistent with the mechanism destabilizing the mRNA of TNFα [80], and thereby reduce TNFα synthesis. Thus, thalidomide and its analogs are excellent candidate agents for use in anti-TNFα therapies in a variety of diseases associated with neuroinflammation, particularly since they act at the levels of TNFα synthesis rather than for scavenge released protein or inhibit its interaction at a receptor level [22]. Infusion of mesenchymal stem cells in a transgenic mouse model of MSA inducing a downregulation of cytokines involved in neuroinflammation suggested a potent effect on immunomodulation and neuroprotection [90].
A recent study investigated the therapeutic efficacy of combining an unconventional anti-inflammatory therapy (lenalidomide, a small thalidomide derivative with immunomodulatory activity and therapeutic effects in multiple myeloma [91,92,93], with inhibition of TNFα production [93, 94]) with an αS-reducing immunotherapeutic approach (CD5-D5 single chain antibody) in a novel transgenic mouse model of MSA pathogenesis. The combined treatment achieved better results than each method alone; it reduced astro- and microgliosis, αS levels, and partially improved deficits in MBP (myelin basic protein)-αS transgenic mice. These effects were associated with an activation of the Akt signaling pathway, which may mediate cytoprotective effects downstream TNFα [75]. Other recent studies demonstrated the neuroprotective and anti-inflammatory activities of allyl isothiocyanate (AITC), an aliphatic isothiocyanate derived from the precursor sinigrin present in vegetables of the Brassica family, on microglial cells through attenuation of JNK/NF-κB/TNFα signaling, which may have significance in neurodegeneration [95]. Combined active humoral and cellular immunization approaches, which are capable of triggering neuroprotective responses of regulatory T cells (Tregs), support the further development of multifunctional (vaccine) approaches for the treatment of synucleinopathies [96]. These results open the door for the design of more complex clinical trials in which a carefully planned combination of therapeutic approaches can complement each other to target multiple aspects of the pathobiology and pathogenesis of MSA and related neurodegenerative disorders.
Conclusions
TNFα-dependent neuroinflammation may play a key role in MSA pathogenesis, and its relevance has been underlined in various models of synucleinopathy. Targeting TNFα with readily available drugs may constitute a promising disease-modifying treatment in this hitherto incurable disease.
Abbreviations
- AD:
-
Alzheimer disease
- BBB:
-
Blood-brain barrier
- DLB:
-
Dementia with Lewy bodies
- GCI:
-
Glial cytoplasmic inclusions
- HSP:
-
Heat shock proteins
- IL-1β:
-
Interleukin-1β
- MPTP:
-
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MSA:
-
Multiple system atrophy
- NCI:
-
Neuronal cytoplasmic inclusions
- NF-κB:
-
Nuclear factor-kappa light chain enhancer of activated B cells
- OS:
-
Oxidative stress
- PD:
-
Parkinson’s disease
- ROS:
-
Reactive oxygen species
- SN:
-
Substantia nigra
- sTNFα:
-
Soluble TNFα
- TACE:
-
Tumor necrosis factor α-converting enzyme
- tmTNFα:
-
Transmembrane tumor necrosis factor
- TNFR:
-
TNFα receptor
- TNFα:
-
Tumor necrosis factor α
- αS:
-
α-Synuclein
References
Fanciulli A, Wenning GK. Multiple-system atrophy. N Engl J Med. 2015;372:1375–6.
Krismer F, Wenning GK. Multiple system atrophy: insights into a rare and debilitating movement disorder. Nat Rev Neurol. 2017;13:232–43.
Jellinger KA, Wenning GK. Multiple system atrophy: pathogenic mechanisms and biomarkers. J Neural Transm (Vienna). 2016;123:555–72.
Jellinger KA. Multiple system atrophy: an oligodendroglioneural synucleinopathy. J Alzheimers Dis. 2018;62:1141–79.
Trojanowski JQ, Revesz T. MSA NWGo: proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol. 2007;33:615–20.
Bleasel JM, Halliday GM, Kim WS. Animal modeling an oligodendrogliopathy--multiple system atrophy. Acta Neuropathol Commun. 2016;4:12.
Wenning GK, Stefanova N, Jellinger KA, Poewe W, Schlossmacher MG. Multiple system atrophy: a primary oligodendrogliopathy. Ann Neurol. 2008;64:239–46.
Dehay B, Vila M, Bezard E, Brundin P, Kordower JH. Alpha-synuclein propagation: new insights from animal models. Mov Disord. 2016;31:161–8.
Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB, Patel S, Oehler A, Lowe JK, Kravitz SN, et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A. 2015;112:E5308–17.
Jellinger KA. Neuropathology and pathogenesis of multiple system atrophy: an update. Curr Trends Neurol. 2015;9:45–54.
Jellinger KA. Multiple system atrophy - a synucleinopathy with specific glioneuronal degeneration. Austin J Clin Neurol. 2015;2:1071.
Ubhi K, Low P, Masliah E. Multiple system atrophy: a clinical and neuropathological perspective. Trends Neurosci. 2011;34:581–90.
Alam Q, Alam MZ, Mushtaq G, Damanhouri GA, Rasool M, Kamal MA, Haque A. Inflammatory process in Alzheimer’s and Parkinson’s diseases: central role of cytokines. Curr Pharm Des. 2016;22:541–8.
Ahmed Z, Asi YT, Sailer A, Lees AJ, Houlden H, Revesz T, Holton JL. The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol Appl Neurobiol. 2012;38:4–24.
Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8:382–97.
McGeer PL, McGeer EG. Glial reactions in Parkinson’s disease. Mov Disord. 2008;23:474–83.
Leal MC, Casabona JC, Puntel M, Pitossi FJ. Interleukin-1β and tumor necrosis factor-α: reliable targets for protective therapies in Parkinson’s Disease? Front Cell Neurosci. 2013;7:53.
Przedborski S. Inflammation and Parkinson’s disease pathogenesis. Mov Disord. 2010;25(Suppl 1):S55–7.
Mutoh T. Current understanding of immunopathogenesis of Parkinson’s disease. Austin J Clin Neurol. 2016;3(3):1097.
Sanchez-Guajardo V, Tentillier N, Romero-Ramos M. The relation between α-synuclein and microglia in Parkinson’s disease: recent developments. Neuroscience. 2015;302:47–58.
Teismann P, Schulz JB. Cellular pathology of Parkinson’s disease: astrocytes, microglia and inflammation. Cell Tissue Res. 2004;318:149–61.
Tweedie D, Sambamurti K, Greig NH. TNF-alpha inhibition as a treatment strategy for neurodegenerative disorders: new drug candidates and targets. Curr Alzheimer Res. 2007;4:378–85.
Vieira BD, Radford RA, Chung RS, Guillemin GJ, Pountney DL. Neuroinflammation in multiple system atrophy: response to and cause of α-synuclein aggregation. Front Cell Neurosci. 2015;9:437.
Lee HJ, Kim C, Lee SJ. Alpha-synuclein stimulation of astrocytes: potential role for neuroinflammation and neuroprotection. Oxidative Med Cell Longev. 2010;3:283–7.
Hoffmann A, Ettle B, Bruno A, Kulinich A, Hoffmann AC, von Wittgenstein J, Winkler J, Xiang W, Schlachetzki JC. Alpha-synuclein activates BV2 microglia dependent on its aggregation state. Biochem Biophys Res Commun. 2016;479:881–6.
Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol. 2013;39:3–18.
Ishizawa K, Komori T, Sasaki S, Arai N, Mizutani T, Hirose T. Microglial activation parallels system degeneration in multiple system atrophy. J Neuropathol Exp Neurol. 2004;63:43–52.
Stefanova N, Schanda K, Klimaschewski L, Poewe W, Wenning GK, Reindl M. Tumor necrosis factor-alpha-induced cell death in U373 cells overexpressing alpha-synuclein. J Neurosci Res. 2003;73:334–40.
Fellner L, Jellinger KA, Wenning GK, Stefanova N. Glial dysfunction in the pathogenesis of α-synucleinopathies: emerging concepts. Acta Neuropathol. 2011;121:675–93.
Stefanova N, Fellner L, Reindl M, Masliah E, Poewe W, Wenning GK. Toll-like receptor 4 promotes α-synuclein clearance and survival of nigral dopaminergic neurons. Am J Pathol. 2011;179:954–63.
Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, Wenning GK, Stefanova N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia. 2013;61:349–60.
Lim S, Chun Y, Lee JS, Lee SJ. Neuroinflammation in synucleinopathies. Brain Pathol. 2016;26:404–9.
Stefanova N, Georgievska B, Eriksson H, Poewe W, Wenning GK. Myeloperoxidase inhibition ameliorates multiple system atrophy-like degeneration in a transgenic mouse model. Neurotox Res. 2012;21:393–404.
Lieberman AP, Pitha PM, Shin HS, Shin ML. Production of tumor necrosis factor and other cytokines by astrocytes stimulated with lipopolysaccharide or a neurotropic virus. Proc Natl Acad Sci U S A. 1989;86:6348–52.
Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201.
Idriss HT, Naismith JH. TNF alpha and the TNF receptor superfamily: structure-function relationship(s). Microsc Res Tech. 2000;50:184–95.
MacEwan DJ. TNF receptor subtype signalling: differences and cellular consequences. Cell Signal. 2002;14:477–92.
Montgomery SL, Bowers WJ. Tumor necrosis factor-alpha and the roles it plays in homeostatic and degenerative processes within the central nervous system. J NeuroImmune Pharmacol. 2012;7:42–59.
Santello M, Volterra A. TNFα in synaptic function: switching gears. Trends Neurosci. 2012;35:638–47.
Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001;4:1116–22.
Madsen PM, Motti D, Karmally S, Szymkowski DE, Lambertsen KL, Bethea JR, Brambilla R. Oligodendroglial TNFR2 mediates membrane TNF-dependent repair in experimental autoimmune encephalomyelitis by promoting oligodendrocyte differentiation and remyelination. J Neurosci. 2016;36:5128–43.
Yang L, Lindholm K, Konishi Y, Li R, Shen Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J Neurosci. 2002;22:3025–32.
Russell AE, Doll DN, Sarkar SN, Simpkins JW. TNF-α and beyond: rapid mitochondrial dysfunction mediates TNF-α-induced neurotoxicity. J Clin Cell Immunol. 2016;7:467.
Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304:1–7.
Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O'Callaghan JP. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: implications for Parkinson’s disease. FASEB J. 2002;16:1474–6.
Streit WJ, Mrak RE, Griffin WS. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. 2004;1:14.
Smith JA, Das A, Ray SK, Banik NL. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. 2012;87:10–20.
Pott Godoy MC, Tarelli R, Ferrari CC, Sarchi MI, Pitossi FJ. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson’s disease. Brain. 2008;131:1880–94.
Stefanova N, Wenning GK. Animal models of multiple system atrophy. Clin Auton Res. 2015;25:9–17.
Stefanova N, Tison F, Reindl M, Poewe W, Wenning GK. Animal models of multiple system atrophy. Trends Neurosci. 2005;28:501–6.
Stefanova N, Reindl M, Neumann M, Kahle PJ, Poewe W, Wenning GK. Microglial activation mediates neurodegeneration related to oligodendroglial alpha-synucleinopathy: implications for multiple system atrophy. Mov Disord. 2007;22:2196–203.
Borrajo A, Rodriguez-Perez AI, Diaz-Ruiz C, Guerra MJ, Labandeira-Garcia JL. Microglial TNF-α mediates enhancement of dopaminergic degeneration by brain angiotensin. Glia. 2014;62:145–57.
Ray A, Sehgal N, Karunakaran S, Rangarajan G, Ravindranath V. MPTP activates ASK1-p38 MAPK signaling pathway through TNF-dependent Trx1 oxidation in parkinsonism mouse model. Free Radic Biol Med. 2015;87:312–25.
Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debré P, Agid Y, Dugas B, Hirsch EC. FcepsilonRII/CD23 is expressed in Parkinson’s disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-alpha in glial cells. J Neurosci. 1999;19:3440–7.
Kaufman E, Hall S, Surova Y, Widner H, Hansson O, Lindqvist D. Proinflammatory cytokines are elevated in serum of patients with multiple system atrophy. PLoS One. 2013;8:e62354.
Croisier E, Graeber MB. Glial degeneration and reactive gliosis in alpha-synucleinopathies: the emerging concept of primary gliodegeneration. Acta Neuropathol. 2006;112:517–30.
Salvesen L, Ullerup BH, Sunay FB, Brudek T, Løkkegaard A, Agander TK, Winge K, Pakkenberg B. Changes in total cell numbers of the basal ganglia in patients with multiple system atrophy - a stereological study. Neurobiol Dis. 2015;74:104–13.
Nagatsu T, Sawada M. Inflammatory process in Parkinson’s disease: role for cytokines. Curr Pharm Des. 2005;11:999–1016.
Hébert G, Arsaut J, Dantzer R, Demotes-Mainard J. Time-course of the expression of inflammatory cytokines and matrix metalloproteinases in the striatum and mesencephalon of mice injected with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, a dopaminergic neurotoxin. Neurosci Lett. 2003;349:191–5.
Ciesielska A, Joniec I, Przybyłkowski A, Gromadzka G, Kurkowska-Jastrzebska I, Członkowska A, Członkowski A. Dynamics of expression of the mRNA for cytokines and inducible nitric synthase in a murine model of the Parkinson’s disease. Acta Neurobiol Exp (Wars). 2003;63:117–26.
Nagatsu T, Mogi M, Ichinose H, Togari A. Changes in cytokines and neurotrophins in Parkinson’s disease. J Neural Transm Suppl. 2000:277–90. https://doi.org/10.1007/978-3-7091-6301-6_19.
Mladenović A, Perović M, Raicević N, Kanazir S, Rakić L, Ruzdijić S. 6-Hydroxydopamine increases the level of TNFalpha and bax mRNA in the striatum and induces apoptosis of dopaminergic neurons in hemiparkinsonian rats. Brain Res. 2004;996:237–45.
McGuire SO, Ling ZD, Lipton JW, Sortwell CE, Collier TJ, Carvey PM. Tumor necrosis factor alpha is toxic to embryonic mesencephalic dopamine neurons. Exp Neurol. 2001;169:219–30.
Leng A, Mura A, Feldon J, Ferger B. Tumor necrosis factor-alpha receptor ablation in a chronic MPTP mouse model of Parkinson’s disease. Neurosci Lett. 2005;375:107–11.
Carvey PM, Chen EY, Lipton JW, Tong CW, Chang QA, Ling ZD. Intra-parenchymal injection of tumor necrosis factor-alpha and interleukin 1-beta produces dopamine neuron loss in the rat. J Neural Transm (Vienna). 2005;112:601–12.
Nishimura M, Kuno S, Kaji R, Kawakami H. Influence of a tumor necrosis factor gene polymorphism in Japanese patients with multiple system atrophy. Neurosci Lett. 2005;374:218–21.
Zhou X, Wang C, Chen Z, Peng Y, Peng H, Hou X, Ye W, Qiu R, Xia K, Tang B, Jiang H. Association of TNF-α rs1799964 and IL-1β rs16944 polymorphisms with multiple system atrophy in Chinese Han population. Int J Neurosci. 2018;128:761–4.
Infante J, Llorca J, Berciano J, Combarros O. Interleukin-8, intercellular adhesion molecule-1 and tumour necrosis factor-alpha gene polymorphisms and the risk for multiple system atrophy. J Neurol Sci. 2005;228:11–3.
Boireau A, Bordier F, Dubédat P, Pény C, Impérato A. Thalidomide reduces MPTP-induced decrease in striatal dopamine levels in mice. Neurosci Lett. 1997;234:123–6.
Ferger B, Leng A, Mura A, Hengerer B, Feldon J. Genetic ablation of tumor necrosis factor-alpha (TNF-alpha) and pharmacological inhibition of TNF-synthesis attenuates MPTP toxicity in mouse striatum. J Neurochem. 2004;89:822–33.
Chertoff M, Di Paolo N, Schoeneberg A, Depino A, Ferrari C, Wurst W, Pfizenmaier K, Eisel U, Pitossi F. Neuroprotective and neurodegenerative effects of the chronic expression of tumor necrosis factor α in the nigrostriatal dopaminergic circuit of adult mice. Exp Neurol. 2011;227:237–51.
Probst-Cousin S, Rickert CH, Schmid KW, Gullotta F. Cell death mechanisms in multiple system atrophy. J Neuropathol Exp Neurol. 1998;57:814–21.
Csencsits-Smith K, Suescun J, Li K, Luo S, Bick DL, Schiess M. Serum lymphocyte-associated cytokine concentrations change more rapidly over time in multiple system atrophy compared to Parkinson disease. Neuroimmunomodulation. 2016;23:301–8.
Valera E, Monzio Compagnoni G, Masliah E. Review: novel treatment strategies targeting alpha-synuclein in multiple system atrophy as a model of synucleinopathy. Neuropathol Appl Neurobiol. 2016;42:95–106.
Valera E, Spencer B, Fields JA, Trinh I, Adame A, Mante M, Rockenstein E, Desplats P, Masliah E. Combination of alpha-synuclein immunotherapy with anti-inflammatory treatment in a transgenic mouse model of multiple system atrophy. Acta Neuropathol Commun. 2017;5:2.
Mandler M, Valera E, Rockenstein E, Mante M, Weninger H, Patrick C, Adame A, Schmidhuber S, Santic R, Schneeberger A, et al. Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol Neurodegener. 2015;10:10.
Thakur P, Nehru B. Inhibition of neuroinflammation and mitochondrial dysfunctions by carbenoxolone in the rotenone model of Parkinson’s disease. Mol Neurobiol. 2015;51:209–19.
Taussig HB. A study of the German outbreak of phocomelia. The thalidomide syndrome. JAMA. 1962;180:1106–14.
Fullerton PM, Kremer M. Neuropathy after intake of thalidomide (distaval). Br Med J. 1961;2:855–8.
Moreira AL, Sampaio EP, Zmuidzinas A, Frindt P, Smith KA, Kaplan G. Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation. J Exp Med. 1993;177:1675–80.
Niwayama S, Turk BE, Liu JO. Potent inhibition of tumor necrosis factor-alpha production by tetrafluorothalidomide and tetrafluorophthalimides. J Med Chem. 1996;39:3044–5.
Marriott JB, Clarke IA, Dredge K, Muller G, Stirling D, Dalgleish AG. Thalidomide and its analogues have distinct and opposing effects on TNF-alpha and TNFR2 during co-stimulation of both CD4(+) and CD8(+) T cells. Clin Exp Immunol. 2002;130:75–84.
D'Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A. 1994;91:4082–5.
Wang Y, Xu J, Zhang X, Wang C, Huang Y, Dai K. TNF-α-induced LRG1 promotes angiogenesis and mesenchymal stem cell migration in the subchondral bone during osteoarthritis. Cell Death Dis. 2017;8:e2715.
Moss ML, Sklair-Tavron L, Nudelman R. Drug insight: tumor necrosis factor-converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat Clin Pract Rheumatol. 2008;4:300–9.
Muller GW, Corral LG, Shire MG, Wang H, Moreira A, Kaplan G, Stirling DI. Structural modifications of thalidomide produce analogs with enhanced tumor necrosis factor inhibitory activity. J Med Chem. 1996;39:3238–40.
Muller GW, Chen R, Huang SY, Corral LG, Wong LM, Patterson RT, Chen Y, Kaplan G, Stirling DI. Amino-substituted thalidomide analogs: potent inhibitors of TNF-alpha production. Bioorg Med Chem Lett. 1999;9:1625–30.
Teubert U, Zwingenberger K, Wnendt S, Eger K. 5′-substituted thalidomide analogs as modulators of TNF-alpha. Arch Pharm (Weinheim). 1998;331:7–12.
Hashimoto Y. Structural development of biological response modifiers based on thalidomide. Bioorg Med Chem. 2002;10:461–79.
Stemberger S, Jamnig A, Stefanova N, Lepperdinger G, Reindl M, Wenning GK. Mesenchymal stem cells in a transgenic mouse model of multiple system atrophy: immunomodulation and neuroprotection. PLoS One. 2011;6:e19808.
Martiniani R, Di Loreto V, Di Sano C, Lombardo A, Liberati AM. Biological activity of lenalidomide and its underlying therapeutic effects in multiple myeloma. Adv Hematol. 2012;2012:842945.
Teo SK. Properties of thalidomide and its analogues: implications for anticancer therapy. AAPS J. 2005;7:E14–9.
Galustian C, Meyer B, Labarthe MC, Dredge K, Klaschka D, Henry J, Todryk S, Chen R, Muller G, Stirling D, et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol Immunother. 2009;58:1033–45.
Zhu YX, Kortuem KM, Stewart AK. Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leuk Lymphoma. 2013;54:683–7.
Subedi L, Venkatesan R, Kim SY. Neuroprotective and anti-inflammatory activities of allyl isothiocyanate through attenuation of JNK/NF-κB/TNF-α signaling. Int J Mol Sci. 2017;18:1423.
Rockenstein E, Ostroff G, Dikengil F, Rus F, Mante M, Florio J, Adame A, Trinh I, Kim C, Overk C, et al. Combined active humoral and cellular immunization approaches for the treatment of synucleinopathies. J Neurosci. 2018;38:1000–14.
Acknowledgements
Not applicable.
Funding
AN, GKW, KAJ, and TB received no funding and have no potential conflicts of interest that relate to the research covered in this review.
KAJ is funded by the Society for Support of Research in Experimental Neurology, Vienna, Austria.
GKW is employed by the Medical University of Innsbruck, Austria.
TB is employed by the Medical University of Vienna, Austria.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
AN, KAJ, and GKW wrote the primary draft. KAJ, TB, and GKW provided comments and proofread the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable as no patients/participants are involved in this review.
Competing interests
The authors declare that they have no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ndayisaba, A., Jellinger, K., Berger, T. et al. TNFα inhibitors as targets for protective therapies in MSA: a viewpoint. J Neuroinflammation 16, 80 (2019). https://doi.org/10.1186/s12974-019-1477-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-019-1477-5