
Abstract
Background
Ethiopia has set a goal for malaria elimination by 2030. Low parasite density infections may go undetected by conventional diagnostic methods (microscopy and rapid diagnostic tests) and their contribution to malaria transmission varies by transmission settings. This study quantified the burden of subpatent infections from samples collected from three regions of northwest Ethiopia.
Methods
Sub-samples of dried blood spots from the Ethiopian Malaria Indicator Survey 2015 (EMIS-2015) were tested and compared using microscopy, rapid diagnostic tests (RDTs), and nested polymerase chain reaction (nPCR) to determine the prevalence of subpatent infection. Paired seroprevalence results previously reported along with gender, age, and elevation of residence were explored as risk factors for Plasmodium infection.
Results
Of the 2608 samples collected, the highest positive rate for Plasmodium infection was found with nPCR 3.3% (95% CI 2.7–4.1) compared with RDT 2.8% (95% CI 2.2–3.5) and microscopy 1.2% (95% CI 0.8–1.7). Of the nPCR positive cases, Plasmodium falciparum accounted for 3.1% (95% CI 2.5–3.8), Plasmodium vivax 0.4% (95% CI 0.2–0.7), mixed P. falciparum and P. vivax 0.1% (95% CI 0.0–0.4), and mixed P. falciparum and Plasmodium malariae 0.1% (95% CI 0.0–0.3). nPCR detected an additional 30 samples that had not been detected by conventional methods. The majority of the nPCR positive cases (61% (53/87)) were from the Benishangul-Gumuz Region. Malaria seropositivity had significant association with nPCR positivity [adjusted OR 10.0 (95% CI 3.2–29.4), P < 0.001].
Conclusion
Using nPCR the detection rate of malaria parasites increased by nearly threefold over rates based on microscopy in samples collected during a national cross-sectional survey in 2015 in Ethiopia. Such subpatent infections might contribute to malaria transmission. In addition to strengthening routine surveillance systems, malaria programmes may need to consider low-density, subpatent infections in order to accelerate malaria elimination efforts.
Similar content being viewed by others
Background
The Global Technical Strategy for Malaria of the World Health Organization (WHO) calls for a world free of malaria [1]. To contribute to this global vision and encouraged by the substantial gains made in malaria control over the last two decades, Ethiopia has embarked on progressively eliminating malaria starting from low malaria transmission areas [2,3,4,5,6]. In the initial phase, 239 woredas (districts) were targeted for malaria elimination by the National Malaria Control and Elimination Programme of Ethiopia [5]. Preparations are underway to shift diagnosis and surveillance approaches from reducing malaria morbidity and mortality to detecting infections and measuring transmission in the selected woredas [2, 5].
Malaria elimination requires the detection and clearing of all Plasmodium infections. A relatively high prevalence of subpatent or low parasite density infections, that are often missed by conventional diagnostic methods (microscopy and rapid diagnostic tests) are being reported especially from low transmission settings [7,8,9,10,11,12,13,14]. Such asymptomatic, subpatent infections could be explained by acquired immunity in higher transmission settings [15]. Even in low transmission settings, asymptomatic and subpatent infections might play a role in transmission dynamics, hindering the progress of malaria elimination [14,15,16,17,18,19].
In Ethiopia, Plasmodium falciparum and Plasmodium vivax are the major reported malaria parasite species with rare reports of Plasmodium malariae and Plasmodium ovale. In 2017, out of a total confirmed malaria cases of 1,530,739, P. falciparum accounted for 1,059,847 cases and P. vivax for 470,892 cases [1, 2].
The current study assessed the magnitude of subpatent infections in northwestern Ethiopia, using samples collected in 2015 from a nationwide household survey of malaria endemic areas.
Methods
Study area
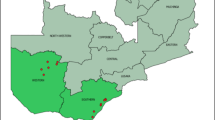
The study used Dried blood spot (DBS) samples collected as part of the National Ethiopian Malaria Indicator Survey conducted in 2015 (EMIS-2015) [20]. The survey was conducted during the peak of the malaria transmission season between September and December, 2015. Sample collection during EMIS-2015 was limited to areas below 2500 m elevation, with 85% of samples collected from areas below 2000 m elevation and the remaining 15% from areas between 2000 and 2500 m elevation. For the present study, samples were selected purposefully from the Amhara, Benishangul-Gumuz and Tigray Regions (Fig. 1). From the EMIS-2015 results, rapid diagnostic test (RDT) prevalence ranged from 10.4% in Benishangul-Gumuz, 1.9% in Tigray, to 1.1% in Amhara. A separate study [21] also found this geographical area to represent a range of malaria transmission from very low to moderate based on seroprevalence. Participants tested by malaria microscopy and RDT that provided DBS samples were selected from the EMIS 2015 samples repository at the Ethiopian Public Health Institute (EPHI) in Addis Ababa, Ethiopia.

Ethiopia and location of the three sample collection regions (shaded grey): Tigray, Amhara and Benishangul-Gumuz
Study population and sample collection
EMIS-2015 was a cross-sectional survey, which used a two-stage cluster sampling methodology [20]. Enumeration Areas (EA), the sampling unit, were selected proportional to population size and 25 households were selected by simple random sampling from each EA. Demographic, socioeconomic, malaria prevention, and malariometric data were also collected in the selected households. All children under 5 years of age in selected households and persons of all ages in every 4th household were eligible for biological sample collection and testing [20]. Whole blood from a single finger prick from consenting individuals (with or without fever) was collected for Plasmodium infection identification by RDT and microscopy, haemoglobin testing (Hemocue Hb 201+, Hemocue AB, Ängelholm, Sweden) and for collection of DBS samples. Whatman 903 (GE Healthcare, Pittsburgh, PA) filter paper cards were used for DBS collection. These were air dried, individually packed in a plastic bag together with a desiccant and stored at − 20 °C at EPHI before they were sent to the U.S. Centers for Disease Control and Prevention (CDC) in, Atlanta, Georgia, for processing.
Plasmodium infection exposure
Serology methods and results for these samples have been previously reported [21]. Briefly, DBS elutions were assayed using bead-based multiplex assays for IgG antibodies for six.
Plasmodium antigens: four human malaria species-specific merozoite surface protein-1 19 kD antigens (MSP-1) and Apical membrane antigen-1 (AMA-1) for P. falciparum and P. vivax.
Parasite identification
Rapid diagnostic tests
The CareStart™ Malaria HRP2/pLDH (Pf/PAN) Combo RDT were used to detect malaria in the field. CareStart™ tests for histidine-rich protein 2 (HRP2) to detect P. falciparum and pan-Plasmodium lactose dehydrogenase (LDH) for P. falciparum, P. vivax, P. ovale and P. malariae.
Microscopy
Thick and thin blood smears were made on the same slide, air-dried, and transported to EPHI. The slides were stained with 3% Giemsa for 10 min and screened for the presence of plasmodial infections. Microscopists were WHO certified and were blinded to the RDT and survey results. A slide was classified as negative if no Plasmodium asexual forms or gametocytes were found after viewing 100 fields. Any positive slide was confirmed by two other microscopists.
Polymerase chain reaction
The DBSs were shipped to CDC at ambient temperature. A total of 2533 RDT-negative and 2 samples without RDT results available from the three regions were pooled and analysed (Fig. 2). An additional 73 RDT-positive samples were analysed and confirmed by PCR individually.

Flow chart showing sampling and pooling analysis procedures
Two-step pooling was used to estimate the prevalence of malaria in the RDT-negative samples, as previously described by Zhou et al. [23]. A pilot was carried out to test for the effect of age (pooled by less than or equal to five and above 5 years of age) and different numbers of sample pooling (five or ten samples per pool), but no major differences by age or pooling size were found. Subsequently, individual samples from the same region were randomized into pools of ten samples.
DNA extraction
A 6-mm disk was punched out from each DBS card and ten disks pooled for DNA extraction. Pooled DBSs were extracted with QIAamp DNA Mini kit as per the manufacturer’s protocol (QIAGEN, Valencia, CA) with slight modification. Samples were left for an overnight incubation in ATL buffer plus Proteinase K to ensure proper lysis during pooling, and the DNA was then extracted following a normal extraction protocol. The purified DNA was tested using nested genus PCR (nPCR). For any pool with a positive PCR result, individual DBSs were extracted following the same protocol as for the pooled samples and species-specific PCR assays were performed.
nPCR detection for 18S ribosome RNA (rRNA)
Purified DNA templates were used for amplification of the 18S rRNA gene subunit using a modified method as previously described [23]. Briefly, the 25 μL PCR reaction contained 1× PCR Master Mix (Promega, Madison, WI) and 100 nM of each PCR primer. For each primary PCR reaction, 5 μL of DNA were used. The reactions were performed under the conditions of initial denaturation at 95 °C for 5 min, 25 cycles at 95 °C for 1 min, annealing at 58 °C for 2 min, and final extension at 72 °C for 2 min, followed by a final extension at 72 °C for 10 min. Species PCR was performed as per primary PCR except for use of 35 cycles and 2.5 μL primary PCR product per reaction. Genus PCR was performed as per species PCR except annealing temperature was set at 64 °C. The amplified DNA products were stained with GelRed (Biotium, Hayward, CA), separated using 2% agarose gel electrophoresis and visualized under UV illumination.
Data analysis
Data were analysed using Stata 13 (Stata Corp. 2015. Stata Statistical Software: Release 14. College Station, TX: Stata Corp LP, USA). Descriptive statistics with 95% confidence interval were estimated for demographic characteristics. Multiple logistic regression was used to assess the association of risk factors (gender, age, elevation, bed net use and seropositivity) with nPCR parasitaemia results. Percent relative difference and concordance for microscopy and RDTs were calculated against PCR results as the reference standard. Values were considered significant at P < 0.05. A Venn diagram was constructed using STATA to visualize the relationship amongst the four diagnostic methods: microscopy, RDTs, nPCR and serology. QGIS 2.18 was used to construct a map of the study area.
Results
Study population
Of the 2669 samples available from the three regions, 2535 samples were pooled for PCR analysis and 73 tested individually: 945 from Amhara, 668 from Benishangul-Gumuz and 922 from Tigray (Fig. 2). A total of 2608 samples had matched assay and survey data, comprising of 2533 RDT-negative, 73 RDT-positive and 2 without RDT results. About 53% (95% CI 50.4–54.2) of the samples tested were from females and 44% (95% CI 42.0–45.8) from children under 5 years of age (Table 1). The mean participant age from the samples was 16.1 years (95% CI 15.4–17.0) (range: 1–88 years). About 80% of the data were collected from areas below 2000 m in elevation. Of the 1354 samples with a paired seroprevalence result, almost half [51.3% (95% CI 48.6–53.9)] were seropositive of which 41.3% (95% CI 38.7–43.9) of the samples were seropositive for P. falciparum (MSP-1 or AMA-1 antigen responses) and 34.0% (95% CI 31.6–36.6) seropositive for P. vivax (MSP-1 or AMA-1). Additionally, 7.0% (95% CI 5.7–8.4) and 4.9% (95% CI 3.8–6.2) of participants were seropositive for P. malariae and P. ovale MSP-1 antigen responses, respectively. Overall, 59.6% (95% CI 57.3–61.9) of respondents providing samples reported having slept under a bed net in the previous night (Table 1).
Prevalence of malaria infection
The detection of Plasmodium infection varied by diagnostic methods: microscopy, RDT and nPCR (Table 2). Of the 2608 samples tested 2.8% (95% CI 2.2–3.5) were found to be positive for Plasmodium using RDTs, comprising of 1.9% (95% CI 1.5–2.5) due to P. falciparum (only HRP2 positive), 0.4% (95% CI 0.2–0.7) due to P. falciparum infection with or without other Plasmodium species (HRP2 and panLDH positive) and 0.5% (95% CI 0.3–0.5) were P. vivax, P. ovale or P. malariae infection (only panLDH positive). Using microscopy, 1.2% (95% CI 0.8–1.7) of the samples were found to be positive for Plasmodium, of which 1.0% (95% CI 0.6–1.4) were due to P. falciparum infection, 0.2% (95% CI 0.0–0.4) were due to P. vivax infection and 0.08% (95% CI 0.0–0.3) were due to mixed P. falciparum and P. vivax infection. RDT positivity was over twofold higher than microscopy positivity (Table 2). Figure 3 shows the relationships and overlap among serology, RDTs, microscopy and nPCR methods.

Venn diagram showing the relationships and overlap among serology (IgG MSP-1 or AMA-1), Rapid Diagnostic Tests (RDTs), Microscopy and nested Polymerase Chain Reaction (nPCR) methods, in 1522 samples with complete results collected from Tigray, Amhara and Benishangul-Gumuz Regions, Ethiopia as part of the Malaria Indicator Survey, 2015
Malaria DNA screening by nPCR detected 3.3% (95% CI 2.7–4.1) positivity for any malaria species. Of this, P. falciparum accounted for 3.1% (95% CI 2.5–3.8) of infection, P. vivax accounted for 0.4% (95% CI 0.2–0.7) of infection, mixed P. falciparum and P. vivax 0.1% (95% CI 0.0–0.4), and mixed P. falciparum and P. malariae 0.1% (95% CI 0.0–0.3). PCR detected 64.4% and 16.1% more positives compared to microscopy and RDT, respectively (P < 0.05). In addition, PCR detected two P. malariae cases mixed with P. falciparum cases. By RDT, these were both appropriately positive for HRP2 and panLDH antigens. Using microscopy, one of the P. malariae positive infection was missed and the other was identified as mixed infection. No P. ovale infection was identified by any diagnostic method (Table 2). Seropositive samples had a very high likelihood of being screened positive for malaria DNA [adjusted OR 10.0 (95% CI 3.2–29.4; P < 0.001)]. While living in lower elevation areas (< 2000 m), sex, age group, or bed net use were not associated with PCR positivity in the study samples (Table 3).
PCR identified 30 additional samples as positive that were negative by RDT (Table 4). Of these, 25 were due to P. falciparum infection, five were due to P. vivax infection, and two were due to a mixed infection of P. falciparum and P. malariae. Half of the additional positive samples (14 of 30), and mixed P. falciparum and P. malariae cases were identified from samples collected in Benishangul-Gumuz, a region of higher malaria transmission in Ethiopia.
The percent concordance of RDTs and microscopy with nPCR detection on sample positivity varied between the three regions (Table 5). Overall, 80.8% (59/73) of the samples identified as positive by RDTs and 83.9% by microscopy were confirmed positive by nPCR. The highest agreement with nPCR data for both RDTs (90.7%) and microscopy (86.7%) was observed in the Benishangul-Gumuz samples where P. falciparum is predominant. In the Amhara and Tigray regions agreement with nPCR diagnosis was lower at 64.3% and 68.8%, respectively.
Discussion
In this study, results of microscopy, RDT and nPCR detection methods were used to examine the prevalence of Plasmodium infection in malaria endemic settings in northwest Ethiopia. Nested PCR identified 30 additional positive malaria infections that were missed by RDTs in the field, and thus not provided treatment at the time of the survey. The prevalence reported by nPCR was 3.3%, 16.1% higher than that detected by RDTs and 64.4% higher than from microscopy. More than 60% of the nPCR positive cases were identified from Benishangul-Gumuz Region, which is one of the higher malaria transmission region in Ethiopia [5]. The region showed a similar high prevalence of malaria by microscopy and RDTs in this study and in a separate serology study of the same samples [21].
The study compared microscopy and RDT detection methods with a pooling nPCR approach that can detect parasite density as low as 0.1–10 parasites/µl of blood [22,23,24,25,26]. Close agreement was observed between both RDT and microscopy results compared to PCR with 80.8% and 83.9% concordance, respectively. Microscopy, considered the gold standard for malaria diagnosis [26], has a variable limit of detection (50–100 parasite/µl of blood) [26,27,28], depending on the skill of the technician and the reagents used [26,27,28]. As per the national malaria diagnosis and treatment guideline [29], Ethiopia uses microscopy in health centres and hospitals and RDTs in health posts (primary health care units). In the current study with slide preparation and RDTs conducted under field conditions where test requirements may not be optimal, high variability was observed in the results across the three methods. This discordance becomes disproportionately higher in these low transmission settings with very few positive samples. The strong association between seropositivity and PCR positivity suggests a role for using multiplex seroprevalence results to more rapidly and inexpensively identify hotspots or confirm lack of Plasmodium infection in areas reporting low incidence.
Although the lower RDT positivity is likely due to low-density infections, the possibility of hrp2/3 gene deleted P. falciparum infections has been raised in Ethiopia with alarming reports originating from Eritrea [29,30,31,32,33]. Although HRP2 and HRP3 deletions were reported recently from a study in Amhara [34], a study of larger geographic scope is currently ongoing in Ethiopia (Sindew Mekasha, EPHI, personal communication).
Several studies have reported asymptomatic Plasmodium infections in low transmission settings using PCR from a range of countries [7,8,9,10,11,12,13,14,15, 18, 22, 35, 36]. The terms asymptomatic, submicroscopic, and subpatent are often used interchangeably to describe a malaria infection that cannot be detected by conventional methods (microscopy and RDTs) but can be detected by more sensitive methods such as PCR. The prevalence of subpatent infection in low transmission setting has ranged from 0.003 to 44%, depending on the sensitivity of the tools used and the sample collection area [36]. Studies in Iran [37] and Sri Lanka [38] reported zero prevalence by the more sensitive methods, indicative of no local transmission and confirmation of malaria elimination. Golassa et al. [14] and Tadesse et al. [15] reported PCR prevalence ranging from 1.7 to 5.8% in southwest Ethiopia, comparable to the 3.3% prevalence reported in the current study.
Subpatent infections, despite the low parasite density, could be infectious to mosquitoes [15, 19, 39]. Studies in The Gambia [40], Thailand [35], Peru [41] and Ethiopia [17] showed even low-density, asymptomatic infections could be infectious to mosquitoes, which may pose a potential, unidentified reservoir for malaria transmission. A recent review [19] summarized that lower density of parasites were seen in low transmission compared to high transmission settings and argued that subpatent infections contribute to the infectious reservoir, could be long-lasting, and predictive of future periods of patent infections.
Several new tools are being developed with higher limits of parasite detection in field settings, such as, ultrasensitive RDTs and loop-mediated isothermal amplification (LAMP) [35, 42]. The current study used a sample pooling methodology that decreases cost and time and is applicable to screening large number of samples, particularly from low transmission settings [22, 43]. The low blood volume eluted from the DBSs in the current study may limit parasite detection as the volume of blood analysed is critical in determining its limit of detection [44, 45]. Although more sensitive molecular methods have been developed [46], the cost and feasibility of the tests in field settings could limit their wide-scale adoption and uptake in resource-limited countries.
Conclusion
The current study reports the presence of subpatent infection in the samples collected as part of a national cross-sectional malaria survey in 2015. Using nPCR, the detection of malaria parasites increased by nearly threefold over rates compared to microscopy. Subpatent infections could potentially be infective to mosquitoes contributing to ongoing malaria transmission. Malaria elimination efforts may need to consider low-density, subpatent infections in managing sources of infectious reservoirs or hotspots, in addition to strengthening routine surveillance and response systems.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AMA-1:
-
Apical membrane antigen-1
- CDC:
-
Centers for Disease Control and Prevention
- DBS:
-
Dried blood spot
- EA:
-
Enumeration Areas
- EMIS:
-
Ethiopian Malaria Indicator Survey
- EPHI:
-
Ethiopian Public Health Institute
- HRP2:
-
Histidine-rich protein 2
- MSP-1:
-
Merozoite surface protein-1
- nPCR:
-
Nested polymerase chain reaction
- pLDH:
-
Plasmodium lactose dehydrogenase
- RDTs:
-
Rapid diagnostic tests
- rRNA:
-
Ribosome RNA
- WHO:
-
World Health Organization
References
WHO. World malaria report 2018. Geneva: World Health Organization; 2018. http://www.who.int/malaria/publications/world-malaria-report-2018/report/en/. Accessed 18 Jan 2019.
Federal Ministry of Health Ethiopia (FMOH). National Malaria strategic plan: 2017–2020. Addis Ababa: Federal Ministry of Health Ethiopia (FMOH); 2017.
WHO. Global Technical Strategy for Malaria 2016–2030. Geneva: World Health Organization; 2019. http://www.who.int/malaria/areas/global_technical_strategy/en/. Accessed 26 Mar 2019.
Moonen B, Cohen JM, Snow RW, Slutsker L, Drakeley C, Smith DL, et al. Operational strategies to achieve and maintain malaria elimination. Lancet. 2010;376:1592–603.
Federal Ministry of Health Ethiopia (FMOH). National malaria elimination roadmap. Addis Ababa: National malaria prevention, control and elimination programme; Disease Prevention and Control Directorate; 2016.
United Nations Sustainable Development. From aspiration to action: what will it take to end malaria? Report by UN and Gates Foundation presents vision for eradicating malaria by 2040; 2015. https://www.un.org/sustainabledevelopment/blog/2015/09/report-by-un-and-gates-foundation-presents-vision-for-eradicating-malaria-by-2040/ Accessed 16 May 2019.
Zaw MT, Thant M, Hlaing TM, Aung NZ, Thu M, Phumchuea K, et al. Asymptomatic and sub-microscopic malaria infection in Kayah State, eastern Myanmar. Malar J. 2017;16:138.
Harris I, Sharrock WW, Bain LM, Gray K-A, Bobogare A, Boaz L, et al. A large proportion of asymptomatic Plasmodium infections with low and sub-microscopic parasite densities in the low transmission setting of Temotu. Malar J. 2010;9:254.
Zhou G, Yewhalaw D, Lo E, Zhong D, Wang X, Degefa T, et al. Analysis of asymptomatic and clinical malaria in urban and suburban settings of southwestern Ethiopia in the context of sustaining malaria control. Malar J. 2016;15:250.
Tsoka-Gwegweni JM, Okafor U. Asymptomatic malaria in refugees living in a non-endemic South African city. PLoS ONE. 2014;9:e107693.
Phommasone K, Adhikari B, Henriques G, Pongvongsa T, Phongmany P, von Seidlein L, et al. Asymptomatic Plasmodium infections in 18 villages of southern Savannakhet Province, Lao PDR (Laos). Malar J. 2019;15:296.
Vasquez Jimenez JM, Arevalo Herrera M, Henao Giraldo J, Molina Gomez K, Arce Plata M, et al. Consistent prevalence of asymptomatic infections in malaria endemic populations in Colombia over time. Malar J. 2016;15:70.
Elbadry MA, Al-Khedery B, Tagliamonte MS, Yowell CA, Raccurt CP, Existe A, et al. High prevalence of asymptomatic malaria infections: a cross-sectional study in rural areas in six departments in Haiti. Malar J. 2015;14:510.
Golassa L, Enweji N, Erko B, Aseffa A, Swedberg G. Detection of a substantial number of sub-microscopic Plasmodium falciparum infections by polymerase chain reaction: a potential threat to malaria control and diagnosis in Ethiopia. Malar J. 2013;12:352.
Tadesse FG, Pett H, Baidjoe A, Lanke K, Sutherland C, Hall T, et al. Submicroscopic carriage of Plasmodium falciparum and Plasmodium vivax in a low endemic area in Ethiopia where no parasitaemia was detected by microscopy or rapid diagnostic test. Malar J. 2015;14:303.
Wirth D, Alonso P. Malaria: Biology in the Era of Eradication. Latest advances in malaria research in free eBook by Cold Spring Harbor Laboratory Press. Cold Spring Harbor Laboratory; 2017 https://www.cshl.edu/latest-advances-malaria-research-free-ebook-cold-spring-harbor-laboratory-press/. Accessed 30 Jan 2019.
Tadesse FG, Slater HC, Chali W, Teelen K, Lanke K, Belachew M, et al. The relative Contribution of symptomatic and asymptomatic Plasmodium vivax and Plasmodium falciparum infections to the infectious reservoir in a low-endemic setting in Ethiopia. Clin Infect Dis. 2018;66:1883–91.
McCreesh P, Mumbengegwi D, Roberts K, Tambo M, Smith J, Whittemore B, et al. Subpatent malaria in a low transmission African setting: a cross-sectional study using rapid diagnostic testing (RDT) and loop-mediated isothermal amplification (LAMP) from Zambezi region, Namibia. Malar J. 2018;17:480.
Slater HC, Ross A, Felger I, Hofmann NE, Robinson L, Cook J, et al. The temporal dynamics and infectiousness of subpatent Plasmodium falciparum infections in relation to parasite density. Nat Commun. 2019;10:1433.
Ethiopian National Malaria indicator Survey 2015. Ethiopian Public Health Institute; 2016. https://www.ephi.gov.et/images/pictures/download2009/MIS-2015-Final-Report-December-_2016.pdf. Accessed 17 Apr 2018.
Assefa A, Ali Ahmed A, Deressa W, Sime H, Mohammed H, Kebede A, et al. Multiplex serology demonstrate cumulative prevalence and spatial distribution of malaria in Ethiopia. Malar J. 2019;18:246.
Zhou Z, Mitchell RM, Gutman J, Wiegand RE, Mwandama DA, Mathanga DP, et al. Pooled PCR testing strategy and prevalence estimation of submicroscopic infections using Bayesian latent class models in pregnant women receiving intermittent preventive treatment at Machinga District Hospital, Malawi, 2010. Malar J. 2014;13:509.
Turki H, Raeisi A, Malekzadeh K, Ghanbarnejad A, Zoghi S, Yeryan M, et al. Efficiency of nested-PCR in detecting asymptomatic cases toward malaria elimination program in an endemic area of Iran. Iran J Parasitol. 2015;10:39–45.
Snounou S, Balbir S. Nested PCR analysis of Plasmodium parasites. In: Doolan DL, editor. Nested PCR analysis of plasmodium parasites. Methods in molecular medicine, vol. 72. Totowa: Humana Press Inc; 2002. p. 189–203.
Snounou G, Viriyakosol S, Jarra W, Thaithong S, Brown KN. Identification of the four human malaria parasite species in field samples by the polymerase chain reaction and detection of a high prevalence of mixed infections. Mol Biochem Parasitol. 1993;58:283–92.
WHO. Guideline for the treatment of malaria. Geneva: World Health Organization; 2015.
Siahaan L. Laboratory diagnostics of malaria. IOP Conf Ser Earth Environ Sci. 2018;125:012090.
WHO. Basic malaria microscopy Part I Learner’s guide. Geneva: World Health Organization; 1991.
Federal Ministry of Health Ethiopia (FMOH). Malaria diagnosis and treatment guidelines for health workers in Ethiopia. Addis Ababa: Federal Ministry of Health Ethiopia; 2004.
Verma AK, Bharti PK, Das A. HRP-2 deletion: a hole in the ship of malaria elimination. Lancet Infect Dis. 2018;18:826–7.
Adu-Gyasi D, Asante KP, Amoako S, Amoako N, Ankrah L, Dosoo D, et al. Assessing the performance of only HRP2 and HRP2 with pLDH based rapid diagnostic tests for the diagnosis of malaria in middle Ghana, Africa. PLoS ONE. 2018; 13: e0203524. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6128572/. Accessed 18 Apr 2019.
Beshir KB, Sepúlveda N, Bharmal J, Robinson A, Mwanguzi J, Busula AO, et al. Plasmodium falciparum parasites with histidine-rich protein 2 (pfhrp2) and pfhrp3 gene deletions in two endemic regions of Kenya. Sci Rep. 2017;7:14718.
Berhane A, Anderson K, Mihreteab S, Gresty K, Rogier E, Mohamed S, et al. Major threat to malaria control programs by Plasmodium falciparum lacking histidine-rich protein 2, Eritrea. Emerging Infect Dis. 2018;24:462–70.
Getie S, Meda AL, Birhanie M, Abere A, Noedl H. High prevalence of Plasmodium falciparum histidine rich protein 2 and 3 gene deletions and their implications for malaria diagnosis in Ethiopia. 66th ASTMH; 2017. p. 97.
Hofmann N, Mwingira F, Shekalaghe S, Robinson LJ, Mueller I, Felger I. Ultra-sensitive detection of Plasmodium falciparum by amplification of multi-copy subtelomeric targets. PLoS Med. 2015;12:e1001788.
Okell LC, Bousema T, Griffin JT, Ouédraogo AL, Ghani AC, Drakeley CJ. Factors determining the occurrence of submicroscopic malaria infections and their relevance for control. Nat Commun. 2012;3:1237.
Zoghi S, Mehrizi AA, Raeisi A, Haghdoost AA, Turki H, Safari R, et al. Survey for asymptomatic malaria cases in low transmission settings of Iran under elimination programme. Malar J. 2012;11:126.
Fernando SD, Abeyasinghe RR, Galappaththy GNL, Rajapaksa LC. Absence of asymptomatic malaria infections in previously high endemic areas of Sri Lanka. Am J Trop Med Hyg. 2009;81:763–7.
Vallejo AF, García J, Amado-Garavito AB, Arévalo-Herrera M, Herrera S. Plasmodium vivax gametocyte infectivity in sub-microscopic infections. Malar J. 2016;17:13.
Okebe J, Bousema T, Affara M, DiTanna G, Eziefula AC, Jawara M, et al. The gametocytocidal efficacy of primaquine in malaria asymptomatic carriers treated with dihydroartemisinin-piperaquine in The Gambia (PRINOGAM): study protocol for a randomised controlled trial. Trials. 2015;16:70.
Waltmann A, Darcy AW, Harris I, Koepfli C, Lodo J, Vahi V, et al. High rates of asymptomatic, sub-microscopic Plasmodium vivax infection and disappearing Plasmodium falciparum malaria in an area of low transmission. PLoS Negl Trop Dis. 2015;9:e0003758.
Cook J, Aydin-Schmidt B, González IJ, Bell D, Edlund E, Nassor MH, et al. Loop-mediated isothermal amplification (LAMP) for point-of-care detection of asymptomatic low-density malaria parasite carriers in Zanzibar. Malar J. 2015;14:43.
Congpuong K, Saejeng A, Sug-Aram R, Aruncharus S, Darakapong A, Meshnick SR, et al. Mass blood survey for malaria: pooling and real-time PCR combined with expert microscopy in north-west Thailand. Malar J. 2012;11:288.
Spring MD, Pichyangkul S, Lon C, Gosi P, Yongvanichit K, Srichairatanakul U, et al. Antibody profiles to Plasmodium merozoite surface protein-1 in Cambodian adults during an active surveillance cohort with nested treatment study. Malar J. 2016;15:17.
Hodgson SH, Douglas AD, Edwards NJ, Kimani D, Elias SC, Chang M, et al. Increased sample volume and use of quantitative reverse-transcription PCR can improve prediction of liver-to-blood inoculum size in controlled human malaria infection studies. Malar J. 2015;14:33.
Imwong M, Hanchana S, Malleret B, Rénia L, Day NPJ, Dondorp A, et al. High-throughput ultrasensitive molecular techniques for quantifying low-density malaria parasitemias. J Clin Microbiol. 2014;52:3303–9.
Acknowledgements
We acknowledge the MIS 2015 Steering Committee, data collectors and study participants for the samples collected. We thank Zewditu Bekele, Fitsum Tesfaye, Hilina Legesse and Hailemariam Difabachew from the Ethiopian Public Health Institute, for sample organization and management.
Funding
This research was made possible through support provided by the Global Fund to Fight AIDS, Tuberculosis and Malaria via the Federal Ministry of Health in Ethiopia. The opinions expressed herein are those of the author(s) and do not necessarily reflect the views of any collaborating institute including the U.S. Centers for Disease Control and Prevention or the U.S. Agency for International Development. JH, HT, and MWM receive salary support from the U.S. President’s Malaria Initiative.
Author information
Authors and Affiliations
Contributions
Conceived the study: AA, AAA, WD, JH, Designed the experiments: AA, YPS, ZZ, BW. Performed the experiments: BW, ER, ZZ, SS, MS. Analysed the data: AA, BW, JH. Assisted study implementation: AA, AK, YPS, AAA, WD, HT, MWM, JH. Critically reviewed and contributed to writing: JH, LS, GW, YPS, BW, ER, ZZ, AAA, MH, HT, MWM, DD WD. Drafted the manuscript: AA. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical clearance was obtained from the Health Science College of Addis Ababa, University (AAUMF 03-008). The EMIS-2015 protocol received ethical clearance from the National Research Ethics Review Committee of Ethiopia. The current survey protocol which included molecular testing underwent human subjects review at CDC and was considered a non-research program evaluation activity.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Assefa, A., Ahmed, A.A., Deressa, W. et al. Assessment of subpatent Plasmodium infection in northwestern Ethiopia. Malar J 19, 108 (2020). https://doi.org/10.1186/s12936-020-03177-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-020-03177-w