
Abstract
Background
RNA-seq is a powerful and cost-effective technology for molecular diagnostics of cancer and other diseases, and it can reach its full potential when coupled with validated clinical-grade informatics tools. Despite recent advances in long-read sequencing, transcriptome assembly of short reads remains a useful and cost-effective methodology for unveiling transcript-level rearrangements and novel isoforms. One of the major concerns for adopting the proven de novo assembly approach for RNA-seq data in clinical settings has been the analysis turnaround time. To address this concern, we have developed a targeted approach to expedite assembly and analysis of RNA-seq data.
Results
Here we present our Targeted Assembly Pipeline (TAP), which consists of four stages: 1) alignment-free gene-level classification of RNA-seq reads using BioBloomTools, 2) de novo assembly of individual targets using Trans-ABySS, 3) alignment of assembled contigs to the reference genome and transcriptome with GMAP and BWA and 4) structural and splicing variant detection using PAVFinder. We show that PAVFinder is a robust gene fusion detection tool when compared to established methods such as Tophat-Fusion and deFuse on simulated data of 448 events. Using the Leucegene acute myeloid leukemia (AML) RNA-seq data and a set of 580 COSMIC target genes, TAP identified a wide range of hallmark molecular anomalies including gene fusions, tandem duplications, insertions and deletions in agreement with published literature results. Moreover, also in this dataset, TAP captured AML-specific splicing variants such as skipped exons and novel splice sites reported in studies elsewhere. Running time of TAP on 100–150 million read pairs and a 580-gene set is one to 2 hours on a 48-core machine.
Conclusions
We demonstrated that TAP is a fast and robust RNA-seq variant detection pipeline that is potentially amenable to clinical applications. TAP is available at http://www.bcgsc.ca/platform/bioinfo/software/pavfinder
Similar content being viewed by others

Background
Advances in second-generation sequencing technologies ushered in the modern era of personalized medicine [1]. In cancer, mutations revealed by clinical sequencing have been shown to be vitally useful in achieving better subtype classification, charting appropriate treatment regimens, and identifying novel drug targets [2,3,4]. One of the well-studied examples is acute myeloid leukemia (AML), for which prognosis and treatment strategies depend on the detection of a wide spectrum of mutations: FLT3 internal tandem application (ITD), MLL partial tandem duplication (PTD), NPM1 insertion, CEBPA insertion/deletions (indels), and gene fusions PML-RARA, RUNX1-RUNX1T and CBFB-MYH11, among others [5].
While the cost of sequencing has decreased dramatically since its introduction, translating whole-genome sequencing methods to the clinical domain remains a challenge due to their sample amount and quality, coverage depth, and turnaround time requirements [6]. With its lower cost and input sample requirements, and faster turnaround times, RNA sequencing (RNA-seq) offers an attractive alternative. Although in the research domain it is primarily used to unveil altered gene expression levels, RNA-seq is increasingly used to capture expressed genomic anomalies such as single nucleotide variants (SNVs) and aberrant transcript structures [7, 8].
Important sequence-based disease markers typically are SNVs, but long-range rearrangements or structural variants (SVs) are also being increasingly appreciated for their important roles in pathogenesis [9]. RNA-seq read sequences allow SNVs and short indels to be readily identified, but SV detection with short reads requires more complex analysis and algorithms. Although the recent development of long-read sequencing technologies has shown promise in facilitating the reconstruction of full-length transcripts and novel isoforms [10, 11], their application in clinical settings remains to be fully explored and assessed for reliability and throughput considerations.
Current state-of-the-art SV detection tools, such as TopHat-Fusion [12] and deFuse [13], discover SVs through interrogating alignments of reads to the reference genome, but ambiguous alignments of short reads limit the sensitivity and specificity of these methods. De novo RNA-seq assembly reconstructs long transcript sequences without relying on alignments of reads to a reference genome, and thus it is widely used in profiling transcriptomes of non-model organisms, reconstructing transcript structures, and detecting novel isoforms [14,15,16,17,18]. Because longer sequences tend to have lower alignment ambiguity than short sequences, SV detection based on alignments of assembled transcripts (instead of short reads) to a reference genome have been shown to be successful in cancer studies [19, 20]. However, analysis of deeply sequenced human transcriptomes remains very resource-intensive, and therefore may not meet the constraints in the clinical domain.
An alternative to analyzing entire genomes or transcriptomes is to focus on target gene sets (or gene panels) that are most relevant for specific diseases [21, 22]. This is effective because for many diseases, in particular cancers, there are many clinically relevant genes to help with disease classification or with the selection of treatment strategies [23]. Here we propose a targeted approach on RNA-seq data analysis called TAP (for Targeted Assembly Pipeline) using de novo assembly for variant identification. TAP offers functionality akin to using data from gene panels, and offers the benefits of a robust sequencing protocol coupled with the flexibility of selecting the genes of interest after data generation, as the clinical question might dictate.
Implementation
TAP detects SVs in four stages described below. It also summarizes other pertinent information, such as the extent of reconstruction of all the targeted genes, and a compilation of all the reconstructed splice junctions (novel or annotated), and their supporting read counts.
Alignment-free extraction of reads for gene targets
The first step of TAP (Fig. 1) is to classify and segregate whole-sample RNA-seq reads into bins corresponding to specific gene targets. Instead of using alignment-based approaches for this purpose, we chose to use a novel multi-index Bloom filter data structure implemented within BioBloomTools (BBT v2.1.0), which is able to achieve sequence classification at comparable accuracy to alignment-based methods, but in a much faster and memory-efficient manner [24]. The inputs to this sequence extraction step are RNA-seq read pairs and transcript sequences of a list of target genes. BBT utilizes a set of five spaced seeds with an allowed miss of two spaced seeds (parameter -a) per k-mer frame evaluated. This allows BBT to better tolerate sequencing errors and variants, and achieve high sensitivity whilst maintaining high specificity. Further, it extracts read pairs when at least one of the pairs is classified as hitting one of the target genes (flag -i), capturing sequences that represent novel splice variants and gene fusions.

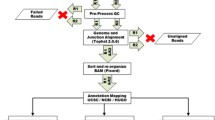
TAP Pipeline. A Bloom filter is generated from reference transcript sequences of a target list and then applied on full transcritpome RNA-seq sequences to extract gene-specific read pairs. Reads classified to each target are segregated into separate bins and assembled using two k-mer values independently in parallel. Contigs from each k-mer assembly of each gene are merged and extracted reads are aligned to them (r2c). Gene-level assemblies are combined into a single file and aligned to the genome (c2g) and transcriptome (c2t). PAVFinder uses the c2g and c2t alignments together with contig sequences and annotation (reference sequences and gene models) to identify structural variant and novel splicing events. r2c alignments are used for determining event support and coverage estimation
De novo reconstruction of transcript sequences
Bins of read pairs belonging to individual genes are assembled independently in parallel using Trans-ABySS (v1.5.4) [14]. De novo assembly is used to reconstruct variant breakpoint-spanning sequences from short reads, and Trans-ABySS has been shown to be successful in capturing such events in various genomic and transcriptomic studies [25,26,27,28,29,30,31,32,33]. To reconstruct transcripts with a range of expression levels and sequence complexity, Trans-ABySS uses a set of overlap lengths (k-mer sizes). Typically, a low k-mer size would be more sensitive to read-to-read overlaps, helping reconstruct low expressed transcripts, while a high k-mer size would be more specific to resolve low complexity sequences.
Alignment of assembled transcripts and extracted reads
Since transcriptomic rearrangements can be complicated and may lead to erroneous alignments, alignments to both reference genome and transcriptome are used in TAP to increase accuracy and sensitivity. Assembled contig sequences are aligned to the reference genome and the reference transcriptome using GMAP (v2014-12-28) [34] and the BWA-MEM algorithm of the BWA package (v0.7.12) [35], respectively. Concurrently, extracted reads are aligned to the assembled contigs to provide support evidence and read counts of SV calls.
Detection of structural variants
As a key module within TAP, we developed PAVFinder (Post Assembly Variant Finder, v0.4.2) to deduce variants from the split or gapped alignments of contigs to the references (Fig. 2a). Based on several criteria (Additional file 1: Table S1, S2), such as the alignment orientation of chimeric sequence fragments, PAVFinder classifies events such as gene fusions, read-throughs, ITDs, PTDs, indels and repeat number changes. It also reports novel splicing events, such as exon skipping, novel exons, novel introns, retained introns, and novel splice donor and acceptor sites by comparing contig-to-genome alignments to reference gene models (Fig. 2b).

PAVFinder detects both (a) structural rearrangements and (b) novel splicing variants. Numbers indicate reference transcript exon numbers. Dotted red lines represent novel adjacencies (joining between non-adjacent transcript sequences) and red blocks represent novel sequences. For splicing variants, canonical splice site motifs are indicated as they are checked for calling potential novel splicing events. Dotted vertical lines depict algorithm for detecting novel splicing variants by aligning contig sequences against annotated gene model
Results
Assessment of the performance of BBT in sequence classification
TAP analyses the sequences selected by BBT. To assess BBT’s performance in sequence classification at different sequencing depths, we simulated Illumina (San Diego, CA) reads with depth of coverage ranging from 10× (229,800 read pairs) to 100× (2,303,019 read pairs) in increments of 10 using pIRS (v1.1.1) [36] from a gene set composed of 580 COSMIC (v77) genes [37] (targets) and an equal number of non-COSMIC genes randomly selected from RefSeq [38]. The non-COSMIC genes were included to mimic non-target genes in the transcriptome. We chose to use pIRS over other RNA-seq simulators because of its simplicity to simulate different read depths and its provision of read-origin information, which readily enables calculation of classification accuracy. We compared the performance of BBT on the COSMIC set against alignment-based classification using BWA-MEM (v0.7.12) to observe that BBT slightly outperformed BWA-MEM in overall sensitivity (BBT 99.9% versus BWA-MEM 98.1%) and both methods show comparable specificity (BBT 99.2% versus BWA-MEM 99.9%) (Additional file 1: Figure S1). However, on a per-gene basis, we found BBT to outperform BWA-MEM in 115 genes, while BWA-MEM outperformed BBT in 66 genes (the remaining 399 are in a virtual tie with their F1 scores within one standard deviation of each other) (Additional file 1: Figure S2). This trend is reverted for the software parameterization used above when we increased the substitution-error rate from 0.37% (default profile of the experimental data) to 1% in the simulation step. For this unusually high error rate, BWA-MEM outperformed BBT in 303 genes, whereas BBT was superior in only 104. The actual overall difference in absolute performance metrics is, however, negligible (within 0.1% in most coverage depths) (Additional file 1: Figure S3). In terms of computation performance, BBT runs faster than BWA-MEM, and scales much better with increasing read depths (Additional file 1: Figure S1).
Assessment of the performance of PAVFinder
We investigated the fusion-calling performance of PAVFinder in relation to sequencing depth, and compared that with two well-established methods in the field [12, 13]. From a published list of gene fusions reported from TCGA RNA-seq experiments [39], 448 “tier-1” (highest level of confidence in the study), inter-chromosomal, and in-frame events with defined breakpoint locations were selected to simulate a titration series consisting of varying sequencing coverages (4× to 20×, increments of 2) of the breakpoint sequences (250 base pair (bp) mean simulation insert size, upstream and downstream of the breakpoint), mixed with whole transcript reference sequences of the fusion genes (803, discounting redundant gene partners) together with a similar number (776) of randomly-selected non-fusion transcripts as background at four different coverage depths (10× to 40×, increments of 10) (Fig. 3a). We used pIRS [36] to simulate 100 bp Illumina reads with a mean insert size of 250 bp for each coverage combination.

Simulation experiment to assess PAVFinder fusion calling performance in relation to sequencing coverage and other software. a. Design of experiment: reads simulated from fusion breakpoints and corresponding reference transcript sequences at different read depths are combined to simulate the titration series. b. Receiver Operating Characteristic (ROC) plots of PAVFinder, Tophat-Fusion [12], and deFuse [13] on 448 fusion events reported on TCGA data [39]
For benchmarking, only events with at least four breakpoint-spanning reads were considered for comparison. A true positive is scored when partners in a detected gene fusion event correspond to one of the 448 input gene pairs. In this experiment, PAVFinder shows high sensitivity (about 95%) at fusion coverage depth of 4×, and this performance reaches 100% at higher fusion coverage depths. The false positive rate remains less than 0.5% throughout the sequencing coverage depths evaluated. When compared with the other tools, PAVFinder has the highest sensitivity and specificity (Fig. 3b).
Assessment of the performance of TAP on real data
The Leucegene Project [40] made available 437 publicly accessible RNA-seq datasets analyzed in several published studies [41,42,43,44]. We leveraged these data (Table 1) for evaluating the performance of TAP due to the wide spectrum of structural variants that are clinically-relevant biomarkers offered by this disease. Using 580 COSMIC cancer genes [37] as our target gene set, we examined fusions and read-throughs of
-
the core-binding factor (CBF) cohort [42], which carries either the CBFB-MYH11 or RUNX1-RUNX1T1 fusion;
-
the NUP98-NSD1 cohort [43], which carries the NUP98-NSD1 fusion; and
-
the MLL fusion (MLL-F) cohort, which carries MLL (a.k.a. KMT2A) fused with different partners [41].
We screened a multitude of samples for MLL-PTD as the sample identities of the MLL-PTD cohort were not disclosed in any of the Leucegene publications. Furthermore, we processed samples from a CEBPA cohort to assess TAP’s ability to detect short indels. For all the processed samples, we also look for the important AML variants FLT3-ITD and NPM1 insertion. Finally, we identified several aberrant splicing events reported in the literature [45, 46] that have potential implications in AML, and checked whether we can detect them in any of the samples we analyzed.
Fusions
We processed 46 RNA-seq samples of the CBF cohort, of which 26 carry the CBFB-MYH11 fusion (inv(16)), and 20 carry the RUNX1-RUNX1T1 fusion (t(8;21)). TAP was successful in detecting all of the fusion events, in agreement with the literature [30]. Two of the CBF-MYH11 cases (03H095 and 12H042) do not have breakpoints at exon boundaries: one presents four extra amino acids at the junction, and the other has a breakpoint internal to the MYH11 exon, both of which nevertheless produce in-frame chimeric transcripts. The PAVFinder module was configured by default to restrict fusion breakpoints to exon boundaries. When this option was turned off, the CBF-MYH11 fusion were identified and reported in TAP. PAVFinder also detected the NUP98-NSD1 fusion in all seven AML samples known to contain the fusion event.
We also processed 31 samples of the MLL-F cohort, which contains MLL fusions involving different partners: CASC5 (1), ENL (MLLT1, 4), ELL (3), GAS7 (1), SEPT9 (2), MLLT (9), MLLT4 (8), MLLT6 (1) and MLLT10 (2) (numbers in brackets indicate number of samples in each case). TAP could detect all nine types of MLL fusions in 30 out of the 31 samples. The only sample we failed to detect any fusion events was 04H080, which was reported to carry the fusion MLL-MLLT3. It was noted in the publication that the fusion was detected by only two reads. We note that the sample contains only 23 million read pairs, only about a quarter of the average size of the other samples. To troubleshoot, we ran the original tool that was used to detect the fusion, TopHat-fusion [12], and still was unable to detect the event.
To benchmark PAVFinder’s relative performance in gene fusion detection, we also processed both the extracted and entire read sets of all the CBF-MYH11, NUP98-NSD1, and MLL-F cohorts with TopHat-Fusion [12] and deFuse [13]. Using mostly default parameters (Additional file 1: Table S5), except with the requirement of at least four reads spanning a breakpoint, TopHat-Fusion failed to report eight MLL-F and one CBF-MYH11 fusions in the reads extracted by BBT. Using the entire read set did not change the results. In two CBF-MYH11 samples, deFuse failed to report the correct fusion in the extracted read set as it had misaligned the MYH11 segment to Nde1. Interestingly, this misalignment was not observed when the entire read set was used. In one MLL-F sample, deFuse failed to detect the MLL fusion in both the extracted and the entire read sets. The samples for which TopHat-Fusion and deFuse missed calling the expected fusion events using the extracted reads were not the same samples, suggesting that it is not the sequence extraction step that causes the false-negatives.
In addition to the signature AML events from the Leucegene study, TAP reported further fusion calls. We assessed these events for their validity by searching for previous reports in the literature. Out of a total of 47 events, about half of them (28) have been reported before (Additional file 1: Figure S4). Notable cases include ETV6-NTRK3, a well-known driver detected in various cancer types including AML [47, 48], and TFG-ADGRG7, a known event reported in healthy individuals [49]. Of the events without any publication reference, one of the partner genes is often found in another fusion event reported in the literature. Examples include fusions involving DDX5, CXCR4, KLF2, and UBC. Fusions in this last category usually exhibit low expression levels, and are apparently promiscuous regarding their fusion partners. Marincevic-Zuniga et al. [50] “blacklisted” these genes, and filtered them out in their detection pipeline, suggesting that although these fusions may indeed be bona fide, their biological significance is dubious or unknown. Amid these “noise” events, however, one novel fusion, PHKB-ATTC, is potentially promising (46 spanning read support) and biologically functional (in-frame fusion of the 5′ of PHKB to the 3’of ATTC), while another, FCGR2C-FCGR2A, is most likely a false positive as a result of mis-assembly due to extensive sequence similarity.
Read-throughs are chimeric transcripts resulting from splicing of two adjacent genes on the same coding strand. They have been found in both normal and neoplastic tissues. An example is SLC45A3-ELK4, which has been detected before in prostate cancer [51, 52]. Interestingly, TAP detected this event in 21 of the Leucegene samples we analyzed. Qin et al. [53] reported that this fusion regulates cell proliferation by its transcript, not through a translated protein. Its presence in AML suggests it may be more widespread in other cancer types than previously thought.
FLT3-ITD
Four NUP98-NSD1, three CBF, and two CEBPA patients were reported to carry a FLT3 ITD. TAP could detect these events and additional alleles from the same samples (Additional file 2: Table S3). In addition, TAP detected FLT3-ITDs in 12 samples analyzed in this study (three from the CBF-cohort, five from the MLL-F cohort, three from the CEBPA cohort, and one from the MLL-PTD sample; see the following paragraph for a description of the latter) that were not reported previously to be positive for this event. Given the fact that the un-reported events all reside in exon 14 (same as all reported cases), and are in-frame, it is highly likely that they are true positives previously missed.
MLL-PTD
Because the 23 sample accessions of the MLL-PTD cohort [41] were not disclosed, we analyzed about 20 samples randomly selected from a list of 377 samples that are not part of the three fusion or CEBPA cohorts (all negative for MLL-PTD), and found one positive candidate. A breakpoint suggesting a tandem duplication of exons 2 to 6, one of the most common MLL-PTD alleles in AML [54], was detected in sample 09H106. Although we cannot provide any precision metric due to missing information, this single positive case nonetheless highlights TAP’s ability to detect PTD events.
CEBPA indels and NPM1 insertion
TAP detected all the reported indels in 12 of the CEBPA samples [44]. Though, these events may be often labeled differently: for example, our pipeline may report them as duplication (06H026) or repeat-expansion events (08H065), instead of an insertion. In one CEBPA (08H082) and one NUP98-NSD1 (11H027) samples, TAP was also able to detect a 4 bp NPM1 insertion, an important AML biomarker, in exon 12 where most reported mutations reside [55].
Novel splicing
Aberrant alternative splicing has been shown to be implicated in AML development [45]. We interrogated the Leucegene dataset for novel splicing events in the genes ANPEP (a.k.a. CD13), NOTCH2, and FLT3, which have been shown to express mis-spliced transcripts in AML patients [45, 46], and TAP detected the different aberrant splicing patterns identified from these studies in various samples (Table 2, Additional file 2: Table S5). Most of the events involve single or multiple exon-skipping events, with or without associated novel splice donor or acceptors. In addition to previously reported novel splicing events in these genes, TAP identifies an additional 11 different novel exon-skipping events in FLT3 and eight different novel exon-skipping events in NOTCH2.
Computational resources and runtime
Using 580 COSMIC genes, TAP processed 100–150 million RNA-seq read pairs within 2 hours using 32 threads on a single Intel Xeon E5–2699 v3 2.30 GHz 36-core machine running CentOS 6. In comparison, TopHat-Fusion and deFuse requires around 30 and 6 hours, respectively, to process the same datasets (Additional file 1: Figure S5).
Discussion
We developed a bioinformatics pipeline, TAP, for analyzing RNA-seq data in a targeted manner, such that anomalies of hundreds of important cancer genes can be identified within a couple of hours, making TAP highly feasible as an analysis tool in clinical genomics applications.
Using COSMIC genes as an example, we demonstrated that BBT is highly accurate and robust at classifying reads with over 99.9% accuracy. Although Bloom filters in BBT were constructed using only the reference sequences of select target genes, BBT could still extract reads containing breakpoints of structural variants. This is particularly useful in detecting gene fusions when one of the two partner genes is unforeseen and thus missing in the target set. An example of this is the promiscuous MLL fusions in AML. We showed in this study that TAP was able to identify the various MLL fusions in the MLL-F Leucegene cohort even when all its partners are intentionally removed from the target set.
De novo sequence assembly has been shown to be a useful approach for detecting structural and splicing variants on both genomic and transcriptomic datasets [25,26,27]. We used AML datasets to demonstrate our pipeline’s versatile ability to identify a diverse spectrum of rearrangements (gene fusions, ITDs, PTDs, indels, etc). To assess PAVFinder performance in fusion calling, we simulated sequences from a set of 448 TCGA fusions, and showed that PAVFinder outperforms two widely used methods based on an alignment-first approach in both sensitivity and specificity. Based on the benchmarking with real AML RNA-seq datasets from Leucegene, PAVFinder captured all the published events but one low coverage event, whereas all other methods evaluated have a number of events not detected. Assembly-based variant detection also offers the advantage of detection of possibly multiple breakpoint alleles of the same event with single base pair resolution. This is evident in the fusion results of all the Leucegene samples we processed (Additional file 2: Table S4), where more than one breakpoint allele of the same event can often be found within the same or among different patients. As it has been reported that different gene fusion products of the same two genes may potentially confer different oncogenic potential [56], the ability to accurately identify different gene fusion alleles could potentially be informative for diagnostics. Another example is FLT3-ITD, which exhibits variability in both length and position, canonically located within exon 14. We showed that PAVFinder was able to handle this variability.
Turnaround time is an important consideration in applying next-generation sequencing for clinical diagnostic applications. With the cost of sequencing rapidly decreasing, the amount of data produced is also increasing at a rate that potentially makes sequence analysis the next bottleneck in result delivery. We demonstrated that analyzing selected disease-relevant genes instead of the entire transcriptome is a viable approach, and showed that all the clinically-relevant structural variants in the target genes can be detected with 100% sensitivity, yet with a much more desirable turnaround time. A typical assembly- or alignment-first variant analysis on an entire transcriptome library of a typical sequencing depth (100 M+ reads) currently takes overnight or longer to finish. With a reduced yet comprehensive dataset (in the context of selected targets), TAP can potentially be extended to additional types of RNA-seq analysis such as SNV detection and expression profiling. Moreover, multiple tools for detecting the same kind of variants can be applied as a complementary approach, and still be feasible timewise because of a reduced data size.
Conclusions
We developed a bioinformatics pipeline, TAP, which assembles and analyses RNA-seq data for detection structural and splicing variants. Applied on a targeted gene set, TAP shows good performance with high sensitivity and specificity with a quick turnaround time, making it a good candidate for downstream analysis on clinical sequencing.
Availability and requirements
Project name: TAP.
Project home page: http://www.bcgsc.ca/platform/bioinfo/software/pavfinder
Operating system(s): Linux.
Programming language: Python 2.7.
Other requirements: None.
License: BCCA (academic use).
Any restrictions to use by non-academics: None.
Abbreviations
- AML:
-
Acute myeloid leukemia
- BBT:
-
BioBloom Tools
- COSMIC:
-
The Catalogue Of Somatic Mutations In Cancer
- indel:
-
Insertion deletion
- ITD:
-
Internal tandem duplication
- PAVFinder:
-
Post Assembly Variant Finder
- PTD:
-
Partial tandem duplication
- RNA-seq:
-
RNA sequencing
- SNV:
-
Single nucleotide variant
- TAP:
-
Targeted Assembly Pipeline
- TCGA:
-
The Cancer Genome Atlas
References
Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793–5.
Rodrigues DN, Boysen G, Sumanasuriya S, Seed G, Marzo AM, de Bono J. The molecular underpinnings of prostate cancer: impacts on management and pathology practice. J Pathol. 2017;241(2):173–82.
Baroy T, Chilamakuri CS, Lorenz S, Sun J, Bruland OS, Myklebost O, Meza-Zepeda LA. Genome analysis of osteosarcoma progression samples identifies FGFR1 overexpression as a potential treatment target and CHM as a candidate tumor suppressor gene. PLoS One. 2016;11(9):e0163859.
Town J, Pais H, Harrison S, Stead LF, Bataille C, Bunjobpol W, Zhang J, Rabbitts TH. Exploring the surfaceome of Ewing sarcoma identifies a new and unique therapeutic target. Proc Natl Acad Sci U S A. 2016;113(13):3603–8.
Ilyas AM, Ahmad S, Faheem M, Naseer MI, Kumosani TA, Al-Qahtani MH, Gari M, Ahmed F. Next generation sequencing of acute myeloid leukemia: influencing prognosis. BMC Genomics. 2015;16(Suppl 1):S5.
Jennings LJ, Arcila ME, Corless C, Kamel-Reid S, Lubin IM, Pfeifer J, Temple-Smolkin RL, Voelkerding KV, Nikiforova MN. Guidelines for validation of next-generation sequencing-based oncology panels: a joint consensus recommendation of the Association for Molecular Pathology and College of American pathologists. J Mol Diagn. 2017;19(3):341–65.
Byron SA, Van Keuren-Jensen KR, Engelthaler DM, Carpten JD, Craig DW. Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nat Rev Genet. 2016;17(5):257–71.
Cummings BB, Marshall JL, Tukiainen T, Lek M, Donkervoort S, Foley AR, Bolduc V, Waddell LB, Sandaradura SA, O'Grady GL, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med. 2017;9(386)
Macintyre G, Ylstra B, Brenton JD. Sequencing structural variants in Cancer for precision therapeutics. Trends Genet. 2016;32(9):530–42.
Vollmers C, Penland L, Kanbar JN, Quake SR. Novel exons and splice variants in the human antibody heavy chain identified by single cell and single molecule sequencing. PLoS One. 2015;10(1):e0117050.
Cavelier L, Ameur A, Haggqvist S, Hoijer I, Cahill N, Olsson-Stromberg U, Hermanson M. Clonal distribution of BCR-ABL1 mutations and splice isoforms by single-molecule long-read RNA sequencing. BMC Cancer. 2015;15:45.
Kim D, Salzberg SL. TopHat-fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12(8):R72.
McPherson A, Hormozdiari F, Zayed A, Giuliany R, Ha G, Sun MG, Griffith M, Heravi Moussavi A, Senz J, Melnyk N, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput Biol. 2011;7(5):e1001138.
Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, Mungall K, Lee S, Okada HM, Qian JQ, et al. De novo assembly and analysis of RNA-seq data. Nat Methods. 2010;7(11):909–12.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52.
Schulz MH, Zerbino DR, Vingron M, Birney E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012;28(8):1086–92.
Peng Y, Leung HC, Yiu SM, Lv MJ, Zhu XG, Chin FY. IDBA-Tran: a more robust de novo de Bruijn graph assembler for transcriptomes with uneven expression levels. Bioinformatics. 2013;29(13):i326–34.
Xie Y, Wu G, Tang J, Luo R, Patterson J, Liu S, Huang W, He G, Gu S, Li S, et al. SOAPdenovo-trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics. 2014;30(12):1660–6.
Fernandez-Cuesta L, Sun R, Menon R, George J, Lorenz S, Meza-Zepeda LA, Peifer M, Plenker D, Heuckmann JM, Leenders F, et al. Identification of novel fusion genes in lung cancer using breakpoint assembly of transcriptome sequencing data. Genome Biol. 2015;16:7.
Verma A, Jiang Y, Du W, Fairchild L, Melnick A, Elemento O. Transcriptome sequencing reveals thousands of novel long non-coding RNAs in B cell lymphoma. Genome Med. 2015;7:110.
Wagle N, Berger MF, Davis MJ, Blumenstiel B, Defelice M, Pochanard P, Ducar M, Van Hummelen P, Macconaill LE, Hahn WC, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012;2(1):82–93.
Choudhary A, Mambo E, Sanford T, Boedigheimer M, Twomey B, Califano J, Hadd A, Oliner KS, Beaudenon S, Latham GJ, et al. Evaluation of an integrated clinical workflow for targeted next-generation sequencing of low-quality tumor DNA using a 51-gene enrichment panel. BMC Med Genet. 2014;7:62.
Tomczak K, Czerwinska P, Wiznerowicz M. The Cancer genome atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol. 2015;19(1A):A68–77.
Chu J, Sadeghi S, Raymond A, Jackman SD, Nip KM, Mar R, Mohamadi H, Butterfield YS, Robertson AG, Birol I. BioBloom tools: fast, accurate and memory-efficient host species sequence screening using bloom filters. Bioinformatics. 2014;30(23):3402–4.
Morin RD, Mungall K, Pleasance E, Mungall AJ, Goya R, Huff RD, Scott DW, Ding J, Roth A, Chiu R, et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood. 2013;122(7):1256–65.
Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr, Laird PW, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74.
Kasaian K, Wiseman SM, Thiessen N, Mungall KL, Corbett RD, Qian JQ, Nip KM, He A, Tse K, Chuah E, et al. Complete genomic landscape of a recurring sporadic parathyroid carcinoma. J Pathol. 2013;230(3):249–60.
Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, Stutz AM, Korshunov A, Reimand J, Schumacher SE, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488(7409):49–56.
Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513(7517):202–9.
Thibodeau ML, Reisle C, Zhao E, Martin LA, Alwelaie Y, Mungall KL, Ch'ng C, Thomas R, Ng T, Yip S, et al. Genomic profiling of pelvic genital type leiomyosarcoma in a woman with a germline CHEK2:c.1100delC mutation and a concomitant diagnosis of metastatic invasive ductal breast carcinoma. Cold Spring Harb Mol Case Stud. 2017;3:5.
Yip S, Butterfield YS, Morozova O, Chittaranjan S, Blough MD, An J, Birol I, Chesnelong C, Chiu R, Chuah E, et al. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J Pathol. 2012;226(1):7–16.
Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, Chen SC, Payne-Turner D, Churchman ML, Harvey RC, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):153–66.
Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45(3):279–84.
Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21(9):1859–75.
Li H, Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. 2010;26(5):589–95.
Hu X, Yuan J, Shi Y, Lu J, Liu B, Li Z, Chen Y, Mu D, Zhang H, Li N, et al. pIRS: profile-based Illumina pair-end reads simulator. Bioinformatics. 2012;28(11):1533–5.
Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer. 2004;4(3):177–83.
O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733–45.
Yoshihara K, Wang Q, Torres-Garcia W, Zheng S, Vegesna R, Kim H, Verhaak RG. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 2015;34(37):4845–54.
The Leucegene Project [https://leucegene.ca/].
Lavallee VP, Baccelli I, Krosl J, Wilhelm B, Barabe F, Gendron P, Boucher G, Lemieux S, Marinier A, Meloche S, et al. The transcriptomic landscape and directed chemical interrogation of MLL-rearranged acute myeloid leukemias. Nat Genet. 2015;47(9):1030–7.
Lavallee VP, Lemieux S, Boucher G, Gendron P, Boivin I, Armstrong RN, Sauvageau G, Hebert J. RNA-sequencing analysis of core binding factor AML identifies recurrent ZBTB7A mutations and defines RUNX1-CBFA2T3 fusion signature. Blood. 2016;127(20):2498–501.
Lavallee VP, Lemieux S, Boucher G, Gendron P, Boivin I, Girard S, Hebert J, Sauvageau G. Identification of MYC mutations in acute myeloid leukemias with NUP98-NSD1 translocations. Leukemia. 2016;30(7):1621–4.
Lavallee VP, Krosl J, Lemieux S, Boucher G, Gendron P, Pabst C, Boivin I, Marinier A, Guidos CJ, Meloche S, et al. Chemo-genomic interrogation of CEBPA mutated AML reveals recurrent CSF3R mutations and subgroup sensitivity to JAK inhibitors. Blood. 2016;127(24):3054–61.
Adamia S, Haibe-Kains B, Pilarski PM, Bar-Natan M, Pevzner S, Avet-Loiseau H, Lode L, Verselis S, Fox EA, Burke J, et al. A genome-wide aberrant RNA splicing in patients with acute myeloid leukemia identifies novel potential disease markers and therapeutic targets. Clin Cancer Res. 2014;20(5):1135–45.
Adamia S, Bar-Natan M, Haibe-Kains B, Pilarski PM, Bach C, Pevzner S, Calimeri T, Avet-Loiseau H, Lode L, Verselis S, et al. NOTCH2 and FLT3 gene mis-splicings are common events in patients with acute myeloid leukemia (AML): new potential targets in AML. Blood. 2014;123(18):2816–25.
Lannon CL, Sorensen PH. ETV6-NTRK3: a chimeric protein tyrosine kinase with transformation activity in multiple cell lineages. Semin Cancer Biol. 2005;15(3):215–23.
Kralik JM, Kranewitter W, Boesmueller H, Marschon R, Tschurtschenthaler G, Rumpold H, Wiesinger K, Erdel M, Petzer AL, Webersinke G. Characterization of a newly identified ETV6-NTRK3 fusion transcript in acute myeloid leukemia. Diagn Pathol. 2011;6:19.
Chase A, Ernst T, Fiebig A, Collins A, Grand F, Erben P, Reiter A, Schreiber S, Cross NC. TFG, a target of chromosome translocations in lymphoma and soft tissue tumors, fuses to GPR128 in healthy individuals. Haematologica. 2010;95(1):20–6.
Marincevic-Zuniga Y, Dahlberg J, Nilsson S, Raine A, Nystedt S, Lindqvist CM, Berglund EC, Abrahamsson J, Cavelier L, Forestier E, et al. Transcriptome sequencing in pediatric acute lymphoblastic leukemia identifies fusion genes associated with distinct DNA methylation profiles. J Hematol Oncol. 2017;10(1):148.
Rickman DS, Pflueger D, Moss B, VanDoren VE, Chen CX, de la Taille A, Kuefer R, Tewari AK, Setlur SR, Demichelis F, et al. SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009;69(7):2734–8.
Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X, Sam L, Barrette T, Palanisamy N, Chinnaiyan AM. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458(7234):97–101.
Qin F, Zhang Y, Liu J, Li H. SLC45A3-ELK4 functions as a long non-coding chimeric RNA. Cancer Lett. 2017;404:53–61.
Aplan PD. Chromosomal translocations involving the MLL gene: molecular mechanisms. DNA repair. 2006;5(9–10):1265–72.
Thiede C, Koch S, Creutzig E, Steudel C, Illmer T, Schaich M, Ehninger G. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood. 2006;107(10):4011–20.
Liu J, Nau MM, Yeh JC, Allegra CJ, Chu E, Wright JJ. Molecular heterogeneity and function of EWS-WT1 fusion transcripts in desmoplastic small round cell tumors. Clin Cancer Res. 2000;6(9):3522–9.
Acknowledgements
The authors would like to thank the Leucegene project for making their RNA-seq data freely available.
Funding
The research presented here was funded by the National Human Genome Research Institute of the National Institutes of Health (under award number R01HG007182), with additional support provided by Genome Canada and Genome British Columbia (under award number BCB2 - 251LRD), and the Canadian Institutes of Health Research. The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other funding organizations.
Availability of data and materials
The Leucegene RNA-seq datasets analyzed in this study are available from the Gene Expression Omnibus under accession codes GSE49642, GSE52656, GSE62190, GSE66917, and GSE67039.
Author information
Authors and Affiliations
Contributions
IB conceived of and directed the project. RC developed the TAP workflow and PAVFinder. RC and KMN are developers of Trans-ABySS. JC is a developer of BBT. RC, KMN, JC, and IB wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Read classification by Bloom-filter vs alignment. Figure S2. Per-gene comparison of classification performance by BBT vs BWA-MEM. Figure S3. Effect of sequencing error rate on performance of read classification. Figure S4. Support level of gene fusions detected in Leucegene samples. Figure S5. Benchmarking of TAP and other fusion callers. Table S1. Alignment features used by PAVFinder for classifying various types of transcriptomic structural variants. Table S2. Block-vs-exon alignment characteristics used by PAVFinder to identify various classes of novel splice variants. Table S3. Software and command lines used in TAP and benchmark experiments. (PDF 1076 kb)
Additional file 2:
Table S4. AML-relevant structural variants detected by TAP in Leucegene. Table S5. Aberrant splicing variants previously identified in AML patients detected by TAP in Leucegene samples. (PDF 978 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chiu, R., Nip, K.M., Chu, J. et al. TAP: a targeted clinical genomics pipeline for detecting transcript variants using RNA-seq data. BMC Med Genomics 11, 79 (2018). https://doi.org/10.1186/s12920-018-0402-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-018-0402-6