
Abstract
Background
Phloroglucinol is an important chemical, and the biosynthesis processes which can convert glucose to phloroglucinol have been established. However, due to approximate 80% of the glucose being transformed into undesirable by-products and biomass, this biosynthesis process only shows a low yield with the highest value of about 0.20 g/g. The industrial applications are usually hindered by the low current productivity and yield and also by the high costs. Generally, several different aspects limit the development of phloroglucinol biosynthesis. The yield of phloroglucinol is one of the most important parameters for its bioconversion especially from economic and ecological points of view. The in vitro biosynthesis of bio-based chemicals, is a flexible alternative with potentially high-yield to in vivo biosynthetic technology.
Results
By comparing the activity of acetyl-CoA synthetase (ACS) from Escherichia coli and Acetobacter pasteurianus, the highly active ACS2 was identified in A. pasteurianus. Acetyl-CoA carboxylase (ACC) from Acinetobacter calcoaceticus and phloroglucinol synthase (PhlD) from Pseudomonas fluorescens pf-5 were expressed and purified. Acetate was successfully transformed into phloroglucinol by the combined activity of above-mentioned enzymes and required cofactor. After optimization of the in vitro reaction system, phloroglucinol was then produced with a yield of nearly 0.64 g phloroglucinol/g acetic acid, which was equal to 91.43% of the theoretically possible maximum.
Conclusions
In this work, a novel in vitro synthetic system for a highly efficient production of phloroglucinol from acetate was demonstrated. The system’s performance suggests that in vitro synthesis of phloroglucinol has some advantages and is potential to become a feasible industrial alternative. Based on the results presented herewith, it is believed that in vitro biosystem will provide a feasible option for production of important industrial chemicals from acetate, which could work as a versatile biosynthetic platform.
Similar content being viewed by others

Background
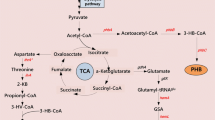
Phloroglucinol and its derivatives have been widely applied in pharmaceuticals, cosmetics, textiles, paints and dyeing industries [1]. At present, the chemical synthetic processes are neither cost-effective nor energy saving. Additionally, the production of phloroglucinol and its derivatives using engineered E. coli has been investigated by many researchers [2, 3]. However, the productivity and yield are currently still low and hence their industrial production is economically unfeasible. Phloroglucinol is toxic to E. coli and its toxicity restrains the further increase of productivity and yield [2]. One of the possible solutions to this problem is to utilize an in vitro biosynthesis technology which does not need the cells and only employs active enzymes. Compared to in vivo living entity-based biosynthetic technology, in vitro synthetic biosystem has broad reaction environment (e.g. high temperature, organic phase catalytic system), free choice of substrate, and potential possibility to solve the problem of the toxicity deriving from the intermediate or final products [4]. The many undesirable side reactions are eliminated in the in vitro biosynthetic processes, thus it becomes possible to achieve about 100% theoretical yield [5]. Moreover, central metabolism which is fundamental to cell survival can be easily re-established through eliminating cellular constraints [6]. Recently, in vitro synthetic biochemistry approaches have been proposed to produce chemicals and proteins [7,8,9]. In theory, phloroglucinol could be also produced by in vitro synthesis system with the precursor of acetate. Acetate could be converted into acetyl coenzyme A (acetyl-CoA), which can further transform into malonyl coenzyme A (malonyl-CoA) and phloroglucinol.
Acetate had been previously utilized as a feedstock for the production of biochemical or biofuel since it could be transformed into acetyl-CoA. Acetate could be derived from a variety of cheap sources, such as (i) products during syngas fermentation [10], hydrolysis under acid or alkali pretreatment [11] and pyrolysis of lignocellulosic biomass [12], (ii) intermediates from anaerobic digestion of organic wastes [13], (iii) production by methanol carbonylation [14], (iv) methane conversion from natural gas or biogas [15]. It has been demonstrated that Cryptococcus curvatus could be able to produce lipid using acetate as a major carbon source [16] and engineered E. coli could convert acetate to succinic acid, fatty acids and β-caryophyllene [17,18,19]. The utilization of acetate as a nontraditional carbon source is becoming one of the most interesting direction in industry biotechnology due to obviously lower cost, sufficient source, no direct competition for food supplies with people, and price stability. However, high concentration of acetate is toxic to cell growth since it damages trans-membrane pH gradients, and which hence destroys internal osmotic pressure, hinders recombinant protein production and inhibits biomass growth [20]. In addition, the formation of the by-products reduces the effective utilization of substrate and also decreases the yield of target product. Considering the advantages of the in vitro biosystem, it is major significant to assimilate acetate via the in vitro synthetic biology.
Acetyl-CoA plays a central role both in catabolism and anabolism, which is a branching point from the substrate carbon into oxidation or biosynthesis pathways [21]. Acetyl-CoA could be also the precursor of many important chemicals, such as fatty acid, succinic acid, 3-hydroxypropionic acid, ethanol, amino acids. Therefore, any biosynthetic process that can utilize acetyl-CoA may become one of the most interesting directions for future biotechnology. It would be important to efficiently convert acetate into acetyl-CoA which is the building block molecule. Until now, there is no report on in vitro biosystem which can make used of acetate as the carbon source to synthetize chemicals.
In this work, we attempted to construct an in vitro system for the biosynthesis of phloroglucinol using acetate as the substrate (Fig. 1). With the aim to enhance the utilization ability of acetate, acetyl-CoA synthetases which derived from two different bacteria were screened with high activity. A. calcoaceticus was used to clone genes of ACC. The fatty acids of A. calcoaceticus is higher than most of the other bacteria, ACC of the bacterium maybe has high activity. P. fluorescens Pf-5 which had been analyzed about phloroglucinol synthesis pathway was a source of PhlD. The in vitro synthesis of phloroglucinol was successfully demonstrated. In addition, the process of multi-enzyme catalytic synthesis of phloroglucinol were observed and optimized.

The synthetic pathway of phloroglucinol. A: In vivo process for phloroglucinol fermentation from glucose; B: In vitro process for the transformation of acetic acid to phloroglucinol. The used enzymes are acetyl-CoA synthetase (ACS), acetyl-CoA carboxylase (ACC), phloroglucinol synthase (PhlD), pyruvate dehydrogenase Complex (PDC)
Methods
Stain, chemicals and culture conditions
All strains and plasmids used in this study are shown in Table 1. E. coli DH5α was used as cloning host and E. coli BL21(DE3) (Tiangen, China) was used as expression of recombinant proteins. A. pasteurianus DSM 3509 and A. calcoaceticus CGMCC 1.6186 were purchased from DSMZ (Brauschweig, Germany) and CGMCC (Beijing, China), respectively. Plasmid pET-28a(+) and Ni-NTA His · Bind Column were purchased from Novagen. PrimeSTAR Max DNA Polymerase (Takara, Japan) was used to amplify genes from plasmid or genomic DNA. Restriction enzymes and T4 DNA ligase (Thermo Scientific, USA) were used for cloning. Commercial enzymes, ATP, coenzyme A and NAD+ were from Sigma. Recombinant proteins were concentrated by using the ultrafiltration membrane (Millipore). Bacteria were cultured in Luria-Bertani (LB) liquid medium or LB agar (50 μg/mL of kanamycin was used as the antibiotic for each recombinant E. coli).
Plasmid construction
ACS gene was amplified from the wild-type E. coli K-12. A. pasteurianus has two putative acetyl-CoA synthetase genes (acs1 and acs2). The genome was extracted from strain A. pasteurianus and was used to amplify ACS1 and ACS2 genes. ACS1 and ACS2 were separately cloned into the plasmid pET-28a(+) to produce expression plasmids with a 6xHis tag for purification. The four subunit (accA, accB, accC, accD) genes of acetyl-CoA carboxylase from A. calcoaceticus CGMCC 1.6186 were cloned into expression vector pET-28a(+), respectively. The gene encoding sequences of phloroglucinol synthase from P. fluorescens Pf-5 were codon-optimized for E. coli expression and synthesized by GeneWiz (Suzhou, China). Then phloroglucinol synthase gene was cloned into pET-28a(+) vector with a 6xHis tag for purification, creating plasmid pET28a-phlD. All the forward and reverse primers for the PCR amplification were provided by GeneWiz.
Enzyme expression and purification
According to the previous protocol [22], all proteins were expressed in E. coli BL21(DE3) and then were purified. The single clone grown on LB agar was chosen and inoculated in 20 mL LB seed medium. Seed cultures were used as 1% inoculum in 1 L LB liquid medium; the cells were grown in medium until OD600 to 0.65 at 37 °C and then induced by 0.25 mM IPTG. Protein expression was performed under the condition of 20 °C for 12 h. Cells were harvested from 1 L of culture by centrifugation at 4 °C, washed twice using PBS buffer (10 mM Na2HPO4, 2 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, pH 7.4), and re-suspended in 15 ml of 50 mM NaH2PO4, 300 mM NaCl, pH 7.5. The cells were lysed by sonication on ice and the suspension was centrifuged at 13,000 g for 30 min at 4 °C in order to remove cell debris. Ni-NTA columns were used to purify protein from clear cell lysate following the manufacturer’s protocol. The eluted protein was desalted by ultrafiltration, and stored in the buffer containing 100 mM Tris-HCl, 100 mM NaCl, pH 7.5 at 4 °C. Bradford assay was used to measure protein concentration using bovine serum albumin (BSA) as a standard. The purified proteins were analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) and visualized through Coomassie Blue staining.
Enzyme activity assays
The specific activity of acetyl-CoA synthetase was measured using Varian Cary50 Bio UV-Visible spectrophotometer at 340 nm according to the previous report [23]. The catalytic activity was determined using the assay mixture at pH 7.4 and 37 °C. The assay mixture (1 mL) contained 1 μmol of NAD+, 0.2 μmol of CoA,10 μmol of MgCl2, 8 μmol of ATP (pH 7.5), 10 μmol of L-malate (pH 7.4), 100 μmol of Tris-HCl (pH 7.4), 3 units of malate dehydrogenase, 6 units of citrate synthase, 100 μmol of potassium acetate (omitted from controls) and 0.02 nmol of purified protein. The reaction was initiated by adding ATP. The Enzyme activities were calculated according to the speed of NADH accumulation. Kinetic data were further analyzed using software Origin 8.0, and the highest activity of ACS was determined.
The activity of acetyl-CoA carboxylase was determined according to the speed of NADPH oxidation [24]. NADPH concentration was monitored at 365 nm using Varian Cary50 Bio UV-Visible spectrophotometer. The assay mixture (1 mL) contained 0.1 mmol of Tricine-KOH (pH 8.0), 1 μmol of dithiothreitol, 1 μmol of ATP, 2.5 μmol of MgCl2, 50 μmol of KCl, 30 μmol of NaHCO3, 0.3 μmol of acetyl-CoA (omitted from controls), MCR-C (C-terminal region of malonyl-CoA reductase) [25] and purified acetyl-CoA carboxylase (weight ratio, accA:accB:accC:accD ≈ 1:1.02:1.66:1.08) [26, 27]. Samples were pre-incubated at 30 °C for 1 min to start the reaction, followed by addition of acetyl-CoA, and then incubated at 30 °C after mixing.
The specific activity of the PhlD was measured using the α-ketoglutarate dehydrogenase (KGDH) according to the reported study [22]. CoASH-dependent oxidation of α-ketoglutarate was accompanied by the reduction of NAD+ to NADH. The rate of NADH (ε = 6220 M-1cm-1) formation was monitored at 340 nm. Assay solutions containing 2 μmol α-ketoglutarate, 100 μmol malonyl-CoA (omitted from controls) were added to a premix of 0.2 U KGDH, 0.3 μmol NAD+, 30 μg PhlD to a final volume of 1 mL in potassium phosphate buffer (50 μmol, pH = 7.0).
In vitro synthesis of phloroglucinol
For the enzymatic synthesis of phloroglucinol from acetate, the assay mixture with the final volume of 1 mL contained 10 mM MgCl2, 10 mM KCl, 30 mM ATP, 3 mM CoA, 30 mM sodium bicarbonate (NaHCO3), 10 mM potassium acetate, 100 mM Tris-HCl pH 7.4, purified ACS2, ACC and phlD, and ampicillin (0.1 mg/mL). To analyze the sensitivity for the concentration of each enzyme, ACS2, ACC, phlD were individually titrated into the reaction system described above. The reactions were executed in 12 mL bottles containing 1 mL of reaction solution. The reaction solution was shaken at 100 rpm in order to exchange substance and energy. The reaction temperature and time were set at 30 °C and 10 h, respectively.
The concentration of acetate and phloroglucinol was analyzed by HPLC. In brief, samples were centrifuged at 12,000 g for 5 min, and the supernatants were filtered using a 0.20 μm nitrocellulose filter and analyzed by a Summit HPLC (Agilent 1200 series, CA, USA) equipped with UV/vis and refractive index detectors. The column (Aminex HPX-87H, 300 mm × 7.8 mm, Bio-Rad, CA, USA) was eluted at 55 °C using 5 mM H2SO4 as the mobile phase at a flow rate of 0.5 mL/min.
The total phloroglucinol amount was calculated based on the measured concentration by HPLC. The phloroglucinol yield was calculated according to acetate consumption based on the following equivalents: 3 acetate ∼ 3 acetyl-CoA ∼ 3 malonyl-CoA ∼ phloroglucinol.
Results and discussion
Kinetic analysis of acetyl-CoA synthetase
Three kinds of acetyl-CoA synthetases were analyzed with Coomassie brilliant blue staining after purification (Fig. 2). The kinetic analyses were carried out in vitro by spectrophotometric method. The catalytic activities of ACS, ACS1 and ACS2 were obtained for 2.2, 1.3 and 4.2 mmol/min/μmol protein, respectively (Fig. 3a), showing that ACS2 has the highest affinity for acetate and higher catalyzing efficiency compared with ACS and ACS1 protein. Acetyl-CoA synthetase can assimilate acetate into acetyl-CoA via two irreversible enzymatic steps [28]. Firstly, acetate reacts with adenosine triphosphate (ATP) to produce acetyl-adenosine monophosphate (AMP). Then the reaction of acetyl-AMP with CoA forms acetyl-CoA-releasing AMP. Acetyl-CoA synthetase has high affinity for acetate, which allows it to function at the low concentration of acetate. Acetobacter aceti has two putative acetyl-CoA synthetase genes (acs1 and acs2). The acs1 gene was up-regulated during ethanol-oxidation phases, while the acs2 gene was significantly up-regulated when cells entered to the acetate-oxidation phase [29, 30]. ACS2 was inferred to play a more important role to assimilate acetate. Many valuable products during biosynthesis pathways can originate from the acetyl-CoA node located in the center of metabolic network, and therefore efficient conversion of acetate to acetyl-CoA is of great significance.

Coomassie brilliant blue-stained SDS-PAGE analysis of purified proteins. Purified ACS, ACS1 and ACS2, Lane M protein molecular weight marker (kD). The positions corresponding to the overexpressed proteins were consistent with their size

Kinetic analysis of acetyl-CoA synthetase. a Enzyme activity comparison of ACS, ACS1, and ACS2. 0.017 nmol (each) of ACS, ACS1, and ACS2 were used to catalyze the reactions at pH 7.4 and 37 °C, respectively. Date represent mean ± standard deviation (N = 3). b Effects of pH on enzymatic activity of His6-tagged ACS2 proteins. The reactions were performed at 37 °C. 100% corresponds to the enzyme activity at the condition of pH7.0 (Tris-HCl). c Effects of temperature on enzymatic activity of His6-tagged ACS2 proteins. The reactions were performed at the condition of pH 7.4 (Tris-HCl). 100% corresponds to the enzyme activity at 37 °C
The optimal pH of ACS2 was tested in Na2HPO4-KH2PO4 and Tris-HCl buffers with different pH (pH 5, 6, 7 and 8 and pH 7, 8 and 9), respectively. The optimal pH for ACS2 activity was about pH 7.0 (Fig. 3b). To determine the optimal temperature for ACS2, the reactions were incubated at a series of temperatures (20, 25, 30, 35, 37, 40, 45, 50 and 55 °C). The results showed that the highest enzyme activity was identified at about 37 °C and enzyme activity was fairly stable over the temperature range of 25-45 °C (Fig. 3c).
Effects of individual protein concentration on the phloroglucinol reactions
The ACS2, accA, accB, accC, accD and PhlD were prepared, and the purified proteins were analyzed by SDS-PAGE. Enzyme activity analysis showed that all the purified enzymes were active (Table 2). A sufficient amount of protein was collected in order to carry out in vitro reaction.
To analyze the sensitivity of the concentration of each enzyme, ACS2, ACC and PhlD were individually titrated into the in vitro system described above. The concentration of phloroglucinol formation in vitro was quantified when the reaction was terminated at 10 h. Titration results revealed that three enzymes in the biosystem were divided into two categories. ACS2 and ACC enhanced the rate of phloroglucinol synthesis in a dose-dependent manner (Fig. 4a and b). ACC had a more remarkably effect on the in vitro synthesis system, whereas increased ACS2 concentration has relatively less effect. The highest achievable concentration of phloroglucinol was about 170 mg/L when the concentration of ACC varied from 1.5 μM to 10 μM. In contrast, PhlD did not obviously influence the rate of phloroglucinol synthesis, although their individual concentration was varied relative to the reference concentration of 1.5 μM (Fig. 4c). Therefore, the catalyzed reaction of ACC is the rate-limiting step during synthesis of phloroglucinol from acetate. The similar result was also observed during fatty acid synthesis in vivo in the previous study which showed that up-regulation of ACC activity can increase the rate of fatty acid biosynthesis [27]. It is reported that the rate of fatty acid biosynthesis can be increased by 100-fold when ACC was titrated into the cell-free extract from E. coli [31], thus high rate of fatty acid synthesis is realizable by increasing the activity or quantity of acetyl-CoA carboxylase. Our results showed the similar importance of acetyl-CoA carboxylase during synthesis of phloroglucinol from acetate in vitro.

Titration of individual enzyme: a ACS2, each assay mixture included 1.5 μM ACC, 1.5 μM PhlD and different concentration of ACS2. b ACC, each assay mixture included 1.5 μM ACS2, 1.5 μM PhlD and different concentration of ACC. c PhlD, each assay mixture included 1.5 μM ACS2, 1.5 μM ACC and different concentration of PhlD. In all the reactions, substrate and cofactor concentrations were as follows: 10 mM potassium acetate, 30 mM ATP, 3 mM CoA
In vitro synthesis of phloroglucinol from acetate
Based on the results of titration studies, an optimal molar ratio of approximately 3 ACS2 : 5 ACC : 1 phlD (6 μM : 10 μM : 2 μM) was observed under the reaction conditions of 30 °C for 10 h. Finally, 10 mM acetate was converted into 2.93 mM (369.18 mg/L) phloroglucinol at the optimized condition (Fig. 5a). Therefore the optimized molar protein ratio is beneficial to in vitro synthesis system.

Production of phloroglucinol at a ACS2:ACC:PhlD molar ratio of 3:5:1 (the optimized condition: 6.0 μM ACS2, 10.0 μM ACC and 2.0 μM PhlD). a Substrate and cofactor concentrations were 10 mM potassium acetate, 30 mM ATP, 3 mM CoA. b Substrate and cofactor concentrations were 20 mM potassium acetate, 60 mM ATP, 6 mM CoA
The synthesis process required ATP consumption and CoA cycle, and thus the ATP and CoA concentrations might influence the in vitro biosynthetic process. ATP and CoA concentrations would be double in order to extend the duration of phloroglucinol production in the reaction system. As shown in Table 3 and Fig. 5a, the results showed that ATP and CoA concentration have little influence on the phloroglucinol synthesis at the concentration of 10 mM potassium acetate. Under the condition of double concentration, the effect of varying the initial acetate concentration was further examined. Finally, 20 mM acetate was converted into 5.87 mM (739.13 mg/L) phloroglucinol at the optimized condition (Table 2, Fig. 5b), while 40 mM acetate was only converted into 4.17 mM (525.42 mg/L) phloroglucinol. As shown in Table 2, the final amount and yield of phloroglucinol decreased when the initial acetate concentration increased to 40 mM. The results indicated that higher substrate may have many negative impacts on the in vitro synthesis system. The further increase of efficiency and productivity will likely require a coordinated increase at the levels of multiple proteins.
In this study, the comparison of the total amounts of transformation and production revealed the high performance of the enzymatic production system. The system shows a much higher yield in comparison to fermentation processes with yields of about 15% [2].
Although the results presented in this work are very promising, several problems should be paid attention. Typically, the productivity was low due to the inhibition of substrate, intermediate or product and the stability of the enzymes was low. The total productivity of the in vitro biosystem is still low (<1 g/L) compared to the reported fermentation processes with productivity of about 4 g/L [2]. The in vitro biosynthetic process for phloroglucinol still needs to further optimize reaction and condition in order to realize the technological applicability. To decrease costs, in vitro biosystem should be operated continuously which can realize the minimal addition of feedstock chemicals. Considering that the consumption of expensive ATP and CoA will increase production costs, low-cost ATP regeneration module need to be designed, and also low-cost and high-stability CoA analogues need to be explored or synthetized [32]. Another possible solution is the construction of efficient enzyme cascade and substrate channeling for in vitro biosynthesis in order to overcome the typical disadvantages for in vitro biotransformation [5, 33] (e.g. the inhibition by the substrate, intermediate, and/or product, and degeneration and diffusive losses of intermediate or cofactors in the reaction). For example, the enzyme cascades were constructed by attaching enzymes to DNA scaffold in the previous work [34], leading to the high reaction rate. It is also important to develop multi-enzyme immobilization cascades and stable enzymes for long-running of in vitro biosynthesis.
Conclusions
This study was the first successful attempt to synthesize phloroglucinol via in vitro system using acetate as the only carbon source. In comparison to fermentation processes from glucose, a novel approach to produce phloroglucinol from acetate was achieved, which showed a high yield of near 65% through the in vitro biosynthetic process. Considering that the proportion of substrate consumption could be 50% or even more of the total production costs, the overall economy primarily depend on the efficiency of substrate utilization in biochemical processes. In this study, the use of acetate as substrate which is a kind of renewable material provides the potential to reduce the costs of phloroglucinol production. However, some problems need to be solved in the batch production of phloroglucinol. Most notable one is that the used enzymes were not cost effective and recycled. Generally, low costs of enzyme can be realized by discovering stable enzymes suitable for long-term production, designing methods for recycling the enzymes, and developing low-cost methods for protein purification. Therefore, it is suggested that the efforts to develop in vitro biotechnology for the synthesis of bio-based chemicals are further needed.
In this work, the simple artificial multi-enzyme cascade reaction was constructed in an in vitro system based on acetate, and other important bio-based chemicals and bioenergy can also be produced based on the in vitro flexible biosynthesis system. Acetyl-CoA is a central metabolite precursor during biological metabolic processes and can be transformed into other industrial chemicals (e.g. 3-hydroxypropionate and fatty acid) [25, 35], and therefore the in vitro biosynthetic technology based on acetate has the potential to work as a versatile synthetic platform. In brief, in vitro synthesis of phloroglucinol from acetate could be an interesting model system for biosynthesis of other chemicals.
Abbreviations
- ACC:
-
Acetyl-CoA carboxylase
- ACS:
-
Acetyl-CoA synthetase
- AMP:
-
Acetyl-adenosine monophosphate
- ATP:
-
Adenosine triphosphate
- IPTG:
-
Isopropyl β-d-thiogalactoside
- KGDH:
-
α-ketoglutarate dehydrogenase
- PhlD:
-
Phloroglucinol synthase
References
Singh IP, Sidana J, Bansal P, Foley WJ. Phloroglucinol compounds of therapeutic interest: global patent and technology status. Expert Opin Ther Pat. 2009;19(6):847–66.
Cao Y, Jiang X, Zhang R, Xian M. Improved phloroglucinol production by metabolically engineered Escherichia coli. Appl Microbiol Biotechnol. 2011;91(6):1545–52.
Achkar J, Xian M, Zhao H, Frost JW. Biosynthesis of phloroglucinol. J Am Chem Soc. 2005;127(15):5332–3.
Zhang YHP, Myung S, You C, Zhu Z, Rollin JA. Toward low-cost biomanufacturing through in vitro synthetic biology: bottom-up design. J Mater Chem. 2011;21(47):18877–86.
Rieckenberg F, Ardao I, Rujananon R, Zeng A-P. Cell-free synthesis of 1, 3-propanediol from glycerol with a high yield. Eng Life Sci. 2014;14(4):380–6.
Opgenorth PH, Korman TP, Bowie JU. A synthetic biochemistry module for production of bio-based chemicals from glucose. Nat Chem Biol. 2016;12(6):393–5.
Gao C, Li Z, Zhang L, Wang C, Li K, Ma C, Xu P. An artificial enzymatic reaction cascade for a cell-free biosystem based on glycerol. Green Chem. 2015;17(2):804–7.
Guterl JK, Garbe D, Carsten J, Steffler F, Sommer B, Reiße S, Kettling U. Cell-Free Metabolic Engineering: Production of Chemicals by Minimized Reaction Cascades. ChemSusChem. 2012;5(11):2165–72.
You C, Shi T, Li Y, Han P, Zhou X, Zhang YHP. An in vitro synthetic biology platform for the industrial biomanufacturing of myo-inositol from starch. Biotechnol Bioeng. 2017. doi:10.1002/bit.26314.
Henstra AM, Sipma J, Rinzema A, Stams AJ. Microbiology of synthesis gas fermentation for biofuel production. Curr Opin Biotechnol. 2007;18(3):200–6.
Ruan Z, Zanotti M, Wang X, Ducey C, Liu Y. Evaluation of lipid accumulation from lignocellulosic sugars by Mortierella isabellina for biodiesel production. Bioresour Technol. 2012;110:198–205.
Zhang YHP. Reviving the carbohydrate economy via multi-product lignocellulose biorefineries. J Ind Microbiol Biotechnol. 2008;35(5):367–75.
Chen Y, Cheng JJ, Creamer KS. Inhibition of anaerobic digestion process: a review. Bioresour Technol. 2008;99(10):4044–64.
Fleisch TH, Sills RA, Briscoe MD. Emergence of the gas-to-liquids industry: a review of global GTL developments. J Nat Gas Chem. 2002;11:1–14.
Periana RA, Mironov O, Taube D, Bhalla G, Jones CJ. Catalytic, oxidative condensation of CH4 to CH3COOH in one step via CH activation. Science. 2003;301(5634):814–8.
Chi Z, Zheng Y, Ma J, Chen S. Oleaginous yeast Cryptococcus curvatus culture with dark fermentation hydrogen production effluent as feedstock for microbial lipid production. Int J Hydrogen Energy. 2011;36(16):9542–50.
Li Y, Huang B, Wu H, Li Z, Ye Q, Zhang YHP. Production of succinate from acetate by metabolically engineered Escherichia coli. ASC Synth Biol. 2016;5(11):1299–307.
Xiao Y, Ruan Z, Liu Z, Wu SG, Varman AM, Liu Y, Tang YJ. Engineering Escherichia coli to convert acetic acid to free fatty acids. Biochem Eng J. 2013;76:60–9.
Yang J, Nie Q. Engineering Escherichia coli to convert acetic acid to β-caryophyllene. Microb Cell Fact. 2016;15:74.
Mazumdar S, Clomburg JM, Gonzalez R. Escherichia coli strains engineered for homofermentative production of D-lactic acid from glycerol. Appl Environ Microbiol. 2010;76(13):4327–36.
Fuchs G, Berg IA. Unfamiliar metabolic links in the central carbon metabolism. J Biotechnol. 2014;192:314–22.
Rao G, Lee JK, Zhao H. Directed evolution of phloroglucinol synthase PhlD with increased stability for phloroglucinol production. Appl Microbiol Biotechnol. 2013;97(13):5861–7.
van den Berg MA, de Jong-Gubbels P, Kortland CJ, van Dijken JP, Pronk JT, Steensma HY. The two acetyl-coenzyme A synthetases of Saccharomyces cerevisiae differ with respect to kinetic properties and transcriptional regulation. J Biol Chem. 1996;271(46):28953–9.
Davis MS, Solbiati J, Cronan JE. Overproduction of acetyl-CoA carboxylase activity increases the rate of fatty acid biosynthesis in Escherichia coli. J Biol Chem. 2000;275(37):28593–8.
Liu C, Ding Y, Zhang R, Liu H, Xian M, Zhao G. Functional balance between enzymes in malonyl-CoA pathway for 3-hydroxypropionate biosynthesis. Metab Eng. 2016;34:104–11.
Choi-Rhee E, Cronan JE. The biotin carboxylase-biotin carboxyl carrier protein complex of Escherichia coli acetyl-CoA carboxylase. J Biol Chem. 2003;278(33):30806–12.
Cronan JE, Waldrop GL. Multi-subunit acetyl-CoA carboxylases. Prog Lipid Res. 2002;41(5):407–35.
Lin H, Castro NM, Bennett GN, San KY. Acetyl-CoA synthetase overexpression in Escherichia coli demonstrates more efficient acetic acid assimilation and lower acetic acid accumulation: a potential tool in metabolic engineering. Appl Microbiol Biotechnol. 2006;71(6):870–4.
Sakurai K, Arai H, Ishii M, Igarashi Y. Transcriptome response to different carbon sources in Acetobacter aceti. Microbiology. 2011;157(3):899–910.
Sakurai K, Arai H, Ishii M, Igarashi Y. Changes in the gene expression profile of Acetobacter aceti during growth on ethanol. J Biosci Bioeng. 2012;113(3):343–8.
Liu T, Vora H, Khosla C. Quantitative analysis and engineering of fatty acid biosynthesis in E. coli. Metab Eng. 2010;12(4):378–86.
Bibart RT, Vogel KW, Drueckhammer DG. Development of a second generation coenzyme A analogue synthon. J Org Chem. 1999;64(8):2903–09.
Ardao I, Hwang ET, Zeng AP. In vitro multienzymatic reaction systems for biosynthesis. In: Zeng AP, editor. Fundamentals and application of new bioproduction systems. Springer: Berlin Heidelberg; 2013. p. 153–84.
Wilner OI, Weizmann Y, Gill R, Lioubashevski O, Freeman R, Willner I. Enzyme cascades activated on topologically programmed DNA scaffolds. Nat Nanotechnol. 2009;4(4):249–54.
Yu X, Liu T, Zhu F, Khosla C. In vitro reconstitution and steady-state analysis of the fatty acid synthase from Escherichia coli. Proc Natl Acad Sci USA. 2011;108(46):18643–8.
Alves J, Westling L, Peters EC, Harris JL, Trauger JW. Cloning, expression, and enzymatic activity of Acinetobacter baumannii and Klebsiella pneumoniae acetyl-coenzyme A carboxylases. Anal biochem. 2011;417(1):103–11.
Acknowledgements
Not applicable.
Funding
This work was sponsored by National Natural Science Foundation of China (21376255 and 21572242) and Taishan Scholars Climbing Program of Shandong (tspd20150210).
Availability of data and materials
The datasets supporting the conclusions of this article are included within the manuscript. Other specific datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Authors’ contributions
MX, HL and RZ developed the idea for the study. RZ and MX designed the research, did the literature review, and prepared the manuscript. RZ, WL, YC and XX did the lab work, plasmid construction, strain cultivation, protein purification, in vitro reaction and product detection. RZ and YC drafted the initial manuscript together and all authors contributed in analyzing the data and writing the draft. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All authors agreed to publish this article.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, R., Liu, W., Cao, Y. et al. An in vitro synthetic biosystem based on acetate for production of phloroglucinol. BMC Biotechnol 17, 66 (2017). https://doi.org/10.1186/s12896-017-0376-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12896-017-0376-z