
Abstract
Background
Long non-coding RNAs (lncRNAs) play a fundamental role in colorectal cancer (CRC) development, however, lncRNA expression profiles in CRC and its precancerous stages remain to be explored. We aimed to study whole genomic lncRNA expression patterns in colorectal adenoma–carcinoma transition and to analyze the underlying functional interactions of aberrantly expressed lncRNAs.
Methods
LncRNA expression levels of colonic biopsy samples (20 CRCs, 20 adenomas (Ad), 20 healthy controls (N)) were analyzed with Human Transcriptome Array (HTA) 2.0. Expression of a subset of candidates was verified by qRT-PCR and in situ hybridization (ISH) analyses. Furthermore, in silico validation was performed on an independent HTA 2.0, on HGU133Plus 2.0 array data and on the TCGA COAD dataset. MiRNA targets of lncRNAs were predicted with miRCODE and lncBase v2 algorithms and miRNA expression was analyzed on miRNA3.0 Array data. MiRNA-mRNA target prediction was performed using miRWALK and c-Met protein levels were analyzed by immunohistochemistry. Comprehensive lncRNA-mRNA-miRNA co-expression pattern analysis was also performed.
Results
Based on our HTA results, a subset of literature-based CRC-associated lncRNAs showed remarkable expression changes already in precancerous colonic lesions. In both Ad vs. normal and CRC vs. normal comparisons 16 lncRNAs, including downregulated LINC02023, MEG8, AC092834.1, and upregulated CCAT1, CASC19 were identified showing differential expression during early carcinogenesis that persisted until CRC formation (FDR-adjusted p < 0.05). The intersection of CRC vs. N and CRC vs. Ad comparisons defines lncRNAs characteristic of malignancy in colonic tumors, where significant downregulation of LINC01752 and overexpression of UCA1 and PCAT1 were found. Two candidates with the greatest increase in expression in the adenoma-carcinoma transition were further confirmed by qRT-PCR (UCA1, CCAT1) and by ISH (UCA1). In line with aberrant expression of certain lncRNAs in tumors, the expression of miRNA and mRNA targets showed systematic alterations. For example, UCA1 upregulation in CRC samples occurred in parallel with hsa-miR-1 downregulation, accompanied by c-Met target mRNA overexpression (p < 0.05).
Conclusion
The defined lncRNA sets may have a regulatory role in the colorectal adenoma-carcinoma transition. A subset of CRC-associated lncRNAs showed significantly differential expression in precancerous samples, raising the possibility of developing adenoma-specific markers for early detection of colonic lesions.
Similar content being viewed by others


Background
The incidence and mortality of colorectal cancer (CRC) are continuously increasing with approximately 1.4 million new CRC cases and 700.000 registered deaths worldwide [1]. Therefore, identification of molecular markers of CRC that might enhance the objective classification or the early detection of the disease remains highly relevant, as CRC is one of the most curable cancers if detected early [2]. Besides the commonly investigated molecular markers, such as DNA mutations, DNA methylation or mRNA expression alterations, interest is growing in an emerging novel class of non-coding RNAs, long non-coding RNAs (lncRNAs) [3,4,5].
LncRNAs are defined as transcripts longer than 200 base pairs without an open reading frame [6]. This class of non-coding RNAs represents a diverse group with known and predicted functions for gene expression regulation [7,8,9]. According to experimental data, lncRNAs can interact with DNA, RNA and also with proteins and can either promote or inhibit transcription [10]. In contrast to miRNA-mediated regulation, the function and mechanism of action of certain lncRNAs can be diverse; lncRNAs are involved in genomic imprinting, transcriptional regulation, protein scaffolding, maintenance of hetero-euchromatin balance, can function as a miRNA sponge, and also mediate disease-derived alterations of mRNAs, miRNAs and proteins [9, 11]. Dysregulated lncRNAs are known to contribute to CRC formation through the disruption of various signaling cascades including Wnt/β-catenin, EGFR/IGF-IR (KRAS and PI3K pathways), TGF-β, p53 and Akt signaling pathways, and also via influencing the epithelial-mesenchymal transition program [12]. To date, 172.216 human lncRNA transcripts have been identified according to NONCODEv5 database [13] and their number continues to increase. Recent studies have demonstrated that several lncRNAs have a key regulatory role in various diseases including CRC [14]. During the carcinogenesis, lncRNA expression alterations affect major biological processes, and therefore. lncRNAs are considered as powerful molecular markers and also potential therapeutic targets in various cancers [3, 15].
In the present study, we aimed to determine the differentially expressed lncRNAs at the whole genome level focusing on the colorectal adenoma-carcinoma transition to identify lncRNAs showing specific alterations only in CRC tissue and common lncRNA patterns characteristic both in benign and malignant colonic neoplasms. Furthermore, we validated the lncRNA expression alterations by qRT-PCR, in situ hybridization, on an independent HTA 2.0 dataset, HGU133 Plus2.0, and The Cancer Genome Atlas (TCGA) Colon adenocarcinoma (COAD) datasets. We also report an association between the dysregulated lncRNAs and mRNA, miRNA and protein expression.
Methods
Sample collection
During routine screening endoscopy examinations biopsy samples were collected from patients with untreated colorectal cancer (n = 20; Astler-Coller modified Dukes B-D), with colorectal adenomas (n = 20; tubulovillous: n = 9, tubular: n = 11; with low-grade dysplasia: n = 18, with high-grade dysplasia: n = 2), and from healthy donors (n = 20). Healthy donors had been referred to the outpatient clinic with constipation, rectal bleeding or chronic abdominal pain. Ileocolonoscopy showed normal macroscopic appearance, and no abnormal histologic changes were detected in biopsy samples. None of the healthy patients had familial history of CRC. Biopsies were immediately put in RNALater stabilization reagent (Qiagen GmbH, Hilden, Germany) and stored at − 80 °C. Written informed consent was provided by all patients. The study was approved by the local ethics committee (Semmelweis University Regional and Institutional Committee of Science and Research Ethics; Nr.: ETT TUKEB 23970/2011). The clinicopathological data for the analyzed sample set are reported in Table 1.
RNA isolation, quality and quantity analyses
Total RNA including the microRNA (miRNA) fraction was isolated with High Pure miRNA isolation kit (Cat no: 05080576001, Roche, Penzberg, Germany) using the one-column protocol according to the manufacturer’s recommendation. RNA quantity was measured on a Qubit fluorometer with the Qubit™ RNA Assay Kit (Life Technologies, Eugene, OR, USA) and also on the NanoDrop-1000 instrument (Thermo Fisher Scientific Inc., Waltham, USA) to determine the purity values (OD260/280, OD260/230). RNA quality analysis was performed on an Agilent Bioanalyzer microcapillary electrophoresis system with the RNA 6000 Pico Kit (Agilent, Santa Clara, CA, USA).
Microarray experiment
For lncRNA expression profiling Human Transcriptome Array 2.0 (HTA 2.0) experiments were performed with 100 ng total RNA sample input according to the manufacturer’s instructions. For single-stranded complementary DNA (sscDNA) synthesis 15 μg complementary RNA (cRNA) was used and 5.5 μg fragmented and labeled sscDNA sample was hybridized to Human Transcriptome Array 2.0 microarrays (Affymetrix, Santa Clara, CA, USA) for 16 h at 45 °C with 60 rpm rotation in the Hybridization Oven (Affymetrix). Microarrays were washed and stained with GeneChip® Hybridization, Wash, and Stain Kit reagents according to the FS450_0001 protocol using the Fluidics Station 450 instrument (Affymetrix). Scanning was performed with GeneChip Scanner 3000 (Affymetrix). The dataset was uploaded to the Gene Expression Omnibus (GEO) data repository: GEO ID GSE100179. Raw CEL file normalization was performed with the Expression Console (Affymetrix, version: 1.4.1.46) with Gene Level – Default: RMA algorithm in order to generate .chp files. LncRNA expression level comparisons were made for adenoma vs. healthy, CRC vs. adenoma and CRC vs. healthy samples (FDR adjusted p < 0.05; log2FC ≤ − 1 or log2FC ≥ 1). LncRNA annotation and classification (with the inclusion of non-coding, 3 prime overlapping ncRNA, antisense, lincRNA, sense intronic, sense overlapping and bidirectional lncRNA subclasses of lncRNAs) were performed using the BioMart data mining tool on the basis of the current Ensembl database (Ensembl release 93 - July 2018 using GRCh38.p12 human genome version) and was further confirmed with the Netaffx database.
qRT-PCR validation
Certain lncRNAs with significant expression alterations in the three comparisons were further studied by qRT-PCR. 500 ng total RNA was reverse transcribed with TaqMan MicroRNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA). Absolute quantification was performed using LightCycler 480 Probes Mastermix (2x), Resolight dye (40x), and primers (200 nM final concentration; Table 2.) with 5 ng cDNA/reaction with the following thermal cycling conditions: enzyme activation: 95 °C for 10 min, 45 cycles of amplification: 95 °C for 10 s, Tannealing (CCAT1: 57 °C, LINC00261: 59 °C, UCA1: 61 °C) for 30 s and signal detection at 72 °C for 1 s and cooling at 40 °C for 30 s. As the normalization of expression during lncRNA quantification is critical, absolute quantification was performed. After PCR amplification of standard samples, amplicons were analyzed by 2% agarose gel electrophoresis and were purified with Agencourt AmpureXP Beads (Beckman Coulter, Brea, USA) according to the standard PCR purification protocol. The expression levels of lncRNAs in the analyzed samples were quantified after establishing standard curves with serial dilution of standard samples (with 2, 10,1 10,2 10,3 3 × 10,3 104 molecules/reaction) calculated based on Qubit fluorometry results. Finally, we validated the standard curves of each amplicon and analyzed the amplification efficiency, R,2 and the slope. LightCycler 480 absolute quantification software was used to calculate copy numbers in the analyzed samples.
In situ hybridization with automated RNAscope and immunofluorescence
The in situ hybridization (ISH) analyses were performed in collaboration with Boye Schnack Nielsen, Bioneer A/S, Hørsholm, Denmark. Five μm thick FFPE sections were processed for RNAscope ISH in a Ventana Discovery Ultra instrument (Roche, Basel, Switzerland) [16]. The following RNAscope probes were obtained from ACD, Biotechne (Newark, CA, USA): UCA1 (Urothelial cancer associated 1, NR_015379.3, target region: 659 − 2289, 20 zz pairs), dapB (a Bacillus subtilis gene, 414 – 862, 10 zz pairs), and PPIB (Cyclophilin B, 139 – 989, 16 zz pairs), and incubated on tissue sections as recommended by the manufacturer. The RNAscope probes were detected using the HRP kit and Discovery-rhodamine substrate (Roche). For cytokeratin immunofluorescence, the AE1/3 mouse monoclonal antibody (Dako-Agilent, Glostrup, Denmark) was used at 1:200 and detected with Alexa-488 conjugated anti-mouse Ig (Jackson Immunoresearch, West Grove, PA). The stained sections were mounted with a DAPI-containing anti-fade solution, ProLong Gold (Thermo Fisher Scientific, Waltham, MA, USA). Digital whole slides were obtained with a Pannoramic Confocal (3DHISTECH Ltd., Budapest, Hungary) slide scanner using a 40x objective, and the localization of expression was examined in these, as well as representative images were acquired from these digital slides.
In silico validation
lncRNA expression on an independent HTA 2.0 dataset
Our HTA 2.0 results were compared with the results of Condorelli et al. from GEO database (GSE73360) [17] of 37 CRC biopsies from 27 patients, and 19 adjacent normal mucosa biopsies (at distance of 3-6 cm from the tumor) that were collected directly after surgical resection. Linear correlation was analyzed between the two datasets.
lncRNA expression on HGU133Plus 2.0 dataset
For in silico validation, expression levels of lncRNAs were also analyzed on GSE37364 HGU133Plus 2.0 microarray (Affymetrix) dataset of 65 human colonic biopsy samples (27 CRCs and 38 normal donors without evidence of disease) [18]. Alignment of the probesets of the different platforms was performed using the BioMart data mining tool based on the current Ensembl database (Ensembl release 93 - July 2018 using GRCh38.p12 human genome version). Among the significant lncRNA expression alterations identified on HTA 2.0 arrays, 11 associated probesets could be found on the HGU133Plus2.0 arrays representing 10 lncRNAs. Linear correlation between the two microarray platforms was also analyzed.
lncRNA expression on TCGA dataset
Selected lncRNAs showing altered expression on HTA 2.0 arrays were analyzed in silico using The Cancer Genome Atlas (TCGA) colon & rectum adenocarcinoma gene expression RNAseq dataset. We have used the COAD (n = 463) subset of the lncRNA dataset from Yan et al. which was compiled from 5.037 human tumor specimens across 13 cancer types in TCGA [19, 20]. Out of the 37 lncRNAs identified in our cohort showing significant under- or overexpression in the CRC vs. N comparison, 16 could be detected in TCGA data. Linear correlation was analyzed between the datasets.
LncRNA-mRNA-miRNA co-expression analysis
The HTA 2.0 microarray provides expression data of lncRNAs and mRNAs allowing the parallel analysis of both from individual samples. In order to assess lncRNA/mRNA relationships, co-expression networks were constructed based on Pearson-correlation calculation. The top 50 negatively and top 50 positively associated, predicted mRNA targets of the significantly (p < 0.05) altered, qRT-PCR validated lncRNAs (UCA1 [TC19000279.hg.1, TC19002012.hg.1] and CCAT1 [TC08001627.hg.1]) were selected. These lncRNAs were visualized with the associated mRNAs by using igraph in the R environment and Gene Ontology (GO) functional analysis was performed using Netaffx database (Affymetrix). On the other hand, MIRCODE, lncBase v2 predicted and lncBase v2 experimental algorithms were used to predict miRNAs associated with UCA1 lncRNA. The expression of selected miRNAs was analyzed on the miRNA 3.0 microarray (GSE83924) dataset from Nagy et al. [21]. The miRWALK database was used for miRNA-mRNA target prediction involving 12 existing miRNA-target prediction algorithms (DIANA-microTv4.0, DIANA-microT-CDS, miRanda-rel2010, mirBridge, miRDB4.0, miRmap, miRNAMap, doRiNA, PicTar2, PITA, RNA22v2, RNAhybrid2.1 and Targetscan6.2) and containing experimentally verified data from existing resources (miRTarBase, PhenomiR, miR2Disease and HMDD) [22].
Immunohistochemistry
6 μm thick slides cut from tissue microarray (TMA) blocks of normal (n = 20), adenoma (n = 20) and CRC (n = 20) patients were deparaffinized and rehydrated. Antigen retrieval was performed in TRIS-EDTA buffer (pH 9.0) using a microwave oven (900 W for 10 min, 340 W for 40 min). Samples were incubated with anti-c-Met antibody (rabbit polyclonal, ab4193, Abcam, Cambridge, UK) diluted 1:800 for 60 min at 37 °C. EnVision + HRP system (Labeled Polymer Anti-Mouse, K4001, Dako) and diaminobenzidine-hydrogen peroxidase–chromogen substrate system (Cytomation Liquid DAB + Substrate Chromogen System, K3468, Dako) were applied followed by hematoxylin counterstaining. Slides were digitally scanned using the Pannoramic 250 Flash instrument (software version 1.11.25.0, 3DHISTECH Ltd., Budapest, Hungary), and analyzed with a Pannoramic Viewer (v. 1.11.43.0. 3DHISTECH Ltd.) digital microscope based on Q-score method; scored by multiplying the percentage of positive cells (P) by the intensity (I: + 3 (strong), + 2 (moderate), + 1 (weak), 0 (no staining)). Formula: Q = P x I; maximum = 300) as described earlier [23].
Statistical analysis
For data distribution analysis the Kolmogorov-Smirnov test was applied. Due to the normal distribution, Student’s t-test was applied for the pairwise comparison with Bonferroni and Hochberg correction. FDR adjusted p-values lower than 0.05 were considered as significant. Pearson-correlation was calculated and lncRNA-mRNA-miRNA co-expression network was constructed by igraph package in the R environment.
Results
Expression of known CRC-associated lncRNAs in the colorectal adenoma and carcinoma samples
As the expression of known CRC-associated lncRNAs has not been studied yet in precancerous adenoma samples, in the present study we aimed to analyze the adenoma-specific alterations with a special focus on adenoma-carcinoma transition.
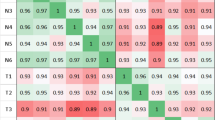
Previous comprehensive studies analyzing healthy colon and CRC tissue samples revealed differentially expressed lncRNAs, so-called CRC-associated lncRNA markers [24,25,26]. First, the expression of these literature-based lncRNA markers was studied on adenoma and CRC samples as part of our whole transcriptome analysis. A subset of lncRNAs from our Human Transcriptome Array 2.0 study (CCAT1, PVT1, CRNDE; LINC01021, LINC-ROR, UCA1, FTX, MEG3, LOC100289019) showed remarkable expression changes already in the precancerous colonic lesions (p < 0.01) (Fig. 1).

CRC-associated lncRNA expression in colorectal adenoma and in CRC samples. Intensity values on the color scale were as follows: red – high intensity, black – intermediate intensity, green – low intensity. A subset of literature-based colorectal cancer-associated lncRNAs showed significant expression difference already between normal and adenoma samples (p < 0.01)
Differentially expressed lncRNAs in the colorectal adenoma-carcinoma sequence progression
On the basis of the HTA 2.0 results, 54 lncRNAs were found to be differentially expressed along the colorectal adenoma-carcinoma sequence (FDR adjusted p-value< 0.05, log2FC ≥ 1 or log2FC ≤ − 1) (Additional file 1: Figure S1, Additional file 2: Table S1). In order to focus on the lncRNA marker candidates, whose dysregulation might be driver in the development of CRC, a group of 17 lncRNAs was identified as overlapping between adenoma vs. normal and CRC vs. normal comparisons showing differential expression early that persisted until CRC formation: LINC02023, MEG8, AC092834.1, AL365361.1, LINC02441, B3GALT5-AS1, THRB-IT1, LINC02535, AC140658.1, AC142086.4, AC019330.1 and LINC01133 were downregulated and CCAT1, CASC19, LINC02163, AC123023.1 and AC021218.1 were upregulated in adenoma and also in CRC samples compared to the healthy controls (FDR adjusted p < 0.05, log2FC ≤ − 1 or log2FC ≥ 1). Three lncRNAs (downregulated LINC01752, and overexpressed UCA1 and PCAT1) were detected in both CRC vs. N and CRC vs. Ad comparisons, suggesting that these changes might be early markers of colonic carcinogenesis (Fig. 2, Table 3).

A) Venn-diagram of the significantly altered lncRNAs in Ad vs. N, CRC vs. N and CRC vs. Ad comparisons (FDR adjusted p-value< 0.05; log2FC ≤ −1 or log2FC ≥ 1). Boxplot representation of the overlapping lncRNAs showing differential expression in B) CRC vs. N and CRC vs. Ad and C) Ad vs. N and CRC vs. N comparisons
The group of transcripts with altered expression only in Ad vs. N comparison contained 2 downregulated (XIST, PP7080) and 9 upregulated lncRNAs (top 5 with the highest logFC: AC124067.4, LINC01594, DLGAP1-AS2, LINC00261. AL365226.2) (Fig. 2a, Additional file 1: Figure S1/A, Additional file 2: Table S1). Further along the adenoma-carcinoma sequence, lncRNAs with altered expression between CRC and adenoma samples can play a role in the transition of dysplasia-carcinoma that contained 8 downregulated lncRNAs (top 5 with the lowest log2FC AL133370.1, LINC00261, AC005833.1, LINC01612, LINC01594) (Fig. 2a, Additional file 1: Figure S1/B, Additional file 2: Table S1). Among the lncRNAs showing significant expression alteration exclusively in the CRC vs. N comparison, 12 downregulated (top 5 with the lowest log2FC: SNHG14, MIR3936HG, AC124312.3, AL928768.1, AL359397.1) and 5 significantly upregulated lncRNAs (AL645939.4, AC126365.1, LINC00152, AC016831.1, LINC02474) were detected (Fig. 2a, Additional file 1: Figure S1/C, Additional file 2: Table S1).
Absolute quantification
By the use of absolute quantification, we were able to confirm the lncRNA expression alterations observed by HTA 2.0 analyses. CCAT1 and UCA1 showed upregulation in tumor samples compared to normal tissues (Fig. 3a). In the case of LINC00261, we observed upregulation in adenomas compared to normal controls and downregulation in CRC samples compared to adenomas, but these expression alterations were not significant (data not shown).

qRT-PCR and ISH analyses of selected lncRNAs. A) UCA1 and CCAT1 qRT-PCR absolute quantification on healthy normal (n = 20), adenoma (n = 20) and CRC (n = 20) biopsy samples *p < 0.05: UCA1: Ad vs. N, CCAT1: Ad vs. N, CRC vs. N; B) Representative UCA1 in situ hybridization results on normal (a-d), adenoma (e-h) and CRC (i-l) FFPE tissue samples. UCA1 expression is seen already in adenoma (full arrowhead) compared to monolayered normal adjacent (NAT) mucosa with no ISH signal (empty arrowhead) (m, n) and UCA1 is focally abundant in CRC (j). White signals represent autofluorescence in blood cells and vessels. Digital microscope slides were scanned with Pannoramic 250 Flash instrument (v1.11.25.0), and analyzed with a Pannoramic Viewer (v1.11.43.0) samples, scale bar: 100 μm (a-m) and 40 μm (n)
In situ hybridization
In normal colonic FFPE tissue samples, no UCA1 ISH signal was detected, whereas UCA1 ISH signal was observed in adenoma tissue and an even stronger signal was detected in colorectal carcinoma samples, in accordance with our qRT-PCR results. The UCA1 ISH signal was localized predominantly in the epithelial cells in adenoma and carcinoma tissue samples (Fig. 3b).
In silico validation on an independent HTA 2.0 dataset
Our HTA 2.0 results were compared with an independent HTA 2.0 dataset [17]. The significantly altered lncRNA set in the CRC vs. normal comparison showed the same tendency, a high positive correlation was found with the independent GSE73360 dataset in the CRC vs. NAT comparison (R2 = 0.7076) (Fig. 4a, Additional file 3: Table S2).

In silico validation of HTA 2.0 data on independent datasets. A) Linear correlation with an independent HTA 2.0 GEO dataset (GSE73360) [16] [LINC02023, AC092834.1, SNHG14, AL365361.1, AC140658.1, AC142086.4, AC019330.1, THRB-IT1, LINC02535, B3GALT5-AS1, LINC02441, MIR3936HG, AC124312.3, AL928768.1, AL359397.2, LINC01752, AL355922.2, AC087379.1, AC011891.1, AC044802.2, LINC01133, MEG8, CDKN2B-AS1, AC025423.4, LINC01268, CCAT1, UCA1, AC123023.1, CASC19, AL645939.4, AC126365.1, LINC00152, AC016831.1, AC021218.1, LINC02474, PCAT1, LINC02163], B) Linear correlation with an independent HGU133 Plus 2.0 dataset (GSE37364) [17] [UCA1, LINC00152, AC016831.1 LINC02023, THRB-IT1, LINC02535, SNHG14, AC124312.3, AL928768.1, CDKN2B-AS1] C) Linear correlation with TCGA COAD dataset [18] [LINC01133, AL365361.1, LINC00152, LINC02023, LINC02163, LINC01268, CASC19, CDKN2B-AS1, AC087379.1, LINC02441, SNHG14, AC124312.3, UCA1, LINC01752, CCAT1, AL928768.1]. Our own HTA 2.0 data (GSE100179) are represented on X-axis
In silico validation on an independent HGU133Plus2.0 microarray dataset
In order to assess the observed altered lncRNA expression using independent hybridization-based results, HTA 2.0 results were compared for lncRNAs available on the HGU133Plus2.0 microarray platform (GSE37364) [18]. According to the above-mentioned analysis, the expression alterations of 3 significantly upregulated (UCA1, LINC00152, AC016831.1) and 7 downregulated (LINC02023, THRB-IT1, LINC02535, SNHG14, AC124312.3, AL928768.1, CDKN2B-AS1) lncRNAs were verified in CRCs compared to the healthy controls (p < 0.01). The expression differences between the HTA 2.0 and HGU133Plus2.0 platforms showed a high positive correlation (R2 = 0.7628) (Fig. 4b, Additional file 3: Table S2).
In silico validation on TCGA COAD RNASeq dataset
Out of the 37 lncRNAs identified in our cohort showing significantly differential expression in the CRC vs. N comparison, 16 were detected on the TCGA COAD dataset [19]. A very high positive correlation (R2 = 0.9029) was observed between the datasets (Fig. 4c, Additional file 3: Table S2).
Co-expression analysis of differentially expressed lncRNAs-mRNAs-miRNAs
mRNAs negatively correlated with CCAT1 are involved in G1/S transition of mitotic cell cycle, G2/M transition of mitotic cell cycle functions, while the positively correlated mRNAs play a role e.g. in cell division and cell cycle regulation. The negatively co-expressed mRNAs with UCA1 have a negative regulatory role of transcription from RNA polymerase II promoter, angiogenesis, DNA methylation, while the positively correlated mRNAs are involved in mitotic cytokinesis and apoptotic processes. The co-expression network of lncRNAs and mRNAs showed certain overlap between the mRNA targets of the selected lncRNAs (Fig. 5 , Additional file 4: Table S3).

Co-expression network of lncRNAs with associated mRNAs. qPCR validated lncRNAs (UCA1 [TC19000279.hg.1, TC19002012.hg.1], CCAT1 [TC08001627.hg.1]) are represented with orange rectangles, target mRNAs are represented with green rectangles in the network, where further associations between the mRNA targets are also represented. The network was visualized with igraph in R environment and Gene Ontology (GO) functional analysis was performed using TAC 4.0 software (Affymetrix)
UCA1 was upregulated in adenoma and also in CRC samples (Fig. 6a) and a potential interaction was predicted between UCA1 and hsa-miR-1 based on independent algorithms. In the GSE83924 dataset [21] hsa-miR-1 was downregulated in adenoma and CRC samples compared to normal colonic samples (p < 0.01) (Fig. 6b). MIRWALK validated target prediction revealed that c-Met mRNA is one of the targets of hsa-miR-1. On the other hand, co-expression analysis showed that c-Met was among the most positively co-expressed mRNAs with both IDs of UCA1 (TC19002012: rho = 0.7811; TC19000279.hg.1: rho = 0.7717).

A) UCA-1 lncRNA expression in healthy colon, colorectal adenoma, and CRC tissue samples; B) hsa-miR-1 expression on GSE83924 miRNA 3.0 array dataset [20] C) C-Met expression quantification and D) C-Met protein levels in the epithelial compartments of normal colon (D/1), adenoma (D/2) and CRC samples (D/3). Digital microscope slides were scanned with Pannoramic 250 Flash instrument (v1.11.25.0), and analyzed with a Pannoramic Viewer (v1.11.43.0), 40x magnification, scale bar: 50 μm) Box plots represent the median and standard deviation of the data (*p < 0.05)
Immunohistochemistry
In normal epithelium the c-Met expression was mild, diffuse and cytoplasmic (main scoring value: + 1; Q-score: 144.00 ± 49.29). Compared to normal samples, adenoma samples showed elevated, but not significantly increased (p = 0.31) epithelial protein expression (scoring values: + 1 and + 2, Q-score: 160.00 ± 29.51). In the epithelial compartment of CRC cases, heterogeneous, moderate/strong (scoring values: + 2 and + 3, Q-score: 201.66 ± 43.02) c-Met protein expression was observed which was significantly higher than in healthy controls (p < 0.05) (Fig. 6c, d).
Discussion
The present study aimed to identify differentially expressed lncRNAs at the whole genome level characteristic of colorectal adenoma and carcinoma tissue samples principally focusing on the adenoma-carcinoma transition. Besides the detection of single lncRNA alterations (by PCR-based methods, Northern blot, RNA-immunoprecipitation and in situ hybridization [27]), genome-wide analysis can be achieved by RNA-Seq [28] and also by microarrays, e.g. by Human Transcriptome Array 2.0, a high-resolution microarray platform detecting lncRNA, miRNA and mRNA levels in parallel from the same sample [29].
Certain lncRNAs were previously analyzed in colorectal diseases, but the focus of most of the studies was limited to one or a small set of lncRNAs [30,31,32]. The strength of our study was the inclusion of colorectal adenoma tissue samples along with the CRC cases in a genome-wide lncRNA expression analysis, in order to identify markers for the early detection of colorectal tumors. Based on the present study, 54 lncRNAs were found to be differentially expressed along the colorectal adenoma-carcinoma sequence that could be validated on independent microarray results and also on TCGA COAD dataset. A subset of lncRNAs already associated with CRC development showed remarkable expression changes in the precancerous colonic adenoma lesions.
The lncRNAs that play a key role during the malignant transformation in CRC development remain to be identified, and therefore, we further focused on the colorectal-adenoma transition. According to our microarray analysis, 12 lncRNAs were downregulated and 5 were upregulated both in adenoma and CRC samples compared to the healthy controls. These lncRNAs might have a key regulatory role during CRC formation, as their dysregulation could be detected already in adenomas, that persisted until the malignant transition. Among these possible key factors, CASC19 overexpression was reported in CRC tissue samples and it was associated with metastasis formation [33]; its knockdown resulted in reduced migration of RKO and Caco2 cells [33]. Upregulation of LINC02163 was also observed in CRC tissue samples by NGS technology [34]. In rectal adenocarcinoma cases Zhang et al. found an association between LINC02163 expression and overall survival [35]. LINC02163 overexpression was documented in gastric cancer tissues and cell lines; knockdown of LINC02163 resulted in reduced cell growth and invasion [36]. Upregulation of AC021218.2 (CRCAL-1) was detected in 3 pairs of CRC compared to their matched normal mucosa by RNA sequencing [37]. According to the current literature in this subset, AL365226.2, LINC02023, AC092834.1, LINC02441, B3GALT5-AS1, THRB-IT1, LINC02535, AC140658.1, and AC142086 have not been reported to be associated with CRC formation and these may serve as novel lncRNA markers of neoplastic processes of the colon.
As in the case of certain presumably ‘driver’ lncRNAs (e.g. MEG8) in the present study, a distinct expression tendency could be detected in adenoma and carcinoma samples, so that their dysregulation may not be a gradual event during cancer formation. It is well-known that up- or downregulation of lncRNAs can be restricted to a certain stage during cancer formation and therefore it is not evident that all differentially expressed RNAs show the same expression tendency in adenoma and carcinoma samples [21]. It is known, that lncRNAs show cell-specific expression in certain tumors [38] and the overall resultant expression found in biopsy samples can be influenced by the epithelial-stromal cell ratio which may differ between colorectal adenomas and adenocarcinomas. On the other hand, similarly to the findings on miRNA [21] and on DNA methylation levels [39], certain lncRNAs showed distinct expression alterations in adenomas and carcinomas that might be due to the counteracting cell-proliferation control pathways in adenomas that are dysregulated in carcinomas.
LncRNAs characteristic only in the malignant state could be identified in the intersection of CRC vs. N and CRC vs. Ad comparisons, where significant downregulation of LINC01752 and overexpression of PCAT1 and UCA1 were observed. Although the altered expression of these lncRNAs are hypothetically associated with malignancy in colonic tumors, due to the relatively low number of CRCs analyzed in the present study, no reliable further conclusion can be formed about the correlation of their expression levels with survival, metastasis or tumor stage data in our cohort. However, on the basis of literature data, overexpression of PCAT1 was suggested to be an independent prognostic factor for CRC [40] and its downregulation inhibited proliferation, blocked cell cycle transition, suppressed cyclin and c-myc expression and induced apoptosis [41]. Silencing of PCAT1 in Caco− 2 and HT-29 cell lines suppressed cell motility, invasiveness, sensitized the cells to 5-FU [42]. Human urothelial carcinoma associated 1 (UCA1) lncRNA has a role in cell proliferation, apoptosis, and cell cycle progression regulation and an increased UCA1 expression level was reported first in urothelial cancer [43] and also reported for breast cancer and CRC patients [44,45,46]. According to Ni et al., CRC patients with high UCA1 expression had a poor prognosis and UCA1 overexpression was found to be an independent predictor of CRC [47]. Exogenous expression of UCA1 enhanced tumorigenicity, invasive potential, and drug resistance in human bladder TCC BLS-211 cells [43]. Silencing of UCA1 in HCT116, HT29, SW480 and LoVo cell lines significantly decreased cell proliferation, while UCA1 overexpression promoted tumorigenicity in LoVo cells [48]. Silencing of UCA1 suppressed proliferation and metastasis and induced apoptosis of oral squamous cell carcinoma cell lines, which may be related to the activation of the WNT/β-catenin signaling pathway [49]. In a recent study, UCA1 expression was also detected in liquid biopsy samples, where Tao et al. reported that the elevated expression level in the tissue samples can also be detected in plasma samples of CRC patients [50]. The above-mentioned functions of UCA1 illustrate the complexity of its mechanism of actions and its diverse role in CRC development [51].
To date, there has been no data reported about UCA1 expression in colorectal adenoma patients. According to our results, UCA1 shows a gradual upregulation in the normal-adenoma-cancer sequence, thus besides its well-established malignancy-associated functions, it holds the possibility to be utilized as an early detection marker for precancerous lesions of the colon, that may contribute to reduce CRC-related deaths in the future.
Interestingly, a slight discrepancy was observed in UCA1 expression between microarray and qRT-PCR methods. By the two probe sets on HTA 2.0 microarray representing UCA1, all 36 transcripts can be detected, while in contrast, qRT-PCR primers used in the present study detect 4 transcripts (namely UCA1-213, UCA1-207, UCA1-201, and UCA1-228). The above-mentioned reason probably contribute to the discrepant expression results observed between the two platforms. Nevertheless, in situ hybridization probes could detect all UCA1 transcripts, and as an added advantage, this approach also provide information regarding lncRNA subcellular localization in colonic tissue samples. Tripathi et al. recently reported moderate UCA1 expression using ISH on colorectal cancer tissue [52]. To the best of our knowledge, our study is the first to report evidence of the upregulation of UCA1 lncRNA in colorectal adenomas and cancers predominantly observed in the epithelial cells adding additional evidence for UCA1 overexpression along colorectal cancer formation.
UCA1 has been reported to regulate miRNAs, such as miR-216b and hsa-miR-1 as a miRNA sponge and as a ceRNA of miR-204-5p among others, influencing growth promotion, apoptosis inhibition and 5-FU resistance [48, 53, 54]. The mutual inhibitory effect and the inverse expression of UCA1 and hsa-miR-1 have already been proven in bladder cancer and one functional interaction site was experimentally confirmed between hsa-miR-1 and UCA1 [54]. Furthermore, after the transfection of pre-miR-1 or following the treatment of UCA1 shRNA, cell proliferation and motility decreased in bladder cancer cell lines in an AGO2-mediated manner [54]. Downregulation of miR-1 in tumors compared to the corresponding normal tissue samples is associated with worse prognosis and its lower expression has been reported to reprogram cancer metabolism via the regulation of tumor glycolysis in colorectal cancer [55]. The interaction between hsa-miR-1 and HGFR (c-Met) is well-known in CRC [56]. On the basis of the comprehensive microarray analysis of Nagy et al. [21] and our present results, the downregulation of hsa-miR − 1 along the adenoma-carcinoma sequence was accompanied by c-MET overexpression. Upregulated UCA1 might exert its effect on c-Met protein and therefore can be associated with metastasis formation and CRC progression. Continuous overexpression of c-Met protein was observed along with the development of CRC [57, 58], which could be confirmed in our cohort, as well. On the basis of our co-expression analysis, c-Met was among the most positively co-expressed mRNAs of UCA1. Taken together, the association of UCA1, hsa-miR-1 and c-Met was predicted by independent algorithms in our study. However, further functional experimental verification is needed to prove the hypothetized interactions in CRC.
Besides the studies aiming at the identification of a signature marker group of lncRNA candidates differentially expressed between CRC and normal samples either on the basis of tissue or plasma samples (e.g. circulating XLOC_006844, LOC152578 and XLOC_000303) [59] or between high-risk and low-risk CRC patients with different overall survival [60], our study identified a subset of lncRNAs potentially playing a key role during the adenoma-carcinoma transition complementing the present knowledge about CRC formation.
Conclusion
In summary, the defined lncRNA sets may have a regulatory role in the adenoma-carcinoma transition. A subset of lncRNAs showed significant differential expression already in precancerous samples that persisted until CRC formation, raising the possibility of developing adenoma-specific markers in order to achieve early detection of colonic lesions.
Availability of data and materials
The dataset used and analyzed in the present study was uploaded to the Gene Expression Omnibus (GEO) data repository: GEO ID GSE100179 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE100179).
Abbreviations
- 5-FU:
-
5-Fluorouracil
- B3GALT5-AS1:
-
B3GALT5 Antisense RNA 1
- BACE2-IT1:
-
Beta-secretase intronic transcript 1
- CASC19:
-
Cancer susceptibility 19
- CCAT1:
-
Colon Cancer Associated Transcript 1
- CDKN2B-AS1:
-
CDKN2B Antisense RNA 1
- CRC:
-
Colorectal cancer
- cRNA:
-
Complementary RNA
- CRNDE:
-
Colorectal neoplasia differentially expressed
- DLGAP1-AS2:
-
DLGAP1 Antisense RNA 2
- GEO:
-
Gene Expression Omnibus
- HTA 2.0:
-
Human Transcriptome Array 2.0
- ISH:
-
In situ hybridization
- lncRNA:
-
Long non-coding RNA
- MEG3:
-
Maternally expressed 3
- MEG8:
-
Maternally expressed 8
- MIR3936HG:
-
MIR3936 Host Gene
- PCAT1:
-
Prostate cancer associated transcript 1
- PVT1:
-
Plasmacytoma variant translocation 1
- qPCR:
-
Quantitative polymerase chain reaction
- rpm:
-
Round per minute
- SNHG14:
-
Small nucleolar RNA host gene 14
- sscDNA:
-
Single-stranded complementary DNA
- TAC:
-
Transcriptome Analysis Console
- TCGA COAD:
-
The Cancer Genome Atlas database, Colon Adenocarcinoma dataset
- THRB-IT1:
-
Thyroid hormone receptor beta-intronic transcript 1
- TNM:
-
Tumor, node and metastasis staging
- TRIS-EDTA:
-
Tris(Hydroxymethyl)aminomethane-ethylenediamine tetraacetic acid
- UCA1:
-
Urothelial cancer associated transcript
- XACT:
-
X Active specific transcript
- XIST:
-
X-Inactive specific transcript
References
Gandomani HS, Yousefi SM, Aghajani M, Mohammadian-Hafshejani A, Tarazoj AA, Pouyesh V, Salehiniya H. Colorectal cancer in the world: incidence, mortality and risk factors. Biomed Res Ther. 2017;4(10):1656–75.
Cheah PY. Recent advances in colorectal cancer genetics and diagnostics. Crit Rev Oncol Hematol. 2009;69(1):45–55.
Qi P, Du X. The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Modern Pathol. 2013;26(2):155–65.
Bolha L, Ravnik-Glavac M, Glavac D. Long noncoding RNAs as biomarkers in Cancer. Dis Markers. 2017;2017:7243968.
Shi T, Gao G, Cao YL. Long Noncoding RNAs as Novel Biomarkers Have a Promising Future in Cancer Diagnostics. Dis Markers. 2016;2016:9085195.
Kornienko AE, Guenzl PM, Barlow DP, Pauler FM. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013;11:59.
Gutschner T, Diederichs S. The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol. 2012;9(6):703–19.
Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009;23(13):1494–504.
Bartonicek N, Maag JL, Dinger ME. Long noncoding RNAs in cancer: mechanisms of action and technological advancements. Mol Cancer. 2016;15(1):43.
Yang Y, Wen L, Zhu H. Unveiling the hidden function of long non-coding RNA by identifying its major partner-protein. Cell Bioscience. 2015;5:59.
Cao J. The functional role of long non-coding RNAs and epigenetics. Biol Procedures Online. 2014;16:11.
Yang Y, Junjie P, Sanjun C, Ma Y. Long non-coding RNAs in colorectal Cancer: progression and future directions. J Cancer. 2017;8(16):3212–25.
Fang SS, Zhang LL, Guo JC, Niu YW, Wu Y, Li H, Zhao LH, Li XY, Teng XY, Sun XH, et al. NONCODEV5: a comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 2018;46(D1):D308–14.
Wang J, Song YX, Ma B, Wang JJ, Sun JX, Chen XW, Zhao JH, Yang YC, Wang ZN. Regulatory roles of non-coding RNAs in colorectal Cancer. Int J Mol Sci. 2015;16(8):19886–919.
Gibb EA, Brown CJ, Lam WL. The functional role of long non-coding RNA in human carcinomas. Mol Cancer. 2011;10:38.
Anderson CM, Zhang B, Miller M, Butko E, Wu X, Laver T, Kernag C, Kim J, Luo Y, Lamparski H, et al. Fully automated RNAscope in situ hybridization assays for formalin-fixed paraffin-embedded cells and tissues. J Cell Biochem. 2016;117(10):2201–8.
Barresi V, Trovato-Salinaro A, Spampinato G, Musso N, Castorina S, Rizzarelli E, Condorelli DF. Transcriptome analysis of copper homeostasis genes reveals coordinated upregulation of SLC31A1,SCO1, and COX11 in colorectal cancer. FEBS Open Bio. 2016;6(8):794–806.
Galamb O, Wichmann B, Sipos F, Spisak S, Krenacs T, Toth K, Leiszter K, Kalmar A, Tulassay Z, Molnar B. Dysplasia-carcinoma transition specific transcripts in colonic biopsy samples. PLoS One. 2012;7(11):e48547.
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7.
Yan X, Hu Z, Feng Y, Hu X, Yuan J, Zhao SD, Zhang Y, Yang L, Shan W, He Q, et al. Comprehensive genomic characterization of long non-coding RNAs across human cancers. Cancer Cell. 2015;28(4):529–40.
Nagy ZB, Wichmann B, Kalmar A, Galamb O, Bartak BK, Spisak S, Tulassay Z, Molnar B. Colorectal adenoma and carcinoma specific miRNA profiles in biopsy and their expression in plasma specimens. Clin Epigenetics. 2017;9:22.
Dweep H, Sticht C, Pandey P, Gretz N. miRWalk - database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J Biomed Inform. 2011;44(5):839–47.
Kalmar A, Peterfia B, Hollosi P, Galamb O, Spisak S, Wichmann B, Bodor A, Toth K, Patai AV, Valcz G, et al. DNA hypermethylation and decreased mRNA expression of MAL, PRIMA1, PTGDR and SFRP1 in colorectal adenoma and cancer. BMC Cancer. 2015;15:736.
Xu MD, Qi P, Du X. Long non-coding RNAs in colorectal cancer: implications for pathogenesis and clinical application. Modern Pathol. 2014;27(10):1310–20.
Xie X, Tang B, Xiao YF, Xie R, Li BS, Dong H, Zhou JY, Yang SM. Long non-coding RNAs in colorectal cancer. Oncotarget. 2016;7(5):5226–39.
Saus E, Brunet-Vega A, Iraola-Guzman S, Pegueroles C, Gabaldon T, Pericay C. Long non-coding RNAs as potential novel prognostic biomarkers in colorectal Cancer. Front Genet. 2016;7:54.
Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15(1):7–21.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–8.
Palermo MDH, Tighe S, Dragon J, Bond J, Shukla A, Vangala M, Vincent J. Expression profiling Smackdown: human transcriptome Array HTA 2.0 vs. RNA-Seq. J Biomol Tech. 2014;25(Suppl):S20–1.
Alaiyan B, Ilyayev N, Stojadinovic A, Izadjoo M, Roistacher M, Pavlov V, Tzivin V, Halle D, Pan H, Trink B, et al. Differential expression of colon cancer associated transcript1 (CCAT1) along the colonic adenoma-carcinoma sequence. BMC Cancer. 2013;13:196.
Liu T, Zhang X, Yang YM, Du LT, Wang CX. Increased expression of the long noncoding RNA CRNDE-h indicates a poor prognosis in colorectal cancer, and is positively correlated with IRX5 mRNA expression. OncoTargets Ther. 2016;9:1437–48.
Graham LD, Pedersen SK, Brown GS, Ho T, Kassir Z, Moynihan AT, Vizgoft EK, Dunne R, Pimlott L, Young GP, et al. Colorectal neoplasia differentially expressed (CRNDE), a novel gene with elevated expression in colorectal adenomas and adenocarcinomas. Genes Cancer. 2011;2(8):829–40.
Wang JJ, Li XM, He L, Zhong SZ, Peng YX, Ji N. Expression and function of long non-coding RNA CASC19 in colorectal Cancer. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2017;39(6):756–61.
Li M, Zhao LM, Li SL, Li J, Gao B, Wang FF, Wang SP, Hu XH, Cao J, Wang GY. Differentially expressed lncRNAs and mRNAs identified by NGS analysis in colorectal cancer patients. Cancer Med. 2018;7(9):4650–4664.
Zhang Z, Wang S, Ji D, Qian W, Wang Q, Li J, Gu J, Peng W, Hu T, Ji B, et al. Construction of a ceRNA network reveals potential lncRNA biomarkers in rectal adenocarcinoma. Oncol Rep. 2018;39(5):2101–13.
Dong L, Hong H, Chen X, Huang Z, Wu W, Wu F. LINC02163 regulates growth and epithelial-to-mesenchymal transition phenotype via miR-593-3p/FOXK1 axis in gastric cancer cells. Artificial Cells Nanomed Biotechnol. 2018:1–9.
Yamada A, Yu P, Lin W, Okugawa Y, Boland CR, Goel A. A RNA-sequencing approach for the identification of novel long non-coding RNA biomarkers in colorectal cancer. Sci Rep. 2018;8(1):575.
Delas MJ, Hannon GJ. lncRNAs in development and disease: from functions to mechanisms. Open Biol. 2017;7(7):170121.
Patai AV, Valcz G, Hollosi P, Kalmar A, Peterfia B, Patai A, Wichmann B, Spisak S, Bartak BK, Leiszter K, et al. Comprehensive DNA methylation analysis reveals a common ten-gene methylation signature in colorectal adenomas and carcinomas. PLoS One. 2015;10(8):e0133836.
Ge X, Chen Y, Liao X, Liu D, Li F, Ruan H, Jia W. Overexpression of long noncoding RNA PCAT−1 is a novel biomarker of poor prognosis in patients with colorectal cancer. Med Oncol. 2013;30(2):588.
Qiao L, Liu X, Tang Y, Zhao Z, Zhang J, Feng Y. Down regulation of the long non-coding RNA PCAT−1 induced growth arrest and apoptosis of colorectal cancer cells. Life Sci. 2017;188:37–44.
Qiao L, Liu X, Tang Y, Zhao Z, Zhang J, Liu H. Knockdown of long non-coding RNA prostate cancer-associated ncRNA transcript 1 inhibits multidrug resistance and c-Myc-dependent aggressiveness in colorectal cancer Caco-2 and HT-29 cells. Mol Cell Biochem. 2018;441(1-2):99–108.
Wang F, Li X, Xie X, Zhao L, Chen W. UCA1, a non-protein-coding RNA up-regulated in bladder carcinoma and embryo, influencing cell growth and promoting invasion. FEBS Lett. 2008;582(13):1919–27.
Ni B, Yu X, Guo X, Fan X, Yang Z, Wu P, Yuan Z, Deng Y, Wang J, Chen D, et al. Increased urothelial cancer associated 1 is associated with tumor proliferation and metastasis and predicts poor prognosis in colorectal cancer. Int J Oncol. 2015;47(4):1329–38.
Huang J, Zhou N, Watabe K, Lu Z, Wu F, Xu M, Mo YY. Long non-coding RNA UCA1 promotes breast tumor growth by suppression of p27 (Kip1). Cell Death Dis. 2014;5:e1008.
Han Y, Yang YN, Yuan HH, Zhang TT, Sui H, Wei XL, Liu L, Huang P, Zhang WJ, Bai YX. UCA1, a long non-coding RNA up-regulated in colorectal cancer influences cell proliferation, apoptosis and cell cycle distribution. Pathology. 2014;46(5):396–401.
Ni B, Yu X, Guo X, Fan X, Yang Z, Wu P, Yuan Z, Deng Y, Wang J, Chen D, et al. Increased urothelial cancer associated 1 is associated with tumor proliferation and metastasis and predicts poor prognosis in colorectal cancer. Int J Oncol. 2015;47(4):1329–38.
Bian Z, Jin L, Zhang J, Yin Y, Quan C, Hu Y, Feng Y, Liu H, Fei B, Mao Y, et al. LncRNA-UCA1 enhances cell proliferation and 5-fluorouracil resistance in colorectal cancer byinhibiting miR-204-5p. Sci Rep. 2016;6:23892.
Yang YT, Wang YF, Lai JY, Shen SY, Wang F, Kong J, Zhang W, Yang HY. Long non-coding RNA UCA1 contributes to the progression of oral squamous cell carcinoma by regulating the WNT/beta-catenin signaling pathway. Cancer Sci. 2016;107(11):1581–9.
Tao K, Yang J, Hu Y, Sun Y, Tan Z, Duan J, Zhang F, Yan H, Deng A. Clinical significance of urothelial carcinoma associated 1 in colon cancer. Int J Clin Exp Med. 2015;8(11):21854–60.
Xue M, Chen W, Li X. Urothelial cancer associated 1: a long noncoding RNA with a crucial role in cancer. J Cancer Res Clin Oncol. 2016;142(7):1407–19.
Tripathi MK, Zacheaus C, Doxtater K, Keramatnia F, Gao C, Yallapu MM, Jaggi M, Chauhan SC. Z Probe, An Efficient Tool for Characterizing Long Non-Coding RNA in FFPE Tissues. Non-coding RNA. 2018;4(3):20.
Wang F, Ying HQ, He BS, Pan YQ, Deng QW, Sun HL, Chen J, Liu X, Wang SK. Upregulated lncRNA-UCA1 contributes to progression of hepatocellular carcinoma through inhibition of miR-216b and activation of FGFR1/ERK signaling pathway. Oncotarget. 2015;6(10):7899–917.
Wang T, Yuan J, Feng N, Li Y, Lin Z, Jiang Z, Gui Y. Hsa-miR−1 downregulates long non-coding RNA urothelial cancer associated 1 in bladder cancer. Tumour Biol. 2014;35(10):10075–84.
Xu W, Zhang Z, Zou K, Cheng Y, Yang M, Chen H, Wang H, Zhao J, Chen P, He L, et al. MiR-1 suppresses tumor cell proliferation in colorectal cancer by inhibition of Smad3-mediated tumor glycolysis. Cell Death Dis. 2017;8(5):e2761.
Reid JF, Sokolova V, Zoni E, Lampis A, Pizzamiglio S, Bertan C, Zanutto S, Perrone F, Camerini T, Gallino G, et al. miRNA profiling in colorectal cancer highlights miR-1 involvement in MET-dependent proliferation. Mol Cancer Res. 2012;10(4):504–15.
Gayyed MF, Abd El-Maqsoud NM, El-Hameed El-Heeny AA, Mohammed MF. C-MET expression in colorectal adenomas and primary carcinomas with its corresponding metastases. J Gastrointest Oncol. 2015;6(6):618–27.
De Oliveira AT, Matos D, Logullo AF, SR DAS, Neto RA, Filho AL, Saad SS. MET is highly expressed in advanced stages of colorectal cancer and indicates worse prognosis and mortality. Anticancer Res. 2009;29(11):4807–11.
Shi J, Li X, Zhang F, Zhang C, Guan Q, Cao X, Zhu W, Zhang X, Cheng Y, Ou K, et al. Circulating lncRNAs associated with occurrence of colorectal cancer progression. Am J Cancer Res. 2015;5(7):2258–65.
Wang YL, Shao J, Wu X, Li T, Xu M, Shi D. A long non-coding RNA signature for predicting survival in patients with colorectal cancer. Oncotarget. 2018;9(31):21687–95.
Acknowledgements
The authors would like to thank Gabriella Kónyáné Farkas (2nd Department of Internal Medicine, Semmelweis University) for her technical assistance in immunohistochemical stainings and Boye S. Nielsen (Bioneer AS) for conducting the ISH experiments. The authors would like to acknowledge Zita Bratu and Zsófia Zsibai, Géza Antalffy and Annamária Csizmadia (3DHISTECH Ltd.) and Tibor Krenács (1st Department of Pathology and Experimental Cancer Research, Semmelweis University) for their work with the digital scanning of the IHC and ISH slides. We would like to thank Theo deVos, Ph.D., Arpad V. Patai, MD, Ph.D., Istvan Furi PharmDr, Ph.D. and Ramani Gopal, Ph.D. for language revision of the manuscript. The results published here are partly based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
Funding
This study was supported by the National Office for Research and Technology, Hungary (KMR 12 − 1-2012-0216; NKVP_16 − 1-2016-0004 grants) for BM and by Novo Nordisk Foundation Interdisciplinary Synergy Programme Grant no. NNF15OC0016584 for IC and AB and by The New National Excellence Program of the Ministry of Human Capacities, Hungary (ÚNKP-17-4-I-SE-56 for AK, ÚNKP-17-4-III-SE-51 for OG). The funders played no role in designing the study, collecting or interpreting the data, or writing the manuscript.
Author information
Authors and Affiliations
Contributions
AK and ZBN performed RNA isolations and microarray experiments, AK, OG, and BKB performed qRT-PCR validation, AB, BW and IC performed in silico analyses and validation, GV analyzed immunohistochemistry results, ZT, PI and BM contributed to the design and critical review of the manuscript. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the local ethics committee (Semmelweis University Regional and Institutional Committee of Science and Research Ethics; Nr.: ETT TUKEB 23970/2011). Written informed consent was provided by all patients.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Differentially expressed lncRNAs in the adenoma-carcinoma sequence. A) Adenoma vs. Normal samples, B) CRC vs. Adenoma samples, C) CRC vs. Normal samples. Intensity values on the color scale are as follows: red – high intensity, black – intermediate intensity, green – low intensity.
Additional file 2: Table S1.
Differentially expressed lncRNAs in the adenoma-carcinoma sequence.
Additional file 3: Table S2.
In silico validation on independent datasets.
Additional file 4 : Table S3.
lncRNA-mRNA co-expression network data, GO analysis.
Additional file 5: Figure S2.
Specific UCA1 in situ hybridization signals using RNAScope probes (red stain) on CRC tissue with merged and single channel captures from ISH of UCA1 (Urothelial cancer associated 1), dapB (a Bacillus subtilis gene, negative control probe), and PPIB (Cyclophilin B, positive control probe). Tissue sections were counter stained with DAPI. Digital microscope samples, scale bar: 100 μm.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kalmár, A., Nagy, Z.B., Galamb, O. et al. Genome-wide expression profiling in colorectal cancer focusing on lncRNAs in the adenoma-carcinoma transition. BMC Cancer 19, 1059 (2019). https://doi.org/10.1186/s12885-019-6180-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-019-6180-5