
Abstract
Long non-coding RNAs (lncRNAs) are emerging as new players in the cancer paradigm demonstrating potential roles in both oncogenic and tumor suppressive pathways. These novel genes are frequently aberrantly expressed in a variety of human cancers, however the biological functions of the vast majority remain unknown. Recently, evidence has begun to accumulate describing the molecular mechanisms by which these RNA species function, providing insight into the functional roles they may play in tumorigenesis. In this review, we highlight the emerging functional role of lncRNAs in human cancer.
Similar content being viewed by others

Introduction
One of modern biology's great surprises was the discovery that the human genome encodes only ~20,000 protein-coding genes, representing <2% of the total genome sequence [1, 2]. However, with the advent of tiling resolution genomic microarrays and whole genome and transcriptome sequencing technologies it was determined that at least 90% of the genome is actively transcribed [3, 4]. The human transcriptome was found to be more complex than a collection of protein-coding genes and their splice variants; showing extensive antisense, overlapping and non-coding RNA (ncRNA) expression [5–10]. Although initially argued to be spurious transcriptional noise, recent evidence suggests that the proverbial "dark matter" of the genome may play a major biological role in cellular development and metabolism [11–17]. One such player, the newly discovered long non-coding RNA (lncRNA) genes, demonstrate developmental and tissue specific expression patterns, and aberrant regulation in a variety of diseases, including cancer [18–27].
NcRNAs are loosely grouped into two major classes based on transcript size; small ncRNAs and lncRNAs (Table 1) [28–30]. Small ncRNAs are represented by a broad range of known and newly discovered RNA species, with many being associated with 5' or 3' regions of genes [4, 31, 32]. This class includes the well-documented miRNAs, RNAs ~22 nucleotides (nt) long involved in the specific regulation of both protein-coding, and putatively non-coding genes, by post-transcriptional silencing or infrequently by activation [33–35]. miRNAs serve as major regulators of gene expression and as intricate components of the cellular gene expression network [33–38]. Another newly described subclass are the transcription initiation RNAs (tiRNAs), which are the smallest functional RNAs at only 18 nt in length [39, 40]. While a number of small ncRNAs classes, including miRNAs, have established roles in tumorigenesis, an intriguing association between the aberrant expression of ncRNA satellite repeats and cancer has been recently demonstrated [41–46].
In contrast to miRNAs, lncRNAs, the focus of this article, are mRNA-like transcripts ranging in length from 200 nt to ~100 kilobases (kb) lacking significant open reading frames. Many identified lncRNAs are transcribed by RNA polymerase II (RNA pol II) and are polyadenylated, but this is not a fast rule [47, 48]. There are examples of lncRNAs, such as the antisense asOct4-pg5 or the brain-associated BC200, which are functional, but not polyadenylated [49–51]. Generally, lncRNA expression levels appear to be lower than protein-coding genes [52–55], and some lncRNAs are preferentially expressed in specific tissues [21]. However, recent findings have suggested novel lncRNAs may contribute a significant portion of the aforementioned 'dark matter' of the human transcriptome [56, 57]. In an exciting report by Kapranov et.al., it was revealed the bulk of the relative mass of RNA in a human cell, exclusive of the ribosomal and mitochondrial RNA, is represented by non-coding transcripts with no known function [57].
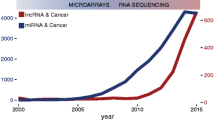
Like miRNAs and protein-coding genes, some transcriptionally active lncRNA genes display histone H3K4 trimethylation at their 5'-end and histone H3K36 trimethylation in the body of the gene [8, 58, 59]. The small number of characterized human lncRNAs have been associated with a spectrum of biological processes, for example, epigenetics, alternative splicing, nuclear import, as structural components, as precursors to small RNAs and even as regulators of mRNA decay [4, 60–70]. Furthermore, accumulating reports of misregulated lncRNA expression across numerous cancer types suggest that aberrant lncRNA expression may be a major contributor to tumorigenesis [71]. This surge in publications reflects the increasing attention to this subject (Figure 1) and a number of useful lncRNA databases have been created (Table 2). In this review we highlight the emerging functional role of aberrant lncRNA expression, including transcribed ultraconserved regions (T-UCRs), within human carcinomas.

Publications describing cancer-associated ncRNAs. Entries are based on a National Library of Medicine Pubmed search using the terms "ncRNA" or "non-coding RNA" or "noncoding RNA" or non-protein-coding RNA" with cancer and annual (Jan.1-Dec.31) date limitations.
Background on Long Non-Coding RNA
Nomenclature and Classification of LncRNA
The definition 'non-coding RNA' is typically used to describe transcripts where sequence analysis has failed to identify an open reading frame. However, one should exercise caution when exploring putative non-coding transcripts, as there are cases where 'non-coding' transcripts were found to encode short, functional peptides [72]. Currently, a universal classification scheme to define lncRNAs does not exist and therefore there are numerous synonyms describing the same type of transcripts. Terms such as large non-coding RNA, mRNA-like long RNA, and intergenic RNA all define cellular RNAs, exclusive of rRNAs, greater than 200 nt in length and having no obvious protein-coding capacity [62]. Consequently, this has led to confusion in the literature as to exactly which transcripts should constitute a lncRNA. For example, one subclass of lncRNAs is called large or long intergenic ncRNAs (lincRNAs). These lncRNAs are exclusively intergenic and are marked by a chromatin signature indicative of transcription [8, 58]. To further compound classification confusion, RNA species that are bifunctional preclude categorization into either group of protein-coding or ncRNAs as their transcripts function both at the RNA and protein levels [73]. In these rare cases, they are classified on a case-by-case basis, reserving the term 'lncRNA' to describe transcripts with no protein-coding capacity. In the meantime, and for the purposes of this review, we will consider lncRNAs as a blanket term to encompass mRNA-like ncRNAs, lincRNAs, as well as antisense and intron-encoded transcripts, T-UCRs and transcribed pseudogenes.
Discovery of LncRNAs
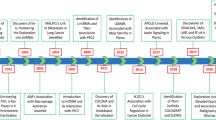
The earliest reports describing lncRNA predated the discovery of miRNAs, although the term 'lncRNA' had not been coined at the time (Figure 2). One of the first lncRNA genes reported was the imprinted H19 gene, which was quickly followed by the discovery of the silencing X-inactive-specific transcript (XIST) lncRNA gene, which plays a critical function in X-chromosome inactivation [74, 75]. However, the discovery of the first miRNA lin-14 dramatically redirected the focus of ncRNA research from long ncRNAs to miRNAs [76]. Moreover, during this time the discovery of miRNAs revealed RNA could regulate gene expression and later that entire gene networks could be affected by ncRNA expression and within the last decade miRNAs were discovered to be associated with cancer (Figure 2) [76–79]. At the time of this writing there are approximately 1049 human miRNAs described in miRBase V16 [80, 81] with the potential of affecting the expression of approximately 60% of protein -coding genes [82, 83]. Conversely, the variety and dynamics of lncRNA expression was not to be fully appreciated until the introduction of whole transcriptome sequencing. With the advent of the FANTOM and ENCODE transcript mapping projects, it was revealed that the mammalian genome is extensively transcribed, although a large portion of this represented non-coding sequences [3, 84]. Coupled with the novel functional annotation of a few lncRNAs, this discovery promoted research focusing on lncRNA discovery and characterization. Recent reports have described new lncRNA classes such as lincRNAs and T-UCRs [8, 58, 85]. Current estimates of the lncRNA gene content in the human genome ranges from ~7000 - 23,000 unique lncRNAs, implying this class of ncRNA will represent an enormous, yet undiscovered, component of normal cellular networks that may be disrupted in cancer biology [62].

Timeline of cancer-associated ncRNA discoveries relative to transcriptome analysis technologies (not drawn to scale).
Emerging Role of Long Non-Coding RNA in Tumorigenesis
A role for differential lncRNA expression in cancer had been suspected for many years, however, lacked strong supporting evidence [86]. With advancements in cancer transcriptome profiling and accumulating evidence supporting lncRNA function, a number of differentially expressed lncRNAs have been associated with cancer (Table 3). LncRNAs have been implicated to regulate a range of biological functions and the disruption of some of these functions, such as genomic imprinting and transcriptional regulation, plays a critical role in cancer development. Here we describe some of the better characterized lncRNAs that have been associated with cancer biology.
Imprinted lncRNA genes
Imprinting is a process whereby the copy of a gene inherited from one parent is epigenetically silenced [87, 88]. Intriguingly, imprinted regions often include multiple maternal and paternally expressed genes with a high frequency of ncRNA genes. The imprinted ncRNA genes are implicated in the imprinting of the region by a variety of mechanisms including enhancer competition and chromatin remodeling [89]. A key feature of cancer is the loss of this imprinting resulting in altered gene expression [90, 91]. Two of the best known imprinted genes are in fact lncRNAs.
H19
The H19 gene encodes a 2.3 kb lncRNA that is expressed exclusively from the maternal allele. H19 and its reciprocally imprinted protein-coding neighbor the Insulin-Like Growth Factor 2 or IGF2 gene at 11p15.5 were among the first genes, non-coding or otherwise, found to demonstrate genomic imprinting [74, 92]. The expression of H19 is high during vertebrate embryo development, but is downregulated in most tissues shortly after birth with the exception of skeletal tissue and cartilage [20, 93, 94]. Loss of imprinting and subsequent strong gene expression has been well-documented in human cancers. Likewise, loss of imprinting at the H19 locus resulted in high H19 expression in cancers of the esophagus, colon, liver, bladder and with hepatic metastases [95–97].
H19 has been implicated as having both oncogenic and tumor suppression properties in cancer. H19 is upregulated in a number of human cancers, including hepatocellular, bladder and breast carcinomas, suggesting an oncogenic function for this lncRNA [97–99]. In colon cancer H19 was shown to be directly activated by the oncogenic transcription factor c-Myc, suggesting H19 may be an intermediate functionary between c-Myc and downstream gene expression [98]. Conversely, the tumor suppressor gene and transcriptional activator p53 has been shown to down-regulate H19 expression [100, 101]. H19 transcripts also serve as a precursor for miR-675, a miRNA involved in the regulation of developmental genes [102]. miR-675 is processed from the first exon of H19 and functionally downregulates the tumor suppressor gene retinoblastoma (RB1) in human colorectal cancer, further implying an oncogenic role for H19[103].
There is evidence suggesting H19 may also play a role in tumor suppression [104, 105]. Using a mouse model for colorectal cancer, it was shown that mice lacking H19 manifested an increased polyp count compared to wild-type [106]. Secondly, a mouse teratocarcinoma model demonstrated larger tumor growth when the embryo lacked H19, and finally in a hepatocarcinoma model, mice developed cancer much earlier when H19 was absent [107]. The discrepancy as to whether H19 has oncogenic or tumor suppressive potential may be due in part to the bifunctional nature of the lncRNA or may be context dependent. In either case, the precise functional and biological role of H19 remains to be determined.
XIST - X-inactive-specific transcript
The 17 kb lncRNA XIST is arguably an archetype for the study of functional lncRNAs in mammalian cells, having been studied for nearly two decades. In female cells, the XIST transcript plays a critical role in X-chromosome inactivation by physically coating one of the two X-chromosomes, and is necessary for the cis-inactivation of the over one thousand X-linked genes [75, 108–110]. Like the lncRNAs HOTAIR and ANRIL, XIST associates with polycomb-repressor proteins, suggesting a common pathway of inducing silencing utilized by diverse lncRNAs. In mice, X inactivation in the extraembryonic tissues is non-random, and the initial expression of Xist is always paternal in origin, followed later by random X inactivation in the epiblast associated with random mono-allelic expression [111]. Intriguingly, regulation of Xist in mouse has been shown to be controlled by interactions amongst additional ncRNAs, including an antisense transcript, Tsix, enhancer-associated ncRNAs (Xite) and the upstream Jpx and Ftx lncRNAs [109, 112–115]. Intriguingly, murine Xist and Tsix duplexes are processed into small RNAs by an apparently Dicer-dependent manner, suggesting a significant overlap between the regulatory networks of both lncRNAs and sRNAs [116]. It is unclear, however, how much of this regulation is conserved in humans, who do not show imprinted X inactivation [117]. While XIST expression levels are correlated with outcome in some cancers, such as the therapeutic response in ovarian cancer [118], the actual role that XIST may play in human carcinomas, if any, is not entirely clear.
There is generally believed to be only a limited developmental window in which X inactivation can occur, and loss of XIST from an inactive X chromosome does not result in reactivation of the X chromosome [119, 120]. Thus, tumors with additional X chromosomes generally keep the inactivation status of the duplicated X. For tumors in which two active X chromosomes are observed, as has been frequently observed in breast cancer, the most common mechanism involves loss of the inactive X and duplication of the active X, often resulting in heterogeneous XIST expression in these tumors [121–123]. Loss of the inactive X and abnormalities of XIST expression may be indicative of more general heterochromatin defects [124]. Analogously, failure to properly reset XIST and X inactivation may serve as a marker of proper resetting of epigenetic marks in stem cells or induced pluripotent stem cells [125, 126]. Correlations between XIST expression and cancer state can also occur spuriously. Notably, in cytogenetically normal cells, XIST is found only in females as males do not have an inactive X. Therefore in cancers correlations can be observed with XIST expression due to differential presence of male or female samples, and many cancers do show different onsets and progressions in males and females. Furthermore, XIST expression will increase with the number of inactive X chromosomes. While it might be anticipated that there would be little advantage to a tumor acquiring inactive chromosomes, it has been shown that approximately 15% of human X-linked genes continue to be expressed from the inactive X chromosome [127].
Involvement in metastasis
HOTAIR - HOX antisense intergenic RNA
Several lncRNAs have been implicated in metastasis. One of the first lncRNAs described to have a fundamental role in cancer was the metastasis-associated HOX Antisense Intergenic RNA (HOTAIR), a 2.2 kb gene located in the mammalian HOXC locus on chromosome 12q13.13 [18]. This lncRNA was found to be highly upregulated in both primary and metastatic breast tumors, demonstrating up to 2000-fold increased transcription over normal breast tissue [68]. High levels of HOTAIR expression were found to be correlated with both metastasis and poor survival rate, linking a ncRNA with cancer invasiveness and patient prognosis [68]. Furthermore, it was demonstrated that if cells expressing HOTAIR were grafted into mouse mammary fat pads, a modest increase in the rate of primary tumor growth was observed [68]. Interestingly, there are reports indicating that numerous lncRNAs are transcribed from the HOX locus, suggesting that HOTAIR may be only one example of a global regulatory phenomena [58].
The spliced and polyadenylated HOTAIR RNA does not encode any proteins but has been demonstrated to be intimately associated with the mammalian polycomb repressive complex 2 (PRC2) which is comprised of the H3K27 methylase EZH2, SUZ12 and EED [68, 128, 129] (Figure 3). Polycomb group proteins mediate repression of transcription of thousands of genes controlling differentiation pathways during development, and have roles in stem cell pluripotency and human cancer [68, 130–133].

Proposed mechanism of HOTAIR mediated gene silencing of 40 kb of the HOXD locus, which is involved in developmental patterning. The HOTAIR lncRNA is transcribed from the HOXC locus and functions in the binding and recruitment and binding of the PRC2 and LSD1 complex to the HOXD locus. For clarity, only the PRC2 complex is indicated in the above figure. Through an undetermined mechanism, the HOTAIR-PRC2-LSD1 complex is redirected to the HOXD locus on chromosome 2 where genes involved in metastasis suppression are silenced through H3K27 methylation and H3K4 demethylation. This drives breast cancer cells to develop gene expression patterns that more closely resemble embryonic fibroblasts than epithelial cells.
HOTAIR binding results in a genome-wide re-targeting of the PRC2 complex. The HOXD locus on chromosome 2 is a PRC2 target (Figure 3). The consequence of PRC2/HOTAIR localization is the transcriptional silencing of a 40 kb region of the HOXD locus which remodels the gene expression pattern of breast epithelial cells to more closely resemble that of embryonic fibroblasts [68]. The HOTAIR RNA appears to act as a molecular scaffold, binding at least two distinct histone modification complexes. The 5' region of the RNA binds the PRC2 complex responsible for H3K27 methylation, while the 3' region of HOTAIR binds LSD1, a histone lysine demethylase that mediates enzymatic demethylation of H3K4Me2 [128, 134]. Although, the precise mechanism of HOTAIR activities remains to be elucidated, it is clear that HOTAIR reprograms chromatin state to promote cancer metastasis.
MALAT1 - Metastasis-associated lung adenocarcinoma transcript 1
The MALAT1 gene, or metastasis-associated lung adenocarcinoma transcript 1, was first associated with high metastatic potential and poor patient prognosis during a comparative screen of non-small cell lung cancer patients with and without metastatic tumors [135]. This lncRNA is widely expressed in normal human tissues [135, 136] and is found to be upregulated in a variety of human cancers of the breast, prostate, colon, liver and uterus [27, 137–139]. Notably, the MALAT1 locus at 11q13.1 has been identified to harbor chromosomal translocation breakpoints associated with cancer [140–142].
Intriguingly, cellular MALAT1 transcripts are subject to post-transcriptional processing to yield a short, tRNA-like molecule mascRNA and a long MALAT1 transcript with a poly(A) tail-like moiety [143] (Figure 4). Using the mouse homologue to the human MALAT1 gene, it was revealed that ribonuclease (RNase) P processing generates the 3' end of the long MALAT1 transcript and the 5' end of the mascRNA. The shorter mascRNA adopts a tRNA clover-leaf structure and is subject to RNaseZ processing and the addition of a CCA to its 3' end before being exported to the cytoplasm. The generation of putatively functional sRNAs by post-transcriptional processing of lncRNAs, such mascRNA from MALAT1, may be reflective of a central, unexplored theme in ncRNA biology [144]. Recent evidence suggests regulated post-transcriptional processing events, such as splicing and post-cleavage capping, may increase the functional diversity of the metazoan transcriptome [145–149]. In contrast, the long MALAT1 transcript is not polyadenylated, but has a poly(A) tail-like sequence that is genome encoded and is putatively present to protect the MALAT1 transcript from degradation. Moreover, MALAT1 localizes to nuclear speckles in a transcription dependent manner [136, 150].

Expression and processing of MALAT1 transcripts. Full length 7.5 kb MALAT1 RNA is processed by RNaseP and RNaseZ to generate the small ncRNA mascRNA, which is then exported to the cytoplasm. The larger MALAT1 RNA is retained in the nuclear speckles where it is thought to have a role in regulating alternative splicing machinery.
Following the correlation between high levels of MALAT1 expression and metastasis, a number of studies have implicated MALAT1 in the regulation of cell mobility. For example, RNA interference (RNAi)-mediated silencing of MALAT1 impaired the in vitro migration of lung adenocarcinoma cells through concomitant regulation of motility regulated genes via transcriptional and/or post-transcriptional means [151]. Similarly, short hairpin RNA inhibition of MALAT1 reduced cell proliferation and invasive potential of a cervical cancer cell line [152]. Collectively, these studies suggest that MALAT1 regulates the invasive potential of metastatic tumor cells. Other examples of cancer-associated lncRNAs subject to post-transcriptional processing include the miR-155 host gene BIC and the miR-17-92 cluster in B-cell lymphoma and neuroblastoma, respectively [153, 154].
Recent efforts have focused on elucidating the molecular role of nuclear speckle localized MALAT1 transcripts, while the function for the mascRNA has yet to be determined. Nuclear speckles are thought to be involved in the assembly, modification and/or storage of pre-mRNA processing machinery [136, 155]. A recent publication revealed MALAT1 RNA strongly associates with serine-arginine rich splicing factor (SR) proteins which are involved in both constitutive and alternative splicing and the levels of MALAT1 regulated the cellular levels of phosphorylated SR proteins [66]. These findings imply that the lncRNA MALAT1 may serve a function in the regulation of alternate splicing by modulating the activity of SR proteins, although precisely how this may contribute to tumorigenesis remains unknown.
LncRNA-mediated tumor suppression by P53 stimulation
MEG3 - Maternally expressed gene 3
The maternally expressed gene 3 (MEG3) was the first lncRNA proposed to function as a tumor suppressor. The MEG3 gene is expressed in many normal human tissues, with the highest expression in the brain and pituitary gland [156, 157]. MEG3 expression was not detectable in various brain cancers, nor in a range of human cancer cell lines implicating a potential role of this lncRNA in suppression of cell growth. Moreover, ectopic expression of MEG3 RNA was found to suppress the growth of several human cancer cell lines, further supporting the role of MEG3 as a tumor suppressor [157]. In clinically nonfunctioning pituitary tumors, it was demonstrated that hypermethylation of the MEG3 regulatory region was associated with the loss of MEG3 expression, providing evidence for a mechanism of MEG3 inactivation [157].
MEG3 is a paternally imprinted, single copy gene comprised of 10 exons [156]. To date, 12 MEG3 isoforms have been detected (MEG3; a-3k) due to alternative splicing [158]. Each isoform contains the common exons 1-3 and 8-10, but varies in the combination of exons 4-7 in the middle of the transcript [158]. Notably, the last intron of MEG3 encodes the evolutionarily conserved miR-770, a miRNA with a number of putative mRNA targets [159]. This arrangement mirrors the presence of miR-675 in H19, except in that instance the miRNA was exon encoded [102]. The originally identified isoform of MEG3 is expressed as a 1.6 kb polyadenylated transcript that is localized to the nucleus where it is associated with chromatin although some cytoplasmic MEG3 transcripts have been detected [156, 160, 161]. All 12 MEG3 isoforms demonstrate three distinct secondary folding motifs designated M1, M2 and M3 [158].
Functionally, MEG3 has been implicated as a top-level regulatory RNA due to its ability to stimulate both p53-dependent and p53-independent pathways [158, 162]. Critically, the MEG3-mediated functional activation of p53 is dependent on the secondary structure of the MEG3 RNA rather than on primary sequence conservation. In an elegant series of experiments, Zhang et al. demonstrated that replacing particular regions of the MEG3 with unrelated sequence had no effect on the activation of p53 provided the original secondary structure was preserved [158]. This observation strengthens the argument that the rapid evolution and relative lack of sequence conservation does not limit the potential functionality of these unique RNA species [163]. Conversely, the novel lincRNA-p21 has been described as a downstream repressor in the p53 transcriptional response, suggesting the complex p53 transcriptional network includes numerous regulatory lncRNAs [164].
Depletion of miRNAs by a lncRNA 'molecular decoy' or 'miRNA sponge'
Expression of miRNAs in cancer can be deregulated by a range of mechanisms, including copy number alterations and epigenetic silencing [165–168]. Two recent examples have demonstrated that lncRNAs can act as natural 'miRNA sponges' to reduce miRNA levels [169].
HULC - Highly Upregulated in Liver Cancer
The most highly upregulated transcript found in a microarray-based study of gene expression in hepatocellular carcinoma was determined to be the ncRNA HULC, or Highly Upregulated in Liver Cancer[170]. Transcribed from chromosome 6p24.3, this lncRNA demonstrates the hallmarks of a typical mRNA molecule, including a single spliced GT-AG intron, canonical polyadenylation signals upstream of the poly(A) tail and nuclear export demonstrating strong localization to the cytoplasm. Although HULC was found to co-purify with ribosomes, no translation product for this lncRNA has been detected, supporting its classification as a non-coding transcript [170]. In addition to liver cancer, HULC was found to be highly upregulated in hepatic colorectal cancer metastasis and in hepatocellular carcinoma cell lines (HCC) producing hepatitis B virus (HBV) [171].
A recent paper began to elucidate the mechanism of upregulation of HULC in liver cancer cells, and to provide a potential mechanism of HULC function [172]. HULC exists as part of an intricate auto-regulatory network, which when perturbed, resulted in increased HULC expression (Figure 5). The HULC RNA appeared to function as a 'molecular decoy' or 'miRNA sponge' sequestering miR-372, of which one function is the translational repression of PRKACB, a kinase targeting cAMP response element binding protein (CREB). Once activated, the CREB protein was able to promote HULC transcription by maintaining an open chromatin structure at the HULC promoter resulting in increased HULC transcription [172].

Proposed mechanism of HULC upregulation in hepatocellular carcinoma. (1) The kinase PRKACB functions as an activator of CREB. (2) Phosphorylated (activated) CREB forms part of the RNA pol II transcriptional machinery to activate HULC expression. (3) Abundant HULC RNA acts as a molecular sponge to sequester and inactivate the repressive function miR-372. (4) PRKACB levels increase, as transcripts are normally translationally repressed by high miR-372 levels.
Pseudogene pairs: PTEN and PTENP1
The discovery that HULC can act as a molecular decoy or 'miRNA sponge' mirrors a recent report describing an intricate relationship between the tumor suppressor phosphatase and tensin homolog (PTEN) and the matching lncRNA phosphatase and tensin homolog pseudogene 1 (PTENP1)[173]. The RNA transcripts of the PTEN/PTENP1 pair share similar 3' untranslated regions (3'UTRs) which both bind the same miRNAs. By binding miRNAs, PTENP1 transcripts reduce the effects of translational repression on PTEN therefore allowing expression of this tumor suppressor. In cancer, specific mutations inactivate these miRNA binding sites in PTENP1, therefore reducing the translation of PTEN and promoting tumor growth [173]. This is especially pertinent as subtle changes in PTEN levels can influence cancer susceptibility [174].
LncRNA-mediated deregulation of the tumor suppressors
ANRIL - Antisense Non-coding RNA in the INK4 Locus
Located as part of the 42kb INK4b-ARF-INK4a locus on chromosome 9p21.3, the Antisense Non-coding RNA in the INK4 Locus (ANRIL) is transcribed by RNA pol II and processed into alternatively spliced isoforms, including an unspliced transcript of 34.8 kb termed p15AS [175, 176]. The INK4b-ARF-INKa locus has an important role in cell cycle control, cell senescence, stem cell renewal and apoptosis through P14ARF-MDM2-P53 and P16Ink4a/p15Ink4b-Cdk4/6-pRb pathways [177–179]. A mouse model suggests that the well-characterized tumor suppressor genes encoded within this locus are regulated by Polycomb proteins [180].
Aberrant expression and single nucleotide polymorphisms (SNPs) within ANRIL have been associated with susceptibility to a range of human diseases, including cancer [181, 182]. Moreover, the INK4b-ARF-INK4a locus is subject to frequent deletion or hypermethylation in cancers, including leukemia, melanoma, lung and bladder cancers [181]. ANRIL has been associated with epigenetic silencing of the tumor suppressor gene p15, although the molecular events leading to this silencing were unclear [175].
Evidence has suggested an intriguing mechanisms for ANRIL-mediated silencing of the INK4b-ARF-INK4a locus. Recently an interaction between chromatin modifying PRC complexes and ANRIL has been described, further elucidating the regulatory mechanism of this lncRNA. Like the lncRNA HOTAIR which binds both the polycomb repressor complex PRC2 and the LSD1 complex, ANRIL binds and recruits two polycomb repressor complexes modifying complexes, PRC1 and PRC2 [183, 184]. This resulted in ANRIL/PRC mediated silencing of the genes in the INK4b-ARF-INK4a locus. While the distinct regions of the HOTAIR lncRNA required for interactions with each protein complex were determined [128], future studies will be necessary to elucidate the structural requirements of lncRNAs such as ANRIL with chromatin regulators such as PRC1/PRC2.
The molecular details of ANRIL-mediated tumor suppression are becoming more clear. However, other mechanisms of lncRNA-mediated suppression of tumor suppressor genes have been reported. For example, a change in the expression ratio of bidirectional genes has been shown to mediate the expression of the tumor suppressor p21 [185]. Collectively, these observations suggest that lncRNA-mediated silencing of tumor suppressor genes may be a major mechanism driving tumorigenesis.
Cancer type specific lncRNA expression
Many of the described lncRNAs are expressed in a variety of cancers, however a select few thus far have been associated with a single cancer type. HOTAIR, for example, has only been described in breast cancer, while three lncRNAs PCGEM1, DD3 and PCNCR1 have been associated solely with prostate cancer [68, 186, 187]. The most recently described of these, Prostate Cancer Non-Coding RNA 1 (PCNCR1) lncRNA, was identified in a 'gene desert' on chromosome 8q24.2 and is associated with susceptibility to prostate cancer. PCNCR1 is expressed as an intronless, ~13 kb transcript with a potential role in trans-activation of androgen receptor (AR), a key player in prostate cancer progression [187]. Likewise, PCGEM1 (Prostate Specific Gene 1) was found to have properties supporting tumorigenesis, as ectopic overexpression of PCGEM1 RNA resulted in increased cell growth and colony formation in cell lines [188]. The lncRNA Differential Display Code 3 (DD3) is also highly over expressed in prostate cancer, yet little is known about the role DD3 may play in prostate cancer progression [189, 190]. Finally, the liver associated lncRNA HULC is highly expressed in primary liver tumors, and in colorectal carcinomas that metastasized to the liver, but not in the primary colon tumors or in non-liver metastases [171].
RNA polymerase III transcription of lncRNA
The lncRNAs described thus far are products of RNA pol II transcription, yet many ncRNAs are transcribed by RNA polymerase III (RNA pol III) [191]. Importantly, RNA pol III is frequently deregulated in cancer cells resulting in increased activity [192, 193]. The molecular mechanisms driving increased RNA pol III activity in tumor cells include overexpression of RNA pol III transcription factors, escape from RNA pol III repressors and direct oncogene-mediated activation [193–195]. Aberrant RNA pol III function may have consequences to the expression of lncRNAs transcribed by this polymerase.
For example, the lncRNA BC200 is a small cytoplasmic lncRNA in the neurons of primate nervous systems and human cancers, but not in non-neuronal organs [20, 50, 51, 196, 197]. Unlike the majority of lncRNAs described thus far, BC200 is transcribed by RNA pol III and shares unique homology with human Alu elements [196, 198]. Similarly, the lncRNA HULC also shares homology with mobile DNA, in this case with a long terminal repeat (LTR) retroelement [170]. The BC200 RNA has been characterized as a negative regulator of eIF4A-dependent translation initiation [199].
Due to the fact that many whole transcriptome sequencing methods were developed to enrich for poly(A) purified transcripts, RNA pol III transcripts may have been excluded from analysis. This suggests that other, yet unidentified RNA pol III lncRNAs over-expressed in cancer may be participating in tumorigenesis.
Aberrant T-UCR expression in human carcinoma
Transcribed ultraconserved Regions (T-UCRs) are evolutionary conserved sequences found in both intergenic and intragenic regions of the human genome [200, 201]. These unique sequences are defined as 481 segments of DNA that are absolutely conserved between orthologous regions of the human, rat and mouse genomes [201, 202]. The transcription products of T-UCRs are 200-779 nt in length and were originally classed into three categories, non-exonic, exonic and possibly exonic, according to their overlap with known protein-coding genes [202]. More recently, T-UCRs have been re-annotated into a more descriptive set of five categories: intergenic (38.7%), intronic (42.6%), exonic (4.2%), partly exonic (5%) or exonic containing (5.6%) (Figure 6) [59].

Genomic locations of the five classes of T-UCRs. The exons of coding genes are indicated by boxes, while the locations of the T-UCR elements are marked by a double-T bar. The five possible positions are as indicated exonic, partly exonic, exon containing, intronic and intergenic.
The high degree of conservation of T-UCRs, combined with their tissue-specific expression, suggests these ncRNAs may play a critical role in cellular metabolism and development [200]. The expression of many T-UCRs is significantly altered in cancer, notably in adult chronic lymphocytic leukemias, colorectal and hepatocellular carcinomas and neuroblastomas [85, 200]. Their aberrant transcription profiles can be used to differentiate types of human cancers and have been linked to patient outcome [85]. Some of the T-UCRs reside in genomic regions associated with specific types of cancer [200]. For example, in colon cancer, the T-UCR uc.73A is one of the most highly upregulated T-UCRs, and this lncRNA has been found to show oncogenic properties by proliferation assays [200]. Similarly, T-UCR uc.338 is significantly upregulated in human hepatocarcinoma tumor and cell lines, and uc.338 was found to be part of a larger transcriptional unit coined TUC338 which is involved in cell growth [203].
The mechanism by which T-UCRs are differentially expressed is unclear. A study profiling T-UCRs in neuroblastomas did not find a consistent association between genomic alterations and T-UCR expression, suggesting aberrant T-UCR expression may be a consequence of epigenetic mechanisms. Like miRNAs and coding genes, T-UCR expression has been shown to be repressed by CpG island hypermethylation [85, 204]. Similarly, miRNAs have been shown to bind to, and downregulate T-UCRs, suggesting a complex regulatory mechanism may exist between ncRNAs in human cancers [85, 200]. Collectively, these reports have suggested that aberrant T-UCR expression may have a yet undetermined biological role in tumorigenesis.
Utility of LncRNAs in Cancer Diagnostics and Therapies
Like their smaller non-coding miRNA counterparts, lncRNAs represent a significant untapped resource in terms of developing diagnostics and therapies. Differential or high level expression of certain cancer type-specific lncRNAs can be exploited for the development of novel biomarkers as lncRNA expression or may potentially correlate with patient response to chemotherapy. Understanding the mechanism(s) by which lncRNAs act will continue to provide novel approaches to regulating genes including the development of mimetics to compete with binding sites for miRNAs, chromatin remodelers, or DNA. It has been suggested that mediating transcriptional gene silencing (TGS) pathways, especially those of tumor suppressors or oncogenes, could be of high therapeutic benefit [205].
It has been widely reported that cancer-specific miRNAs are detectable in the blood, sputum and urine of cancer patients [206–210]. Likewise, lncRNAs have demonstrated utility as fluid-based markers of specific cancers. For example, the prostate specific lncRNA DD3 has been developed into highly specific, nucleic acid amplification-based marker of prostate cancer, which demonstrated higher specificity than serum prostate-specific antigen (PSA) [211, 212]. Similarly, the highly expressed hepatocarcinoma-associated lncRNA HULC is detectable in the blood of hepatocarcinoma patients by conventional PCR methods [170].
The use of lncRNAs as therapeutic agents is only beginning to be explored [213]. Although our understanding of the molecular mechanisms of lncRNA function is limited, some features of lncRNAs make them ideal candidates for therapeutic intervention. Many lncRNAs appear to have protein-binding or functional potential that is dependent on secondary structure, this may provide a means of intervention [214]. Preventing the interactions of HOTAIR with the PRC2 or LSD1 complexes, for example, may limit the metastatic potential of breast cancer cells [215].
The tumor expression of certain lncRNAs provides a source of regulatory regions that can be used to reduce the risk of affecting normal tissues during transgene-mediated treatment. For example, H19 is strongly expressed in embryonic cells and in a wide-range of human cancers [20, 93, 94]. As such, a plasmid-based system has been developed to exploit the tumor-specific expression of H19, primarily tested in treating bladder cancer. A plasmid construct, harboring a diptheria toxin gene driven by H19 specific regulatory sequences, is either administered via intratumoral injection as naked DNA, or complexed to the cationic polymer polyethylenimine (PEI) to form a polyplex vector [216]. PEI-complexed plasmid is thought to increase the efficiency of DNA uptake via clathrin-dependent and -independent (cholesterol-dependent) pathways [217]. Upon uptake, high levels of diptheria toxin are expressed in the tumor, resulting in a reduction in tumor size in human trials [218–221]. Collectively, these advances indicate the potential in developing lncRNA mediated diagnostics and therapies.
Conclusions
Differential expression of lncRNAs is becoming recognized as a hallmark feature in cancer, however the functional role for the vast majority of these unique genes is still in question. In this review, we highlight characterized lncRNAs described to play a functional role in cancer-associated processes, such as metastasis and loss of imprinting. Aberrant lncRNA expression participates in carcinogenesis by disrupting major biological processes, such as redirecting chromatin remodeling complexes or inactivating major tumor suppressor genes. We also describe the potential role of dysregulated T-UCRs in cancer, and place these unique RNAs in the lncRNA category. Finally, we note the potential utility of lncRNAs in cancer as diagnostic and prognostic markers, as well as the potential of developing lncRNA mediated therapy.
Authors' information
EAG is a research fellow at the BC Cancer Agency Research Centre, specializing in non-coding RNA gene structure and function. CJB is a professor of Medical Genetics at the University of British Columbia. Her research focuses on human X chromosome inactivation, in particular the role of XIST, a gene she first described during postdoctoral studies in the Willard lab. WLL is a professor of Pathology and Laboratory Medicine at the University of British Columbia. His team invented tiling path array technologies for whole genome and methylome analyses.
Abbreviations
- ANRIL :
-
Antisense Non-coding RNA in the INK4 Locus
- AR:
-
androgen receptor
- CREB:
-
cAMP response element binding protein
- DD3 :
-
Differential Display Code 3
- HBV:
-
hepatitis B virus
- HCC:
-
hepatocellular carcinoma cell lines
- HOTAIR :
-
HOX Antisense Intergenic RNA
- HULC :
-
Highly Upregulated in Liver Cancer
- IGF2:
-
Insulin-Like Growth Factor 2
- kb:
-
kilobase
- lincRNA:
-
long or large intergenic non-coding RNA
- lncRNA:
-
long non-coding RNA
- LTR:
-
long terminal repeat
- MALAT1 :
-
Metastatis-Associated Lung Adenocarcinoma Transcript 1
- MEG3 :
-
Maternally Expressed Gene 3
- miRNA:
-
microRNA
- ncRNA:
-
non-coding RNA
- nt:
-
nucleotide
- PCNCR1 :
-
Prostate Cancer Non-Coding RNA 1
- PEI:
-
polyethylenimine
- PRC:
-
polycomb repressive complex
- PTEN :
-
phosphatase and tensin homolog
- PTENP1 :
-
phosphatase and tensin homolog pseudogene 1
- RNase:
-
ribonuclease
- RNA pol II:
-
RNA polymerase II
- RNA pol III:
-
RNA polymerase III
- RNAi:
-
RNA interference
- SR:
-
serine-arginine rich splicing proteins
- TGS:
-
transcriptional gene silencing
- tiRNA:
-
transcription initiation RNAs
- T-UCR:
-
transcribed ultraconserved region
- UTR:
-
untranslated region
- XIST :
-
X-Inactive-Specific Transcript.
References
Stein LD: Human genome: end of the beginning. Nature. 2004, 431 (7011): 915-916.
Ponting CP, Belgard TG: Transcribed dark matter: meaning or myth?. Hum Mol Genet. 2010, 19 (R2): R162-168.
Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE: Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007, 447 (7146): 799-816.
Costa FF: Non-coding RNAs: Meet thy masters. Bioessays. 2010, 32 (7): 599-608.
Kapranov P, Willingham AT, Gingeras TR: Genome-wide transcription and the implications for genomic organization. Nat Rev Genet. 2007, 8 (6): 413-423.
Frith MC, Pheasant M, Mattick JS: The amazing complexity of the human transcriptome. Eur J Hum Genet. 2005, 13 (8): 894-897.
Khachane AN, Harrison PM: Mining mammalian transcript data for functional long non-coding RNAs. PLoS One. 2010, 5 (4): e10316-
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP: Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009, 458 (7235): 223-227.
Mattick JS, Makunin IV: Non-coding RNA. Hum Mol Genet. 2006, 15 (1): R17-29.
Washietl S, Hofacker IL, Lukasser M, Huttenhofer A, Stadler PF: Mapping of conserved RNA secondary structures predicts thousands of functional noncoding RNAs in the human genome. Nat Biotechnol. 2005, 23 (11): 1383-1390.
Wang J, Zhang J, Zheng H, Li J, Liu D, Li H, Samudrala R, Yu J, Wong GK: Mouse transcriptome: neutral evolution of 'non-coding' complementary DNAs. Nature. 2004, 431 (7010): 1-p following 757; discussion following 757,
Struhl K: Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat Struct Mol Biol. 2007, 14 (2): 103-105.
Ebisuya M, Yamamoto T, Nakajima M, Nishida E: Ripples from neighbouring transcription. Nat Cell Biol. 2008, 10 (9): 1106-1113.
van Bakel H, Nislow C, Blencowe BJ, Hughes TR: Most "dark matter" transcripts are associated with known genes. PLoS Biol. 2010, 8 (5): e1000371.
Wilusz JE, Sunwoo H, Spector DL: Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009, 23 (13): 1494-1504.
Ponting CP, Oliver PL, Reik W: Evolution and functions of long noncoding RNAs. Cell. 2009, 136 (4): 629-641.
Mercer TR, Dinger ME, Mattick JS: Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009, 10 (3): 155-159.
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E: Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007, 129 (7): 1311-1323.
Loewer S, Cabili MN, Guttman M, Loh YH, Thomas K, Park IH, Garber M, Curran M, Onder T, Agarwal S: Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet. 2010, 42 (12): 1113-1117.
Castle JC, Armour CD, Lower M, Haynor D, Biery M, Bouzek H, Chen R, Jackson S, Johnson JM, Rohl CA: Digital genome-wide ncRNA expression, including SnoRNAs, across 11 human tissues using polyA-neutral amplification. PLoS One. 2010, 5 (7): e11779-
Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS: Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci USA. 2008, 105 (2): 716-721.
Wu SC, Kallin EM, Zhang Y: Role of H3K27 methylation in the regulation of lncRNA expression. Cell Res. 2010, 20 (10): 1109-1116.
Taft RJ, Pang KC, Mercer TR, Dinger M, Mattick JS: Non-coding RNAs: regulators of disease. J Pathol. 2010, 220 (2): 126-139.
Maruyama R, Shipitsin M, Choudhury S, Wu Z, Protopopov A, Yao J, Lo PK, Bessarabova M, Ishkin A, Nikolsky Y: Breast Cancer Special Feature: Altered antisense-to-sense transcript ratios in breast cancer. Proc Natl Acad Sci USA. 2010,
Silva JM, Perez DS, Pritchett JR, Halling ML, Tang H, Smith DI: Identification of long stress-induced non-coding transcripts that have altered expression in cancer. Genomics. 2010, 95 (6): 355-362.
Perez DS, Hoage TR, Pritchett JR, Ducharme-Smith AL, Halling ML, Ganapathiraju SC, Streng PS, Smith DI: Long, abundantly expressed non-coding transcripts are altered in cancer. Hum Mol Genet. 2008, 17 (5): 642-655.
Guffanti A, Iacono M, Pelucchi P, Kim N, Solda G, Croft LJ, Taft RJ, Rizzi E, Askarian-Amiri M, Bonnal RJ: A transcriptional sketch of a primary human breast cancer by 454 deep sequencing. BMC Genomics. 2009, 10: 163-
Brosnan CA, Voinnet O: The long and the short of noncoding RNAs. Curr Opin Cell Biol. 2009, 21 (3): 416-425.
Mattick JS: Non-coding RNAs: the architects of eukaryotic complexity. EMBO Rep. 2001, 2 (11): 986-991.
Costa FF: Non-coding RNAs: new players in eukaryotic biology. Gene. 2005, 357 (2): 83-94.
Okamura K, Chung WJ, Ruby JG, Guo H, Bartel DP, Lai EC: The Drosophila hairpin RNA pathway generates endogenous short interfering RNAs. Nature. 2008, 453 (7196): 803-806.
Kawaji H, Hayashizaki Y: Exploration of small RNAs. PLoS Genet. 2008, 4 (1): e22.
Bartel DP: MicroRNAs: target recognition and regulatory functions. Cell. 2009, 136 (2): 215-233.
Carthew RW, Sontheimer EJ: Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009, 136 (4): 642-655.
Krol J, Loedige I, Filipowicz W: The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010, 11 (9): 597-610.
Fabian MR, Sonenberg N, Filipowicz W: Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010, 79: 351-379.
Chekulaeva M, Filipowicz W: Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr Opin Cell Biol. 2009, 21 (3): 452-460.
Kim VN, Han J, Siomi MC: Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009, 10 (2): 126-139.
Taft RJ, Glazov EA, Cloonan N, Simons C, Stephen S, Faulkner GJ, Lassmann T, Forrest AR, Grimmond SM, Schroder K: Tiny RNAs associated with transcription start sites in animals. Nat Genet. 2009, 41 (5): 572-578.
Taft RJ, Simons C, Nahkuri S, Oey H, Korbie DJ, Mercer TR, Holst J, Ritchie W, Wong JJ, Rasko JE: Nuclear-localized tiny RNAs are associated with transcription initiation and splice sites in metazoans. Nat Struct Mol Biol. 2010, 17 (8): 1030-1034.
Farazi TA, Spitzer JI, Morozov P, Tuschl T: miRNAs in human cancer. J Pathol. 2011, 223 (2): 102-115.
Fabbri M, Calin GA: Epigenetics and miRNAs in human cancer. Adv Genet. 2010, 70: 87-99.
Iorio MV, Croce CM: MicroRNAs in cancer: small molecules with a huge impact. J Clin Oncol. 2009, 27 (34): 5848-5856.
Chang LS, Lin SY, Lieu AS, Wu TL: Differential expression of human 5S snoRNA genes. Biochem Biophys Res Commun. 2002, 299 (2): 196-200.
Liao J, Yu L, Mei Y, Guarnera M, Shen J, Li R, Liu Z, Jiang F: Small nucleolar RNA signatures as biomarkers for non-small-cell lung cancer. Mol Cancer. 2010, 9: 198.
Ting DT, Lipson D, Paul S, Brannigan BW, Akhavanfard S, Coffman EJ, Contino G, Deshpande V, Iafrate AJ, Letovsky S: Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science. 2011, 331 (6017): 593-596.
Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, Long J, Stern D, Tammana H, Helt G: Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005, 308 (5725): 1149-1154.
Wu Q, Kim YC, Lu J, Xuan Z, Chen J, Zheng Y, Zhou T, Zhang MQ, Wu CI, Wang SM: Poly A- transcripts expressed in HeLa cells. PLoS One. 2008, 3 (7): e2803.
Hawkins PG, Morris KV: Transcriptional regulation of Oct4 by a long non-coding RNA antisense to Oct4-pseudogene 5. Transcr. 2010, 1 (3): 165-175. 10.4161/trns.1.3.13332.
Chen W, Bocker W, Brosius J, Tiedge H: Expression of neural BC200 RNA in human tumours. J Pathol. 1997, 183 (3): 345-351.
Iacoangeli A, Lin Y, Morley EJ, Muslimov IA, Bianchi R, Reilly J, Weedon J, Diallo R, Bocker W, Tiedge H: BC200 RNA in invasive and preinvasive breast cancer. Carcinogenesis. 2004, 25 (11): 2125-2133.
Ramskold D, Wang ET, Burge CB, Sandberg R: An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput Biol. 2009, 5 (12): e1000598.
Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C: Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol. 2010, 28 (5): 503-510.
Babak T, Blencowe BJ, Hughes TR: A systematic search for new mammalian noncoding RNAs indicates little conserved intergenic transcription. BMC Genomics. 2005, 6: 104.
Bono H, Yagi K, Kasukawa T, Nikaido I, Tominaga N, Miki R, Mizuno Y, Tomaru Y, Goto H, Nitanda H: Systematic expression profiling of the mouse transcriptome using RIKEN cDNA microarrays. Genome Res. 2003, 13 (6B): 1318-1323.
Yang L, Duff MO, Graveley BR, Carmichael GG, Chen LL: Genomewide characterization of non-polyadenylated RNAs. Genome Biol. 2011, 12 (2): R16.
Kapranov P, St Laurent G, Raz T, Ozsolak F, Reynolds CP, Sorensen PH, Reaman G, Milos P, Arceci RJ, Thompson JF: The majority of total nuclear-encoded non-ribosomal RNA in a human cell is 'dark matter' un-annotated RNA. BMC Biol. 2010, 8: 149.
Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A: Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA. 2009, 106 (28): 11667-11672.
Mestdagh P, Fredlund E, Pattyn F, Rihani A, Van Maerken T, Vermeulen J, Kumps C, Menten B, De Preter K, Schramm A: An integrative genomics screen uncovers ncRNA T-UCR functions in neuroblastoma tumours. Oncogene. 2010, 29 (24): 3583-3592.
Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q: Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010, 143 (1): 46-58.
Chen LL, Carmichael GG: Decoding the function of nuclear long non-coding RNAs. Curr Opin Cell Biol. 2010, 22 (3): 357-364.
Lipovich L, Johnson R, Lin CY: MacroRNA underdogs in a microRNA world: evolutionary, regulatory, and biomedical significance of mammalian long non-protein-coding RNA. Biochim Biophys Acta. 2010, 1799 (9): 597-615.
Costa FF: Non-coding RNAs, epigenetics and complexity. Gene. 2008, 410 (1): 9-17.
Yang PK, Kuroda MI: Noncoding RNAs and intranuclear positioning in monoallelic gene expression. Cell. 2007, 128 (4): 777-786.
Heard E, Disteche CM: Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes Dev. 2006, 20 (14): 1848-1867.
Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA: The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010, 39 (6): 925-938.
Goodrich JA, Kugel JF: Non-coding-RNA regulators of RNA polymerase II transcription. Nat Rev Mol Cell Biol. 2006, 7 (8): 612-616.
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL: Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010, 464 (7291): 1071-1076.
Prasanth KV, Spector DL: Eukaryotic regulatory RNAs: an answer to the 'genome complexity' conundrum. Genes Dev. 2007, 21 (1): 11-42.
Gong C, Maquat LE: lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3' UTRs via Alu elements. Nature. 2011, 470 (7333): 284-288.
Huarte M, Rinn JL: Large non-coding RNAs: missing links in cancer?. Hum Mol Genet. 2010, 19 (R2): R152-161.
Kondo T, Plaza S, Zanet J, Benrabah E, Valenti P, Hashimoto Y, Kobayashi S, Payre F, Kageyama Y: Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science. 2010, 329 (5989): 336-339.
Ulveling D, Francastel C, Hube F: When one is better than two: RNA with dual functions. Biochimie. 2010,
Brannan CI, Dees EC, Ingram RS, Tilghman SM: The product of the H19 gene may function as an RNA. Mol Cell Biol. 1990, 10 (1): 28-36.
Brown CJ, Ballabio A, Rupert JL, Lafreniere RG, Grompe M, Tonlorenzi R, Willard HF: A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature. 1991, 349 (6304): 38-44.
Lee RC, Feinbaum RL, Ambros V: The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993, 75 (5): 843-854.
Lau NC, Lim LP, Weinstein EG, Bartel DP: An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001, 294 (5543): 858-862.
Reinhart BJ, Weinstein EG, Rhoades MW, Bartel B, Bartel DP: MicroRNAs in plants. Genes Dev. 2002, 16 (13): 1616-1626.
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K: Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002, 99 (24): 15524-15529.
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ: miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008, 36 Database issue: D154-158.
Griffiths-Jones S: miRBase: microRNA sequences and annotation. Curr Protoc Bioinformatics. 2010, Chapter 12:Unit 12 19 11-10,
Berezikov E, Plasterk RH: Camels and zebrafish, viruses and cancer: a microRNA update. Hum Mol Genet. 2005, 14 (2): R183-190.
Friedman RC, Farh KK, Burge CB, Bartel DP: Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19 (1): 92-105.
Kawai J, Shinagawa A, Shibata K, Yoshino M, Itoh M, Ishii Y, Arakawa T, Hara A, Fukunishi Y, Konno H: Functional annotation of a full-length mouse cDNA collection. Nature. 2001, 409 (6821): 685-690.
Scaruffi P, Stigliani S, Moretti S, Coco S, De Vecchi C, Valdora F, Garaventa A, Bonassi S, Tonini GP: Transcribed-Ultra Conserved Region expression is associated with outcome in high-risk neuroblastoma. BMC Cancer. 2009, 9: 441.
Rainier S, Johnson LA, Dobry CJ, Ping AJ, Grundy PE, Feinberg AP: Relaxation of imprinted genes in human cancer. Nature. 1993, 362 (6422): 747-749.
Wood AJ, Oakey RJ: Genomic imprinting in mammals: emerging themes and established theories. PLoS Genet. 2006, 2 (11): e147.
Feil R, Berger F: Convergent evolution of genomic imprinting in plants and mammals. Trends Genet. 2007, 23 (4): 192-199.
Wan LB, Bartolomei MS: Regulation of imprinting in clusters: noncoding RNAs versus insulators. Adv Genet. 2008, 61: 207-223.
Lim DH, Maher ER: Genomic imprinting syndromes and cancer. Adv Genet. 2010, 70: 145-175.
Monk D: Deciphering the cancer imprintome. Brief Funct Genomics. 2010, 9 (4): 329-339.
Gabory A, Jammes H, Dandolo L: The H19 locus: role of an imprinted non-coding RNA in growth and development. Bioessays. 2010, 32 (6): 473-480.
Poirier F, Chan CT, Timmons PM, Robertson EJ, Evans MJ, Rigby PW: The murine H19 gene is activated during embryonic stem cell differentiation in vitro and at the time of implantation in the developing embryo. Development. 1991, 113 (4): 1105-1114.
Lustig O, Ariel I, Ilan J, Lev-Lehman E, De-Groot N, Hochberg A: Expression of the imprinted gene H19 in the human fetus. Mol Reprod Dev. 1994, 38 (3): 239-246.
Hibi K, Nakamura H, Hirai A, Fujikake Y, Kasai Y, Akiyama S, Ito K, Takagi H: Loss of H19 imprinting in esophageal cancer. Cancer Res. 1996, 56 (3): 480-482.
Fellig Y, Ariel I, Ohana P, Schachter P, Sinelnikov I, Birman T, Ayesh S, Schneider T, de Groot N, Czerniak A: H19 expression in hepatic metastases from a range of human carcinomas. J Clin Pathol. 2005, 58 (10): 1064-1068.
Matouk IJ, DeGroot N, Mezan S, Ayesh S, Abu-lail R, Hochberg A, Galun E: The H19 non-coding RNA is essential for human tumor growth. PLoS One. 2007, 2 (9): e845.
Barsyte-Lovejoy D, Lau SK, Boutros PC, Khosravi F, Jurisica I, Andrulis IL, Tsao MS, Penn LZ: The c-Myc oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Res. 2006, 66 (10): 5330-5337.
Berteaux N, Lottin S, Monte D, Pinte S, Quatannens B, Coll J, Hondermarck H, Curgy JJ, Dugimont T, Adriaenssens E: H19 mRNA-like noncoding RNA promotes breast cancer cell proliferation through positive control by E2F1. J Biol Chem. 2005, 280 (33): 29625-29636.
Dugimont T, Montpellier C, Adriaenssens E, Lottin S, Dumont L, Iotsova V, Lagrou C, Stehelin D, Coll J, Curgy JJ: The H19 TATA-less promoter is efficiently repressed by wild-type tumor suppressor gene product p53. Oncogene. 1998, 16 (18): 2395-2401.
Farnebo M, Bykov VJ, Wiman KG: The p53 tumor suppressor: a master regulator of diverse cellular processes and therapeutic target in cancer. Biochem Biophys Res Commun. 2010, 396 (1): 85-89.
Cai X, Cullen BR: The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA. 2007, 13 (3): 313-316.
Tsang WP, Ng EK, Ng SS, Jin H, Yu J, Sung JJ, Kwok TT: Oncofetal H19-derived miR-675 regulates tumor suppressor RB in human colorectal cancer. Carcinogenesis. 2010, 31 (3): 350-358.
Yoshimizu T, Miroglio A, Ripoche MA, Gabory A, Vernucci M, Riccio A, Colnot S, Godard C, Terris B, Jammes H: The H19 locus acts in vivo as a tumor suppressor. Proc Natl Acad Sci USA. 2008, 105 (34): 12417-12422.
Hao Y, Crenshaw T, Moulton T, Newcomb E, Tycko B: Tumour-suppressor activity of H19 RNA. Nature. 1993, 365 (6448): 764-767.
Colnot S, Niwa-Kawakita M, Hamard G, Godard C, Le Plenier S, Houbron C, Romagnolo B, Berrebi D, Giovannini M, Perret C: Colorectal cancers in a new mouse model of familial adenomatous polyposis: influence of genetic and environmental modifiers. Lab Invest. 2004, 84 (12): 1619-1630.
Leighton PA, Saam JR, Ingram RS, Stewart CL, Tilghman SM: An enhancer deletion affects both H19 and Igf2 expression. Genes Dev. 1995, 9 (17): 2079-2089.
Kanduri C, Whitehead J, Mohammad F: The long and the short of it: RNA-directed chromatin asymmetry in mammalian X-chromosome inactivation. FEBS Lett. 2009, 583 (5): 857-864.
Leeb M, Steffen PA, Wutz A: X chromosome inactivation sparked by non-coding RNAs. RNA Biol. 2009, 6 (2): 94-99.
Payer B, Lee JT: X chromosome dosage compensation: how mammals keep the balance. Annu Rev Genet. 2008, 42: 733-772.
Kay GF, Barton SC, Surani MA, Rastan S: Imprinting and X chromosome counting mechanisms determine Xist expression in early mouse development. Cell. 1994, 77 (5): 639-650.
Lee JT, Davidow LS, Warshawsky D: Tsix, a gene antisense to Xist at the X-inactivation centre. Nat Genet. 1999, 21 (4): 400-404.
Tian D, Sun S, Lee JT: The long noncoding RNA, Jpx, is a molecular switch for X chromosome inactivation. Cell. 2010, 143 (3): 390-403.
Chureau C, Chantalat S, Romito A, Galvani A, Duret L, Avner P, Rougeulle C: Ftx is a non-coding RNA which affects Xist expression and chromatin structure within the X-inactivation center region. Hum Mol Genet. 2010,
Ogawa Y, Lee JT: Xite, X-inactivation intergenic transcription elements that regulate the probability of choice. Mol Cell. 2003, 11 (3): 731-743.
Ogawa Y, Sun BK, Lee JT: Intersection of the RNA interference and X-inactivation pathways. Science. 2008, 320 (5881): 1336-1341.
Migeon BR: Is Tsix repression of Xist specific to mouse?. Nat Genet. 2003, 33 (3): 337; author reply 337-338. 10.1038/ng0303-337a.
Huang KC, Rao PH, Lau CC, Heard E, Ng SK, Brown C, Mok SC, Berkowitz RS, Ng SW: Relationship of XIST expression and responses of ovarian cancer to chemotherapy. Mol Cancer Ther. 2002, 1 (10): 769-776.
Wutz A, Jaenisch R: A shift from reversible to irreversible X inactivation is triggered during ES cell differentiation. Mol Cell. 2000, 5 (4): 695-705.
Brown CJ, Willard HF: The human X-inactivation centre is not required for maintenance of X-chromosome inactivation. Nature. 1994, 368 (6467): 154-156.
Vincent-Salomon A, Ganem-Elbaz C, Manie E, Raynal V, Sastre-Garau X, Stoppa-Lyonnet D, Stern MH, Heard E: X inactive-specific transcript RNA coating and genetic instability of the X chromosome in BRCA1 breast tumors. Cancer Res. 2007, 67 (11): 5134-5140.
Sirchia SM, Tabano S, Monti L, Recalcati MP, Gariboldi M, Grati FR, Porta G, Finelli P, Radice P, Miozzo M: Misbehaviour of XIST RNA in breast cancer cells. PLoS One. 2009, 4 (5): e5559.
Sirchia SM, Ramoscelli L, Grati FR, Barbera F, Coradini D, Rossella F, Porta G, Lesma E, Ruggeri A, Radice P: Loss of the inactive X chromosome and replication of the active X in BRCA1-defective and wild-type breast cancer cells. Cancer Res. 2005, 65 (6): 2139-2146.
Pageau GJ, Hall LL, Ganesan S, Livingston DM, Lawrence JB: The disappearing Barr body in breast and ovarian cancers. Nat Rev Cancer. 2007, 7 (8): 628-633.
Tchieu J, Kuoy E, Chin MH, Trinh H, Patterson M, Sherman SP, Aimiuwu O, Lindgren A, Hakimian S, Zack JA: Female human iPSCs retain an inactive X chromosome. Cell Stem Cell. 2010, 7 (3): 329-342.
Silva SS, Rowntree RK, Mekhoubad S, Lee JT: X-chromosome inactivation and epigenetic fluidity in human embryonic stem cells. Proc Natl Acad Sci USA. 2008, 105 (12): 4820-4825.
Carrel L, Willard HF: X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005, 434 (7031): 400-404.
Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY: Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010, 329 (5992): 689-693.
Simon JA, Kingston RE: Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol. 2009, 10 (10): 697-708.
Morey L, Helin K: Polycomb group protein-mediated repression of transcription. Trends Biochem Sci. 2010, 35 (6): 323-332.
Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT: Genome-wide Identification of Polycomb-Associated RNAs by RIP-seq. Mol Cell. 2010, 40 (6): 939-953.
Zhang Z, Jones A, Sun CW, Li C, Chang CW, Joo HY, Dai Q, Mysliwiec MR, Wu LC, Guo Y: PRC2 Complexes with JARID2, and esPRC2p48 in ES Cells to Modulate ES Cell Pluripotency and Somatic Cell Reprogramming. Stem Cells. 2010,
Simon JA, Lange CA: Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008, 647 (1-2): 21-29.
Hayami S, Kelly JD, Cho HS, Yoshimatsu M, Unoki M, Tsunoda T, Field HI, Neal DE, Yamaue H, Ponder BA: Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int J Cancer. 2011, 128 (3): 574-586.
Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E: MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003, 22 (39): 8031-8041.
Hutchinson JN, Ensminger AW, Clemson CM, Lynch CR, Lawrence JB, Chess A: A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics. 2007, 8: 39.
Yamada K, Kano J, Tsunoda H, Yoshikawa H, Okubo C, Ishiyama T, Noguchi M: Phenotypic characterization of endometrial stromal sarcoma of the uterus. Cancer Sci. 2006, 97 (2): 106-112.
Lin R, Maeda S, Liu C, Karin M, Edgington TS: A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene. 2007, 26 (6): 851-858.
Luo JH, Ren B, Keryanov S, Tseng GC, Rao UN, Monga SP, Strom S, Demetris AJ, Nalesnik M, Yu YP: Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology. 2006, 44 (4): 1012-1024.
Davis IJ, Hsi BL, Arroyo JD, Vargas SO, Yeh YA, Motyckova G, Valencia P, Perez-Atayde AR, Argani P, Ladanyi M: Cloning of an Alpha-TFEB fusion in renal tumors harboring the t(6;11)(p21;q13) chromosome translocation. Proc Natl Acad Sci USA. 2003, 100 (10): 6051-6056.
Kuiper RP, Schepens M, Thijssen J, van Asseldonk M, van den Berg E, Bridge J, Schuuring E, Schoenmakers EF, van Kessel AG: Upregulation of the transcription factor TFEB in t(6;11)(p21;q13)-positive renal cell carcinomas due to promoter substitution. Hum Mol Genet. 2003, 12 (14): 1661-1669.
Rajaram V, Knezevich S, Bove KE, Perry A, Pfeifer JD: DNA sequence of the translocation breakpoints in undifferentiated embryonal sarcoma arising in mesenchymal hamartoma of the liver harboring the t(11;19)(q11;q13.4) translocation. Genes Chromosomes Cancer. 2007, 46 (5): 508-513.
Wilusz JE, Freier SM, Spector DL: 3' end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell. 2008, 135 (5): 919-932.
Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermuller J, Hofacker IL: RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007, 316 (5830): 1484-1488.
Mercer TR, Wilhelm D, Dinger ME, Solda G, Korbie DJ, Glazov EA, Truong V, Schwenke M, Simons C, Matthaei KI: Expression of distinct RNAs from 3' untranslated regions. Nucleic Acids Res. 2010,
Mercer TR, Dinger ME, Bracken CP, Kolle G, Szubert JM, Korbie DJ, Askarian-Amiri ME, Gardiner BB, Goodall GJ, Grimmond SM: Regulated post-transcriptional RNA cleavage diversifies the eukaryotic transcriptome. Genome Res. 2010, 20 (12): 1639-1650.
Ni T, Corcoran DL, Rach EA, Song S, Spana EP, Gao Y, Ohler U, Zhu J: A paired-end sequencing strategy to map the complex landscape of transcription initiation. Nat Methods. 2010, 7 (7): 521-527.
Otsuka Y, Kedersha NL, Schoenberg DR: Identification of a cytoplasmic complex that adds a cap onto 5'-monophosphate RNA. Mol Cell Biol. 2009, 29 (8): 2155-2167.
Post-transcriptional processing generates a diversity of 5'-modified long and short RNAs. Nature. 2009, 457 (7232): 1028-1032.
Bernard D, Prasanth KV, Tripathi V, Colasse S, Nakamura T, Xuan Z, Zhang MQ, Sedel F, Jourdren L, Coulpier F: A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 2010, 29 (18): 3082-3093.
Tano K, Mizuno R, Okada T, Rakwal R, Shibato J, Masuo Y, Ijiri K, Akimitsu N: MALAT-1 enhances cell motility of lung adenocarcinoma cells by influencing the expression of motility-related genes. FEBS Lett. 2010, 584 (22): 4575-4580.
Guo F, Li Y, Liu Y, Wang J, Li G: Inhibition of metastasis-associated lung adenocarcinoma transcript 1 in CaSki human cervical cancer cells suppresses cell proliferation and invasion. Acta Biochim Biophys Sin (Shanghai). 2010, 42 (3): 224-229.
Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE: Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci USA. 2005, 102 (10): 3627-3632.
Mestdagh P, Bostrom AK, Impens F, Fredlund E, Van Peer G, De Antonellis P, von Stedingk K, Ghesquiere B, Schulte S, Dews M: The miR-17-92 microRNA cluster regulates multiple components of the TGF-beta pathway in neuroblastoma. Mol Cell. 2010, 40 (5): 762-773.
Zhao R, Bodnar MS, Spector DL: Nuclear neighborhoods and gene expression. Curr Opin Genet Dev. 2009, 19 (2): 172-179.
Miyoshi N, Wagatsuma H, Wakana S, Shiroishi T, Nomura M, Aisaka K, Kohda T, Surani MA, Kaneko-Ishino T, Ishino F: Identification of an imprinted gene, Meg3/Gtl2 and its human homologue MEG3, first mapped on mouse distal chromosome 12 and human chromosome 14q. Genes Cells. 2000, 5 (3): 211-220.
Zhang X, Zhou Y, Mehta KR, Danila DC, Scolavino S, Johnson SR, Klibanski A: A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J Clin Endocrinol Metab. 2003, 88 (11): 5119-5126.
Zhang X, Rice K, Wang Y, Chen W, Zhong Y, Nakayama Y, Zhou Y, Klibanski A: Maternally expressed gene 3 (MEG3) noncoding ribonucleic acid: isoform structure, expression, and functions. Endocrinology. 2010, 151 (3): 939-947.
Hagan JP, O'Neill BL, Stewart CL, Kozlov SV, Croce CM: At least ten genes define the imprinted Dlk1-Dio3 cluster on mouse chromosome 12qF1. PLoS One. 2009, 4 (2): e4352.
Schuster-Gossler K, Bilinski P, Sado T, Ferguson-Smith A, Gossler A: The mouse Gtl2 gene is differentially expressed during embryonic development, encodes multiple alternatively spliced transcripts, and may act as an RNA. Dev Dyn. 1998, 212 (2): 214-228.
Mondal T, Rasmussen M, Pandey GK, Isaksson A, Kanduri C: Characterization of the RNA content of chromatin. Genome Res. 2010, 20 (7): 899-907.
Zhou Y, Zhong Y, Wang Y, Zhang X, Batista DL, Gejman R, Ansell PJ, Zhao J, Weng C, Klibanski A: Activation of p53 by MEG3 non-coding RNA. J Biol Chem. 2007, 282 (34): 24731-24742.
Pang KC, Frith MC, Mattick JS: Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet. 2006, 22 (1): 1-5.
Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M: A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010, 142 (3): 409-419.
Zhang L, Huang J, Yang N, Greshock J, Megraw MS, Giannakakis A, Liang S, Naylor TL, Barchetti A, Ward MR: microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci USA. 2006, 103 (24): 9136-9141.
Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M: Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA. 2004, 101 (9): 2999-3004.
Weber B, Stresemann C, Brueckner B, Lyko F: Methylation of human microRNA genes in normal and neoplastic cells. Cell Cycle. 2007, 6 (9): 1001-1005.
Lehmann U, Hasemeier B, Christgen M, Muller M, Romermann D, Langer F, Kreipe H: Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J Pathol. 2008, 214 (1): 17-24.
Ebert MS, Neilson JR, Sharp PA: MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007, 4 (9): 721-726.
Panzitt K, Tschernatsch MM, Guelly C, Moustafa T, Stradner M, Strohmaier HM, Buck CR, Denk H, Schroeder R, Trauner M: Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology. 2007, 132 (1): 330-342.
Matouk IJ, Abbasi I, Hochberg A, Galun E, Dweik H, Akkawi M: Highly upregulated in liver cancer noncoding RNA is overexpressed in hepatic colorectal metastasis. Eur J Gastroenterol Hepatol. 2009, 21 (6): 688-692.
Wang J, Liu X, Wu H, Ni P, Gu Z, Qiao Y, Chen N, Sun F, Fan Q: CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 2010, 38 (16): 5366-5383.
Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP: A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010, 465 (7301): 1033-1038.
Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, Salmena L, Sampieri K, Haveman WJ, Brogi E: Subtle variations in Pten dose determine cancer susceptibility. Nat Genet. 2010, 42 (5): 454-458.
Yu W, Gius D, Onyango P, Muldoon-Jacobs K, Karp J, Feinberg AP, Cui H: Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature. 2008, 451 (7175): 202-206.
Folkersen L, Kyriakou T, Goel A, Peden J, Malarstig A, Paulsson-Berne G, Hamsten A, Hugh W, Franco-Cereceda A, Gabrielsen A: Relationship between CAD risk genotype in the chromosome 9p21 locus and gene expression. Identification of eight new ANRIL splice variants. PLoS One. 2009, 4 (11): e7677.
Serrano M, Hannon GJ, Beach D: A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993, 366 (6456): 704-707.
Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV, Stockert E, Day RS, Johnson BE, Skolnick MH: A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994, 264 (5157): 436-440.
Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ: Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997, 91 (5): 649-659.
Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M: The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999, 397 (6715): 164-168.
Popov N, Gil J: Epigenetic regulation of the INK4b-ARF-INK4a locus: in sickness and in health. Epigenetics. 2010, 5 (8): 685-690.
Cunnington MS, Santibanez Koref M, Mayosi BM, Burn J, Keavney B: Chromosome 9p21 SNPs Associated with Multiple Disease Phenotypes Correlate with ANRIL Expression. PLoS Genet. 2010, 6 (4): e1000899.
Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, Gil J, Walsh MJ, Zhou MM: Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. 2010, 38 (5): 662-674.
Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, Kitagawa M, Xiong Y: Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene. 2010,
Morris KV, Santoso S, Turner AM, Pastori C, Hawkins PG: Bidirectional transcription directs both transcriptional gene activation and suppression in human cells. PLoS Genet. 2008, 4 (11): e1000258.
Szell M, Bata-Csorgo Z, Kemeny L: The enigmatic world of mRNA-like ncRNAs: their role in human evolution and in human diseases. Semin Cancer Biol. 2008, 18 (2): 141-148.
Chung S, Nakagawa H, Uemura M, Piao L, Ashikawa K, Hosono N, Takata R, Akamatsu S, Kawaguchi T, Morizono T: Association of a novel long non-coding RNA in 8q24 with prostate cancer susceptibility. Cancer Sci. 2011, 102 (1): 245-252.
Petrovics G, Zhang W, Makarem M, Street JP, Connelly R, Sun L, Sesterhenn IA, Srikantan V, Moul JW, Srivastava S: Elevated expression of PCGEM1, a prostate-specific gene with cell growth-promoting function, is associated with high-risk prostate cancer patients. Oncogene. 2004, 23 (2): 605-611.
Bussemakers MJ, van Bokhoven A, Verhaegh GW, Smit FP, Karthaus HF, Schalken JA, Debruyne FM, Ru N, Isaacs WB: DD3: a new prostate-specific gene, highly overexpressed in prostate cancer. Cancer Res. 1999, 59 (23): 5975-5979.
de Kok JB, Verhaegh GW, Roelofs RW, Hessels D, Kiemeney LA, Aalders TW, Swinkels DW, Schalken JA: DD3(PCA3), a very sensitive and specific marker to detect prostate tumors. Cancer Res. 2002, 62 (9): 2695-2698.
Dieci G, Fiorino G, Castelnuovo M, Teichmann M, Pagano A: The expanding RNA polymerase III transcriptome. Trends Genet. 2007, 23 (12): 614-622.
White RJ: RNA polymerases I and III, non-coding RNAs and cancer. Trends Genet. 2008, 24 (12): 622-629.
White RJ: RNA polymerase III transcription and cancer. Oncogene. 2004, 23 (18): 3208-3216.
Lockwood WW, Chari R, Coe BP, Thu KL, Garnis C, Malloff CA, Campbell J, Williams AC, Hwang D, Zhu CQ: Integrative genomic analyses identify BRF2 as a novel lineage-specific oncogene in lung squamous cell carcinoma. PLoS Med. 2010, 7 (7): e1000315.
Marshall L, White RJ: Non-coding RNA production by RNA polymerase III is implicated in cancer. Nat Rev Cancer. 2008, 8 (12): 911-914.
Watson JB, Sutcliffe JG: Primate brain-specific cytoplasmic transcript of the Alu repeat family. Mol Cell Biol. 1987, 7 (9): 3324-3327.
Tiedge H, Chen W, Brosius J: Primary structure, neural-specific expression, and dendritic location of human BC200 RNA. J Neurosci. 1993, 13 (6): 2382-2390.
Martignetti JA, Brosius J: BC200 RNA: a neural RNA polymerase III product encoded by a monomeric Alu element. Proc Natl Acad Sci USA. 1993, 90 (24): 11563-11567.
Lin D, Pestova TV, Hellen CU, Tiedge H: Translational control by a small RNA: dendritic BC1 RNA targets the eukaryotic initiation factor 4A helicase mechanism. Mol Cell Biol. 2008, 28 (9): 3008-3019.
Calin GA, Liu CG, Ferracin M, Hyslop T, Spizzo R, Sevignani C, Fabbri M, Cimmino A, Lee EJ, Wojcik SE: Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell. 2007, 12 (3): 215-229.
Katzman S, Kern AD, Bejerano G, Fewell G, Fulton L, Wilson RK, Salama SR, Haussler D: Human genome ultraconserved elements are ultraselected. Science. 2007, 317 (5840): 915.
Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D: Ultraconserved elements in the human genome. Science. 2004, 304 (5675): 1321-1325.
Braconi C, Valeri N, Kogure T, Gasparini P, Huang N, Nuovo GJ, Terracciano L, Croce CM, Patel T: Expression and functional role of a transcribed noncoding RNA with an ultraconserved element in hepatocellular carcinoma. Proc Natl Acad Sci USA. 2010,
Lujambio A, Portela A, Liz J, Melo SA, Rossi S, Spizzo R, Croce CM, Calin GA, Esteller M: CpG island hypermethylation-associated silencing of non-coding RNAs transcribed from ultraconserved regions in human cancer. Oncogene. 2010, 29 (48): 6390-6401.
Morris KV: RNA-directed transcriptional gene silencing and activation in human cells. Oligonucleotides. 2009, 19 (4): 299-306.
Scholer N, Langer C, Dohner H, Buske C, Kuchenbauer F: Serum microRNAs as a novel class of biomarkers: a comprehensive review of the literature. Exp Hematol. 2010, 38 (12): 1126-1130.
Xie Y, Todd NW, Liu Z, Zhan M, Fang H, Peng H, Alattar M, Deepak J, Stass SA, Jiang F: Altered miRNA expression in sputum for diagnosis of non-small cell lung cancer. Lung Cancer. 2010, 67 (2): 170-176.
Xing L, Todd NW, Yu L, Fang H, Jiang F: Early detection of squamous cell lung cancer in sputum by a panel of microRNA markers. Mod Pathol. 2010, 23 (8): 1157-1164.
Kosaka N, Iguchi H, Ochiya T: Circulating microRNA in body fluid: a new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 2010, 101 (10): 2087-2092.
Hanke M, Hoefig K, Merz H, Feller AC, Kausch I, Jocham D, Warnecke JM, Sczakiel G: A robust methodology to study urine microRNA as tumor marker: microRNA-126 and microRNA-182 are related to urinary bladder cancer. Urol Oncol. 2010, 28 (6): 655-661.
Tinzl M, Marberger M, Horvath S, Chypre C: DD3PCA3 RNA analysis in urine--a new perspective for detecting prostate cancer. Eur Urol. 2004, 46 (2): 182-186. discussion 187,
Hessels D, Klein Gunnewiek JM, van Oort I, Karthaus HF, van Leenders GJ, van Balken B, Kiemeney LA, Witjes JA, Schalken JA: DD3(PCA3)-based molecular urine analysis for the diagnosis of prostate cancer. Eur Urol. 2003, 44 (1): 8-15. discussion 15-16,
Costa FF: Non-coding RNAs and new opportunities for the private sector. Drug Discov Today. 2009, 14 (9-10): 446-452.
Hung T, Chang HY: Long noncoding RNA in genome regulation: Prospects and mechanisms. RNA Biol. 2010, 7 (5):
Tsai MC, Spitale RC, Chang HY: Long Intergenic Noncoding RNAs: New Links in Cancer Progression. Cancer Res. 2011, 71 (1): 3-7.
Smaldone MC, Davies BJ: BC-819, a plasmid comprising the H19 gene regulatory sequences and diphtheria toxin A, for the potential targeted therapy of cancers. Curr Opin Mol Ther. 2010, 12 (5): 607-616.
Midoux P, Breuzard G, Gomez JP, Pichon C: Polymer-based gene delivery: a current review on the uptake and intracellular trafficking of polyplexes. Curr Gene Ther. 2008, 8 (5): 335-352.
Amit D, Hochberg A: Development of targeted therapy for bladder cancer mediated by a double promoter plasmid expressing diphtheria toxin under the control of H19 and IGF2-P4 regulatory sequences. J Transl Med. 2010, 8 (1): 134.
Ohana P, Gofrit O, Ayesh S, Al-Sharef W, Mizrahi A, Birman T, Schneider T, Matouk I, de Groot N, Tavdy E: Regulatory sequences of the H19 gene in DNA based therapy of bladder cancer. Gene Therapy and Molecular Biology. 2004, 8: 181-192.
Scaiewicz V, Sorin V, Fellig Y, Birman T, Mizrahi A, Galula J, Abu-Lail R, Shneider T, Ohana P, Buscail L: Use of H19 Gene Regulatory Sequences in DNA-Based Therapy for Pancreatic Cancer. J Oncol. 2010, 2010: 178174.
Mizrahi A, Czerniak A, Levy T, Amiur S, Gallula J, Matouk I, Abu-lail R, Sorin V, Birman T, de Groot N: Development of targeted therapy for ovarian cancer mediated by a plasmid expressing diphtheria toxin under the control of H19 regulatory sequences. J Transl Med. 2009, 7: 69.
Phizicky EM, Hopper AK: tRNA biology charges to the front. Genes Dev. 2010, 24 (17): 1832-1860.
Hannan KM, Hannan RD, Rothblum LI: Transcription by RNA polymerase I. Front Biosci. 1998, 3: d376-398.
Stults DM, Killen MW, Pierce HH, Pierce AJ: Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res. 2008, 18 (1): 13-18.
Lin H: piRNAs in the germ line. Science. 2007, 316 (5823): 397.
Kapranov P, Ozsolak F, Kim SW, Foissac S, Lipson D, Hart C, Roels S, Borel C, Antonarakis SE, Monaghan AP: New class of gene-termini-associated human RNAs suggests a novel RNA copying mechanism. Nature. 2010, 466 (7306): 642-646.
Kiss T: Small nucleolar RNA-guided post-transcriptional modification of cellular RNAs. EMBO J. 2001, 20 (14): 3617-3622.
Filipowicz W, Pogacic V: Biogenesis of small nucleolar ribonucleoproteins. Curr Opin Cell Biol. 2002, 14 (3): 319-327.
Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, Young RA, Sharp PA: Divergent transcription from active promoters. Science. 2008, 322 (5909): 1849-1851.
Valadkhan S: snRNAs as the catalysts of pre-mRNA splicing. Curr Opin Chem Biol. 2005, 9 (6): 603-608.
Faulkner GJ, Carninci P: Altruistic functions for selfish DNA. Cell Cycle. 2009, 8 (18): 2895-2900.
Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, Irvine KM, Schroder K, Cloonan N, Steptoe AL, Lassmann T: The regulated retrotransposon transcriptome of mammalian cells. Nat Genet. 2009, 41 (5): 563-571.
Kandhavelu M, Lammi C, Buccioni M, Dal Ben D, Volpini R, Marucci G: Existence of snoRNA, microRNA, piRNA characteristics in a novel non-coding RNA: x-ncRNA and its biological implication in Homo sapiens. Journal of Bioinformatics and Sequence Analysis. 2009, 1 (2): 31-40.
Christov CP, Gardiner TJ, Szuts D, Krude T: Functional requirement of noncoding Y RNAs for human chromosomal DNA replication. Mol Cell Biol. 2006, 26 (18): 6993-7004.
Li Z, Kim SW, Lin Y, Moore PS, Chang Y, John B: Characterization of viral and human RNAs smaller than canonical MicroRNAs. J Virol. 2009, 83 (24): 12751-12758.
Parrott AM, Mathews MB: snaR genes: recent descendants of Alu involved in the evolution of chorionic gonadotropins. Cold Spring Harb Symp Quant Biol. 2009, 74: 363-373.
Parrott AM, Tsai M, Batchu P, Ryan K, Ozer HL, Tian B, Mathews MB: The evolution and expression of the snaR family of small non-coding RNAs. Nucleic Acids Res. 2011, 39 (4): 1485-1500.
Stadler PF, Chen JJ, Hackermuller J, Hoffmann S, Horn F, Khaitovich P, Kretzschmar AK, Mosig A, Prohaska SJ, Qi X: Evolution of vault RNAs. Mol Biol Evol. 2009, 26 (9): 1975-1991.
Zheng D, Frankish A, Baertsch R, Kapranov P, Reymond A, Choo SW, Lu Y, Denoeud F, Antonarakis SE, Snyder M: Pseudogenes in the ENCODE regions: consensus annotation, analysis of transcription, and evolution. Genome Res. 2007, 17 (6): 839-851.
Zhang ZD, Frankish A, Hunt T, Harrow J, Gerstein M: Identification and analysis of unitary pseudogenes: historic and contemporary gene losses in humans and other primates. Genome Biol. 2010, 11 (3): R26.
Zheng R, Shen Z, Tripathi V, Xuan Z, Freier SM, Bennett CF, Prasanth SG, Prasanth KV: Polypurine-repeat-containing RNAs: a novel class of long non-coding RNA in mammalian cells. J Cell Sci. 2010, 123 (Pt 21): 3734-3744.
Louro R, Smirnova AS, Verjovski-Almeida S: Long intronic noncoding RNA transcription: expression noise or expression choice?. Genomics. 2009, 93 (4): 291-298.
Rearick D, Prakash A, McSweeny A, Shepard SS, Fedorova L, Fedorov A: Critical association of ncRNA with introns. Nucleic Acids Res. 2010,
Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J: Antisense transcription in the mammalian transcriptome. Science. 2005, 309 (5740): 1564-1566.
Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, Jensen TH: RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008, 322 (5909): 1851-1854.
Zhang Y, Guan DG, Yang JH, Shao P, Zhou H, Qu LH: ncRNAimprint: a comprehensive database of mammalian imprinted noncoding RNAs. RNA. 2010, 16 (10): 1889-1901.
Szymanski M, Erdmann VA, Barciszewski J: Noncoding RNAs database (ncRNAdb). Nucleic Acids Res. 2007, 35 (Database issue): D162-164.
Mituyama T, Yamada K, Hattori E, Okida H, Ono Y, Terai G, Yoshizawa A, Komori T, Asai K: The Functional RNA Database 3.0: databases to support mining and annotation of functional RNAs. Nucleic Acids Res. 2009, 37 (Database issue): D89-92.
Liu C, Bai B, Skogerbo G, Cai L, Deng W, Zhang Y, Bu D, Zhao Y, Chen R: NONCODE: an integrated knowledge database of non-coding RNAs. Nucleic Acids Res. 2005, 33 (Database issue): D112-115.
Amaral PP, Clark MB, Gascoigne DK, Dinger ME, Mattick JS: lncRNAdb: a reference database for long noncoding RNAs. Nucleic Acids Res. 2011, 39 (Database issue): D146-151.
Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC, Finn RD, Griffiths-Jones S, Eddy SR: Rfam: updates to the RNA families database. Nucleic Acids Res. 2009, 37 (Database issue): D136-140.
Dinger ME, Pang KC, Mercer TR, Crowe ML, Grimmond SM, Mattick JS: NRED: a database of long noncoding RNA expression. Nucleic Acids Res. 2009, 37 (Database issue): D122-126.
Erdmann VA, Szymanski M, Hochberg A, Groot N, Barciszewski J: Non-coding, mRNA-like RNAs database Y2K. Nucleic Acids Res. 2000, 28 (1): 197-200.
Wu T, Wang J, Liu C, Zhang Y, Shi B, Zhu X, Zhang Z, Skogerbo G, Chen L, Lu H: NPInter: the noncoding RNAs and protein related biomacromolecules interaction database. Nucleic Acids Res. 2006, 34 (Database issue): D150-152.
Fellenberg J, Bernd L, Delling G, Witte D, Zahlten-Hinguranage A: Prognostic significance of drug-regulated genes in high-grade osteosarcoma. Mod Pathol. 2007, 20 (10): 1085-1094.
Koshimizu TA, Fujiwara Y, Sakai N, Shibata K, Tsuchiya H: Oxytocin stimulates expression of a noncoding RNA tumor marker in a human neuroblastoma cell line. Life Sci. 2010, 86 (11-12): 455-460.
Ariel I, Sughayer M, Fellig Y, Pizov G, Ayesh S, Podeh D, Libdeh BA, Levy C, Birman T, Tykocinski ML: The imprinted H19 gene is a marker of early recurrence in human bladder carcinoma. Mol Pathol. 2000, 53 (6): 320-323.
Yballe CM, Vu TH, Hoffman AR: Imprinting and expression of insulin-like growth factor-II and H19 in normal breast tissue and breast tumor. J Clin Endocrinol Metab. 1996, 81 (4): 1607-1612.
Tanos V, Ariel I, Prus D, De-Groot N, Hochberg A: H19 and IGF2 gene expression in human normal, hyperplastic, and malignant endometrium. Int J Gynecol Cancer. 2004, 14 (3): 521-525.
Douc-Rasy S, Barrois M, Fogel S, Ahomadegbe JC, Stehelin D, Coll J, Riou G: High incidence of loss of heterozygosity and abnormal imprinting of H19 and IGF2 genes in invasive cervical carcinomas. Uncoupling of H19 and IGF2 expression and biallelic hypomethylation of H19. Oncogene. 1996, 12 (2): 423-430.
Arima T, Matsuda T, Takagi N, Wake N: Association of IGF2 and H19 imprinting with choriocarcinoma development. Cancer Genet Cytogenet. 1997, 93 (1): 39-47.
Tanos V, Prus D, Ayesh S, Weinstein D, Tykocinski ML, De-Groot N, Hochberg A, Ariel I: Expression of the imprinted H19 oncofetal RNA in epithelial ovarian cancer. Eur J Obstet Gynecol Reprod Biol. 1999, 85 (1): 7-11.
Berteaux N, Lottin S, Adriaenssens E, Van Coppenolle F, Leroy X, Coll J, Dugimont T, Curgy JJ: Hormonal regulation of H19 gene expression in prostate epithelial cells. J Endocrinol. 2004, 183 (1): 69-78.
Kondo M, Takahashi T: [Altered genomic imprinting in the IGF2 and H19 genes in human lung cancer]. Nippon Rinsho. 1996, 54 (2): 492-496.
Pasic I, Shlien A, Durbin AD, Stavropoulos DJ, Baskin B, Ray PN, Novokmet A, Malkin D: Recurrent focal copy-number changes and loss of heterozygosity implicate two noncoding RNAs and one tumor suppressor gene at chromosome 3q13.31 in osteosarcoma. Cancer Res. 2010, 70 (1): 160-171.
Srikantan V, Zou Z, Petrovics G, Xu L, Augustus M, Davis L, Livezey JR, Connell T, Sesterhenn IA, Yoshino K: PCGEM1, a prostate-specific gene, is overexpressed in prostate cancer. Proc Natl Acad Sci USA. 2000, 97 (22): 12216-12221.
Fu X, Ravindranath L, Tran N, Petrovics G, Srivastava S: Regulation of apoptosis by a prostate-specific and prostate cancer-associated noncoding gene, PCGEM1. DNA Cell Biol. 2006, 25 (3): 135-141.
Wang XS, Zhang Z, Wang HC, Cai JL, Xu QW, Li MQ, Chen YC, Qian XP, Lu TJ, Yu LZ: Rapid identification of UCA1 as a very sensitive and specific unique marker for human bladder carcinoma. Clin Cancer Res. 2006, 12 (16): 4851-4858.
Wang F, Li X, Xie X, Zhao L, Chen W: UCA1, a non-protein-coding RNA up-regulated in bladder carcinoma and embryo, influencing cell growth and promoting invasion. FEBS Lett. 2008, 582 (13): 1919-1927.
Tsang WP, Wong TW, Cheung AH, Co CN, Kwok TT: Induction of drug resistance and transformation in human cancer cells by the noncoding RNA CUDR. RNA. 2007, 13 (6): 890-898.
Korneev SA, Korneeva EI, Lagarkova MA, Kiselev SL, Critchley G, O'Shea M: Novel noncoding antisense RNA transcribed from human anti-NOS2A locus is differentially regulated during neuronal differentiation of embryonic stem cells. RNA. 2008, 14 (10): 2030-2037.
Pasmant E, Laurendeau I, Heron D, Vidaud M, Vidaud D, Bieche I: Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. 2007, 67 (8): 3963-3969.
Mourtada-Maarabouni M, Pickard MR, Hedge VL, Farzaneh F, Williams GT: GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene. 2009, 28 (2): 195-208.
Leygue E: Steroid receptor RNA activator (SRA1): unusual bifaceted gene products with suspected relevance to breast cancer. Nucl Recept Signal. 2007, 5: e006.
Chooniedass-Kothari S, Hamedani MK, Troup S, Hube F, Leygue E: The steroid receptor RNA activator protein is expressed in breast tumor tissues. Int J Cancer. 2006, 118 (4): 1054-1059.
Zhu Y, Yu M, Li Z, Kong C, Bi J, Li J, Gao Z: ncRAN, a Newly Identified Long Noncoding RNA, Enhances Human Bladder Tumor Growth, Invasion, and Survival. Urology. 2010, 77 (2): 510 e511-515.
Yu M, Ohira M, Li Y, Niizuma H, Oo ML, Zhu Y, Ozaki T, Isogai E, Nakamura Y, Koda T: High expression of ncRAN, a novel non-coding RNA mapped to chromosome 17q25.1, is associated with poor prognosis in neuroblastoma. Int J Oncol. 2009, 34 (4): 931-938.
Acknowledgements
We would like to thank Chad Malloff and Gavin Wilson for their insightful comments. This work was supported by funds from the Canadian Institutes for Health Research (MOP 86731, MOP 77903 to WLL and MOP 13690 to CJB), Canadian Cancer Society (CCS20485), NCI Early Detection Research Network (5U01 CA84971-10) and Department of Defense (CDMRP W81XWH-10-1-0634).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
EAG wrote the first draft of the article, CJB and WLL finalized the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Gibb, E.A., Brown, C.J. & Lam, W.L. The functional role of long non-coding RNA in human carcinomas. Mol Cancer 10, 38 (2011). https://doi.org/10.1186/1476-4598-10-38
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-4598-10-38