
Abstract
Background
Genetic testing is becoming an essential tool for breast cancer (BC) diagnosis and treatment pathway, and particularly important for early detection and cancer prevention. The purpose of this study was to explore the diagnostic yield of targeted sequencing of the high priority BC genes.
Methods
We have utilized a cost-effective targeted sequencing approach of high priority actionable BC genes (BRCA1, BRCA2, ERBB2 and TP53) in a homogeneous patient cohort from Bangladesh (n = 52) by using tumor and blood samples.
Results
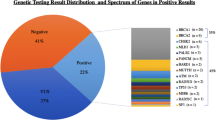
Blood derived targeted sequencing revealed 25.58% (11/43) clinically relevant mutations (both pathogenic and variants of uncertain significance (VUS)), with 13.95% (6/43) of samples carrying a pathogenic mutations. We have identified and validated five novel pathogenic germline mutations in this cohort, comprising of two frameshift deletions in BRCA2, and missense mutations in BRCA1, BRCA2 and ERBB2 gene respectively. Furthermore, we have identified three pathogenic mutations and a VUS within three tumor samples, including a sample carrying pathogenic mutations impacting both TP53 (c.322dupG; a novel frameshift insertion) and BRCA1 genes (c.116G > A). 22% of tissue samples had a clinically relevant TP53 mutation. Although the cohort is small, we have found pathogenic mutations to be enriched in BRCA2 (9.30%, 4/43) compare to BRCA1 (4.65%, 2/43). The frequency of germline VUS mutations found to be similar in both BRCA1 (4.65%; 2/43) and BRCA2 (4.65%; 2/43) compared to ERBB2 (2.32%; 1/43).
Conclusions
This is the first genetic study of BC predisposition genes in this population, implies that genetic screening through targeted sequencing can detect clinically significant and actionable BC-relevant mutations.
Similar content being viewed by others
Background
Breast cancer (BC) is the most common type of cancer among women, impacting 2 million new cases and causes over 600,000 deaths worldwide [1]. While the prevalence of BC is increasing globally, it is critical to screen known BC genes to improve breast cancer survival and early detection, especially in developing countries where majority of women with BC are diagnosed at an advanced stage. The five-year survival rate is below 40% in low-income countries, 60% in middle-income countries and 80% or over in North America, Sweden and Japan where early detection and different treatment options are available [2].
Traditionally, breast self-examination, clinical breast assessment and mammography all have been used alone or in combination to screen BC and facilitate early detection of potentially malignant breast lesions. In recent years, genetic screening has become a critical tool for BC assessment, diagnosis and in guiding treatment choices [3, 4].Mutation profiling for BC has been an integral part of clinical care since the discovery of the BRCA1 and BRCA2 genes [5,6,7].Cases with BRCA1 and BRCA2 pathogenic mutations have a significantly increased risk of developing BC before the age of 50 years [8,9,10,11]. Breast cancer risk for late onset cases (above 70 years old) who carry pathogenic mutation in BRCA1 and BRCA2 is 57 and 50%, respectively [12]. These numbers are higher than any other studied mutations associated with hereditary (familial) BC. Importantly, the frequency distribution of some of the BRCA mutations varies by population studied, suggesting a population specific mutational profile.
Another important gene is TP53 that was originally identified as a risk factor for Li–Fraumeni syndrome. TP53 mutations are the most frequent genetic abnormalities in BC tumors. Approximately 30% of all BC tumors reported to have a mutation in TP53 and mutation within this gene is also associated with poor prognosis [13]. The mutation frequency varies depending on the tumor subtypes, with mutations in 26% luminal, 50% in HER2 amplified tumors, and 88% in basal-like subtypes [13].
It has been reported that theTP53 mutation status may influence the patient’s response to treatment, and determine resistance to several chemotherapy drugs [14,15,16]. Although genetic mutation screening is becoming an essential test for BC diagnosis and therapeutics, the cost associated with whole genome sequencing is still high and not a feasible option for clinical practice in developing countries. Given these challenges, we explored the diagnostic yield of targeted sequencing of the high priority BC genes BRCA1, BRCA2, TP53 and ERBB2.
Methods
Study subjects
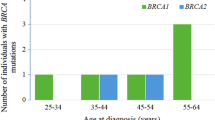
The cohort comprised of 52 individuals (43 blood and 9 breast tissue samples) with age ranges between 30 and 70 years, including 30.77% (16/52, 13 blood and 3 tissue samples) between 30 and 39 years, 36.54% (19/52, 16 blood and 3 tissue samples) between 40 and 49 years and 32.69% (17/52, 14 blood and 3 tissue samples) between 50 and 70 years (Table 3). Of the 52, 76.92% (40/52, 38 blood and 2 tissue samples) had a positive family history of breast cancer and 61.53% (32/52, 23 blood and 9 tissue samples) were diagnosed with breast cancer. 30.77% (16/52, all are blood samples) had symptoms of breast lump, pain and swelling but were, as yet, undiagnosed. Although the rest 7.70% (4/52) had no symptoms, these individuals were included into the cohort due to positive family history. Of the 32 diagnosed breast cancer patients, 15.62% (5/32, all are blood samples) were in stage I, 53.13% (17/32, 12 blood and 5 tissue samples) were in stage II, 25% (8/32, 6 blood and 2 tissue samples) were in stage III and 6.25% (2/32, all are tissue samples) did not provide stage information. These cases went through clinical assessment for breast cancer at multiple centers between January, 2017 to August, 2018 in Dhaka, Bangladesh. They were prospectively recruited from Dhaka Medical College and Hospital, Holy Family Red Crescent Medical College and Hospital, Oncology Center and General Hospital, and IbnSina Diagnostic and Imaging Center of Bangladesh. The study was approved by the Institutional Review Board of Holy Family Red Crescent Medical College, and all samples were collected with written informed consent.
DNA extraction and amplification
DNA was extracted from breast tissue and blood sample using GeneJET Genomic DNA Purification Kit (Thermo Fisher Scientific, USA) and ReliaPrep™ Blood gDNA isolation kit (Promega, USA) respectively according to manufacturer protocols. The concentration and quality of DNA was measured using NanoPhotometer C40 (Implen, Germany).We have developed a panel comprised of four high impact genes in breast cancer using high throughput sequencing technology. We have designed (using Primer 3 plus software, IDT and UCSC Genome Browser) 52 sets of primers targeting all exons and splicing junctions of BRCA1, BRCA2, TP53 and ERBB2 genes (Additional file 1). In total, 13 sets of multiplex PCR were carried out to amplify all the 52 amplicons (Additional file 1: Table S1-S5) using GoTaq® Hot Start Colorless Master Mix and GoTaq Long PCR Master Mix (Promega, USA). The amplicons were visually confirmed by 0.8% agarose gel electrophoresis. After confirmation, amplicons were purified using the Agencourt AMPure XP PCR purification bead (Beckman Coulter, Pasadena, CA) and quantified using the QuantiFluor® ONE dsDNA System (Promega, USA). Then 1 ng target amplicons were used for library preparation. Nextera XT library preparation kit (Illumina, Inc., San Diego, CA), using the manufacturer’s recommended protocol, was used for library preparation.
DNA sequencing and analysis
The barcoded libraries were subsequently sequenced at NeuroGen Technologies Ltd. genetics and genomics laboratory using the MiniSeq sequencer (Illumina, Inc. USA) with miniseq mid output kit, which generated 150 base paired-end sequence reads. The run was set up in local run manager that is an integrated computer software platform of MiniSeq, which uses a Burrows-Wheeler Aligner (BWA) [17] and the Genome Analysis ToolKit (GATK 4.0.11.0) [18] for converting raw sequence reads to Binary Alignment/Map (BAM) and Variant Call Format (VCF) v4.1 files. Sequencing proceeded on the platform for 24 h with default settings within the MiniSeq system. For quality control, a Q-score of 30 was used as a threshold for each identified variant, corresponding to a 1:1000 error rate. The average coverage is 91.42% for all four genes (for 52 primer sets) and 96% of the amplicons have a mean sequencing coverage of 50X.We have used ANNOVAR (2018Apr16 version) for functional annotation of the variants. For genomic annotations, we have also used GenomeArc, a custom genetic annotation tool. Mutation classification analysis was conducted based on American College of Medical Genetics (ACMG) guidelines [19]. Sanger sequencing was used to confirm the novel mutations [20].
Sanger sequencing
All novel variants identified using MiniSeq (Illumina, Inc. USA) were validated by Sanger sequencing. For this purpose, PCR was performed using genomic DNA as a template and primer pairs flanking the deleterious variant sites. We have designed primers (Additional file 1: Table S6) for this validation using Primer 3 plus software, IDT and UCSC Genome Browser. The PCR products were visually confirmed by 2.0% agarose gel electrophoresis. The products were then purified using the Wizard® SV Gel and PCR Clean-Up System (Promega, USA) according to the manufacturer’s instructions. Cycle sequencing was performed using purified PCR products as template and BigDye® Terminator v3.1 (Applied Biosystem, USA). Then sequencing was performed using 3500 DNA Analyzer (Applied Biosystem, USA). Subsequently Sanger sequencing data were analyzed using Sequence Scanner v2.0 (Applied Biosystem, USA).
Results
We have used nine tumor samples to sequence four bonafide BC genes and identified three pathogenic mutations and a VUS within three tumor samples (Table 1, Table 3 and Additional file 1: Table S7). The first patient (Table 1), carries a TP53 (c.733 G > A) missense pathogenic mutation in exon 7 that impacts the DNA binding domain of TP53 protein. The second patient (Table 1) carries 2 pathogenic mutations in both BRCA1 (c.116 G > A) and TP53 (c.322dupG) gene. A variant of uncertain significance (VUS) in BRCA2 (c. 2459A > G) was identified in the third patient (Table 1).
The overall clinical yield from the 43 blood specimens was 11 clinically relevant (pathogenic or VUS) mutations (25.58%), including 13.95% (6/43) patients with clinically pathogenic mutations (Table 2, Table 3 and Additional file 1: Table S7). Among these 6 variants, 4 are novel pathogenic germline mutations (Table 2) comprising 2 frameshift deletions in BRCA2 (Table 2), 1 missense mutation impacting BRCA2 (Table 2) and 1 missense mutation impacting BRCA1 (Table 2) gene. Two novel frameshift sequence variants are c.1301_1308delAAAGAAAG in exon 10 and c.351_352delTC in exon 4 of BRCA2. These mutations truncated the protein at amino acid positions of 118 and 436. In BRCA2 gene, another novel missense variant c.6451 G > A, was found in exon 11. Another two novel mutations c.5011 T > C and c.2272 G > C were found in BRCA1 and ERBB2, respectively (Table 2, Table 3 and Additional file 1: Table S7). All the novel mutations (c.322dupG, c.351_352delTC, c.1301_1308delAAAGAAAG, c.5011 T > C, c.6451 G > A and c.2272 G > C) were further validated by Sanger sequencing (Fig. 1).

Sequence chromatograms of all novel mutations detected in BRCA1, BRCA2, TP53, and ERBB2 genes. a and b Forward and reverse strands sequence of pathogenic frameshift insertion mutation c.322dupG in TP53 gene. c and (d) Forward and reverse strands sequence of pathogenic missense mutation c.5011 T > C in BRCA1 gene. e and f Forward and reverse strands sequence of pathogenic frameshift deletion mutations c.351_352delTC in BRCA2 gene. g and h Forward and reverse strands sequence of pathogenic frameshift deletion mutationc.1301_1308del AAAGAAAG in BRCA2 gene. i and j Reverse strand sequence of missense mutation c.6451G > A and c.2272G > Cin BRCA2 and ERBB2 genes respectively
In this cohort, we have also identified 6 known clinically relevant variants that include 2 known pathogenic variants, c.7722 G > A in exon 16 of BRCA2 and c.1058 G > A in exon 10 of BRCA1 (Table 2) and the remaining 4 mutations are missense. Out of 4, 2 are located in exon 10 of BRCA2 gene and 2 are located in exon 10 of BRCA1 gene (Table 2).
Discussion
Although large scale targeted sequencing has identified new BC mutations in the developed countries [21], data on the mutational architecture in low and middle income countries, such as Bangladesh, remains limited [22]. The recent increase of BC prevalence and the detection of bonafide causal genes imply the exploration of the genomic landscape of BC in countries such as Bangladesh to facilitate early diagnosis and screening to target treatments appropriately. We have designed a targeted gene-sequencing panel for known high-risk breast cancer genes, namely BRCA1, BRCA2, TP53 and ERBB2, which was after proper quality control incorporated into the diagnostic pathway.
The incidence and prevalence of BC in Bangladesh is mostly unknown due to the lack of population-based cancer registries either locally or centrally. In Bangladesh, most patients are diagnosed at an advanced stage of the disease and suffer from worse treatment outcome due to lack of breast cancer awareness, inadequate access to healthcare and excessive treatment related cost. Genetic screening for BRCA1/2 and TP53 genes and identification of novel mutation and variants serves as key roles for timely diagnosis, treatment, counseling, follow-up of patients and management of disease [23]. BRCA1/2 carriers now can have targeted therapies that apply parp-inhibitor to facilitate DNA repair process in tumor cell. It was found that the oral PARP inhibitor olaparib has antitumour activity in those patients who have lost BRCA1 or BRCA2-associated DNA repair [24]. Similarly, for TP53 mutations multiple targeted therapies shown promising result to improve the survival rate for TP53 mutation carriers [25].
We have identified 25.58% (11/43) germline (blood derived DNA) clinically relevant mutations with familial cancer history. Results obtained in our cohort corroborate the previously reported studies that investigated only BRCA genes within familial patients [26]. Of the 11 germline mutations, we identified 10 sequence variants in BRCA1 and BRCA2 genes, including 13.95% (6/43) cases carrying a pathogenic mutation and the frequency correlate strongly with Cyprus study that reported a similar clinical yield of 13% for BRCA genes [27]. Despite of small cohort size, we have found germline pathogenic mutation impact on BRCA2 (9.30%, 4/43) which is approximately 2-fold higher than BRCA1 (4.65%, 2/43), suggesting that BRCA2 is frequently mutated or altered in our cohort. This is also consistent with a Chinese cohort where BRCA2 was shown to have a higher prevalence compared to BRCA1 [28]. The frequency of germline VUS mutations are same in both BRCA1 (4.65%; 2/43) and BRCA2 (4.65%; 2/43) compared to ERBB2 (2.32%; 1/43).
Of the 11germline mutations, we have found 2 novel pathogenic frameshift deletions (Table 2) in exon 10 and 4 of BRCA2 (Table 2) gene respectively. These mutations truncated the BRCA2 protein at amino acid positions of 118 and 436. The truncated protein lacks BRCA2 functional domains (RAD51 and a DNA binding domain) that plays an important role in the homologous recombination (HR) repair of damaged DNA in cells [29, 30]. In the exon 10 of BRCA2 gene we have also identified 2 known missense mutations (Table 2). Among these two, one is p.Asn372His (rs144848) which is a common non-synonymous polymorphism in exon 10 of the BRCA2 gene [31]. The change from A to C in the rs144848 polymorphism results in an asparagine-to-histidine transition (p.N372H) which may affect BRCA2 structure at residues 290–453, a region responsible to interact with the histone acetyl transferase P/CAF prior to transcriptional activation of target genes [32]. Multiple independent studies have identified varying degree of association of rs144848 p.N372H polymorphism in cancer risk, but the susceptibility to breast cancer is still inconclusive [33,34,35,36,37,38]. Another variant (p.Thr598Ala) within exon 10 was found to have conflicting pathogenicity within literature [39]. We have identified another novel missense variant p.Val2151Ile (Table 2) in exon 11 of BRCA2 gene. We have also identified a known pathogenic mutation p.Trp2574Ter (Table 2) in the exon 16 of BRCA2 gene. This variant likely to cause the protein to be abnormally truncated at the amino acid position 2574. It is located within the DNA-binding domain (DBD: belongs to 2481–3186 amino acids) of BRCA2 gene that binds single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA). The DBD contains five components: a 190-amino-acid α-helical domain, three oligonucleotide binding (OB) folds that are ssDNA-binding modules, and a tower domain (TD) that protrudes from OB2 and binds dsDNA [40]. The helical domains, OB1 and OB2 also associate with deleted in split-hand/split-foot syndrome (DSS1), which has been linked to BRCA2 protein stabilization [40,41,42,43]. This variant is pathogenic and was found in the patients of Hereditary Breast and Ovarian Cancer syndrome [39].
Out of 10 sequence variants in BRCA1 and BRCA2 gene, we have found 1 novel missense mutation p.Met1671Val (Table 2) in exon 16 of BRCA1 gene. This mutation is located within the phosphoprotein-binding C-terminal BRCT domain which is critical for the tumor suppression function of BRCA1 gene [44]. We have also identified 3 known mutations in the exon 10 of BRCA1 gene. Among these mutations, 2 are missense mutations p.Arg762Ser and p.Gly1332Val (Table 2) and another 1 is stopgain mutation p.Trp353Ter (Table 2). The missense mutations were previously identified in patients with breast cancer and hereditary cancer-predisposing syndrome and were defined as VUS [39, 45]. The stopgain mutation was previously identified in the patients of hereditary breast and ovarian cancer syndrome and was defined as pathogenic [39, 45, 46].
We have also identified a novel missense mutation p.Val758Leu (Table 2) in the exon 24 of ERBB2 gene. ERBB2 kinase domain mutation occurs in human cancers such as gastric, breast, and colorectal cancers, and suggested that alterations of ERBB2-mediated signaling pathway by ERBB2 mutations alone or together with K-RAS mutations may contribute to the development of human cancers [47].
We also identified four mutations in 3 of the 9 breast tumor tissue samples, including 22% (2/9) with pathogenic mutations in TP53 gene, comparable to data from the International Agency for Research on Cancer (IARC) [48]. The patient carrying this frameshift mutation also has another pathogenic mutation, c.116 G > A in the exon 3 of BRCA1 gene which was previously identified in the patients of hereditary breast and ovarian cancer syndrome, neoplasm of the breast and hereditary cancer-predisposing syndrome [39, 49]. The majority of the mutations occur in the hot-spots exons 5, 7 and 8 [50, 51] of TP53 gene. Mazars et al found that among patients with ovarian cancer, all p53 mutations clustered in exons 5 and 7 [52]. Somatic TP53 gene alterations are frequent in most human cancers, ranging from 5 to 80% depending on the type, stage, and etiology of tumors [53]. BC tumors also are impacted by frequent TP53 mutations and based on the mutation type and location, cancer subtype can be classified based on treatment and prognosis. Hence, TP53 mutated warrant targeted treatment depending on the TP53 mutation status [13]. Inherited TP53 mutations predispose to a wide spectrum of early-onset cancers and are associated with Li-Fraumeni and Li-Fraumeni-like syndrome (LFS and LFL), respectively [54]. We also identified another missense mutation p.Asp820Gly (Table 1) in the exon 11 of BRCA2 gene that was previously identified in the patients of familial breast-ovarian cancer syndrome [39, 55].
Conclusion
In this study, we have designed a cost effective targeted gene panel to investigate the mutational landscape of 4 high impact breast cancer genes (BRCA1, BRCA2, ERBB2 and TP53) in breast tumor and in blood. This is the first paper on breast cancer mutation screening on Bangladeshi population that is ethnically a homogenous population. We have identified 5 novel mutations that are extremely rare in other populations, as well as known pathogenic mutations for breast cancer. The accumulation of more genomic data will be able to quantitate their true prevalence, as well as their association with other disease phenotypes. We have also observed clinically relevant mutation enrichment within BRCA2 genes. Our list of detected variants of unknown significance will also provide the opportunity to conduct further research to clarify ambiguous pathogenicity. In recent years, breast cancer becoming a major issue in developing countries due to low survival rate compared to developed countries. The identification of novel mutations from a homogenous population will add great value to breast cancer genetics. Moreover, this paper will bring broader community awareness on cancer genetic tests and the implementation of precision medicine in general for the country.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. Confidential patient data are not shared.
Abbreviations
- ACMG:
-
American College of Medical Genetics
- BAM:
-
Binary Alignment/Map
- BC:
-
Breast Cancer
- BWA:
-
Burrows-Wheeler Aligner
- DBD:
-
DNA-Binding Domain
- dsDNA:
-
double-stranded DNA
- DSS1:
-
Deleted in Split-hand/Split-foot Syndrome
- GATK:
-
Genome Analysis ToolKit
- HR:
-
Homologous Recombination
- IARC:
-
International Agency for Research on Cancer
- IERC:
-
Institutional Ethical Review Committee
- OB:
-
Oligonucleotide Binding
- ssDNA:
-
single-stranded DNA
- TD:
-
Tower Domain
- VCF:
-
Variant Call Format
- VUS:
-
Variant of Uncertain Significance
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Coleman MP, Quaresma M, Berrino F, Lutz JM, De Angelis R, Capocaccia R, Baili P, Rachet B, Gatta G, Hakulinen T, Micheli A. Cancer survival in five continents: a worldwide population-based study (CONCORD). Lancet Oncol. 2008;9(8):730–56.
Mavaddat N, Pharoah PD, Michailidou K, Tyrer J, Brook MN, Bolla MK, Wang Q, Dennis J, Dunning AM, Shah M, Luben R. Prediction of breast cancer risk based on profiling with common genetic variants. JNCI: J Nat Cancer Inst. 2015;107(5).
Rebbeck TR, Mitra N, Wan F, Sinilnikova OM, Healey S, McGuffog L, Mazoyer S, Chenevix-Trench G, Easton DF, Antoniou AC, Nathanson KL. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. Jama. 2015;313(13):1347–61.
Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. Lancet. 1994;343(8899):692–5.
Breast Cancer Linkage Consortium. Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91(15):1310–6.
King MC, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302(5645):643–6.
Newman B, Austin MA, Lee M, King MC. Inheritance of human breast cancer: evidence for autosomal dominant transmission in high-risk families. Proc Natl Acad Sci. 1988 May 1;85(9):3044–8.
Langston AA, Malone KE, Thompson JD, Daling JR, Ostrander EA. BRCA1 mutations in a population-based sample of young women with breast cancer. N Engl J Med. 1996;334(3):137–42.
Thorlacius S, Olafsdottir G, Tryggvadottir L, Neuhausen S, Jonasson JG, Tavtigian SV, et al. A single BRCA2 mutation in male and female breast cancer families from Iceland with varied cancer phenotypes. Nat Genet. 1996;13(1):117.
FitzGerald MG, MacDonald DJ, Krainer M, Hoover I, O'neil E, Unsal H, Silva-Arrieto S, Finkelstein DM, Beer-Romero P, Englert C, Sgroi DC. Germ-line BRCA1 mutations in Jewish and non-Jewish women with early-onset breast cancer. N Engl J Med. 1996;334(3):143–9.
Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol Off J Am Soc Clin Oncol. 2007;25(11):1329.
Bertheau P, Lehmann-Che J, Varna M, Dumay A, Poirot B, Porcher R, et al. p53 in breast cancer subtypes and new insights into response to chemotherapy. Breast. 2013;22:S27–9.
Ungerleider NA, Rao SG, Shahbandi A, Yee D, Niu T, Frey WD, et al. Breast cancer survival predicted by TP53 mutation status differs markedly depending on treatment. Breast Cancer Res. 2018;20(1):115.
Islam SS, Uddin M, Noman AS, Akter H, Dity NJ, Basiruzzman M, et al. Antibody-drug conjugate T-DM1 treatment for HER2+ breast cancer induces ROR1 and confers resistance through activation of hippo transcriptional coactivator YAP1. EBioMedicine. 2019.
Noman AS, Uddin M, Chowdhury AA, Nayeem MJ, Raihan Z, Rashid MI, et al. Serum sonic hedgehog (SHH) and interleukin-(IL-6) as dual prognostic biomarkers in progressive metastatic breast cancer. Sci Rep. 2017;7(1):1796.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform bioinformatics 2009;25(14):1754–1760.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405.
Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 1975;94(3):441–8.
Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534(7605):47.
Kim JY, Lee E, Park K, Park WY, Jung HH, Ahn JS, et al. Clinical implications of genomic profiles in metastatic breast cancer with a focus on TP53 and PIK3CA, the most frequently mutated genes. Oncotarget. 2017;8(17):27997.
Richter S, Haroun I, Graham TC, Eisen A, Kiss A, Warner E. Variants of unknown significance in BRCA testing: impact on risk perception, worry, prevention and counseling. Ann Oncology. 2013;24(suppl_8):viii69–74.
Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly (ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–44.
Bykov VJ, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. 2018;18(2):89.
Easton DF. How many more breast cancer predisposition genes are there? Breast Cancer Res. 1999;1(1):14.
Loizidou MA, Hadjisavvas A, Pirpa P, Spanou E, Delikurt T, Tanteles GA, et al. BRCA1 and BRCA2 mutation testing in Cyprus; a population based study. Clin Genet. 2017;91(4):611–5.
Wang YA, Jian JW, Hung CF, Peng HP, Yang CF, Cheng HC, et al. Germline breast cancer susceptibility gene mutations and breast cancer outcomes. BMC Cancer. 2018;18(1):315.
Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7(2):263–72.
Wong AK, Pero R, Ormonde PA, Tavtigian SV, Bartel PL. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J Biol Chem. 1997;272(51):31941–4.
Fuks F, Milner J, Kouzarides T. BRCA2 associates with acetyltransferase activity when bound to P/CAF. Oncogene. 1998;17(19):2531.
Healey CS, Dunning AM, Teare MD, Chase D, Parker L, Burn J, Chang-Claude J, Mannermaa A, Kataja V, Huntsman DG, Pharoah PD. A common variant in BRCA2 is associated with both breast cancer risk and prenatal viability. Nat Genet. 2000;26(3):362.
Auranen A, Spurdle AB, Chen X, Lipscombe J, Purdie DM, Hopper JL, et al. BRCA2 Arg372Hispolymorphism and epithelial ovarian cancer risk. Int J Cancer. 2003;103(3):427–30.
Agalliu I, Kwon EM, Salinas CA, Koopmeiners JS, Ostrander EA, Stanford JL. Genetic variation in DNA repair genes and prostate cancer risk: results from a population-based study. Cancer Causes Control. 2010;21(2):289–300.
Hill DA, Wang SS, Cerhan JR, Davis S, Cozen W, Severson RK, et al. Risk of non-Hodgkin lymphoma (NHL) in relation to germline variation in DNA repair and related genes. Blood. 2006;108(9):3161–7.
Freedman ML, Penney KL, Stram DO, Le Marchand L, Hirschhorn JN, Kolonel LN, et al. Common variation in BRCA2 and breast cancer risk: a haplotype-based analysis in the multiethnic cohort. Hum Mol Genet. 2004;13(20):2431–41.
Li Q, Guan R, Qiao Y, Liu C, He N, Zhang X, et al. Association between the BRCA2 rs144848 polymorphism and cancer susceptibility: a meta-analysis. Oncotarget. 2017;8(24):39818.
Gruber K. BRCA2 variant aerodigestive cancer risk. Lancet Oncol. 2015;16(5):e204.
Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2013;42(D1):D980–5.
Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thomä NH, et al. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297(5588):1837–48.
Li J, Zou C, Bai Y, Wazer DE, Band V, Gao Q. DSS1 is required for the stability of BRCA2. Oncogene. 2006;25(8):1186.
Kojic M, Yang H, Kostrub CF, Pavletich NP, Holloman WK. The BRCA2-interacting protein DSS1 is vital for DNA repair, recombination, and genome stability in Ustilagomaydis. Mol Cell. 2003;12(4):1043–9.
Kristensen CN, Bystol KM, Li B, Serrano L, Brenneman MA. Depletion of DSS1 protein disables homologous recombinational repair in human cells. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 2010;694(1–2):60–64.
Tischkowitz M, Hamel N, Carvalho MA, Birrane G, Soni A, Van Beers EH, Joosse SA, et al. Pathogenicity of the BRCA1 missense variant M1775K is determined by the disruption of the BRCT phosphopeptide-binding pocket: a multi-modal approach. Eur J Hum Genet. 2008;16(7):820.
Saleem M, Ghazali MB, Wahab MA, Yusoff NM, Mahsin H, Seng CE, et al. The BRCA1 and BRCA2 genes in early-onset breast Cancer patients. Adv Exp Med Biol. 2018.
Szabo C, Masiello A, Ryan JF. BIC consortium, Brody LC. The breast cancer information core: database design, structure, and scope. Hum Mutat. 2000;16(2):123–31.
Lee JW, Soung YH, Seo SH, Kim SY, Park CH, Wang YP, et al. Somatic mutations of ERBB2 kinase domain in gastric, colorectal, and breast carcinomas. Clin Cancer Res. 2006;12(1):57–61.
Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat. 2002;19(6):607–14.
Kais Z, Chiba N, Ishioka C, Parvin JD. Functional differences among BRCA1 missense mutations in the control of centrosome duplication. Oncogene. 2012;31(6):799.
Børresen AL, Hovig E, Smith-Sørensen B, Malkin D, Lystad S, Andersen TI, et al. Constant denaturant gel electrophoresis as a rapid screening technique for p53 mutations. Proc Natl Acad Sci. 1991;88(19):8405–9.
Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hosteller R, Cleary K, et al. Mutations in the p53 gene occur in diverse human tumour types. Nature. 1989;342(6250):705.
Mazars R, Pujol P, Maudelonde T, Jeanteur P, Theillet C. p53 mutations in ovarian cancer: a late event? Oncogene. 1991;6(9):1685–90.
Hainaut P, Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res. Vol. 77. Academic Press, 1999. 81–137.
Malkin D, Li FP, Strong LC, Fraumeni JF, Nelson CE, Kim DH, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250(4985):1233–8.
Mucaki EJ, Ainsworth P, Rogan PK. Comprehensive prediction of mRNA splicing effects of BRCA1 and BRCA2 variants. Hum Mutat. 2011;32(7):735–42.
Acknowledgements
We thank all the patients and participants of this study. We graciously thank Mohammed Salah Uddin Danis, Managing Director, NeuroGen Technologies Ltd. and Mohammad Moshfiqur Rahman, Chief Operating Officer (COO), NeuroGen Technologies Ltd. Dhaka, Bangladesh for helping us at NeuroGen conducting these experiments.
Funding
This project has been funded through the NeuroGen Research Grant for targeted sequencing and Sanger sequencing validation. Research grant from College of Medicine, Mohammed Bin Rashid University of Medicine and Health Sciences was used to analyze, interpret and writing the manuscript.
Author information
Authors and Affiliations
Contributions
The following authors contributed to: Conception and design of study (M.U., B.K.B.,H.A.); Designing primers and sequencing (N.J.D., M.A.R., H.A., N.M., A.S.); recruiting patients (N.S., N.M., A.S., A.S., R.H., M.R.A., M.A.B., K.M.F.U., R.A.S); conducted surgical procedure (A.S., R.H.); analysis and interpretation of data (B.K.B.,M.B., H.A., B.S.S., M.W-S, M.U.); drafting and revising the manuscript (H.A., B.K.B.,M.W-S, S.S.I., B.B. M.U.). All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was conducted according to the Declaration of Helsinki and was approved by the Institutional Ethical Review Committee (IERC) of Holy Family Red Crescent Medical College, and all samples were collected with written informed consent. The reference number is IERC/21/Res//Jul/2018/28.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Supplementary Information. Table S1. Detail of BRCA1 primers used. Table S2. Detail of BRCA2 primers used. Table S3. Detail of ERBB2 primers used. Table S4. Detail of TP53 primers used. Table S5. Detail of multiplex PCR. Table S6. Detail of primers for Sanger sequencing. Table S7. Detail of the history of study cases who have mutations. (DOCX 41 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Akter, H., Sultana, N., Martuza, N. et al. Novel mutations in actionable breast cancer genes by targeted sequencing in an ethnically homogenous cohort. BMC Med Genet 20, 150 (2019). https://doi.org/10.1186/s12881-019-0881-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-019-0881-0