
Abstract
Background
Elytrigia lolioides (Kar. et Kir.) Nevski, which is a perennial, cross-pollinating wheatgrass that is distributed in Russia and Kazakhstan, is classified into Elytrigia, Elymus, and Lophopyrum genera by taxonomists on the basis of different taxonomic classification systems. However, the genomic constitution of E. lolioides is still unknown. To identify the genome constitution and evolution of E. lolioides, we used single-copy nuclear genes acetyl-CoA carboxylase (Acc1) and elongation factor G (EF-G), multi-copy nuclear gene internal transcribed space (ITS), chloroplast gene trnL-F together with fluorescence and genomic in situ hybridization.
Results
Despite the widespread homogenization of ITS sequences, two distinct lineages (genera Pseudoroegneria and Hordeum) were identified. Acc1 and EF-G sequences suggested that in addition to Pseudoroegneria and Hordeum, unknown genome was the third potential donor of E. lolioides. Data from chloroplast DNA showed that Pseudoroegneria is the maternal donor of E. lolioides. Data from specific FISH marker for St genome indicated that E. lolioides has two sets of St genomes. Both genomic in situ hybridization (GISH) and fluorescence in situ hybridization (FISH) results confirmed the presence of Hordeum genome in this species. When E genome was used as the probe, no signal was found in 42 chromosomes. The E-like copy of Acc1 sequences was detected in E. lolioides possibly due to the introgression from E genome species. One of the H chromosomes in the accession W6–26586 from Kazakhstan did not hybridize H genome signals but had St genome signals on the pericentromeric regions in the two-color GISH.
Conclusions
Phylogenetic and in situ hybridization indicated the presence of two sets of Pseudoroegneria and one set of Hordeum genome in E. lolioides. The genome formula of E. lolioides was designed as StStStStHH. E. lolioides may have originated through the hybridization between tetraploid Elymus (StH) and diploid Pseudoroegneria species. E and unknown genomes may participate in the speciation of E. lolioides through introgression. According to the genome classification system, E. lolioides should be transferred into Elymus L. and renamed as Elymus lolioidus (Kar. er Kir.) Meld.
Similar content being viewed by others


Background
The taxonomic history of Triticeae mainly includes three stages, that is, artificial classification, natural or phonetic classification, and phylogenetic classification [1, 2]. Löve [3] divided Triticeae species into 37 genera according to the genomic system of classification, in which different species with the same genome or genome constitution were classified into one genus, although the justifiability of some genera remains controversial up to now. Many Triticeae species were reclassified into different genera on the basis of their genome constitutions [4,5,6,7]. However, the genome constitutions of many species with high ploidy in the genera Elytrigia, Elymus, and Roegneria in Triticeae still remain unknown or controversial.
Elytrigia lolioides (Kar. et Kir.) Nevski is a perennial, cross-pollinating wheatgrass that is distributed in Russia and Kazakhstan, has strong rhizomatous, and generally grows in stony mountain slopes, sandy land, and steppe [3, 8]. According to its morphological characteristics, E. lolioides is classified into different genera, including Triticum, Agropyron, Elytrigia, and Elymus, according to different classification systems [8, 9]. Cytological studies indicated that the chromosome number of E. lolioides is either 42 or 58 [10, 11]. It was suggested that this species contains St, E, and J genomes that are derived from Pseudoroegneria and Lophopyrum elongatum and L. bessarabicum, respectively, and is classified into Elytrigia [3]. Dewey [9] supported this treatment but indicated that E. lolioides has St and unknown genomes, and the genome formula was designed as StX (X is undetermined genome). Yen and Yang [8] speculated that E. lolioides should be classified into genus Lophopyrum with E genome. Tao and Lin [12] suggested that E. lolioides contains StE genome. Therefore, the genome constitution and origin of E. lolioides remain controversial. According to the Dewey taxonomic principle, Elytrigia genus has five species, including E. repens, E. lolioides, E. pycnantha, E. pungens, and E. elongatiformis [9]. Genome constitution of E. repens、E. pycnantha, and E. pungens were reported and these three species were classified into other genera [8]. Therefore, investigating genome constitution of E. lolioides will be useful to investigate the taxonomic status of Elytrigia.
Chromosome pairing at meiosis in artificial hybrid is commonly used to detect the genome constitution of species [13,14,15,16]. Chromosome pairing at the metaphase I of meiosis is convincing step in determining the genome constitution of the target species. However, interpreting chromosome pairing at high ploidy levels is difficult because of the difficulty in distinguishing autosyndetic and homoeologous pairing in meiosis [17]. Therefore, genomic in situ hybridization (GISH), specific molecular marker for genome or chromosome and phylogenetic analysis were used to investigate the genome constitution and origin of target species [18,19,20,21]. GISH is a fast and valuable tool to detect genome constitution and chromosomal translocation in species [22, 23]. Some genome-specific molecular and FISH markers were developed. These markers are stable and visual in detecting genome constitution and tracing target chromatin in wheat breeding program [24,25,26]. Phylogenetic analyses can identify the genome donors and introgression of polyploid. Single- or low-copy nuclear gene, multi-copy nuclear gene and cytoplasm gene have been successfully used to investigate parental and maternal origins [27,28,29]. Although undergoing concerted evolution, internal transcribed space (ITS) is a useful marker to conclude the genome origin of polyploid [30]. Acetyl-CoA carboxylase (Acc1) gene, elongation factor G (EF-G), ITS, and the space between tRNA-Leu and tRNA-Phe gene (trnL-trnF region) sequences have been used to examine the phylogenetic relationship, hybridization events, parental donor, and maternal origin [31,32,33,34,35].
In the present study, the specific molecular markers for St genome, GISH, single-copy nuclear genes Acc1 and EF-G, multi-copy nuclear gene ITS, and chloroplast DNA trnL-F were used to investigate the genome constitution of E. lolioides. The objectives are as follows: (1) to detect the genome constitution and taxonomic treatment of E. lolioides, (2) to identify the maternal donor of E. lolioides, and (3) to clarify the origin of E. lolioides.
Results
Phylogenetic analyses of ITS sequence
The length of E. lolioides ITS sequences ranged from 588 bp to 602 bp, and that of most sequences was ~ 601 bp. After multiple sequence alignments, the 13 bp deletion from 174 to 186 bp in the ITS1 region was found in an ITS sequence from E. lolioides (W6–26586, Fig. 1). The ITS data matrix of 79 sequences was analyzed based on maximum likelihood (ML) by using the GTR + I + G model (−Ln likelihood = 4280.8823). Brachypodium sylvaticum and Avena longiglumis were used as the outgroup. A total of 626 characters were used for phylogenetic analysis, in which 351 were constants, 112 were parsimony uninformative, and 163 were parsimony informative. Finally, a single phylogenetic tree was yielded with the following assumed nucleotide frequencies: A = 0.22610, C = 0.28870, G = 0.27680, T = 0.20840. The trees generated by Bayesian analysis and ML were similar to each other. The ML tree with bootstrap support (BS) values (above the branch) and Bayesian posterior probability (PP, below the branch) is displayed in Fig. 2.

The deletion in ITS1 region from Elytrigia lolioides. The 13 bp deletion from 176 to 188 bp in ITS1 region of one ITS sequence from Elytrigia lolioides (W6–26586)

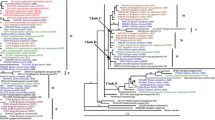
Strict consensus tree generated from ITS sequence data. Numbers above the branches were bootstrap support (BS) values and below the branches were Bayesian posterior probability (PP) values. The bold indicated sequences from three accessions of Elytrigia lolioides. The same superscript indicated the sequences from same accession
The multiple copies of sequences from each E. lolioides accession were separated into two distinct clades. One clade contained 10 sequences each from E. lolioides and diploid Pseudoroegneria species (PP = 0.78). The other clade contained two sequences each from E. lolioides and diploid Hordeum species (BS = 98%, PP = 1.00).
Phylogenetic analyses of Acc1 sequence
The 31 and 38 positive clones were sequenced for the three accessions of E. lolioides. The length of these sequences ranged from 1400 bp to 1495 bp, and that of most sequences was ~ 1440 bp. All sequences contained eight exons and seven introns. These findings are similar to the results in a previous study [36]. After multiple sequence alignments, 10 bp deletion was found in 43 sequences (17/31 in PI 440059, 14/38 in W6–26586, 12/28 in W6–26567) from the 110–119 position in intron 1 region. A 67 bp insertion at the 1015–1081 position in intron 5 region was found in two sequences from the two accessions of E. lolioides (PI440059 and W6–26586, Fig. 3). Blast search indicated that the 67 bp insertion belongs to Tc1 DNA transposon. Termination codon was found in the exon 5 of the two sequences from accession W6–26586, and these sequences were excluded in the phylogenetic analysis.

The deletion and insertion in Acc1 sequnces from Elytrigia lolioides. The 10 bp deletion from 110 to 119 bp and 67 bp insertion from 1015 to 1081 bp were found in intron 1 and intron 5 regions of Acc1 sequences from Elytrigia lolioides, respectively
The phylogenetic analysis of the 82 Acc1 sequences was performed using Bromus inermis as the outgroup. The data matrix contained 1885 characters, 1353 of which were constants, 257 were parsimony uninformative, and 275 were parsimony informative. A signal phylogenetic tree generating by maximum likelihood analysis using the GTR + I + G model (−Ln likelihood = 8780.6729; assumed nucleotide frequencies: A = 0.25250, C = 0.18430, G = 0.21920, T = 0.34400) is shown in Fig. 4 with the BS from ML and PP values from Bayesian analyses.

Strict consensus tree generated from Acc1 sequence data. Numbers above the branches were bootstrap support values (BS) and below the branches were Bayesian posterior probability (PP) values. The bold indicated sequences from three accessions of Elytrigia lolioides
Three distinct Acc1 copies of the sequences from each three accessions of E. lolioides were grouped into three different clades. The first clade was St clade, which included five diploid Pseudoroegneria species, tetraploid Elymus and Roegneria species, hexaploid species of Campeiostachys and E. lolioides (BS = 69%, PP = 1.00). In the St clade, Pseudoroegneria tauri, P. strigosa, P. stipifolia, Elymus sibiricus, Ely. trachycaulus, Roegneria caucasica, and three E. lolioides formed a paraphyletic grade. The second clade was the H genome clade, which contained the diploid species of Hordeum, tetraploid species of Elymus, hexaploid species of Campeiostachys and E. lolioides (BS = 100%, PP = 1.00). In the second clade, five E. lolioides sequences, two tetraploid species of Elymus (Ely. sibiricus and Ely. confuses) and Campeiostachys kamoji formed a subclade (PP = 56%, BS = 1.00). However, the third clade only contained sequences from E. lolioides without any diploid species (BS = 100%, PP = 1.00). One sequence each from the accession PI440059 of E. lolioides and L. bessarabicum was grouped together (BS = 56%, PP = 1.00). The Acc1 data displayed an evident Y genome clade (BS = 100%, PP = 1.00), and no sequences from E. lolioides were grouped in this clade.
Phylogenetic analysis of EF-G sequence
The EF-G matrix contained 71 taxa and 870 characters, 590 of which were constants, 127 were parsimony uninformative, and 153 were parsimony informative. HKY + G, as the best-fit model (−Ln likelihood = 3898.0823), was used in phylogenetic analysis. A single phylogenetic tree was yielded and the assumed nucleotide frequencies: A = 0.26340, C = 0.19760, G = 0.21780, T = 0.32120. The tree generated by Bayesian analyses and ML were similar to each other. The ML tree with BS values (above the branch) and Bayesian PP (below the branch) is displayed in Fig. 5.

Strict consensus tree generated from EF-G sequence data. Numbers above the branches were bootstrap support values (BS) and below the branches were Bayesian posterior probability (PP) values. The bold indicated sequences from three accessions of Elytrigia lolioides
The phylogenetic analyses of the EF-G sequence distinctly separated the three copies of sequences from the three accessions of E. lolioides into three different clades. The first clade was St clade, which included five Pseudoroegneria species, seven tetraploid Elymus species, five tetraploid Roegneria species and E. lolioides (PP = 93%, BS = 1.00). The second clade was the H clade, which contained the diploid of Hordeum species, seven tetraploid Elymus species and E. lolioides (PP = 99%, BS = 1.00). However, in the third clade, all sequences were only from E. lolioides that were grouped together (PP = 85%, BS = 1.00) and sisters to the sequences from diploid Aegilops, Triticum, Lophopyrum, Taeniatherum, and Crithopsis species. All these sequences had 7 bp deletion and 8 bp insertion in 164–172 and 215–224 bp positions, respectively (Fig. 6). Meanwhile, EF-G data displayed an evident Y genome clade (BS = 100%, PP = 1.00) and without any sequence from E. lolioides, which is similar to the results of the Acc1 phylogenetic tree.

The deletion and insertion in EF-G sequnces from Elytrigia lolioides. The 7 bp deletion and 8 bp insertion in position 164–172 bp and 215–224 bp were found in EF-G sequences from Elytrigia lolioides, respectively
Phylogenetic analyses of trnL-F sequence
A total of 42 trnL-F sequences were selected for ML analysis. B. tectorum was used as the outgroup. The data matrix contained 1013 characters, 884 of which were constants, 56 were parsimony uninformative, and 73 were parsimony informative. HKY + G as the best-fit model (−Ln likelihood = 2435.1624) was used in phylogenetic analysis, and a single phylogenetic tree was yielded. The assumed nucleotide frequencies were A = 0.33724, C = 0.15580, G = 0.13259, T = 0.37437. The tree generated by Bayesian analyses was similar to ML tree. The ML tree with BS values (above the branch) and Bayesian PP (below the branch) is shown in Fig. 7.

Strict consensus tree generated from trnL-F sequence data. Numbers above the branches are bootstrap support values and below the branches were Bayesian posterior probability (PP) values. The bold indicated sequences from three accessions of Elytrigia lolioides
All trnL-F sequences were distinctly divided into five clades. The sequences from E. lolioides were grouped with diploid Pseudoroegneria, Lophopyrum, and Dasypyrum species (BS = 59%, PP = 0.99). In this clade, sequences from Lophopyrum species which contained E genome, formed a subclade (PP = 0.80).
In situ hybridization
Chromosome number analysis results indicated that E. lolioides is a hexaploid (2n = 6x = 42) wheatgrass. St2–80 is a FISH marker for St genome. Signals produced by St2–80 were present on the entire arm of the St genome chromosomes, except at the centromeric region. However, signals produced by St2–80 were present in the terminal region of the E and H genome chromosomes [26]. This marker was used to detect St genome in the E. lolioides chromosomes. The signal on 28 chromosomes was displayed St type (Fig. 8a, b, and c). This result was confirmed by GISH, wherein 28 chromosomes were hybridized with the St probe from Pseudoroegneria libanotica (Fig. 8d). To detect the 14 other chromosomes, E (from Lophopyrum bessarabicum) and H (from Hordeum bogdanii) genomes were used as probes. After two-color GISH, no any E genome signals were displayed on the chromosomes (Fig. 8e). However, the chromosomes that did not hybridize with St genome displayed intense signals when probed by the H genome (Fig. 8f). Then, the test was performed using H genomes as probes in the three E. lolioides accessions (Fig. 8g, h, and i). Minor disparity was displayed in chromosome of W6–26586 that one chromosome which belongs to H genome did not hybridized H genome signals on the pericentromeric regions (Fig. 8i).

Results of FISH and GISH in Elytrigia lolioides. a and g: PI 440059, b and h: W6–26567, c-f and i: W6–26586. a-c: Used St2–80 as probe, 28 chromosomes were labeled as St type (arrows). The rest of the chromosomes were labeled as non-St type in three accessions of E. lolioides, respectively. d: Total genomic DNA of Pseudoroegneria libanotica was labeled with fluorescein-12-dUTP (green) as probe, 28 chromosomes were hybridized with P. libanotica probe (St genome). e: Total genomic DNA of P. libanotica labeled with fluorescein-12-dUTP (green) and total genomic DNA of Lophopyrum bessarabicum labeled with Texas-red-5-dCTP (red) as probes, 28 chromosomes were hybridized with the P. libanotica probe (St genome) and no any E genome signals were displayed on the chromosomes. f: Total genomic DNA of P. libanotica labeled with fluorescein-12-dUTP (green) and total genomic DNA of Hordeum bogdanii labeled with Texas-red-5-dCTP (red) as probes, 28 chromosomes were hybridized with the P. libanotica probe (St genome) and the rest were hybridized with the H. bogdanii probe (H genome). g-i: Total genomic DNA of H. bogdanii labeled with fluorescein-12-dUTP (green), 14 chromosomes were hybridized with the H. bogdanii probe (H genome) in three accessions of E. lolioides, respectively. However, in accession of W6–26586, one chromosome which belongs to H genome did not hybridized H genome signals on the pericentromeric regions (arrow). Insets showed the magnified view of the signal on this chromosome
Discussion
Maternal donor of E. lolioides
The chloroplast DNA (cpDNA) is maternally inherited in grasses [37]. Several sequences, including space and coding regions (e.g., trnL-F, trnD-T, trnH-psbA, matK, Rps16, and rbcL) were used to identify the maternal donor of wheatgrass or genera in Triticeae [29, 32, 34, 38, 39]. In the present study, all sequences from E. lolioides with the sequences from the diploid Pseudoroegneria (St) and Dasypyrum (V) and the diploid and tetraploid Lophopyrum (E) formed a clade (BS = 59%, PP = 0.99) in trnL-F data. It seems to suggest that Pseudoroegneria, Dasypyrum, and Lophopyrum were the potential maternal donors of E. lolioides. In a previous study, close relationships among Pseudoroegneria, Dasypyrum, and Lophopyrum were discovered using the cpDNA data from diploid species in Triticeae [33, 40]. In contrast to the trnL-F data, Acc1 sequences from Pseudoroegneria, Dasypyrum, Lophopyrum, Hordeum, and other diploids were distinctly separated into different clades, and sequences from E. lolioides were placed into Pseudoroegneria, Lophopyrum, and Hordeum clades. No chromosomes that were hybridized with E probe were derived from L. bessarabicum in GISH. Thus, we infer Pseudoroegneria is a maternal donor of E. lolioides. Numerous studies indicated that Pseudoroegneria is a maternal donor of polyploid species containing St genome in Triticeae [41,42,43,44]. The female species in Triticeae carrying St genome are successful in terms of distant hybridization [45].
Origin of E. lolioides
Our results indicated that Pseudoroegneria is the major genome donor for E. lolioides. GISH and FISH indicated that E. lolioides had two sets of St genome. In strict consensus tree generated from the Acc1 sequence, several Acc1 sequences from the three accessions of E. lolioides formed a clade without any sequence from the diploid species in Triticeae. Meanwhile, all of these sequences had a 10 bp deletion that is absent in the other published Acc1 sequences of Triticeae species from the 110 to 119 positions in intron 1 region. Some non-synonymous substitutions were discovered in several of these sequences. One sequence that was placed into the St clade also had this deletion. The preliminary results in our laboratory also showed that the same deletion was found in Acc1 sequences from E. pungens, Psammopyrum athericum, and E. elongatiformis, which were collected from the Middle East (unpublished). Two of these sequences had 67 bp insertions at the 1015–1081 bp positions in intron 5. And in strict consensus tree generated from the EF-G sequence, three sequences from the three accessions of E. lolioides that formed a clade without other sequences were observed. Similarly as Acc1 sequences, these sequences also contained special insert fragments. Therefore, sequences with special indel may be derived from an independent diploid species, which is extinct or unknown. This hypothesis will be validated by checking whether Acc1 and EF-G sequences with special indel were obtained from Triticeae diploid species in the Middle East.
The contribution from Hordeum to the accessions of E. lolioides was indicated by the data of Acc1, EF-G, and ITS sequences with high support and also confirmed by GISH. However, the Hordeum-like copy of ITS sequence was not obtained from accessions PI 440059 and W6–26567 possibly due to concerted evolution. In the Acc1 data, E. lolioides and Elymus species (StH) formed a subclade without the diploid Hordeum species in H clade. It can be concluded that the tetraploid Elymus species was the direct donor during E. lolioides speciation. One chromosome had intensive hybridization signals derived from H. bogdanii on the arms but had clear signals derived from P. libanotica on the pericentromeric regions in GISH. Previous studies showed that these mosaic chromosomes are observed in Psammopyrum athericum (StEP/LEP) [46], E. pungens (StStEP/StLEP) [47], Elymus repens (StStH) [19], Thinopyrum intermedium (StEE) [20], and T. ponticum (StStEEE/EEEEE) [48] by genomic in situ hybridization. Thus, the chromosome rearrangement between St and the other genomes (i.e., E, H, and P) or the retrotransposon activity of St genome may lead to mosaic chromosomes after allopolyploidization. Interestingly, most of these mosaic chromosomes have the same character that St genome signal appeared on the centromeric or pericentromeric region of the other genomes. Redinbaugh et al. [45] found that the female species carrying the St genome are more successful in terms of distant hybridization. Those rearrangement or retrotransposon insertion may contribute to the stability of different genomes after allopolyploidization.
In the Acc1 data, one sequence from E. lolioides (PI440059) and L. bessarabicum formed a clade with moderate support (BS = 56%, PP = 1.00). Tao and Lin [12] produced a specific SCAR marker, which was used to detect StE genome in Triticeae, thereby indicating that E. lolioides contain StE genomes. However, parental donor from E genome was undiscovered by EF-G, ITS data, and GISH. The Lophopyrum-like Acc1 copy that was obtained from E. lolioides is likely caused by introgression [49]. L. bessarabicum is a diploid species that is distributed in Mediterranean Sea, Azov Sea, and Euxine Sea. Therefore, overlapping the geographical area between L. bessarabicum and E. lolioides can increase the possibility for introgression.
The hypothetical scenarios of the origin of E. lolioides origin were suggested by combining the data of the four sequences and in situ hybridization. E. lolioides may have originated through the hybridization between tetraploid Elymus (StH) and diploid of Pseudoroegneria species, and then followed by whole genome duplication. After hybridization and polyploidy, the transposon or retrotransposon activation and E and unknown genome introgression may participate in E. lolioides speciation (Fig. 9).

A hypothetical scenario about the origin of Elytrigia lolioides inferred from the three sequences data and in situ hybridization. Contribution of major genome constituents (Pseudoroegneria and Hordeum) were displayed with solid lines and hypothetical donor was displayed with dashed lines
Genomic constitution of E. lolioides
E. lolioides is a perennial wheatgrass that was first reported by Karelin and Kiriloff in 1841. The chromosome number of E. lolioides is controversial because different ploidy levels were reported. Schulz-Schaeffer and Jurasits [11] reported that the accession PI 223325 of Agropyron lolioides (= Elytrigia lolioides) has 58 chromosomes. However, when this accession was rechecked in the US National Plant Germplasm System, it was named as Elymus repens subsp. elongatiformis (https://npgsweb.ars-grin.gov/gringlobal/accessiondetail.aspx?id=1180280), which is an octaploid grass and distributed in Central Asia and southern Russia [50]. Considering the report of Löve [10] and our present results, E. lolioides (2n = 42) is a hexaploid wheatgrass. In the present study, Pseudoroegneria and Hordeum were regarded as the major progenitors for E. lolioides as indicated by the data of the three nuclear gene (Acc1, EF-G, and ITS) sequences. Both GISH and FISH results indicated that E. lolioides had two sets of St genome and one set of H genome. However, in the strict consensus tree generated from EF-G and Acc1 sequences, several sequences from the three accessions of E. lolioides formed a clade without any sequence from the diploid species in Triticeae. All these sequences had a special deletion or insertion that is not present in other published sequences for Triticeae species. These sequences may be contributed by unknown species, which were extinct or undiscovered. The contribution from Lophopyrum to E. lolioides was only indicated by the Acc1 data with moderate support. However, parental donor from E genome was undiscovered by EF-G, ITS data, and GISH. The Lophopyrum-like Acc1 copy that was obtained from E. lolioides was likely caused by introgression. Our results indicated that E. lolioides contained two sets of St genome and one set of H genome, and the genome formula was designed as StStStStHH.
Classification status of E. lolioides
The main morphological characteristics of E. lolioides included long, creeping rhizome, long anther, apex subobtuse of lemmas, spikelet sessile, and one spikelet per node of rachis. The mid-nerve of lemma slightly extended and formed an extremely short point. According to the morphological characteristics, this species was successively classified into Agropyron, Elytrigia, and Elymus by an agrostologist with different classification systems [9]. The problem on the classification of E. lolioides was mainly caused by the taxonomic status of Elytrigia genus that was established by Desvaux in 1810, and then treated as a genus, sectional status within Agropyron, subgenus of Agropyron or section of Elymus by different taxonomists, respectively [8] According to genome constitution, E. lolioides was classified into Elytrigia genus that genome constitution of either SJE [3] or SX [9]. Yen and Yang [8] indicated that this species may be classified into Lophopyrum with E genome. Our results suggested that the genomic constitution of E. lolioides was StStStStHH. According to the genome classification system, this species should be transferred into Elymus L. and renamed as Elymus lolioidus (Kar. er Kir.) Meld..
Conclusions
According to our study, E. lolioides is a hexaploid wheatgrass. Pseudoroegneria and Hordeum are major genome donors. E and unknown genomes may participate in the speciation of E. lolioides through introgression. This species should be transferred into Elymus L by combining four sequences data and in situ hybridization. The results of this study will help in investigating the taxonomic status of Elytrigia genus.
Methods
Plant materials and DNA extraction
The seeds of the three E. lolioides accessions from Former Soviet Union and Kazakhstan were provided by the Germplasm Resources Information Network of the United States Department of Agriculture (USDA). The total genomic DNA was extracted from fresh leaf tissues by using the CTAB method [51]. Three nuclear genes (Acc1, EF-G, and ITS) and one chloroplast trnL-F sequence from E. lolioides were amplified and sequenced. Acc1, EF-G, ITS, and trnL-F sequences from polyploid and diploid species representing A, S, D, E, W, St, V, K, Xp, Q, H, I, Ns, F, P, and Xe genomes in Triticeae were downloaded from GenBank (http://www.ncbi.nlm.nih.gov) and included in phylogenetic analysis. The basic information about these sequences, including genomic constitutions and GenBank identification numbers, are listed in Additional file 1: Table S1. The voucher specimens of E. lolioides were deposited in the Herbarium of Triticeae Research Institute, Sichuan Agricultural University, China.
Amplification and sequencing
Low-copy and multi-copy nuclear genes (Acc1, EF-G and ITS) and the chloroplast gene trnL-F sequences were amplified via polymerase chain reaction (PCR) by using the primers of cMWG699T3–2 and CMWG699T7–2 [52], AccF1 and AccF2 [53], ITSL and ITS4 [54], and c and f [55], respectively. The primers and PCR profiles for the Acc1, EF-G, ITS, and trnL-F genes are listed in Table 1. Sequences were amplified in a 25 μL reaction mixture containing 50 ng template DNA, 1× reaction buffer, 2 mM MgCl2, 0.4 μM of each primer, 200 μM dNTP, and 1.5 U ExTaq. In addition, 8% dimethylsulfoxide (DMSO) was added to avoid influence of higher GC content during ITS sequence amplification [28]. The PCR products were detected on 1.0% agarose gels and then cloned into a PMD19-T vector according to the manufacturer’s instructions (TaKaRa, China). After white-blue plaque selection, 30–40 randomly selected clones of Acc1, EF-G, and ITS and 5 clones of trnL-F for each accession were sequenced in both directions by Shanghai Sangon Biological Engineering and Technology Service Ltd. (Shanghai, China).
Data analysis
Multiple sequence alignments were made using MAFFT 7.3 [56] and adjusted manually. The phylogenetic analyses of Acc1, EF-G, ITS, and trnL-F data by using the ML method was performed using PAUP*4.0bet10 (Swofford DL, Sinauer Associates, http://www.sinauer.com). The best-fit evolutionary model for phylogenetic analysis was determined using ModelTest v3.7 with Akaike information criterion [57]. ML heuristic searches were performed with 100 random addition sequence replications and Tree Bisection-Reconnection (TBR) branch swapping algorithm. As a measurement of the robustness of tree clades, the BS values were calculated with 1000 replications and displayed in figure (above the branch) if the BS values were > 50% [58].
In addition to ML analysis, Bayesian analyses were also performed using MrBayes 3.1 [59]. The evolutionary model selected for Bayesian analyses was same as ML analysis. Two sets of four chains were run 3.3 million generations for Acc1 data, 4.2 million generations for EF-G data, 6.5 million generations for ITS data, and 0.54 million generations for trnL-F data, and samples were taken and saved every 100 generation under best-fit model. After discarding the first 25% samples as “burn-in”, a majority rule consensus tree with PP value (under the branch) was obtained.
Chromosome preparation, fluorescence, and multicolor genomic in situ hybridization
Rapidly growing roots were collected from adult plants. The roots were treated with N2O for 2 h at 0.1 MPa and then fixed for 5 min with 90% glacial acetic acid. Chromosomes were prepared for analysis by using a previously reported method [60]. Plasmid DNA with St2–80 sequence was extracted using the EndoFree Plasmid Mini Kit (Tiangen, China), which was used to distinguish the chromosomes of St genome and others, including A, B, D, E, H, P, and Y genomes [26], according to the manufacturer’s instructions. The total genomic DNA of P. libanotica and plasmid (contained St2–80 sequence) were labeled with fluorescein-12-dUTP, and the genomic DNA of L. bessarabicum and H. bogdanii were labeled with Texas-red-5-dCTP by using the nick translation method. Hybridization procedure was performed according to the previously reported method [61]. Slides were detected under an Olympus BX53 fluorescence microscope with camera. At least five metaphase cells for each accession were analyzed. Adobe Photoshop was used to proceed the color images.
Abbreviations
- Acc1 :
-
Acetyl-CoA carboxylase 1
- AIC:
-
Akaike information criterion
- BS:
-
Bootstrap support
- cpDNA:
-
Chloroplast DNA
- DMSO:
-
Dimethylsulfoxide
- EF-G:
-
elongation factor G
- FISH:
-
Fluorescence in situ hybridization
- GISH:
-
Genomic in situ hybridization
- ITS:
-
Internal transcribed space
- PCR:
-
Polymerase chain reaction
- PP:
-
Posterior probability
- rDNA:
-
Ribosomal DNA
- SCAR:
-
Sequence characterized amplified region
- TBR:
-
Tree bisection-reconnection
- TrnL-F :
-
Intergenic spacer between transfer RNA gene trnL (UAA) and trnF (GAA)
References
Barkworth ME. Taxonomy of the Triticeae: a historical perspective. Hereditas. 1992;116:1–14.
Al-saghir MG. Taxonomy and phylogeny in Triticeae: a historical review and current status. Adv Plants Agr Res. 2016;3:00108.
Löve Á. Conspectus of the Triticeae. Feddes Repert. 1984;95:425–521.
Lu BR. Biosystematic investigations of Asiatic wheatgrasses - Elymus L. (Triticeae: Poaceae). Alnarp: Swedish Agricultural University; 1993.
Zhang XQ, Yen C, Yang JL, Yen Y. Cytogenetic analyses in Kengyilia laxiflora (Poaceae, Triticeae ). Plant Syst Evol. 1998;212:79–86.
Zhang HQ, Zhou YH. Meiotic analysis of the interspecific and intergeneric hybrids between Hystrix patula Moench and H. duthiei ssp. longearistata, Pseudoroegneria, Elymus, Roegneria, and Psathyrostachys species (Poaceae, Triticeae). Bot J Linn Soc. 2007;153:213–9.
Yang CR, Baum BR, Chen WH, Zhang HQ, Liu XY, Fan X, et al. Genomic constitution and taxonomy of the Chinese hexaploids Elymus cylindricus and E. breviaristatus (Poaceae: Triticeae). Bot J Linn Soc. 2016;182:650–7.
Yen C, Yang JL. Biosystematics of Triticeae (Vol 5). Beijing: China Agriculture Press; 2013.
Dewey DR. The genomic system of classification as a guide to intergeneric hybridization with the perennial Triticeae. In: Gustafson JP, editor. Gene manipulation in plant improvement. New York: Plenum Publishing Corporation; 1984. p. 563–70.
Löve Á. Chromosome number report XC. Taxon. 1986;35:195–8.
Schulz-Schaeffer J, Jurasits P. Biosystematic investigations in the genus Agropyron. I. Cytological studies of species karyotypes. Am J Bot. 1962;49:940–54.
Tao ZH, Yin L. Identification of EeSt-genome species in Pseudoroegneria and Elytrigia (Poaceae: Triticeae) by using SCAR markers from ITS sequences. Genet Mol Res. 2015;14:815–22.
Jensen KB. Cytology and taxonomy of Elymus kengii, E. grandiglumis, E. alatavicus, and E. batalinii (Poaceae: Triticeae). Genome. 1990;33:668–73.
Jarvie JK, Barkworth ME. Morphological variation and genome constitution in some perennial Triticeae. Bot J Linn Soc. 1992;108:167–80.
Torabinejad J, Mueller RJ. Genome constitution of the Australian hexaploid grass Elymus scabrus (Poaceae: Triticeae). Genome. 1993;36:147–51.
Zhou YH, Yen C, Yang JL, Zheng YL. Biosystematic study of Roegneria tenuispica, R. ciliaris and R. pendulina (Poaceae: Triticeae). Plant Syst Evol. 1999;217:215–20.
Liu ZW, Wang RRC. Genome analysis of Elytrigia caespitosa, Lophopyrum nodosum, Pseudoroegneria geniculata ssp. scythica, and Thinopyrum intermedium (Triticeae: Gramineae). Genome. 1993;36:102–11.
Zhang HQ, Yang RW, Dou QW, Tsujimoto H, Zhou YH. Genome constitutions of Hystrix patula, H. duthiei ssp. duthiei and H. duthiei ssp. longearistata (Poaceae: Triticeae) revealed by meiotic pairing behavior and genomic in-situ hybridization. Chromosom Res. 2006;14:595–604.
Mahelka V, Kopecký D. Gene capture from across the grass family in the allohexaploid Elymus repens (L.) Gould (Poaceae, Triticeae) as evidenced by ITS, GBSSI, and molecular cytogenetics. Mol Biol Evol. 2010;27:1370–90.
Mahelka V, Kopecký D, Baum BR. Contrasting patterns of evolution of 45S and 5S rDNA families uncover new aspects in the genome constitution of the agronomically important grass Thinopyrum intermedium (Triticeae). Mol Biol Evol. 2013;30:2065–86.
Yang CR, Zhang HQ, Chen WH, Kang HY, Wang Y, Sha LN, et al. Genomic constitution and intergenomic translocations in the Elymus dahuricus complex revealed by multicolor GISH. Genome. 2017;60:510–7.
Chester M, Leitch AR, Soltis PS, Soltis DE. Review of the application of modern cytogenetic methods (FISH/GISH) to the study of reticulation (polyploidy/hybridisation). Genes. 2010;1:166–92.
Dou QW, Wang RRC, Lei YT, Yu F, Li Y, Wang HQ, et al. Genome analysis of seven species of Kengyilia (Triticeae: Poaceae) with FISH and GISH. Genome. 2013;56:641–9.
Gustafson P, Okito P, Mott IW, Wu Y, Wang RRC. A Y genome specific STS marker in Pseudoroegneria and Elymus species (Triticeae: Gramineae). Genome. 2009;52:391–400.
Hu LJ, Li GR, Zeng ZX, Chang ZJ, Liu C, Yang ZJ. Molecular characterization of a wheat -Thinopyrum ponticum partial amphiploid and its derived substitution line for resistance to stripe rust. J Appl Genet. 2011;52:279–85.
Wang L, Shi QH, Su HD, Wang Y, Sha LN, Fan X, et al. St2-80: a new FISH marker for St genome and genome analysis in Triticeae. Genome. 2017;60:553–63.
Yan C, Sun GL. Multiple origins of allopolyploid wheatgrass Elymus caninus revealed by RPB2, PepC and TrnD/T genes. Mol Phylogenet Evol. 2012;64:441–51.
Fan X, Liu J, Sha LN, Sun GL, Hu ZQ, Zeng J, et al. Evolutionary pattern of rDNA following polyploidy in Leymus (Triticeae: Poaceae). Mol Phylogenet Evol. 2014;77:296–306.
Yan C, Hu Q, Sun G, McIntyre CL. Nuclear and chloroplast DNA phylogeny reveals complex evolutionary history of Elymus pendulinus. Genome. 2014;57(2):97–109.
Liu QL, Ge S, Tang HB, Zhang XL, Zhu GF, Lu BR. Phylogenetic relationships in Elymus (Poaceae: Triticeae) based on the nuclear ribosomal internal transcribed spacer and chloroplast trnL-F sequences. New Phytol. 2006;170:411–20.
Fan X, Sha LN, Dong ZZ, Zhang HQ, Kang HY, Wang Y, et al. Phylogenetic relationships and Y genome origin in Elymus L. sensu lato (Triticeae; Poaceae) based on single-copy nuclear Acc1 and Pgk1 gene sequences. Mol Phylogenet Evol. 2013;69:919–28.
Dong ZZ, Fan X, Sha LN, Wang Y, Zeng J, Kang HY, et al. Phylogeny and differentiation of the St genome in Elymus L. sensu lato (Triticeae; Poaceae) based on one nuclear DNA and two chloroplast genes. BMC Plant Biol. 2015;15:179.
Ma WJ, Wu DC, He DM, Gu HL, Wu PP, Yi X, et al. Origin and evolution of allopolyploid wheatgrass Elymus fibrosus (Schrenk) Tzvelev (Poaceae: Triticeae) reveals the effect of its origination on genetic diversity. PLoS One. 2016;11:e0167795.
Sha LN, Fan X, Wang XL, Dong ZZ, Zeng J, Zhang HQ, et al. Genome origin, historical hybridization and genetic differentiation in Anthosachne australasica (Triticeae; Poaceae), inferred from chloroplast rbcL, trnH-psbA and nuclear Acc1 gene sequences. Ann Bot. 2017;119:95–107.
Sun GL, Komatsuda T. Origin of the Y genome in Elymus and its relationship to other genomes in Triticeae based on evidence from elongation factor G (EF-G) gene sequences. Mol Phylogenet Evol. 2010;56:727–33.
Fan X, Sha LN, Wang XL, Zhang HQ, Kang HY, Wang Y, et al. Phylogeny and molecular evolution of the Acc1 gene within the StH genome species in Triticeae (Poaceae). Gene. 2013;529:57–64.
Middleton CP, Senerchia N, Stein N, Akhunov ED, Keller B, Wicker T, et al. Sequencing of chloroplast genomes from wheat, barley, rye and their relatives provides a detailed insight into the evolution of the Triticeae tribe. PLoS One. 2014;9:e85761.
Mahelka V, Kopecky D, Pastova L. On the genome constitution and evolution of intermediate wheatgrass (Thinopyrum intermedium: Poaceae, Triticeae). BMC Evol Biol. 2011;11:127.
Hu QN, Sun DF, Sun GL. Molecular phylogeny revealed distinct origin of the Y and St genome in Elymus longearistatus (Triticeae: Poaceae). Mol Phylogenet Evol. 2015;85:141–9.
Mason-Gamer KEA. Chloroplast DNA analysis of the monogenomic Triticeae: phylogenetic implications and genome-specific markers. Boca Raton. Florida: CRC press; 1995.
Mason-Gamer R. Reticulate evolution, introgression, and intertribal gene capture in an allohexaploid grass. Syst Biol. 2004;53:25–37.
Liu QL, Lu BR, Zhang NN, Liu J, Yu Y, Wang HG, et al. Polyploidy origin of wheatgrass Douglasdeweya wangii (Triticeae, Poaceae): evidence from nuclear ribosomal DNA internal transcribed spacer and chloroplast trnL-F sequences. Dev Genes Evol. 2010;220:173–8.
Sha LN, Fan X, Yang RW, Kang HY, Ding CB, Zhang L, et al. Phylogenetic relationships between Hystrix and its closely related genera (Triticeae; Poaceae) based on nuclear Acc1, DMC1 and chloroplast trnL-F sequences. Mol Phylogenet Evol. 2010;54:327–35.
Badger Jonathan H, Mason-Gamer RJ. Phylogeny of a genomically diverse group of Elymus (Poaceae) allopolyploids reveals multiple levels of reticulation. PLoS One. 2013;8:e78449.
Redinbaugh MG, Jones TA, Zhang Y. Ubiquity of the St chloroplast genome in St-containing Triticeae polyploids. Genome. 2000;43:846–52.
Refoufi A, Jahier J, Esnault MA. Genome analysis of Elytrigia pycnantha and Thinopyrum junceiforme and of their putative natural hybrid using the GISH technique. Genome. 2001;44:708–15.
Ellneskog-Staam P, Salomon B, Rv B, Anamthawat-Jónsson K. The genome composition of hexaploid Psammopyrum athericum and octoploid Psammopyrum pungens (Poaceae: Triticeae). Genome. 2003;46:164–9.
Chen Q, Conner RL, Laroche A, Thomas JB. Genome analysis of Thinopyrum intermedium and Th. ponticum using genomic in situ hybridization. Genome. 1998;41:580–6.
Mason-Gamer RJ, Burns MM, Naum M. Polyploidy, introgression, and complex phylogenetic patterns within Elymus. Czech J Genet Plant. 2005;41:21–6.
Dewey DR. Morphological, cytological, and taxonomic relationships between Agropyron repens and A. elongatiforme (Gramineae). Syst Bot. 1980;5:61–70.
Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–5.
Komatsuda T, Nakamura I, Takaiwa F, Oka S. Development of STS markers closely linked to the vrs1 locus in barley, Hordeum vulgare. Genome. 1998;41:680–5.
Huang S, Sirikhachornkit A, Faris JD, Su X, Gill BS, Haselkorn R, et al. Phylogenetic analysis of the acetyl-CoA carboxylase and 3-phosphoglycerate kinase loci in wheat and other grasses. Plant Mol Biol. 2002;48:805–20.
Hsiao C, Chatterton NJ, Asay KH, Jensen KB. Phylogenetic relationships of the monogenomic species of the wheat tribe, Triticeae (Poaceae), inferred from nuclear rDNA (internal transcribed spacer) sequences. Genome. 1995;38:211–23.
Taberlet P, Gielly L, Pautou G, Bouvet J. Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol. 1991;17:1105–9.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–8.
Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–91.
Ronquist F, Huelsenbeck JP. MRBAYES 3: Bayesian phylogenetic inference under mixed model. Bioinformatics. 2003;19:1572–4.
Han FP, Liu B, Fedak G, Liu ZH. Genomic constitution and variation in five partial amphiploids of wheat-Thinopyrum intermedium as revealed by GISH, multicolor GISH and seed storage protein analysis. Theor Appl Genet. 2004;109:1070–6.
Guo X, Han FP. Asymmetric epigenetic modification and elimination of rDNA sequences by polyploidization in wheat. Plant Cell. 2014;26:4311–27.
Acknowledgements
We are very thankful to the US National Plant Germplasm System for providing the seeds, and sincerely thanks to Prof. Fangpu Han from Chinese Academy of Sciences for technology support for GISH and FISH.
Funding
This project was supported by the National Natural Science Foundation of China (Grant Nos. 31470305, 31270243, and 31200252), the National Key Research and Development of China (2016YFD0102000), and the fund from the Science and Technology Bureau and Education Bureau of Sichuan Province, China. Funds were used to design of the study and collection, analysis, and in writing the manuscript.
Availability of data and materials
All sequences data from this study were deposited in National Center for Biotechnology Information (NCBI) and the accession number from MK721792 to MK721830.
Author information
Authors and Affiliations
Contributions
ZYH designed the study, and revised the manuscript; WL carried out most of the experiments and data analyses, and wrote the manuscript; JYY participated in the writing and carried out the English modification; ZL and SGL helped to draft the manuscript and participated in language editing; SQH carried out parts of experiments; WY, SLN and FX participated in the data analyses and gave very important suggestions in the writing; KHY and ZHQ collected seed materials and gave very important suggestions in the experiments; All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. The related species in Triticeae used in this study. (XLSX 21 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, L., Jiang, Y., Shi, Q. et al. Genome constitution and evolution of Elytrigia lolioides inferred from Acc1, EF-G, ITS, TrnL-F sequences and GISH. BMC Plant Biol 19, 158 (2019). https://doi.org/10.1186/s12870-019-1779-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-019-1779-x