
Abstract
Background
Homologous transformation sterility-1 (HTS-1) is a novel wheat mutant that exhibits pistillody, the transformation of stamens into pistils or pistil-like structures. More extreme phenotypes of this mutation can have six pistils or pistil-like structures without any stamens in a floret. Thus, HTS-1 is highly valuable for studies of wheat hybrid breeding and flower development. Previous studies have shown that two major genes (Pis1 and hts) control pistillody in HTS-1. The Pis1 gene controls the three-pistil trait in the three-pistil wheat mutant and has been mapped on chromosome 2D, but the hts gene has not been mapped or identified. To do so, we crossed HTS-1 with CM28TP (three-pistil mutant) and constructed a high-density linkage map with the F2 population (200 individuals).
Results
The map covered 2779.96 cM, and the genetic distance per chromosome ranged from 37.59 cM to 318.95 cM. The average distance between markers was 1.04 cM. We then mapped hts between GBS-SNP markers 4A_109 and 4A_119, separated by 2.0 cM and 5.2 Mb. To find the candidate genes, the hts region was enlarged to 7.2 Mb, encompassing 752 protein-coding genes. We identified TaWin1 as a possible candidate gene after comparing the 752 genes with 206 common differentially expressed genes between pistillody stamens (PS) versus normal stamens (S) and pistils (P) versus S. Real-time PCR indicated that TaWin1 was highly expressed in HTS-1 during the pistil-and-stamen-differentiating stage, at levels approximately 120 times greater than those in CM28TP. Further analysis indicated that TaWin1 was mainly expressed in HTS-1 PS, supporting its status as a candidate gene of hts. Thus, TaWin1 overexpression probably leads to the transformation of stamens into pistils in wheat.
Conclusions
The results of this study provide a foundation for further research on stamen and pistil development, with implications for wheat-hybrid breeding programs.
Similar content being viewed by others


Background
Common wheat (Triticum aestivum L.) is a staple food crop worldwide, and approximately 11.02% of the global wheat cultivation area (22.16 million ha annually) is in China [1]. Current wheat yield in China averages approximately 7000 kg/ha and is far lower than that of rice or corn yield [1]. Crossbreeding to generate hybrids with superior traits (heterosis) is a promising method to improve wheat yield. Although progress has been made in developing hybrid wheat over the past few decades, the results have not been applied to production because a suitable sterile male line is currently lacking. Homologous transformation sterility-1 (HTS-1) is a novel wheat male-sterile mutant that was observed in the offspring of a cross of the three-pistil (TP) mutant with Chinese Spring (CS) [2]. HTS-1 and Chinese Spring three pistils (CSTP) are sib lines that show the three-pistil trait because they carry the Pis1 gene. However, the HTS-1 phenotype is characterized by pistillody, the partial or complete transformation of stamens into pistils or pistil-like structures [3,4,5]. Some HTS-1 mutants with more extreme phenotypes even exhibit six pistils and lack stamens entirely, and their seed-setting rate is often very low, 15.3% on average, under the natural pollination condition [2]. HTS-1 differs from other pistillody mutants, such as alloplasmic line of wheat cultivar Norin 26 (N26) and (cr)-CSdt7BS, because the phenotype is caused by nuclear-cytoplasm interactions in the latter two lines [6, 7], with the MADS-box gene TaAGL2 being a potential contributor to the trait [8]. In contrast, the pistillody trait in HTS-1 is determined by two recessive karyogenes [2]. We hypothesized that one of these genes is Pis1 because the mutant exhibits TP traits. Unfortunately, the identification and fine mapping of functional genes in wheat has proven more difficult than that in diploid species, such as rice, because its genome is enormous (17 Gb) and very complex (allohexaploid: 2n = 6X = 42, with A, B, D genomes) [9].
The development of next-generation, high-throughput sequencing has allowed the sequencing of the entire wheat genome [9,10,11], accelerating the efficiency of wheat breeding using molecular marker-assisted selection (MAS) and providing valuable tools for gene mapping. Notably, genotyping-by-sequencing (GBS) is a highly accurate, cost-effective, reliable, and rapid method for assessing such large, complex genomes because genome complexity is reduced through multiple enzyme digestions [12]. Furthermore, GBS does not rely on prior genome information for genetic linkage map construction [13], and multiplexed GBS can reduce the cost per sample when used to generate high-density linkage maps for gene identification and fine mapping [14,15,16,17,18]. Indeed, we previously used GBS to map Pis1 on chromosome 2D, between SNP markers M70 (3 cM away) and M71 (1.1 cM away) [19]. However, the second gene, hts, which controls pistillody in HTS-1, remains unidentified to date.
In the present study, we used GBS-SNP markers to construct a high-density linkage map of wheat on the basis of an F2 population derived from crossing HTS-1 with Chuanmai 28 three pistils (CM28TP). We used this linkage map to map another major gene (hts) controlling the pistillody trait. We expected our work to lay a foundation for the study of the development of stamen and pistil and hybrid breeding in wheat.
Materials and methods
Plant materials
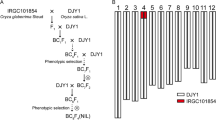
HTS-1 is a novel wheat male-sterile mutant that was selected in the offspring of a cross of TP mutant with CS [2]. CM28TP was derived from (Chuanmai 28/TP mutant)//Chuanmai 28 by conventional breeding [20]. HTS-1 exhibits both three pistils (Pis1) and pistillody (hts), whereas CM28TP only exhibits three pistils (Pis1) (Fig. 1). HTS-1 and CM28TP were crossed to produce an F2 population of 200 individuals. To map hts, we first counted pistil-like stamens per floret for all 200 F2 individuals during the flowering period. Plants were categorized as pistillody mutant if any pistil-like stamens were present and plants were wild type otherwise. The following nulli-tetrasomic (NT) lines derived from Chinese Spring (CS) were used for verifying mapping results: N1AT1B, N1BT1A, N1DT1B, N2AT2D, N2BT2D, N2DT2A, N3AT3B, N3BT3D, N3DT3A, N4AT4B, N4BT4A, N4DT4B, N5AT5D, N5BT5A, N5DT5A, N6AT6B, N6BT6D, N6DT6B, N7AT7B, N7BT7A, and N7DT7A [21]. With the exception of NT lines from the United States National Plant Germplasm System (NPGS), all other experimental materials have been cultivated by our laboratory. All materials are stored at China West Normal University in Nanchong, China.

Morphology of the florets in CM28TP and HTS-1. A: Floral organs of CM28TP. CM28TP carrying Pis1 gene, and showing three pistils and three stamens. B: Floral organs of HTS-1. HTS-1 carrying Pis1 gene and hts gene, and exhibit that transforms all or parts of stamen into pistils or pistil-like structures. P: pistil; S: stamen; PS: pistillody stamen
DNA and RNA extraction
Total genomic DNA was extracted from the fresh leaves of F2, parental, and NT lines using a Plant Genomics DNA Kit (Tiangen Biotech, China) following the manufacturer’s protocol. Expression analysis of candidate genes was performed on young CM28TP and HTS-1 spikes at various developmental stages: double ridge to floret differentiation (spike length, 2–5 mm), pistil-and-stamen differentiation (spike length, 5–7 mm), as well as anther-lobe formation (spike length, 7–10 mm). Pistillody stamens (PS), pistils (P), and stamens (S) in HTS-1 at the heading stage were also used. Total RNA was isolated from young spikes, PS, P, and S using an EASYspin Plus Plant RNA Extraction Kit (Axygen, USA) following the manufacturer’s protocol. DNA and RNA quality were assessed via 1% agarose gel electrophoresis. Nucleic acid concentrations were determined in an ND-2000 spectrophotometer (Thermo Fisher Scientific, USA) by measuring the absorbance ratio of 260/280 nm.
GBS library construction and SNP genotype calling
Genomic DNA (100–1000 ng) from the 200 F2 plants and two parental plants (HTS-1 and CM28TP) was subjected to GBS following published methods [19, 22]. Two restriction enzymes (MseI, NlaIII) were used. Digested fragments were ligated to barcoded forward adapters and common reverse adapters. For library construction, samples were PCR-amplified using Illumina primers with sequences complimentary to the adapters. Amplicons were sequenced on an Illumina Hi-seq 2000 platform at Beijing Novogene Bioinformatics Technology Co., Ltd. One library was double-loaded onto two lanes of the Illumina flow cell as technical replicates. All sequences obtained in this study were submitted to NCBI (accession number: SRP127844). The bioinformatics pipeline UNEAK was used to call SNPs from GBS results [23]. To ensure linkage map quality, GBS-SNPs were not used for final mapping if they exhibited significant distortion (chi-square 1:2:1 test, P < 0.001) and > 25% missing data.
Construction of high-density genetic maps and mapping of hts
The genetic linkage map was constructed in JoinMap version 4.0 [24], excluding markers with obvious distortion from expected Mendelian segregation ratios of 1:2:1. Independent logarithm of the odds (LOD) thresholds of 3.0 were used to position markers on linkage groups. Linkage analysis and marker-order assignment were performed with regression mapping. Recombination fractions between markers were converted to map distances (cM) using the Kosambi mapping function [25]. Overly similar markers were considered degenerate bin markers [26]. Linkage maps were drawn in MapChart 2.2 [27]. All bin markers were BLASTn-searched against the bread-wheat genome, IWGSC1 + popseq.31.pep (ftp://ftp.ensemblgenomes.org/pub/plants/release-31/fasta/triticum_aestivum/pep/). A co-linear analysis was performed on the genetic maps. The phenotypic data of the F2 individuals and the SNP markers on chromosome 4A were combined to map the hts gene using JoinMap version 4.0 [24].
Verification of hts mapping results and possible candidate gene identification
Mapping results were verified by hybridizing HTS-1 with 21 NT lines to check for pseudodominance in F1 and to determine the chromosomal location of hts. Sequences of markers flanking hts were aligned to IWGSC1 + popseq.31.pep, and all genes found in between were considered candidates. Candidate genes were compared with 206 common differentially expressed genes (DEGs) across PS versus S and P versus S. These DEGs had been previously identified in an RNA-seq study of HTS-1 [28].
Cloning of the possible candidate gene from CM28TP and HTS-1
Specific PCR primers were designed using Primer Premier 5.0 on the basis of the possible candidate gene sequence. The primer sequences were as follows: TaWin1–1F: 5′-CTGGCTAACCATCAGCAGTCC-3′ and TaWin1–1R: 5′-GGTCAAATCAAATCAAGAGGGAGT-3′. PCR amplification was done in a T-100 Thermal Cycler (Bio-Rad, USA) and reactions consisting of 100 ng template DNA, 25 μL 2 × PCR Mix (Tiangen Biotech, China), and 0.5 μM of each primer in a final reaction volume of 50 μL.
The PCR cycling conditions were as follows: initial denaturation at 94 °C for 3 min, followed by 40 cycles at 94 °C for 45 s, 58 °C for 45 s, 72 °C for 60 s, and a final extension at 72 °C for 10 min. The resulting amplified products were visualized by gel electrophoresis in 1.2% agarose gels. The specific DNA band was recovered using AxyPrep DNA Gel extraction kit (Axygen, USA). Purified PCR products were cloned into the pMD-20 T vector (TaKaRa, China), according to the manufacturer’s instructions. Transformants were plated on Luria-Bertani agar containing ampicillin. Fifteen positive clones for each material were screened and sequenced by Sangon BiotechCo., Ltd. (Shanghai, China).
Expression analysis of the possible candidate gene by real-time PCR
The real-time PCR Primers were designed using Beacon Designer version 6.0 to amplify the 82 bp TaWin1 (wound-induced protein 1) fragment (TaWin1-2F: 5′-TGGCTAACCATCAGCAGTCCC-3′ and TaWin1-2R: 5′-AGGCGCAGCACGAGGAACT-3′). Real-time assays were performed with SsoFast EvaGreen (BIO-RAD, USA) in the Bio-Rad CFX96 real-time PCR platform. All samples were analyzed in three biological replicates and the fold change in RNA transcripts was calculated using the 2ΔΔct method [29]. Wheat ubiquitin (DQ086482) and actin (AB181911) were selected as reference genes [20].
Results
Sequencing of the parental lines and the F2 population
Using GBS, CM28TP and HTS-1 were sequenced at effective sequencing depths of 44.7-fold and 54.96-fold, resulting in 74,503,950 and 49,266,501 clean reads mapped to the bread wheat genome, respectively (mapping rates: 99.08 and 98.51%). The average MseI enzyme capture rate was high (98.2%) across GBS data of the F2 population, validating digestion quality. We obtained 939,256,850 clean reads from F2, averaging 4,696,284 reads per individual. The average GC content was 42.5%, with a Q20 score of 96.8%. We screened 1,457,623 SNPs (homozygous: 543,979; hybrid: 913,644) from CM28TP and HTS-1. As the parental lines are homozygous, only the aa × bb genotype (53,352 SNPs) was used for further analysis. After filtering the low-coverage (< 75%) sequences from F2, 3316 candidate SNPs were obtained. Significantly distorted SNPs were filtered out (χ2 test, P < 0.001) to yield 3108 SNPs for determining bin markers.
Genetic linkage map with GBS-SNP markers
We mapped 2684 GBS-SNP bin markers to 21 linkage maps (Additional file 1: Fig. S1). The genetic map was 2779.96 cM long, and the average distance between two markers was 1.04 cM. Of all mapped chromosomes, 2A contained 368 markers, the highest percentage (13.7%), while 4D had the lowest with 11 markers (Table 1). Chromosomal genetic distances ranged from 37.59 cM (5B) to 318.95 cM (2A), and their maximum gaps ranged from 10.08 cM (1A) to 18.21 cM (6B) (Table 1). Among the 2684 markers, 1158 covered 1163.49 cM in wheat genome A, whereas 1119 covered 1043.35 cM in genome B. Only 407 were located in genome D, covering 573.12 cM (Table 1). We observed 137 gaps of > 5 cM among all 21 chromosomes: 55 were in genome A, 46 in B, and 36 in D.
A BLASTn search against the wheat genome verified all 2684 GBS-SNP markers in the linkage map (Additional file 1: Table S1). Additionally, a co-linearity analysis showed that all markers were consistent with the reference wheat genome, indicating highly accurate inference of the genetic recombination rate (Additional file 1: Figure S2). Overall, the genetic map constructed with GBS-SNP markers had sufficient coverage over the wheat genome, and most SNPs occurred in the same order as their corresponding chromosomes on the physical wheat-genome map.
Mapping of hts
Among the F2 population, 45 individuals showed the pistillody trait and 155 individuals showed normal stamens, the pistillody-to-normal fit to Mendelian segregation ratio 1:3 (χ2 = 0.66, P > 0.05), indicating that a recessive effect gene controls the pistillody trait in HTS-1 × CM28TP plants (Additional file 1: Table S2). The genetic linkage map of chromosome 4A was constructed using stamen phenotypic (normal or pistillody) and SNP data. Two SNP markers were found to be tightly linked to the hts gene, namely, 4A_109 and 4A_119, with genetic distances of 0.9 cM and 1.1 cM from hts, respectively. These two markers are separated by a genetic distance of 2.0 cM and a physical distance of 5.2 Mb (Fig. 2). The chromosomal location of hts was further confirmed when all 11 F1 individuals of the HTS-1 × N4AT4B cross exhibited pistillody.

The genetic linkage map of the hts gene. Genetic distances are indicated on the left side of linkage group in centiMorgans (cM), and the marker names are shown on the right side. The physical distances are shown in the brackets
Candidate gene prediction
Candidate genes were identified from an enlarged 7.2 Mb region (5.2-Mb interval and outer regions extending 1 Mb from GBS-SNP markers 4A_109 and 4A_119) encompassing 752 protein-coding genes (Additional file 1: Table S3). Comparison of the candidate genes with the DEGs showed that a possible candidate gene, Traes_4AS_F516F49FA.4 (physical distance: 162551918–162,552,664), shared 100% sequence identity with DEG comp83842. The latter was upregulated in PS and P, with log2 fold-change values of 5.75 and 5.46 for PS versus S and P versus S, respectively. A search against the NCBI nucleotide database (http://www.ncbi.nlm.nih.gov) revealed that Traes_4AS_F516F49FA.4 shared 96% sequence identity with Win1 of Aegilops tauschii (GenBank accession number: XM 020340959.1), leading us to tentatively designate the candidate possible gene as TaWin1.
PCR using DNA from fresh leaves of CM28TP and HTS-1 and the TaWin1–1 primer pair yielded a fragment of approximately 900 bp in length. The sequencing result indicated that the length of the TaWin1 gene in CM28TP and HTS-1 was 883 bp and 885 bp, respectively. The open reading frame (ORF) was 408 bp and had no intron. The sequence similarity of TaWin1 in CSTP and HTS-1 was 99.77%, with the only differences being two thymine (T) nucleotides inserted downstream of the ORF in HTS-1 (Fig. 3).

Alignment of the TaWin1 sequence in CM28TP and HTS-1. The ORF region and variation site are indicated by solid line frame and dotted line frame
Real-time PCR demonstrated that during the pistil-and-stamen-differentiating stages, TaWin1 was expressed in HTS-1 at 120 times the level in CM28TP (Fig. 4a). TaWin1 expression was much lower in other stages of HTS-1 and CM28TP development. Additionally, TaWin1 expression in HTS-1 PS was about 3.6-fold and 2.7-fold higher than in P and S, respectively (Fig. 4b).

TaWin1 gene expression assessed by real-time PCR. A: Relative levels of TaWin1 gene in young spikes of CM28TP and HTS-1 at various development stages. B: Relative levels of TaWin1 gene in different tissues of HTS-1. The transcript levels are shown as relative values and columns represent the means ± SEM of three replicates. P: pistil, S: stamen; PS: pistillody stamens
Discussion
The main objective of plant breeding is to develop high-yielding varieties to increase crop productivity to feed a growing human population. In rice and maize, the shift to hybrid breeding has facilitated large increases in yield [30]. In recent years, the potential of hybrid breeding in wheat has received renewed interest and is considered as a potential strategy to increase yield and to enhance yield stability [31]. Despite the progress made in hybrid wheat breeding, it is still in its infancy and a number of issues remain to be solved. Among them, the most important issue is the lack of a suitable, maintainable male-sterile line in wheat. HTS-1 is a novel pistillody mutant in wheat, and its stamens partially or completely transform into pistils or pistil-like structures [2]. Although the seed-setting rate of HTS-1 is 15.3% on average under natural pollination condition [2], our recent studies have shown that pistillody in HTS-1 is controlled not only genetically, but also by the environment. HTS-1 can be made completely male sterile by appropriately elevating the temperature at the booting stage (data not shown). Therefore, we speculate that HTS-1 is a male-sterile line that is controlled genetically as well as by temperature, and this can be applied in wheat-hybrid breeding. Using HTS-1 plants as female parents to cross with normal wheat material Chinese Spring, the seed-setting rate was reached about 30% by hand pollination [2].
Previous studies indicated that the Pis1 and hts genes contribute to the pistillody trait in HTS-1 [2], and Pis1 has been fine mapped on chromosome 2D of wheat [19]. In this study, we used GBS to map the hts gene, one of the two major genes controlling pistillody in HTS-1. This technique addresses issues with previous attempts using simple sequence repeats (SSR), which did not uncover markers linked to hts (data not shown). This failure was likely because too few SSR markers are present on the wheat chromosomes; for example, only 53 SSR markers (covering 88 cM) are located on chromosome 4A [32]. GBS is a highly accurate, cost-effective, reliable, and rapid method for assessing large, complex genomes, such as those of wheat and barley, for SNP discovery, and for genotyping [12]. The greatest advantage of GBS is that it does not rely on prior genome information for genetic linkage map construction [13], although imputation of SNPs can become more accurate in bi-parental mapping populations when a reference genome is available for the tested plants [33]. Thus, we took advantage of the availability of a wheat genome sequence in this study using CM28TP and HTS-1 as the parental lines. The effective sequencing depths of the two parents were 44.7-fold and 54.96-fold, respectively, with a sequencing error rate of only 0.03%, suggesting high-quality genome sequences. Furthermore, the relatively high error rates of low-coverage sequence data did not noticeably affect genotype-calling accuracy [33].
The two parental mutant lines were cultivated in our laboratory to create an appropriate mapping population for constructing a high-density linkage map [2, 34]. The originating line of HTS-1, CSTP, belongs to a population subgroup distinct from CM28TP, based on an analysis with sequence-related amplified polymorphism (SRAP) markers [34]. Thus, the founder parents were expected to yield F1 hybrids sufficiently heterozygous for generating an informative F2 mapping population. Additionally, Pis1 interference can be excluded when mapping hts because both parents possess Pis1. We screened 1,457,623 SNPs from CM28TP and HTS-1; from these, 2684 polymorphic bin markers were identified for high-density linkage map construction. Bin-marker distribution across 21 linkage groups corresponded to the base chromosome number (n = 21) of common wheat. The total map length (2779.96 cM) was longer than a previous CM28 × CM28TP linkage map (2371.4 cM) constructed on the basis of 200 F2 plants and using 1987 bin markers [19]. The 2684 SNPs markers were identified across the three wheat genomes A, B, and D. The highest number of markers was found in the A genome, while the lowest was found in the D genome. In particular, only 11 markers were found for 4D. This poor representation of the D genome indicates a lower genetic diversity due to the low frequency of recombination and polymorphism [35, 36].
Using the constructed linkage map, we were able to identify and map hts, locating it on chromosome 4A between GBS-SNP markers 4_109 and 4A_119 (genetic distance: 2.0 cM, physical distance: 5.2 Mb). We then identified a gene, TaWin1, upregulated in HTS-1 PS and P. The sequence of TaWin1 showed high similarity in CM28TP and HTS-1, in particular, their ORF sequences were identical, with only two T nucleotides inserted downstream of the ORF in HTS-1. However, this gene had unusually high expression during the pistil and stamen-differentiating stages of HTS-1 and was primarily expressed in PS. Therefore, we speculate that the pistillody trait is not caused by a change in the amino acid sequence of TaWIN1 but may be related to the overexpression of the TaWin1 gene in wheat. It remains to be studied whether the insertion of the two T nucleotides downstream of the ORF in HTS-1 is the cause of TaWin1 overexpression.
The Win gene was first isolated from potato and Win is mainly involved in wound-healing mechanisms. Ethylene upregulates Win-encoded mRNA [37, 38]. Numerous studies have shown that exogenous ethylene induces male sterility (cytoplasmic, nuclear, or thermosensitive) in many crops, including wheat, specifically through altering endogenous ethylene content [39,40,41]. However, we have no direct data on the role of Win in flower development. Therefore, future studies should examine the relationship between ethylene, pistillody, and Win genes in detail.
Conclusion
In the present study, we constructed a high-density linkage map using the F2 population of a cross between HTS-1 and CM28TP. Using this linkage map, we mapped hts between GBS-SNP markers 4A_109 and 4A_119, separated by a genetic distance of 2.0 cM and a physical distance of 5.2 Mb. Of the 752 protein-coding genes around the two markers, we identified TaWin1 as the possible candidate gene for hts. Real-time PCR supported this conclusion through demonstrating extremely high expression of TaWin1 gene during the pistil-and-stamen-differentiating stage of HTS-1, as well as preferential expression in PS.Therefore, overexpression of TaWin1 in the wheat mutant likely results in the transformation of stamens into pistils.
Abbreviations
- CM28TP:
-
Chuanmai28 three pistils
- CS:
-
Chinese Spring
- CSTP:
-
Chinese Spring three pistils
- DEGs:
-
Differentially expressed genes (DEGs)
- GBS:
-
Genotyping-by-sequencing
- HTS-1:
-
Homologous transformation sterility-1
- MAS:
-
Marker-assisted selection
- NT:
-
Nulli-tetrasomic
- P:
-
Pistils
- PS:
-
Pistillody stamens
- S:
-
Stamens
- TP:
-
Three-pistil
References
FAO. FAOSTAT, http://www.fao.org/faostat/zh/#data/QC. 2014.
Peng ZS, Yang ZJ, Ouyang ZM, Yang H. Characterization of a novel pistillody mutant in common wheat. Aust J Crop Sci. 2013;7:159–64.
Goto K, Meyerowitz EM. Function and regulation of the Arabidopsis floral homeotic gene pistillata. Genes Dev. 1994;8:1548–60. https://doi.org/10.1101/gad.8.13.154.
Sommer H, Beltrán JP, Huijser P, Pape H, Lönnig WE, Saedler H. Deficiens, a homeotic gene involved in the control of flower morphogenesis in Antirrhinum majus: the protein shows homology to transcription factors. EMBO J. 1990;9:605–13. https://doi.org/10.1002/j.1460-2075.1990.tb08152.x.
Zhang Q, Xu J, Li Y, Xu P, Zhang H, Wu X. Morphological, anatomical and genetic analysis for a rice mutant with abnormal hull. Acta Genet Sin. 2007;34:519–26. https://doi.org/10.1016/S1673-8527(07)60057-5.
Murai K, Tsunewaki K. Photoperiod-sensitive cytoplasmic male sterility in wheat with Aegilops crassa cytoplasm. Euphytica. 1993;67:41–8. https://doi.org/10.1007/BF00022723.
Murai K, Takumi S, Koga H, Ogihara Y. Pistillody, homeotic transformation of stamens into pistil-like structures, caused by nuclear-cytoplasm interaction in wheat. Plant J. 2002;29:169–81. https://doi.org/10.1046/j.0960-7412.2001.01203.x.
Yamada K, Saraike T, Shitsukawa N, Hirabayashi C, Takumi S, Murai K. Class B and Bsister MADS-box genes are associated with ectopic ovule formation in the pistil-like stamens of alloplasmic wheat (Triticum aestivum L.). Plant Mol Biol. 2009;71:1–14. https://doi.org/10.1007/s11103-009-9504-z.
Zimin AV, Puiu D, Hall R, Kingan S, Clavijo BJ, Salzberg SL. The first near-complete assembly of the hexaploid bread wheat genome, Triticum aestivum. Gigascience. 2017;6:1–7. 0.1093/gigascience/gix097.
Brenchley R, Spannagl M, Pfeifer M, Barker GLA, D’Amore R, Allen AM, et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature. 2012;491:705–10. https://doi.org/10.1038/nature11650.
Jia J, Zhao S, Kong X, Li Y, Zhao G, He W, et al. Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature. 2013;496:91–5. https://doi.org/10.1038/nature12028.
Poland JA, Brown PJ, Sorrells ME, Jannink JL. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS One. 2012;7:251–64. https://doi.org/10.1371/journal.pone.0032253.
Chen Z, Wang B, Dong X, Liu H, Ren L, Chen J. An ultra-high density bin-map for rapid QTL mapping for tassel and ear architecture in a large F2 maize population. BMC Genomics. 2014;15:433–42. https://doi.org/10.1186/1471-2164-15-433.
Alipour H, Bihamta MR, Mohammadi V, Peyghambari SA, Bai G, Zhang G. Genotyping-by-sequencing (GBS) revealed molecular genetic diversity of iranian wheat landraces and cultivars. Front Plant Sci. 2017;8:1293–316. https://doi.org/10.3389/fpls.2017.01293.
He J, Zhao X, Laroche A, Lu ZX, Liu H, Li Z. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front Plant Sci. 2014;5:484–91. https://doi.org/10.3389/fpls.2014.00484.
Guajardo V, Solís S, Sagredo B, Gainza F, Muñoz C, Gasic K. Construction of high density sweet cherry (Prunus avium L.) linkage maps using microsatellite markers and SNPs detected by genotyping-by-sequencing (GBS). Plos One. 2015;10:1–17. https://doi.org/10.1371/journal.pone.0127750.
Jiang B, Liu W, Xie D, Peng Q, He X, Lin Y. High-density genetic map construction and gene mapping of pericarp color in wax gourd using specific-locus amplified fragment (SLAF) sequencing. BMC Genomics. 2015;16:1035–44. https://doi.org/10.1186/s12864-015-2220-y.
Zhao L, Yuanda L, Caiping C, Xiangchao T, Xiangdong C, Wei Z. Toward allotetraploid cotton genome assembly: integration of a high-density molecular genetic linkage map with DNA sequence information. BMC Genomics. 2012;13:539–55. https://doi.org/10.1186/1471-2164-13-539.
Yang ZJ, Chen ZY, Peng ZS, Yu Y, Liao ML, Wei SH. Development of a high-density linkage map and mapping of the three-pistil gene (pis1) in wheat using gbs markers. BMC Genomics. 2017;18:567–74. https://doi.org/10.1186/s12864-017-3960-7.
Yang ZJ, Peng ZS, Yang H, Yang J, Wei SH, Cai P. Suppression subtractive hybridization identified differentially expressed genes in pistil mutations in wheat. Plant Mol Bio Rep. 2011;29:431–9. https://doi.org/10.1007/s11105-010-0249-2.
Sears ER. The aneuploids of common wheat. Mo Agric Exp Res Bull. 1954;572:1–8.
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6:1–10. https://doi.org/10.1371/journal.pone.0019379.
Lu L, Luo G, Xiao F. A retrospective survey of the quality of reports and their correlates among randomized controlled trials of immunotherapy for guillain-Barre syndrome. Immunotherapy. 2013;5:829–36. https://doi.org/10.2217/imt.13.75.
Van Oojjen JW. MapQTL 6, software for the mapping of quantitative trait loci in experimental populations of diploid species. Wageningen: Kyazma BW; 2009.
Kosambi D. The estimation of map distances from recombination values. Ann Eugenics. 1994;12:172–5.
Wang S, Chen J, Zhang W, Hu Y, Chang L, Fang L. Sequence-based ultra-dense genetic and physical maps reveal structural variations of allopolyploid cotton genomes. Genome Biol. 2015;16:108–25. https://doi.org/10.1186/s13059-015-0678-1.
Voorrips R. MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered. 2002;93:77–8. https://doi.org/10.1093/jhered/93.1.77.
Yang ZJ, Peng ZS, Wei SH, Liao ML, Yan Y, Jang ZY. Pistillody mutant reveals key insights into stamen and pistil development in wheat (Triticum aestivum, l.). BMC Genomics. 2015;16:211–20. https://doi.org/10.1186/s12864-015-1453-0.
Livbak K, Schmittgen TD. Analysis of relative gene expression data using real time quantitative PCR and the 22ΔΔct method. Methods. 2001;25:402–8. https://doi.org/10.1006/meth.2001.1262.
Duvick DN. Heterosis: feeding people and protecting natural resources. In: Coors JG, Pandey S, editors. The Book: the genetics and exploitation of heterosis in crops. 677 South Segoe Road, Madison: ASA-CSSA-SSSA; 1999. p. 53711.
Longin CFH. Future of wheat breeding is driven by hybrid wheat and efficient strategies for pre-breeding on quantitative traits. J Bot Sci. 2016;S3:32–3.
Somers DJ, Isaac P, Edwards K. A high-density microsatellite consensus map for bread wheat (Triticum aestivum L.). Theor Appl Genet. 2004;109:1105–14. https://doi.org/10.1007/s00122-004-1740-7.
Huang Y, Wu J, Ma J, Beaty TH, Sull JW, Zhu L. Association between IRF6 SNPs and oral clefts in West China. J Dent Res. 2009;88:715–8. https://doi.org/10.1177/0022034509341040.
Yang ZJ, Peng ZS, Zhou YH, Peng LJ, Wei SH. Evaluation on the genetic background of wheat near isogentic lines for three pistils character by SRAP markers. J Nucle Agricul Sci. 2012;26:22–7.
Chao S, Zhang W, Akhunov E, Sherman J, Ma Y, Luo MC. Analysis of gene-derived SNP marker polymorphism in US wheat (Triticum aestivum L.) cultivars. Mol Breed. 2009;23:23–33. https://doi.org/10.1007/s11032-008-9210-6.
Eltaher S, Sallam A, Belamkar V, Emara HA, Nower AA, Salem KFM, Poland J, Baenziger PS. Genetic diversity and population structure of F3:6 Nebraska winter wheat genotypes using genotyping-by-sequencing. Front Genet. 2018;9:76–86. https://doi.org/10.3389/fgene.2018.00076.
Stanford A, Bevan M, Northcote D. (1989). Differential expression within a family of novel wound-induced genes in potato. Mol Gen Genet. 1989;215:200–8.
Harris N, Taylor JE, Roberts JA. Characterization and expression of an mRNA encoding a wound-induced (win) protein from ethylene-treated tomato leaf abscission zone tissue 1. J Exp Bot. 1997;48:1223–7.
Keyes G, Sorrells ME. Mutations blocking sensitivity to gibberellic acid promote ethylene-induced male sterility in wheat. Euphytica. 1990;48:129–39. https://doi.org/10.1007/BF00037192.
Liu HW, Zhang GS, Wang JW, Wang XL, Fang ZW. Relation between ethylene of young spike and male sterility induced by genesis in wheat. J Northwest Sci-Tech Univer Agricul Forest. 2003;31:39–42.
Tian C. Relationship between ethylene and the occurrence of cytoplasmic male sterility in rice. Rice Sci. 2000;8:9–9.
Acknowledgments
The authors thank by Editage, a company dedicated to helping international researchers publish their findings in the best English language, for editing the language.
Funding
This work was financially supported by the National Natural Science Foundation of China (grant number 31760425), Foundation of Science & Technology department of Sichuan Province, China (grant number 16JC022), National General Cultivation Project of China West Normal University (grant number 17C043), and the Innovation Team Project of Education Department of Sichuan Province (Grant No. 16TD0020). Each of the funding bodies granted the funds based on a research proposal. They had no role in the design of the study; collection, analysis, and interpretation of data; or in writing the manuscript.
Availability of data and materials
The raw sequence data have been deposited in the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/sra/SRP127844). All other relevant supplementary data is provided within this manuscript as Additional file 1: Figures S1, S2. Tables S1, S2 and S3.
Author information
Authors and Affiliations
Contributions
QY and HT contributed to the field experiments and GBS library construction; ZY designed the research and wrote the manuscript with input from all authors; YY and ZC analysed the data; SW and QS performed gene cloning and real-time PCR analysis. ZP edited the submitted version. All authors approve the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. Distribution map of linkage groups. x-axis, chromosome number; y-axis, genetic distance (in cM); and blue, bin marker. Figure S2. The genetic linkage map and physical map. The genetic map is shown in red, while the physical map is shown in blue, and the green line indicated the position of each marker on the genetic map and the physical map. Table S1. GBS-SNP markers and their BLAST hit information. Table S2. The phenotypic of the F2 individuals in HTS-1 × CM28TP. Table S3. Genes located in the intervals of hts. (DOCX 623 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yang, Q., Yang, Z., Tang, H. et al. High-density genetic map construction and mapping of the homologous transformation sterility gene (hts) in wheat using GBS markers. BMC Plant Biol 18, 301 (2018). https://doi.org/10.1186/s12870-018-1532-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-018-1532-x