
Abstract
Background
Modifications in root morphology are important strategies to maximize soil exploitation under phosphorus starvation in plants. Here, we used two multiple interval models to map QTLs related to root traits, biomass accumulation and P content in a maize RIL population cultivated in nutrient solution. In addition, we searched for putative maize homologs to PSTOL1, a gene responsible to enhance early root growth, P uptake and grain yield in rice and sorghum.
Results
Based on path analysis, root surface area was the root morphology component that most strongly contributed to total dry weight and to P content in maize seedling under low-P availability. Multiple interval mapping models for single (MIM) and multiple traits (MT-MIM) were combined and revealed 13 genomic regions significantly associated with the target traits in a complementary way. The phenotypic variances explained by all QTLs and their epistatic interactions using MT-MIM (23.4 to 35.5 %) were higher than in previous studies, and presented superior statistical power. Some of these QTLs were coincident with QTLs for root morphology traits and grain yield previously mapped, whereas others harbored ZmPSTOL candidate genes, which shared more than 55 % of amino acid sequence identity and a conserved serine/threonine kinase domain with OsPSTOL1. Additionally, four ZmPSTOL candidate genes co-localized with QTLs for root morphology, biomass accumulation and/or P content were preferentially expressed in roots of the parental lines that contributed the alleles enhancing the respective phenotypes.
Conclusions
QTL mapping strategies adopted in this study revealed complementary results for single and multiple traits with high accuracy. Some QTLs, mainly the ones that were also associated with yield performance in other studies, can be good targets for marker-assisted selection to improve P-use efficiency in maize. Based on the co-localization with QTLs, the protein domain conservation and the coincidence of gene expression, we selected novel maize genes as putative homologs to PSTOL1 that will require further validation studies.
Similar content being viewed by others


Background
The increasing demand for agricultural production poses a global challenge to improve the phosphorus (P) use efficiency in plants due to its low availability in a large proportion of arable lands [1, 2]. Plants uptake phosphorus from the soil in the orthophosphate forms (Pi), which are available at low concentration in the soil solution [3]. In a large fraction of soils, P is tightly fixed to the clay’s surface, which requires high amounts of phosphate fertilizers for high-yielding farming systems, increasing production costs and hampering soil fertility management [3–5]. However, low-input farmers have limited access to phosphate fertilizer, which is the second most used fertilizer for plant growth [6]. Maize is the most common grain produced worldwide and a major staple food in Africa and Latin America [7], where soils often show limited P availability. Thus, improving maize P-use efficiency is expected to increase yield stability and, consequently, food security [1, 4, 8].
Plants have evolved two major strategies to overcome P limitation in the soil, which are P internal utilization and P uptake [3]. P internal utilization mechanisms involve transport, partitioning and remobilization of P within the plant, whereas the mechanisms that increase P uptake are associated with alterations in the root system, interactions with microorganisms, and chemical modifications of the rhizosphere [3]. Indeed, P acquisition efficiency has been considered from two to three times more important than P internal utilization to explain the variability for P-use efficiency in tropical maize genotypes evaluated in low- and high-P soils [9]. Considering the limited mobility and low P concentration in the soil, mechanisms related to P acquisition are greatly dependent of the proximity of this nutrient to the root system [3, 10]. Thus, a well-developed root system should be an important adaptation mechanism to maximize soil exploitation, enabling plants to improve P acquisition efficiency [11–13]. Studies have shown that plants that are more efficient in P acquisition presented higher root:shoot dry weight ratios [14, 15], reduced root diameters [16], longer and denser root hairs [17], increased lateral roots [18], greater lateral branching and shallower basal roots [17, 19]. These changes in root morphology are key strategies used by plants to improve soil exploitation at a minimal metabolic cost [5, 20].
Root morphology is controlled by multiple genes in maize [13, 21], but only a few of them such as roothairless (Rth1) [22], brittle stalk-2-like protein 3 (Bk2l3) [23], and rootless concerning crown and seminal roots (Rtcs) [24] have been cloned and characterized. However, an appropriate strategy to dissect these traits is through quantitative trait loci (QTL) mapping. Indeed, several QTLs were mapped for root traits under contrasting conditions of P availability in nutrient solution [25–27], in glasshouse [28] and in the field [29–32]. These QTLs individually explained from 1 to 14 % of the phenotypic variation, confirming the genetic complexity of root traits. These studies were carried out using composite interval mapping (CIM) strategy, however, multiple interval mapping (MIM) [33] offers a significant improvement on both statistical power and precision for detecting main and epistatic QTLs over CIM. This happens because MIM utilizes the estimated positions of QTLs as cofactors in the multiple regression model, whereas CIM utilizes the nearest markers to the estimated QTL as cofactors [34]. Besides all statistical advantages achieved by MIM, it still cannot capture the genetic correlation that might exist between traits. The multiple-trait multiple interval mapping (MT-MIM) method [35], which is an extension of MIM, applies multiple regression on a multiple dimensional (traits) space context, which enables it to capture information that might be available from the existing genetic correlation between traits, therefore, boosting the precision and power to detect QTLs [35]. To the best of our knowledge, this method has never been applied to map QTLs with effects on root morphology traits in maize.
In rice, a major QTL controlling phosphorus uptake (Pup1) was mapped to chromosome 12, explaining approximately 80 % of the phenotypic variance of this trait [36]. Rice near isogenic lines (NILs) carrying the Pup1 QTL showed a three-fold increase in P uptake and enhanced root surface area when grown in P-deficient soil [36, 37]. Additionally, irrigated and upland rice varieties introgressed with Pup1 showed a significant improvement in grain yield in different low-P soils compared to their parents [38, 39]. The gene underlying the Pup1 locus was identified and named Phosphorus-starvation tolerance 1 (PSTOL1), which encodes a serine/threonine kinase of the LRK10L-2 subfamily [40]. The overexpression of PSTOL1 in two transgenic rice varieties enhanced the grain yield over 60 % under low-P conditions due to larger root system (i.e., root length, and total root surface area), which also improved the uptake of P and other nutrients [40]. Furthermore, Hufnagel et al. [41] showed that sorghum homologs to OsPSTOL1 were associated with enhanced P uptake and grain yield in sorghum grown in a low-P soil due modifications on root system morphology and architecture. A remarkable conservation of protein-encoding genes among maize, sorghum and rice has been confirmed in silico based on genome sequencing comparison, once approximately 89 % of the 11,892 maize gene families predicted in the B73 genome were shared with rice and sorghum [42]. Of these, genes encoding important adaptive traits are expected to be conserved among these grass species.
In order to better understand the genetic basis of root morphology and P acquisition related traits, as well as the relationship between these traits, a path analysis and a QTL mapping study were carried out in a maize RILs in nutrient solution cultivated under low P. We also integrated the QTL mapping, sequence comparison and expression analysis to identify putative homologs to PSTOL1 in maize.
Results
Transgressive segregation of phenotypic traits in RILs
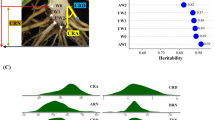
Significant genetic variation in root morphology traits, biomass accumulation and phosphorus content in the seedlings were observed for the RIL population with high broad sense heritability estimates, which ranged from 0.65 for root:shoot dry weight ratio to 0.82 for root length (Additional file 1: Table S1). The P-efficient line, L3, tended to present superior phenotypic measurements for all traits compared with the P-inefficient line L22, with the exception of root diameter and root:shoot ratio (Fig. 1a). The range of the phenotypic variation in the RILs was larger than both parents, suggesting transgressive segregation for all phenotypic traits (Fig. 1a). The RILs showing extreme root systems in comparison with their parental lines were highlighted in the Fig. 1b. The parental lines belonged to distinct heterotic groups (L3, flint and L22, dent) [43], were genetically divergent based on SNP markers [44] and contrasted for grain yield in low-P soil [45] and for root morphology traits in nutrient solution [46].

a Frequency distribution of traits evaluated in 145 maize RILs grown in low-P (2.5 μM). The P-efficient (L3) and the P-inefficient (L22) parental lines are indicated by arrows. b Root system of the parental lines (L3 and L22) and two extreme RILs (RIL 66 and 113) after 13 days cultivated in nutrient solution under low-P
Surface area is an important root trait contributing to seedling dry weight and P content
Root length, root surface area and surface area of fine roots were high and positively correlated with each other (Table 1). The correlation coefficients among these traits exhibited comparable magnitudes to those observed in a sorghum diversity panel composed of 287 accessions [41] and in a group of 30 maize lines [46]. These root traits also showed strong correlation coefficients with total seedling dry weight (0.77 to 0.86), moderate correlations with total P content in the seedling (0.31 to 0.48), and negative correlations with root:shoot ratio (−0.29 to −0.38) (Table 1). In contrast, root diameter was negatively correlated with root length, root surface area and total seedling dry weight, but no significant correlation was found with total P content. The negative correlation between root length and root diameter (−0.62) was similar to the coefficients obtained for root diameter with lateral (−0.65) and non-lateral (−0.68) roots in temperate maize RILs [18].
To further investigate the relative importance of root traits on seedling dry weight and P content, we performed a path analysis, using the root traits as explanatory variables and the total seedling dry weight and P content as dependent variables. As P content was the product of total seedling dry weight and P concentration as previously proposed [30, 31], both dependent variables were significantly correlated (0.58) and were evaluated separately. The partitioning of the correlation coefficients revealed that root length had the lowest direct effect on total seedling dry weight (−4.831) and P content (−0.997), but had a strong indirect effect through root surface area (5.816 and 1.565, respectively) (Table 2). Thus, the negative direct effect of root length was counterbalanced by the indirect effect via root surface area, probably due to the high positive correlation between these traits (r = 0.98). A similar pattern was observed for surface area of fine roots, which was also positively correlated with root surface area (r = 0.8). This trait exerted a minor negative direct influence on total seedling dry weight (−0.681) and P content (−0.117), which were mitigated by the positive indirect effect via root surface area (4.748 and 1.277, respectively). Root diameter also played a more important effect indirectly via root surface area on both variables, masking its direct contribution, which corroborates with the negative correlation between these root traits (r = −0.48). Therefore, root surface area contributed the highest direct effect on total seedling dry weight (5.935) and P content (1.597) and mediated an important proportion of the indirect effects of the other root traits on the dependent variables.
Thus, the path analysis clarified the direct and indirect importance of a greater root surface area, which is a combination of longer roots with smaller diameters, to improve total dry weight and P content in the seedlings under P deficiency. This root morphology also promoted an additional advantage for shoot over root development, confirming that the investment in root growth was beneficial to P acquisition as discussed by Zhu and Lynch [18].
Two distinct QTL mapping strategies reveal complementary results
A linkage map was constructed using 292 markers that covered 1787.5 cM of the maize genome, with an average interval of 6.1 cM between adjacent markers (Additional file 2: Figure S1). In addition to SSR and SNP markers, six ZmPSTOL candidate genes and three genes previously associated with root morphology (Rth1, Bk2l3, and Rtcs) were mapped to their predicted physical positions. Multiple interval mapping models for single (MIM) and multiple traits (MT-MIM) provided statistical evidence for 13 genomic regions harboring QTLs on all maize chromosomes, with the exception of chromosome 5 (Tables 3 and 4). The QTL regions were named using the trait initials if they were detected through single trait analysis or as “multi” if they were detected by multiple trait analysis, followed by their genetic position in bin (Fig. 2). A bin is the interval of approximately 20 cM between two core markers previously defined and mapped in maize [47], which are designated with the chromosome number followed by a two-digit decimal.

QTLs identified for root traits, seedling dry weight and P content using single and multiple traits MIM analyses. The markers are represented as vertical traces along the horizontal lines, which represent the chromosomes and are numbered in centiMorgans (cM). The candidate genes are depicted below the red vertical traces. QTL profiles for single trait MIM are shown as colored lines according to the legend for root length (RL), root average diameter (RD), root surface area (SA), surface area of fine roots (SA2), root:shoot ratio (R:S), total seedling dry weight (TDW) and total P content (Pcont). Multi trait QTL profiles are shown as black line. The QTL peaks are depicted with an inverted triangle colored according to the legend followed by the bin. The confidence interval (95 %) for each QTL is represented by a horizontal line above the chromosomes colored according to the legend
MIM models for individual traits detected seven QTLs controlling root length, root diameter, surface area of fine roots, and root:shoot ratio. The proportion of the phenotypic variance explained by each QTL (R2) ranged from 6.84 % (qRD4.05) to 15.12 % (qSA2_10.03). The magnitude of QTL effects ranged from 0.270 standard deviations from the progeny mean (sd) to −0.393 sd (Table 3).
The MT-MIM analysis revealed the presence of ten QTLs with R2 ranging from 2.04 % (qMulti8.02 for R:S) up to 15.17 % (qMulti10.03 for SA2). The highest additive effect was also observed for qMulti10.03 for SA2 (−0.403 sd) (Table 4).
LOD estimates for the MT-MIM model were higher and the confidence intervals were narrower than those for the individual MIM models, suggesting superior statistical power of MT-MIM compared with the MIM models applied to each trait individually (Fig. 2). Despite these differences, the MT-MIM and MIM models were coincident in revealing QTLs at bins 1.03, 1.07, 3.06, and 10.03, whereas QTLs at bins 1.06, 2.08, 3.04, 6.06, 8.02, and 9.04 were only revealed using the MT-MIM model. Conversely, QTLs at regions 4.05, 7.02, and 8.05 were only detected by MIM models. Therefore, combining the results of MIM and MT-MIM analyses conveyed the most accurate information regarding the genetic architecture of the traits under investigation in this particular study.
Using a simulation, Silva et al. [35] showed that when a QTL affects only a small subset of the traits, the MT-MIM model might have a lower power than MIM models to identify this QTL due to a greater genome-wide threshold for the MT-MIM model. This may be the reason why MT-MIM failed to identify QTLs at regions 4.05, 7.02, and 8.05. Although the MT-MIM LOD profile revealed peaks at these regions, the values were not statistically significant according to the score threshold employed.
The additive main effect of QTLs detected by MIM and MT-MIM had both positive and negative signs, confirming the contribution of favorable alleles coming from both parental lines for most of the traits analyzed (Tables 3 and 4). Additionally, five epistatic interactions were identified using the MT-MIM model, including some with magnitudes comparable to the main additive effects (Table 5). No epistatic effect was detected based on single trait analysis. Taken together, the additive and epistatic effects on the MT-MIM model explained between 23.41 % and 35.54 % of the phenotypic variance for each trait (Tables 4 and 5).
ZmPSTOL predicted proteins share a conserved serine/threonine kinase domain with OsPSTOL1
Using OsPSTOL1 [GenBank: BAK26566] as a query, six predicted proteins were selected on the maize genome, sharing more than 55 % amino acid sequence identity. The genes encoding these proteins were predicted to be located on chromosomes 3, 4 and 8, and named according to their genetic position in bin (Table 6).
A phylogenetic analysis revealed that the six predicted ZmPSTOL proteins clustered together with PSTOL1 from rice, SNC4 and PR5 from Arabidopsis (circled in Fig. 3), which were classified as LRK10L-2 subfamily of serine/threonine receptor-like kinases by Gamuyao et al. [40]. In a detailed alignment of structural predictions, all maize PSTOL-like proteins shared conserved ATP-binding and serine/threonine protein kinase domains with OsPSTOL1 (Additional file 3: Figure S2). A distinct glycosyl hydrolase domain was predicted for ZmPSTOL4.05. The maize proteins ZmPSTOL4.05, ZmPSTOL8.02, ZmPSTOL8.05_1 and ZmPSTOL8.05_2 were classified as receptor-like kinases (RLKs), which are characterized by the presence of a transmembrane domain for signal perception and an intracellular kinase domain [48, 49]. In contrast, the proteins ZmPSTOL3.04 and ZmPSTOL3.06 contained the intracellular kinase domain but lacked the transmembrane domain similarly to OsPSTOL1 [40], and thus were classified as receptor-like cytoplasmic kinases (RLCKs) [49].

Phylogenetic tree of predicted serine/theronine receptor-like kinases from maize, rice and Arabidopsis thaliana. The rice PSTOL1, the six maize proteins sharing more than 55 % sequence identity to OsPSTOL1, PR5K and SNC4 from Arabidopsis thaliana were grouped separately from other rice kinases, and are highlighted. Numbers on branches are bootstrap values for the percentage of coincidence (%) inferred from 1,000 replicates. Only percentage values higher than 50 % are shown
ZmPSTOL candidate genes have distinct expression patterns
The expression analyses revealed that ZmPSTOL4.05, ZmPSTOL8.02 and ZmPSTOL8.05_1 were highly and consistently expressed in the roots of the P-inefficient genotype (L22) under low (2.5 μM) and high (250 μM) P conditions, but were not responsive to P in either L22 or L3 (Fig. 4). ZmPSTOL3.06 was preferentially expressed in the roots of the P-efficient line (L3) with lower expression under high-P compared to the low-P concentration, and induced by high-P in roots of L22 (Fig. 4). The expression of ZmPSTOL3.04 and ZmPSTOL8.05_2 was induced in the root and repressed in the shoot of L22 under high-P concentration, but were not differentially expressed in L3 (Fig. 4). Additionally, the expression pattern of ZmPSTOL3.04 and ZmPSTOL8.05_2 in roots may reflect the total P content in the seedling, whereas the expression in shoots could be negatively associated with the total P content in L22 (Additional file 4: Table S2).

Expression profiles of the ZmPSTOL genes. The expression of the maize candidate genes are presented as relative gene expression (RQ) evaluated in roots and shoots of maize seedlings of the two parental lines L3 (P-efficient) and L22 (P-inefficient) grown under two levels of P (2.5 and 250 μM) after 13 days of treatment. Error bars indicate the standard errors of three technical replicates composed of three seedlings each
Discussion
Complex inheritance of root traits, seedling dry weight and P content in maize
Two distinct and powerful statistical models for QTL mapping (MIM and MT-MIM) were applied to dissect root morphology traits, total seedling dry weight, root:shoot ratio and P content in tropical maize RILs. The QTLs identified using the MIM model explained from 6 to 15 % of the total phenotypic variance for each trait, which was similar to the QTLs previously mapped for root traits and P efficiency indices in maize [25–27, 29, 31]. However, the proportion of phenotypic variance explained by all QTLs and their epistatic interactions using MT-MIM ranged from 23.4 to 35.5 %, which was higher than in previous studies. To the best of our knowledge, the present study demonstrates the first QTL mapping of root morphology traits, seedling biomass and P content in maize using the MT-MIM model, confirming that complementary information can be generated when this strategy is combined with single trait MIM analyses, as previously suggested by Silva et al. [35].
The genetic complexity of these traits was highlighted by the epistatic interactions among QTLs that showed effects of magnitudes comparable to those of main effects. The significant contribution of epistatic interactions was also detected by other authors for root traits in nutrient solution [25–27, 50] and for P-use efficiency indices under field conditions [30, 51].
QTL mapping revealed that both parents contributed favorable alleles for most of the traits evaluated, which possibly leads to transgressive segregation. The parents L3 and L22 were also shown to donate favorable alleles for P acquisition efficiency and P-use efficiency based on grain yield, when these RILs were backcrossed with both parental lines and evaluated in low-P soil [51]. The occurrence of segregating progenies with extreme phenotypes, out of the parental range, has been detected in plants subjected to different abiotic stresses under field or nutrient solution [50, 52, 53]. Transgressive segregation was also observed in maize RILs derived from a cross between Mo17 and B73 for the length and number of lateral and seminal roots [25, 27] and for root hair length [26].
The QTL mapping results strongly reflected the phenotypic correlations among the target traits. The high correlation between root length and root surface area (r = 0.98) reflected the coincidence in position and sign of the QTLs detected based on MT-MIM (qMulti1.07, 3.04, 8.02 and 10.03; Table 4). Additionally, two significant QTLs for root diameter were either mapped to unique regions or with opposite signs compared with other root traits, corroborating the negative correlations of these traits. Moreover, the importance of the root surface area to seedling dry weight and P content was supported by the presence of multi-trait QTLs that significantly affected these traits (qMulti1.07, 8.02 and 10.03; Table 4), which could be a result of determinant genes with pleiotropic effects or the presence of linked genes.
Coincidence of QTLs for root morphology in the seedling stage with QTLs for grain yield
Three genes previously associated with root morphology in maize (Rth1, Bk2l3 and Rtcs) were mapped to chromosome 1, but did not overlap with any mapped QTLs (Fig. 2). Even though early root growth enhancement has not always led to superior yield performance in the field [54], a large number of QTL studies have indicated that some genomic regions consistently affect root morphology traits during the seedling stage and agronomic performance under different environments. A similar situation was also found for three QTL regions on chromosome 1 in our current study, which were coincident with QTLs previously reported for early root traits and for yield components in the field.
qMulti1.03 associated with root diameter, total seedling dry weight and root:shoot ratio overlapped with the QTL influencing seminal root length and weight [50], primary axile root diameter [55], daily elongation rate of axile roots [56] and total length of second-order lateral roots [57] in nutrient solution. This region was coincident with QTLs for drought tolerance index [50], grain yield, kernel number and weight in low-P soil [58]. This genomic region was also detected in a meta-analysis for low-P tolerance in maize as the consensus cQTL1-2 [59].
qMulti1.06 was detected based on the multi-trait MIM model and was significantly associated only with the total P content in the seedling (Table 4 and Fig. 2). A major QTL at bin 1.06 was associated with root traits in nutrient solution, grain yield under well-watered and water-stressed conditions [50], and root-pulling resistance in adult plants [60]. Due to the consistency in the effects of this genomic region on root traits and grain yields in different studies, this QTL was named root-yield-1.06, and validated as constitutively affecting roots, agronomic features and grain yield under different water regimens [61]. A QTL for P utilization efficiency (qPUTIL1) based on grain yield under low-P soil also overlapped in this region [51].
The third QTL region on chromosome 1 was mapped at bin 1.07 spanning from 214 to 223 Mbp (qMulti1.07), which was associated with root length, root surface area, surface area of fine roots, total seedling dry weight and root:shoot ratio. This region was coincident with a cluster of QTLs named Ax-2 that controlled the root numbers and lengths identified in a meta-analysis combining 15 QTL studies [54]. QTLs for grain yield and drought tolerance index were also mapped to this genomic region [50].
The association of root morphology QTLs in early stages of plant development with yield performance, including the validation of the root-yield-1.06, suggested that at least some of these genomic regions can be further used in marker-assisted selection to improve yield stability under drought and other mineral stresses in maize.
ZmPSTOL genes co-localized with QTLs for root morphology, biomass accumulation and P content
The QTL mapped using MT-MIM (qMulti8.02) that was associated with root length, root surface area, root:shoot ratio and P content co-localized with ZmPSTOL8.02 (Fig. 2). This ZmPSTOL candidate gene was highly expressed in the roots of L22 (Fig. 4), the donor line of the favorable QTL alleles for all traits mentioned above. In rice, the overexpression of OsPSTOL1 enhanced total root length and root surface area in transgenic seedlings in nutrient solution as well as grain yield of transgenic varieties cultivated in P-deficient soils [40]. According to these authors, the larger root system contributed to a significant increase in the uptake of nutrients such as phosphorus, nitrogen and potassium in transgenic rice lines overexpressing OsPSTOL1. The sequence similarity and conserved domains of these protein kinases from rice and maize combined with the evidences shown here suggest that ZmPSTOL8.02 could be one of the genes underlying qMulti8.02, sharing similar functions in root development and P acquisition efficiency in maize to OsPSTOL1 in rice. Additionally, QTLs associated with seminal root number in high-P levels [27], shoot dry weight [32] and primary root length [56] overlapped with qMulti8.02, confirming that genes controlling root and shoot development in this genomic region are also expressed in other genetic backgrounds.
The other three ZmPSTOL genes co-localized with single trait QTLs for root length (ZmPSTOL8.05_1), root diameter (ZmPSTOL4.05), and root:shoot ratio (ZmPSTOL3.06). These genes were preferentially expressed in the roots of the donor line that contributes positive alleles for the respective QTLs under low- and high-P conditions.
ZmPSTOL8.05_1 was mapped to 117 cM on chromosome 8, flanking qRL8.05. Additionally, a weak LOD peak for total P content was coincident with this candidate gene (Fig. 2). The MIM model based on score as a significance threshold was not able to detect this QTL, but a minor effect QTL was detected using the Bayesian Information Content threshold with LOD 1.8 and explaining 6.5 % of the total variance for the total P content in the seedling (data not shown). This genomic region also harbored QTLs explaining 5 to 6 % of the phenotypic variance for P acquisition efficiency based on grain yield [51], indicating that this genomic region consistently contributed to P acquisition during the seedling and adult plant stages. Additionally, QTLs in this genomic region were mapped for root length and grain yield under field conditions [32], root length and root dry weight in nutrient solution [26, 50, 62]. The coincidence of QTLs for root traits and for grain yield from different studies associated with the superior expression of ZmPSTOL8.05_1 in roots under low-P are highly compatible with the role of its putative homolog (OsPSTOL1) in rice.
ZmPSTOL3.06 was mapped to 169.6 cM on chromosome 3 within the confidence interval of qRS3.06 for root:shoot ratio (Fig. 2). This genomic region spans bin 3.06 that harbored QTLs for root traits in a meta-analysis using 15 QTL studies in nine maize mapping populations [54]. This candidate gene was highly expressed in roots cultivated with both P levels of the P-efficient line L3 (Fig. 4), which contributed with positive alleles for the root:shoot ratio QTL. ZmPSTOL3.06 had the lowest e-value with OsPSTOL1 (Table 6) and its predicted protein lacks the transmembrane domain, similarly to OsPSTOL1 (Additional file 3: Figure S2). This combined information makes this predicted gene also a candidate to OsPSTOL1 homolog in maize.
ZmPSTOL4.05 was coincident with the qRD4.05, with L22 donating the allele that reduced the root diameter. ZmPSTOL4.05 was highly expressed only in roots of the donor line, L22, cultivated in both P availability. Under P starvation conditions the root diameter decreases, while the root surface area increases, enabling the root system to explore a larger volume of soil [11]. Thus, as observed in the path analysis, fine roots are an important component to improve the root surface area, which played a strong contribution to total seedling dry weight and to P content in maize seedlings cultivated in low-P conditions. A QTL controlling the plasticity of lateral root number (i.e., change in the lateral root number in response to P availability) in hydroponics [26] was also mapped at this same region, suggesting that genes harboring in this genomic region may control root traits across different populations.
The finding that ZmPSTOL genes were preferentially expressed in the roots of the lines that contributed the allelic enhancing root traits, seedling dry weight and P content indicate that at least ZmPSTOL3.06, ZmPSTOL4.05, ZmPSTOL8.02 and ZmPSTOL8.05_1 may have a functional relationship with root morphology and/or with P acquisition in maize. Considering the role of PSTOL1 genes in rice and sorghum, it could be expected that genes encoding important adaptive traits would be shared among rice, sorghum and maize, such as the case for the major Al tolerance gene in sorghum (SbMATE) [63] that was found to be functionally conserved in maize (ZmMATE1) [64] and rice (OsFRDL4) [65].
Conclusions
Comprehensive QTL analyses revealed important regions associated with root traits, seedling dry weight and P content in maize under low-P concentration. Using the multiple trait-multiple interval mapping model, these QTLs explained a larger extent of the phenotypic variance for the target traits compared with previous studies. The complementary genomic regions identified using both models jointly offered putative targets for molecular breeding aiming to improve P acquisition efficiency in maize. Additionally, this study identified new maize candidate genes sharing high identity with OsPSTOL1 that were preferentially expressed in the roots and co-localized with QTLs for root morphology and P acquisition related traits.
Material and methods
Mapping population
The segregating population was composed by 145 maize recombinant inbred lines (RILs) derived from a bi-parental cross of lines L3 (P-efficient) and L22 (P-inefficient). F1 plants were self-pollinated and individual F2 plants were advanced for seven cycles of selfing by single seed descent, after which seeds were bulked for multiplication. The parental lines and the population were developed at Embrapa Maize and Sorghum (Brazil, latitude 19_270S and 716 m above sea level). The parental lines were previously characterized as contrasting for P-use efficiency under low and high P conditions in the field [45] and for root morphology traits [46].
Quantitative analysis of root traits, seedling biomass accumulation and P content using a paper pouch system
The mapping population and parents were evaluated in randomized complete block design with four biological replicates, each composed by three plants per pouch. Each biological replicate was evaluated in an identical but independent experiment performed on a seven-day interval. Maize seeds were surface sterilized with 0.5 % (v/v) sodium hypochlorite for 5 min and germinated in moistened germination paper rolls. After four days, uniform seedlings were transferred to moist blots in paper pouches after removing the endosperm to eliminate seed reserves [46]. A modified Magnavaca nutrient solution [66] containing 2.5 μM P was replaced every three days and the pH was maintained at 5.65. Each container was filled with 5 l of nutrient solution with the bottom 3 cm of the pouches immersed in the solution. The containers were maintained in a growth chamber with a 12 h photoperiod at 27/20 °C day/night temperatures and 330 μmol m−2 s−1 of light intensity. After 13 days, root images were captured using a digital photography setup and analyzed using RootReader2D (http://www.plantmineralnutrition.net/rr2d.php) and WinRHIZO (http://www.regent.qc.ca/assets/winrhizo_about.html) software according to de Sousa et al. [46]. The total root system, which includes all together primary, seminal and initial adventitious roots, was evaluated for total root length (RL) (cm), average root diameter (RD) (cm3), total root surface area (SA) (cm2) and surface area of fine roots (SA2) (1.0 < d ≤ 2.0 mm) (cm2).
Root and shoot tissues were dried separately at 65 °C in a forced-air oven until a constant weight was obtained to determine the root:shoot dry weight ratio (R:S) and total seedling dry weight (TDW). For P analysis, root and shoot tissues were subjected to nitric perchloric acid digestion [67]. The total P content in the seedling (Pcont) was calculated as the sum of the P content in each seedling component, which was the product of the dry weight and the P concentration in the root and shoot. As maize absorbs phosphate in its orthophosphate form (Pi), the P in the nutrient solution refers to phosphate, whereas the total P content in the seedling comprises both organic and inorganic P forms.
Analysis of variance (ANOVA), correlations between pairs of traits and path analysis were performed using the GENES software [68]. The phenotypic correlations were calculated based on the mean values. Broad sense heritability was estimated as \( {h}^2={\widehat{\sigma}}_G^2/\left({\widehat{\sigma}}_G^2+{\widehat{\sigma}}_E^2\right) \) with \( {\widehat{\sigma}}_G^2 = \left(M{S}_G\mathit{\hbox{-}}\ M{S}_E\right)/r \) and \( {\widehat{\sigma}}_E^2=M{S}_E \), where \( {\widehat{\sigma}}_G^2 \) and \( {\widehat{\sigma}}_E^2 \) are the estimates of genetic and error variance, respectively; MS G and MS E are the genetic and error mean squares, respectively, and r is the number of replications.
For the path analysis [69], the five root traits (total root length, root average diameter, surface area and surface area of fine roots) were considered as independent variables x i (i = 1, 2, …, 5). The total seedling dry weight and phosphorus content were considered as dependent variables y j (j = 1, 2) in two distinct path analyses. The estimated path coefficient (P ij ) was considered as the direct effect of variable x i on y j . Indirect effects of x i on y j mediated by variable x i′ were calculated by multiplying the correlation between x i and x i′ (r ii′ ) by P i′j . Root:shoot ratio was excluded from the analysis due to its contribution to both dependent variables.
Linkage map
DNA was isolated from young leaves using the CTAB method [70]. Initially, 60 polymorphic SSR markers were genotyped in the RIL population according to [71]. A total of 332 SNPs (Single Nucleotide Polymorphisms) were genotyped in the population using Kompetitive Allele-Specific PCR or the KASPTM assay (LGC Genomics, Teddington, UK). The sequences, genetic and genomic locations of SSR and SNP markers are available at the Maize Genetics and Genomics Database (http://www.maizegdb.org/data_center/locus).
The markers were tested for the expected segregation ratio of 1:1 using chi-square statistics (p < 0.05) corrected for multiple tests based on Bonferroni’s method. The linkage map was constructed using MapMaker/EXP 3.0 [72] considering a minimum LOD of 3.0 and a maximum frequency of recombination (r) of 0.4. The mapping function Kosambi [73] was used to convert recombination fractions into map distances. The final linkage map was drawn using Windows QTL Cartographer v 2.5 (http://statgen.ncsu.edu/qtlcart/WQTLCart.htm).
QTL mapping
Phenotypic fitted values were obtained from the following statistical model adjusted for each single trait:
where y ij is the phenotypic observation from the i th (i = 1, …, 4) replication on the j th genotype (j = 1, …, 145); μ is the phenotypic average; B i is the effect of the i th block; G j is the j th genotype; and ε ij is the residual associated with the y ij observation. We tested the fit of two models by first assuming that the residuals were normally distributed with constant variance, ε ij ~ N(0, σ 2 ) and by second considering the heteroscedasticity, ε ij ~ N(0, σ j 2 ). Both models were fitted using the gls function from the nlme R package [74] and compared using the ANOVA function from the same package. For each trait, the fitted phenotypic values were extracted from the model with the best fit. Each replicate was composed by three plants that were bulked for all laboratorial analyses.
Due to the large variability in the absolute values, all phenotypic fitted values were standardized to achieve unity as standard deviations and zero means as follows:
where z it is the standardized observation of trait t (t = 1, 2, …, 7) on subject i; y it is the observation of trait t on subject i; μ t is the average of trait t; and σ t is the standard deviation of trait t.
For the joint QTL analysis, a multiple trait-multiple interval mapping (MT-MIM) model was evaluated as previously described [35]. The complete model was fitted using the following equation:
where z ti is the standardized observation of trait t on subject i; μ t is the intercept for trait t; the parameter β tr has the genetic interpretation of the additive effect of QTL r on trait t (r = 1, 2, …, m QTLs included in the model); and the regressive variables x ir represent the contrast coefficients codified according to the Cockerham model [33, 75] (i.e., x ir is 1 for the dominant and −1 for the recessive homozygous). The third component on the right side of the model refers to a subset of the p pairwise interactions among QTLs previously included in the model, where w trl is the epistatic effect between QTL r and QTL l on trait t and the random error ε ti was assumed to be independent and identically distributed according to a multivariate normal distribution, with a mean vector of zero and a positive definite symmetric variance-covariance matrix ∑ ε , i.e., ε ti = MVN(0, ∑ ε ).
Multi-trait QTL mapping was initiated with a forward search for the main effect QTL using a grid of 1 cM in the genome and a 15 % genome-wide significance level. After three rounds of QTL search, the positions of all QTLs in the model were re-estimated as along with all other parameters in the model. After the inclusion of the main effects in the model, the forward search for epistasis was performed by testing all pairwise interactions among QTLs already included in the model, employing a 5 % genome-wide significance level. Only the epistatic effects that displayed at least one significant marginal effect were kept in the final model. The Haley-Knott regression [76] was used to estimate the model parameters, and the resampled score statistics [35, 77] were employed to obtain the empirical genome-wide threshold for the QTL mapping analysis. Using this approach, a final model was selected to calculate the proportion of phenotypic variance explained by all QTLs as the ratio between the genotypic variance of the QTL effect to the phenotypic variance times 100 (coefficient of determination R2), and the LOD profile along the chromosomes. The R2 values were estimated using the fitted full model, including non-significant QTL effects. The QTL confidence intervals were obtained using the drop 1.5-LOD support interval method with approximately 0.95 confidence levels [78].
Multiple interval mapping (MIM) analysis was performed for each single trait [33, 79] in a similar procedure to that performed for the joint analysis, considering t = 1. All QTL analyses were performed using R software (version 2.15.2) and a QTL mapping package named OneQTL that is under development by L.C. Silva.
Identification of maize PSTOL1 homologs
Using the OsPSTOL1 amino acid sequence [GenBank: BAK26566] we performed searches against the maize genome database (http://ensembl.gramene.org/Zea_mays/Info/Index) using BLASTp. Six predicted maize proteins with more than 55 % sequence identity to rice PSTOL1 were selected and aligned using ClustalX software version 1.83 [80]; the alignment included OsPSTOL1 and the Arabidopsis protein kinases SNC4 [81] and PRK5 [82]. The phylogenetic tree was constructed based on the maximum likelihood method with 1000 bootstraps [83] using MEGA software [84]. The protein domains were identified using the CDART (Conservative Domain Architecture Retrieval Tool (http://www.ncbi.nlm.nih.gov/Structure/lexington/lexington.cgi).
Mapping candidate genes
Specific primers for the maize candidate genes were designed using Primer Blast (ncbi.nlm.nih.gov/tools/primer-blast/index.cgi). PCR reactions were performed using 30 ng of DNA, 0.2 mM of each dNTP, 2 mM of MgCl2, 10 ρmols of each primer, 5 % (v/v) dimethyl sulfoxide (DMSO) and 1 U of Taq DNA polymerase (Invitrogen, Carlsbad, CA). The amplification profile included an initial step of 95 °C for 1 min, followed by 35 cycles of denaturing at 94 °C for 1 min, annealing at 58 to 60 °C for 30 s, depending on the primers, and extension at 72 °C for 1 min. The amplification products were treated with ExoSAP-IT reagent (USB Corporation, Cleveland, OH) and sequenced using the BigDye Terminator v3.1 cycle sequencing kit on an ABI PRISM 3100 genetic analyser (Applied Biosystems, Foster City, CA) to identify polymorphisms between the parental lines.
Sequence-tagged site (STS) markers were developed to map genes previously associated with root morphology in maize. For the roothairless gene (Rth1) [22], a 22-bp indel was amplified using the primers 5′-TTGCCCACGGCTGGCAAGAG-3′ and 5′- GGCTCTGTAGCACGCCCCTC - 3′ and resolved on a silver-stained polyacrylamide gel [85]. The same strategy was used for the brittle stalk-2-like protein 3 gene (Bk2l3) [23], which was revealed after the amplification of a 15-bp indel using the primer pair: 5′-GCTGGTTAGATCCCCCGCCCA-3′ and 5′-GCACTGGAGCCACCGACACTG-3′. The rootless concerning crown and seminal roots gene (Rtcs) [24] was genotyped as a CAPS marker obtained after digestion of the amplification product of genomic DNA with the primers 5′-CGCGCCATAGCCCGCAGTAA-3′ and 5′-GATTGGCACGGGCCGGTCAG-3′ with the restriction enzyme AciI and was visualized on a silver-stained polyacrylamide gel [85].
Cleaved amplified polymorphic sequence (CAPS) markers were developed for the other candidate genes. For ZmPSTOL3.04 the PCR product amplified using the primers 5′-ACGGGGCTTGGAGGCACATG-3′ and 5′-TGAGACCGCGTGGGGAAGGG-3′ was digested with the restriction enzyme StuI. The polymorphism for ZmPSTOL8.02 was obtained after digestion with RsaI of the genomic fragment amplified with the primers 5′-TGACTGGTGCCAGAGGTACGC-3′ and 5′-TGCATACAAGGGACTGCTTCGGA-3′. CAPS markers were resolved on silver stained polyacrylamide gels [85]. The images were digitally captured using a Nikon digital camera.
ZmPSTOL3.06 was mapped based on the presence and absence of the amplification product using the primers 5′-AAGGGCGTCCAACCGCCTTG-3′ and 5′-TTGTTGGCCGGTCCGTTGGG-3′ on a 1 % (w/v) agarose gel stained with ethidium bromide.
A G/A SNP was revealed after sequencing the amplified product obtained using the primer pair 5′-CCGCTACGCCTTGGTTGCCA-3′ and 5′-CGCCGTAGTTAGCGGAGCCG-3′ to map ZmPSTOL4.05, the primer pair 5′-AGCCTCCACGATGGCCGACA-3′ and 5′-TGCATTTGTGTGACCTGGAA-3′ to map ZmPSTOL8.05_1, and the primers 5′- TCCACGGCCGACAGGTAGCA-3′ and 5′-GCTCAAGAGAACTCAGGGTGGC-3′ to map ZmPSTOL8.05_2.
Gene expression analysis
The expression profiles of the candidate genes were assessed in the roots and shoots of the L3 and L22 genotypes harvested after 13 days in modified Magnavaca’s nutrient solution containing low (2.5 μM) and high (250 μM) P. Total RNA was extracted from a bulk of three plants using the RNeasy Plant Mini kit (Qiagen, Valencia, CA), and 1 μg of total RNA pretreated with DNase I was used for cDNA synthesis using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. Gene expression was determined by quantitative PCR (qPCR-RT) using SYBR Green I and TaqMan assays in the ABI Prism 7500 Fast System (Applied Biosystems, Foster City, CA). Primers were designed for all target genes using Primer Express Software (Applied Biosystems, Foster City, CA), and 18S rRNA was used as an endogenous constitutive control (Additional file 5: Table S3). With the exception of ZmPSTOL4.05, two primers pair were designed for each gene to confirm their expression pattern. However, only the expression profile obtained with the primer pairs highlighted in bold in the Additional file 5 are shown. The relative gene expression was calculated using the 2-ΔΔCT method [86], with three technical replicates and L3 roots under low-P conditions as a calibrator. Variance analysis of the gene expression and total P content was performed using GENES software [68].
Availability of supporting data
Abbreviations
- BIC:
-
Bayesian information content
- bp:
-
Base pair
- CAPS:
-
Cleaved amplified polymorphic sequence
- CDART:
-
Conservative domain architecture retrieval tool
- CI:
-
Confidence interval
- cM:
-
centiMorgan
- CTAB:
-
Cetyl trimethyl ammonium bromide
- ICP:
-
Inductively-coupled argon
- indel:
-
Insertion/deletion
- KASP:
-
Kompetitive allele-specific PCR
- LOD:
-
Likelihood of odds
- Mbp:
-
Mega base pairs
- MIM:
-
Single trait-multiple interval mapping
- MT-MIM:
-
Multiple traits-multiple interval mapping
- μM:
-
micro Molar
- mM:
-
mili Molar
- Pcont:
-
Total phosphorus content in the seedling
- PCR:
-
Polymerase chain reaction
- P:
-
Phosphorus
- PSTOL1:
-
Phosphorus-starvation tolerance 1
- qPCR-RT:
-
Quantitative real-time PCR
- QTL:
-
Quantitative trait loci
- RD:
-
Average of root diameter
- RIL:
-
Recombinant inbred line
- RL:
-
Total root length
- R:S:
-
Root:shoot dry weight ratio
- SA:
-
Total root surface area
- SA2:
-
Surface area of fine roots
- sd:
-
Standard deviations from the progeny mean
- SNP:
-
Single nucleotide polymorphism
- SSR:
-
Simple sequence repeats
- STS:
-
Sequence-tagged site
- TDW:
-
Total seedling dry weight
- v/v:
-
Volume per volume
- w/v:
-
Weight per volume
References
Cordell D, Drangert J-O, White S. The story of phosphorus. Global food security and food for thought. Global Environ Chang. 2009;19:292–305.
Dawson CJ, Hilton J. Fertilizer availability in a resource-limited world: production and recycling of nitrogen and phosphorus. Food Policy. 2011;36:S14–22.
Vance C, Uhde-Stone C, Allan DL. Phosphorus acquisition and use: critical adaptations by plants for securing a nonrenewable resource. New Phytol. 2003;157:423–47.
Shen J, Yuan L, Zhang J, Li H, Bai Z, Chen X, et al. Phosphorus dynamics: from soil to plant. Plant Physiol. 2011;156:997–1005.
Lynch JP, Brown KM. New roots for agriculture: exploiting the root phenome. Phil Trans R Soc B. 2012;367:1598–604.
Batten GD. A review of phosphorus efficiency in wheat. Plant Soil. 1992;146:163–8.
Awika JM. Major cereal grains production and use around the world. In: Awika JM, Piironen V, Bean S, editors. Advances in cereal science: implications to food processing and health promotion. Washington: American Chemical Society; 2011. p. 1–13.
Calderón-Vázquez C, Sawers RJH, Herrera-Estrella L. Phosphate deprivation in maize: genetics and genomics. Plant Physiol. 2011;156:1067–77.
Parentoni SN, Souza Jr CL. Phosphorus acquisition and internal utilization efficiency in tropical maize genotypes. Pesqui Agropecu Bras. 2008;43:893–901.
Lambers H, Raven JA, Shaver GR, Smith SE. Plant nutrient-acquisition strategies change with soil age. Trends Ecol Evol. 2008;23:95–103.
Raghothama KG. Phosphate acquisition. Annu Rev Plant Phys. 1999;50:665–93.
Horst WJ, Kamh M, Jibrin JM, Chude VO. Agronomic measures for increasing P availability to crops. Plant Soil. 2001;237:211–23.
Ramaekers L, Remans R, Rao IM, Blair MW, Vanderleyden J. Strategies for improving phosphorus acquisition efficiency of crop plants. Field Crop Res. 2010;117:169–76.
Mollier A, Pellerin S. Maize root system growth and development as influenced by phosphorus deficiency. J Exp Bot. 1999;50:487–97.
Hermans C, Hammond JP, White PJ, Verbruggen N. How do plants respond to nutrient shortage by biomass allocation? Trends Plant Sci. 2006;11:610–7.
Silberbush M, Barber SA. Sensitivity of simulated phosphorus uptake to parameters used by a mechanistic-mathematical model. Plant Soil. 1983;100:93–100.
Lynch JP. Roots of the second green revolution. Aust J Bot. 2007;55:493–512.
Zhu J, Lynch JP. The contribution of lateral rooting to phosphorus acquisition efficiency in maize (Zea mays) seedlings. Func Plant Biol. 2004;31:949–58.
Lynch JP, Brown KM. Topsoil foraging—an architectural adaptation of plants to low phosphorus availability. Plant Soil. 2001;237:225–37.
York LM, Nord EA, Lynch JP. Integration of root phenes for soil resource acquisition. Fron Plan Sci. 2013;4:1–15.
Hochholdinger F, Tuberosa R. Genetic and genomic dissection of maize root development and architecture. Curr Opin Plant Biol. 2009;12:1–6.
Wen T-S, Hochholdinger F, Sauer M, Bruce W, Schnable PS. The roothairless1 gene of maize encodes a homolog of sec3, which is involved in polar exocytosis. Plant Physiol. 2005;138:1637–43.
Brady S, Song S, Dhugga KS, Rafalski JA, Benfey PN. Combining expression and comparative evolutionary analysis. The COBRA gene family. Plant Physiol. 2007;143:172–87.
Taramino G, Sauer M, Stauffer Jr JL, Multani D, Niu X, Sakai H, et al. The maize (Zea mays L.) RTCS gene encodes a LOB domain protein that is a key regulator of embryonic seminal and post-embryonic shoot-borne root initiation. Plant J. 2007;50:649–59.
Zhu J, Kaeppler SM, Lynch JP. Mapping of QTL controlling root hair length in maize (Zea mays L.) under phosphorus deficiency. Plant Soil. 2005;270:299–310.
Zhu J, Kaeppler SM, Lynch JP. Mapping of QTLs for lateral root branching and length in maize (Zea mays L.) under differential phosphorus supply. Theor Appl Genet. 2005;111:688–95.
Zhu J, Mickelson SM, Kaeppler SM, Lynch JP. Detection of quantitative trait loci for seminal root traits in maize (Zea mays L.) seedlings grown under differential phosphorus levels. Theor Appl Genet. 2006;113:1–10.
Kaeppler SM, Parke JL, Mueller SM, Senior L, Stuber C, Tracy WF. Variation among maize inbred lines and detection of quantitative trait loci for growth at low phosphorus and responsiveness to arbuscular mycorrhizal fungi. Crop Sci. 2000;40:358–64.
Chen J, Xu L, Cai Y, Xu J. QTL mapping of phosphorus efficiency and relative biologic characteristics in maize (Zea mays L.) at two sites. Plant Soil. 2008;313:251–66.
Chen J, Xu L, Cai Y, Xu J. Identification of QTLs for phosphorus utilization efficiency. Euphytica. 2009;167:245–52.
Chen J, Xu L. The candidate QTLs affecting phosphorus absorption efficiency. Front Agric China. 2011;5:456–62.
Cai H, Chen F, Mi G, Zhang F, Maurer HP, Liu W, et al. Mapping QTLs for root system architecture of maize (Zea mays L.) in the field at different developmental stages. Theor Appl Genet. 2012;125:1313–24.
Kao C-H, Zeng Z-B, Teasdale RD. Multiple interval mapping for quantitative trait loci. Genetics. 1999;152:1203–16.
E Silva LC, Zeng Z-B. Current progress on statistical methods for mapping quantitative trait loci from inbred line crosses. J Biopharm Stat. 2010;20:454–81.
Silva LDCE, Wang S, Zeng Z-B. Multiple trait multiple interval mapping of quantitative trait loci from inbred line crosses. BMC Genet. 2012;13:67.
Wissuwa M, Wegner J, Ae N, Yano M. Substitution mapping of Pup1: a major QTL increasing phosphorus uptake of rice from a phosphorus-deficient soil. Theor Appl Genet. 2002;105:890–7.
Wissuwa M, Ae N. Further characterization of two QTLs that increase phosphorus uptake of rice (Oryza sativa L.) under phosphorus deficiency. Plant Soil. 2001;237:275–86.
Chin JH, Lu X, Haefele SM, Gamuyao R, Ismail A, Wissuwa M, et al. Development and application of gene-based markers for the major rice QTL Phosphorus uptake 1. Theor Appl Genet. 2010;120:1073–86.
Chin JH, Gamuyao R, Dalid C, Bustaman M, Prasetiyono J, Moeljopawiro S, et al. Developing rice with high yield under phosphorus deficiency: Pup1 sequence to application. Plant Physiol. 2011;156:1202–16.
Gamuyao R, Chin JH, Pariasca-Tanaka J, Pesaresi P, Catausan S, Dalid C, et al. The protein kinase Pstol1 from traditional rice confers tolerance of phosphorus deficiency. Nature. 2012;488:535–9.
Hufnagel B, de Sousa SM, Assis L, Guimaraes CT, Leiser W, Azevedo GC, et al. Duplicate and conquer: Multiple homologs of PHOSPHORUS-STARVATION TOLERANCE1 enhance phosphorus acquisition and sorghum performance on low-phosphorus soils. Plant Physiol. 2014;166:659–77.
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–5.
Parentoni SN, Magalhaes JV, Pacheco CAP, Santos MX, Abadie T, Gama EEG, et al. Heterotic groups based on yield-specific combining ability data and phylogenetic relationship determined by RAPD markers for 28 tropical maize open pollinated varieties. Euphytica. 2001;121:197–208.
Lu Y, Yan J, Guimaraes CT, Taba S, Hao Z, Gao S, et al. Molecular characterization of global maize breeding germplasm based on genome-wide single nucleotide polymorphisms. Theor Appl Genet. 2009;120:93–115.
Parentoni SN, de Souza Jr CL, de Carvalho Alves VMC, Gama EEG, Coelho AM, de Oliveira AC, et al. Inheritance and breeding strategies for phosphorus efficiency in tropical maize (Zea mays L.). Maydica. 2010;55:1–15.
de Sousa SM, Clark RT, Mendes FF, de Oliveira AC, Vasconcelos MJV, Parentoni SN, et al. A role for root morphology and related candidate genes in P acquisition efficiency in maize. Func Plant Biol. 2012;39:925–35.
Gardiner JM, Coe EH, Melia-Hancock S, Hoisington DA, Chao S. Development of a core RFLP map in maize using an immortalized F2 population. Genetics. 1993;134:917–30.
Shiu SH, Bleecker AB. Receptor-like kinases from Arabidopsis form a monophyletic gene family related to animal receptor kinases. P Natl Acad Sci USA. 2001;98:10763–8.
Vij S, Giri J, Dansana PK, Kapoor S, Tyagi AK. The receptor-like cytoplasmic kinase (OsRLCK) gene family in rice : organization, phylogenetic relationship, and expression during development and stress. Mol Plant. 2008;1:732–50.
Tuberosa R, Sanguineti MC, Landi P, Giuliani MM, Salvi S, Conti S. Identification of QTLs for root characteristics in maize grown in hydroponics. Plant Mol Biol. 2002;48:697–712.
Mendes FF, Guimarães LJM, Souza JC, Guimarães PEO, Magalhaes JV, Garcia AAF, et al. Genetic architecture of phosphorus use efficiency in tropical maize cultivated in a low-P soil. Crop Sci. 2014;54:1–9.
Li M, Guo X, Zhang M, Wang X, Zhang G, Tian Y, et al. Mapping QTLs for grain yield and yield components under high and low phosphorus treatments in maize (Zea mays L.). Plant Sci. 2010;178:454–62.
Ruta N, Liedgens M, Fracheboud Y, Stamp P, Hund A. QTLs for the elongation of axile and lateral roots of maize in response to low water potential. Theor Appl Genet. 2010;120:621–31.
Hund A, Fracheboud Y, Soldati A, Frascaroli E, Salvi S, Stamp P. QTL controlling root and shoot traits of maize seedlings under cold stress. Theor Appl Genet. 2004;109:618–29.
Hund A, Reimer R, Messmer R. A consensus map of QTLs controlling the root length of maize. Plant Soil. 2011;344:143–58.
Trachsel S, Messmer R, Stamp P, Hund A. Mapping of QTLs for lateral and axile root growth of tropical maize. Theor Appl Genet. 2009;119:1413–24.
Burton AL, Johnson JM, Foerster JM, Hirsch CN, Buell CR, Hanlon MT, et al. QTL mapping and phenotypic variation for root architectural traits in maize (Zea mays L.). Theor Appl Genet. 2014;127:2293–311.
Cai H, Chu Q, Gu R, Yuan L, Liu J, Zhang X, et al. Identification of QTLs for plant height, ear height and grain yield in maize (Zea mays L.) in response to nitrogen and phosphorus supply. Plant Breed. 2012;131:502–10.
Zhang H, Uddin MS, Zou C, Xie C, Xu Y, Li W-X. Meta-analysis and candidate gene mining of low-phosphorus tolerance in maize. J Int Plant Biol. 2014;56:262–70.
Landi P, Sanguineti MC, Darrah LL, Giuhani MM, Salvi S, Conti S, et al. Detection of QTLs for vertical root pulling resistance in maize and overlap with QTLs for root traits in hydroponics and for grain yield under different water regimes. Maydica. 2002;47:233–43.
Landi P, Giuliani S, Salvi S, Ferri M, Tuberosa R, Sanguineti MC. Characterization of root-yield-1.06, a major constitutive QTL for root and agronomic traits in maize across water regimes. J Exp Bot. 2010;61:3553–62.
Liu J, Li J, Chen F, Zhang F, Ren T, Zhuang Z, et al. Mapping QTLs for root traits under different nitrate levels at the seedling stage in maize (Zea mays L.). Plant Soil. 2008;305:253–65.
Magalhaes JV, Liu J, Guimaraes CT, Lana UGP, Alves VMC, Wang YH, et al. A gene in the multidrug and toxic compound extrusion (MATE) family confers aluminum tolerance in sorghum. Nat Genet. 2007;39:1156–61.
Maron LG, Piñeros MA, Guimaraes CT, Magalhaes JV, Pleiman JK, Mao C, et al. Two functionally distinct members of the MATE (multi-drug and toxic compound extrusion) family of transporters potentially underlie two major aluminum tolerance QTLs in maize. Plant J. 2010;61:728–40.
Yokosho K, Yamaji N, Ma JF. An Al-inducible MATE gene is involved in external detoxification of Al in rice. Plant J. 2011;68:1061–9.
Magnavaca R, Gardner CO, Clark RB. Evaluation of maize inbred lines for aluminum tolerance in nutrient solution. In: Gabelman HW, Longhman BC, editors. Genetic aspects of plant mineral nutrition. Dordrecht: Martinus Nijhoff Publishers; 1987. p. 255–65.
da Silva FC. Manual de análises químicas de solos, plantas e fertilizantes. Brasília: Embrapa Informação Tecnológica; 2009.
Cruz CD. GENES—a software package for analysis in experimental statistics and quantitative genetics. Acta Sci. 2013;35:271–6.
Wright S. Correlation and causation. J Agric Res. 1921;20:557–85.
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW. Ribosomal DNA spacer length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. P Natl Acad Sci USA. 1984;81:8014–8.
Ninamango-Cárdenas FE, Guimaraes CT, Martins PR, Parentoni SN, Carneiro NP, Lopes MA, et al. Mapping QTLs for aluminum tolerance in maize. Euphytica. 2003;130:223–32.
Lander ES, Green P, Abrahamon J, Barlow A, Daly M, Lincoln SE, et al. MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics. 1987;1:174–81.
Kosambi DD. The estimation of map distances from recombination values. Ann Eugenic. 1944;12:172–5.
Pinheiro J, Bates D, Debrov S. R-CORE-TEAM: nlme: Linear and nonlinear mixed effects models. 2014. http://cran.r-project.org/package=nlme.
Kao C-H, Zeng Z-B. Modeling epistasis of quantitative trait loci using Cockerham’s model. Genetics. 2002;160:1243–61.
Haley CS, Knott SA. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity. 1992;69:315–24.
Zou F, Fine JP, Hu J, Lin DY. An efficient resampling method for assessing genome-wide statistical significance in mapping quantitative trait loci. Genetics. 2004;168:2307–16.
Lynch M, Walsh B. Genetics and analysis of quantitative traits. Sunderland: Sinauer Associates; 1998.
Zeng ZB, Kao CH, Basten C. Estimating the genetic architecture of quantitative traits. Genet Res. 1999;74:279–89.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–82.
Bi D, Cheng YT, Li X, Zhang Y. Activation of plant immune responses by a gain-of-function mutation in an atypical receptor-like kinase. Plant Physiol. 2010;153:1771–9.
Wang X, Zafian P, Choudhary M, Lawton M. The PR5K receptor protein kinase from Arabidopsis thaliana is structurally related to a family of plant defense proteins. P Natl Acad Sci USA. 1996;93:2598–602.
Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci. 1992;8:275–82.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9.
Creste S, Neto AT, Figueira A. Detection of single sequence repeat polymorphisms in denaturing polyacrylamide sequencing gels by silver staining. Plant Mol Biol Rep. 2001;19:299–306.
Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008;3:1101–8.
Acknowledgments
The authors would like to acknowledge funding from the Generation Challenge Programme (project G7010.03.01), the Embrapa Macroprogram, FAPEMIG and CNPq/Embrapa REPENSA. PhD fellowship for GCA was granted by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES/Embrapa) and BH was supported by Fundação de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG). JVM and AAFG received fellowships from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). We are thankful to Dr. Sigrid Heuer (ACPFG, Australia) for sharing information about the rice PSTOL1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived and designed experiments: CTG, SMS, JVM. Performed experiments: GCA, BFN, BH, UGPL. Analyzed the data: GCA, ACG, LCS, AAFG. Wrote and revised the paper: GCA, ACG, LCS, JVM, AAFG, SMS, CTG. All authors read and approved the final manuscript.
Additional files
Additional file 1: Table S1.
Trait means, coefficients of variation (CV), genetic variances (σ 2 G ), environmental variances (σ 2 E ) and heritability estimates (h 2) in 145 RILs derived from a cross between maize lines L3 and L22.
Additional file 2: Figure S1.
Genetic linkage map including 292 markers in a maize RIL population derived from a cross between L3 and L22. For each chromosome (chr), marker names are indicated on the right and the genetic distance in centimorgans (cM) is on the left. The colored bars indicate the position of QTLs for root traits, total plant dry weight, root:shoot ratio and P content (for details see Fig. 3, Tables 3 and 4).
Additional file 3: Figure S2.
Alignment of the OsPSTOL1 and six maize serine/theronine receptor-like kinases highlighting the predicted domains. Letters in green indicate the glycosil hydrolase domain and in red, the transmembrane domain. The kinase domain is represented by a gray background, whereas the ATP-binding site is highlighted with black background with white letters and the Serine/Threonine protein kinase active site with yellow background.
Additional file 4: Table S2.
Total P content in the seedling and relative expression of ZmPSTOL genes in shoots and roots of both parental lines in low- and high-P concentration.
Additional file 5: Table S3.
Primers used for gene expression analyses.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Azevedo, G.C., Cheavegatti-Gianotto, A., Negri, B.F. et al. Multiple interval QTL mapping and searching for PSTOL1 homologs associated with root morphology, biomass accumulation and phosphorus content in maize seedlings under low-P. BMC Plant Biol 15, 172 (2015). https://doi.org/10.1186/s12870-015-0561-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-015-0561-y