
Abstract
Background
Daqu, the saccharification, fermentation, and aroma-producing agents for Baijiu brewing, is prepared using a complex process. Aging is important for improving the quality of Daqu, but its impact has rarely been studied. This study investigated changes in the physicochemical properties, flavor compounds, and microbial communities during aging of Daqu with a roasted sesame-like flavor.
Results
The physicochemical properties changed continuously during aging to provide a high esterifying activity. Aging removed unpleasant flavor compounds and helped to stabilize the flavor compounds in mature Daqu. A high-throughput sequencing approach was used to analyze the changing composition of the microbial communities during aging. Aging helped to modify the microbial population to produce better Baijiu by eliminating low-abundance microbial communities and optimizing the proportion of predominant microbial communities. Nine genera of prokaryotic microbes formed the core microbiota in Daqu after aging. Regarding eukaryotic microbes, Zygomycota, the predominant community, increased in the first 2 months, then decreased in the third month of aging, while Ascomycota, the subdominant community, showed the opposite behavior. Absidia, Trichocomaceae_norank and Rhizopus were the predominant genera in the mature Daqu.
Conclusions
Significant correlations between microbiota and physicochemical properties or flavor compounds were observed, indicating that optimizing microbial communities is essential for aging Daqu. This study provides detailed information on aging during Daqu preparation.
Similar content being viewed by others


Background
Baijiu, one of the most famous traditional distilled spirits in the world, is produced by the spontaneous solid-state fermentation of a sorghum, wheat, and/or rice mixed-culture, a process which mainly involves Daqu manufacture and grain fermentation [1, 2]. Daqu, the saccharifying and fermenting agent used for brewing Baijiu, is critical for obtaining the final flavor characteristics of Baijiu [3]. Daqu acts not only as a resource for the brewing process, but also provides important crude enzymes, complex microbial flora, and even aroma precursors that will determine the flavor characteristics of the final liquor product [4]. Therefore, the contribution of Daqu to Baijiu manufacture has been summarized by brewers in the expression “better Daqu, better Baijiu” [5].
Daqu is known as the heart of Baijiu owing to its importance in the process. Similar to Baijiu, Daqu is made from wheat, barley, and/or peas and formulated through a complex solid-state fermentation in an open environment. The process includes preparation of the ingredients, grinding, mixing, shaping, incubation, spontaneous solid fermentation for 1 month, and a long aging period (drying and ripening) to allow the Daqu to reach maturity (Fig. 1) [6]. Each of these stages has a significant impact on Daqu quality. Owing to differences in these stages and production environments, different types of Daqu with different characteristics are produced, such as Jiang-, Nong-, Qing-, and roasted sesame-like flavor Daqu, characterized by flavor, or low-temperature, medium-temperature, and high-temperature Daqu, characterized by the maximum incubation temperature [7,8,9]. Each type of Daqu has a unique combination of microbiota dynamics, functional enzymes, and special flavor that are essential in Baijiu production as saccharification, fermentation, and aroma-producing agents [2, 9, 10]. For example, Bacillus species are predominant in Nong- and Jiang-like flavor Daqu, while lactic acid bacteria (LAB) are dominant in all types of Daqu [10]. Furthermore, Thermomyces lanuginosus is dominant in Jiang-like flavor Daqu, while Saccharomycopsis fibuligera and Lichtheimia ramose occur in Qing- and Nong-like flavor Daqu [11]. In other words, no single genus has been found in all types of Daqu. For example, Aspergillus, Gilmaniella, Monascus, Penicillum, Mucor, Trichoderma, Rhizomucor, Absidia and Rhizopus are common fungi in Daqu, but are not present in all types of Daqu, while each type of Daqu does not contain all the above fungi [9]. The differences in microbial communities between different types of Daqu are related to the environmental conditions and production processes [2, 7].

Process flow diagram for the production of Daqu and the diagrammatic drawing of sampling for Daqu samples
Aging, the last step in the process of Daqu preparation, seems to be simple procedure, in which fermented Daqu is placed somewhere open, cool, well-ventilated, and dry for a given period of time. In fact, from thousands of years of Daqu production experience, aging is known to plays a particularly important role in achieving high-quality Daqu. After fermentation, Daqu is usually stored for at least 2 months before being used to make Baijiu [12]. During this time, according to experienced production workers, Daqu is “purified” by “removing discordant” substances, and its “fragrance increases” [13]. Only Daqu that has been aged for a reasonable period can produce high-quality Baijiu. With recent developments in analytical technologies, such as high-throughput sequencing (HTS) and headspace solid-phase microextraction–gas chromatography–mass spectrometry (HS-SPME-GC-MS), researchers have discovered that the aging process when making Daqu is not simply affected by storage time, but also by a very complicated process in which the microbial flora constantly change with changes in the external environment, such as moisture and temperature. The microorganisms in Daqu after fermentation also remain active, causing changes in its enzyme and metabolite contents. Therefore, aging can encourage the enriched microbes to interact further, such that the mixed microbial communities achieve a balance that helps to stabilize the quality of the final Baijiu product [14]. Aging is critical for making Daqu, as it affects its quality, which, in turn, influences the quality of Baijiu. Accordingly, only well-aged Daqu can be used to make Baijiu, making the end point of Daqu storage an important control point in Daqu preparation.
Presently, decisions regarding whether the quality of Daqu is sufficient for use in Baijiu production depends mainly on sensory evaluation according to accumulated production experience and partly on its physicochemical properties, which can be subjective and uncertain. This leads to an unstable level of quality in the Baijiu produced. With the increase in Baijiu production and a reduction in the number of workers engaged in its production, Daqu manufacture has evolved from the original simple artisanal scale into a large-scale industrial process to keep up with demand [15]. During this transformation process, it is necessary to clearly understand the internal mechanisms during each stage of Daqu production. However, the effects of aging during Daqu manufacture have yet to be explained scientifically, which is a problem that needs to be solved to promote a successful transformation from the artisanal to large-scale production of Daqu, as well as its effect on Baijiu production.
The process of Daqu preparation involves collecting functional microbes suitable for Baijiu production, such that the composition of the microbial communities and their abundance can undergo complex changes during the entire production process. Changes in microbial activity also cause changes in the physicochemical properties (especially enzymatic activity) and flavor substances of Daqu. Therefore, fully understanding the changing role of the microbial community, physicochemical properties, and flavor substances might provide a more comprehensive scientific explanation of the necessity of each step during the Daqu production process. The process of aging is important for microbial growth and the flavor compounds produced in Daqu. However, few studies have analyzed the dynamics of the microbiota, flavor compounds, and physicochemical properties of Daqu at this stage, with no scientific methods available for determining whether Daqu was mature. Therefore, analyzing these dynamics can provide a better understanding of the contribution of aging to the quality of Daqu, especially in providing a scientific basis for judging when Daqu reaches maturity during storage.
Therefore, this study used roasted sesame-like flavor (RSF) Daqu as the specific research object to investigate changes in its microbial communities and the content of flavor substances during aging, using HTS and HP-SPME-GC-MS, respectively. The physicochemical properties will be investigated to determine the potential role of the microorganisms in the Daqu. To our knowledge, this is the first comprehensive study to report the effects of aging during RSF Daqu manufacture on its physicochemical properties, flavor substances, and microbial flora.
Results and discussion
Physicochemical properties
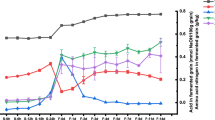
As shown in Table 1, all physicochemical parameters of Daqu changed during aging, except for fermenting activity. During aging, the moisture content of Daqu decreased as aging proceeded. This was caused by the Daqu being stored in a relatively open well-ventilated environment with dry, cool air (Fig. 1). The contents of protein and starch in the Daqu tended to increase during storage. Two possible explanations for this were that moisture in the Daqu decreased significantly during aging, which increased the contents in terms of concentration, or that enrichment of the microbial communities and changes in some microbes were partly responsible [14]. For example, in the subsequent analysis of microbial strains, the numbers of Cyanobacteria increased with storage time, which might lead to the increase in protein and starch contents, because this genus synthesized starch and protein through photosynthesis [16]. Owing to the microbial activity, especially in acid-producing microorganisms such as Acetobacter and Lactobacillus, the acidity and pH fluctuated during aging (Table 1), After 2–3 months, the acidity of the Daqu became more stable. Presently, acidity is an important criterion used to judge whether Daqu is mature, and is the only objective standard used during the current Daqu preparation process [17]. After aging for 3 months, the saccharifying and liquefying activities had reduced significantly from the initial values, while the esterifying activity had increased. This was probably caused by changes in the microbial flora during aging. Some microorganisms that produce lipase and other enzymes related to ester synthesis increased, while others mainly producing amylase and glucoamylase decreased. These differences in enzyme production might have led to the changes in temperature during the process of brewing high quality Baijiu, which conforms to the principle of “slow rise in the early stage, rapid rise in the middle, and slow fall in the later stage”, as summarized in the Baijiu brewing process [18]. Interestingly, the fermenting activity did not change significantly during aging, mainly due to the balance of interactions between the various microorganisms.
Flavor compounds in Daqu
In the present study, a total of 34 volatile compounds, namely, six alcohols, seven esters, six aldehydes, three benzodiazepines, one nitrogen-containing compound, five ketones, one phenol, four alkanes, and one alkene, were identified in Daqu using HS-SPME-GC-MS (Additional file 1: Table S1). In general, the relative abundance of the volatile compounds changed during aging because of the changes in microbial flora and their activity [19]. Overall, the alcohols, aldehydes, nitrogen-containing compounds, and alkenes increased slightly in concentration, while the concentrations of benzodiazepines, ketones, phenols, and alkanes decreased. However, the concentrations of the esters remained relatively unchanged. Similar to previous results, the concentrations of various flavor compounds had changed, but they all tended to be stable during aging [14, 20]. This might provide a scientific basis for determining the appropriate aging treatment of Daqu through sensory evaluation [17]. Regarding a single important group of flavor substances, the content of phenethyl alcohol with its rose scent, 1-octen-3-ol with its roasted mushroom aroma, and some biologically active compounds, such as ledol, tetramethylpyrazine, and caryophyllene, had increased during aging. Therefore, the aging stage of making Daqu has played an essential role in removing discordant flavors from Daqu and forming the unique full-bodied “Daqu-flavor” [20].
Overall structure and diversity of prokaryotic communities
For prokaryotic microbes, 587,885 valid sequencing reads with an average read length of 452 bp were obtained from all Daqu samples (Table 2). Rarefaction analysis indicated that all prokaryotic communities were well-represented at the sequencing depth, as the rarefaction curves approached a clear-cut asymptotic plateau. A total of 40 phyla and 733 genera were obtained based on 97% similarity as a cutoff in the 16S rRNA sequences. The richness and evenness of prokaryotic communities in Daqu were evaluated using α-diversity metrics, including the Chao 1, ACE, Shannon and Simpson indices. The richness estimators (Chao 1 and ACE) were consistent with the Shannon indices (Table 2). All indices showed that the microbial richness of Daqu decreased during the first month of aging, increased after 2 months, and then stabilized until the end of aging, indicating that aging-time-related changes had occurred at the OTU level. Based on the relative abundances of OTUs and taxonomic ranks from genus to OTU level, the prokaryotic communities in the four Daqu samples formed three clusters, namely, group I, sample A0; group II, sample A1; and group III, samples A2 and A3 (Fig. 2). Similarly, UniFrac clustered analysis and PCoA analysis showed that the prokaryotic communities could be clustered into three groups (Additional file 2: Figs. S1 and S2). These results showed that the differences between Daqu samples decreased during aging.

Taxonomic classification of sequences from prokaryotic communities of four samples (A0, at the beginning of aging; A1, after 1 month of aging; A2, after 2 months of aging; A3, after 3 months of aging) at genus level. The relative abundance was calculated by dividing the number of classified tags by the total tags number of each sample. The top 50 genera detected in RSF Daqu are shown. The relative abundance of each genus was indicated by color intensity in a heat map
The taxonomic distribution at the phylum level is shown in Fig. 3a. More than 95% of the total abundances in each Daqu sample was precisely assigned to nine known bacterial phyla, namely, Firmicutes, Proteobacteria, Cyanobacteria, Bacteroidetes, Actinobacteria, Acidobacteria, Verrucomicrobia, Chloroflexi, and Gemmatimonadetes (Fig. 3a). Overall, during the Daqu aging process, the bacterial structure became optimized and the bacterial flora gradually stabilized. Specifically, Firmicutes, the predominant phylum accounting for 43.44% of the total prokaryotic reads before aging, increased to 60.73% after 1 month, then became stable at around 60% as aging progressed. Meanwhile, the proportion of reads for the subdominant phylum, Proteobacteria, fell slightly from 24.53 to 19.38%, before stabilizing. Bacteroidetes, the third most dominant phylum, accounted for 11.71% of total prokaryotic sequences at the beginning of aging, which decreased sharply as aging proceeded, accounting for only 0.01% after 3 months of aging. Other phyla, such as Actinobacteria, Acidobacteria, Verrucomicrobia, Chloroflexi, Gemmatimonadetes, Planctomycetes, Fusobacteria, Saccharibacteria, Parcubacteria, Nitrospirae, Latescibacteria, and Euryarchaeota, also decreased clearly. However, the increase in Cyanobacteria from 0.50% at the beginning of aging to 19.45% after 3 months of aging was more obvious, becoming the third most dominant phylum by the end of aging. This might explain why the starch content and protein content increased during aging. During aging, many low-abundance phyla were gradually eliminated, which could further increase the abundance of the dominant bacteria (Fig. 3). In short, aging is a process for further rebalancing the microorganisms in Daqu.

Analysis of prokaryotic communities composition in four samples (A0, at the beginning of aging; A1, after 1 month of aging; A2, after 2 months of aging; A3, after 3 months of aging) using high-throughput sequencing. Results of taxonomy at the phylum level and genus level are shown in (a) and (b), respectively. The relative abundance defines sequence percentages in samples as depicted by the colors in the bar chart. The top 20 phyla and genera detected in RSF Daqu are shown in (a) and (b), respectively. The rest of phyla and genera detected in all samples were classified as others
In general, the structure of bacteria in Daqu at the genus level at the beginning of aging was quite different to that after aging, but tended to be consistent with different aging times (Fig. 3b). At the beginning of aging, Unclassified (7.60%), Thermoactinomyces (7.57%), Bacteroides (6.84%), and Faecalibacterium (5.66%) were regarded as the predominant genera, with a relative abundance greater than 5%. Subdoligranulum (3.93%), Lactobacillus (3.61%), Pseudobutyrivibrio (2.43%), Bifidobacterium (1.89%), Lachnospiraceae_norank (1.81%), Escherichia-Shigella (1.60%), Akkermansia (1.37%), Streptococcus (1.36%), Megamonas (1.31%), Sphingomonas (1.29%), Acinetobacter (1.27%), and Lachnoclostridium (1.13%) were the subdominant genera (1% < subdominant < 5%) (Fig. 3b). As aging proceeded, the proportion of all above genera, except Lactobacillus, tended to decrease. After aging for a month, Weissella, Mitochondria_norank, Lactobacillus, and Staphylococcus increased to become the predominant genera, while Pediococcus increased, and Unclassified and Thermoactinomyces decreased, to become the subdominant genera. Compared with Daqu aged for 1 month, Kroppenstedtia, Acetobacter, and Enterobacteriaceae_norank increased to become the subdominant genera, while Thermoactinomyces decreased to become a subdominant genus at the end of aging. The proportions of the other genera decreased with aging time. Therefore, during aging, the microbial flora of Daqu were decontaminated by removing unwanted genera such as Bdellovibrio, and the proportion of functional microbes, which have an important role to play in the quality of Baijiu, was optimized. This might explain why the flavor compounds in Daqu were purified during aging.
Only properly aged Daqu can be used in Baijiu brewing, meaning that the composition of microorganisms in aged Daqu plays an important role in Baijiu manufacture. The core microbiota, comprising the dominant microbial genera, are generally considered to play the most important role in the process of Baijiu manufacture. In the present study, we identified nine genera of prokaryotic microbes, including Weissella, Mitochondria_norank, Staphylococcus, Lactobacillus, Kroppenstedtia, Pediococcus, Unclassified, Acetobacter, and Enterobacteriaceae_norank as the core microbiota in mature Daqu. Among these, three were LAB genera (Weissella, Lactobacillus and Pediococcus), meaning that LAB were one of the dominant bacterial genera in Daqu, consistent with previous studies [21]. LABs play a significant role in improving Baijiu quality, not only transform glucose or starch into lactic acid and keeping Daqu acidic, but also greatly contributing to providing substrates for the esterification of yeasts forming ester compounds [21, 22]. This may be important for explaining the increase in the esterifying activity of Daqu during aging. A previous study has shown that the contents of important esters in Baijiu are positively correlated with the numbers of LAB, indicating that a lag in fermentation and a lower ester content during Baijiu production could be caused by a lack of LAB [1]. Previous studies have shown that Staphylococcus is dominated in Daqu, with the potential to form amino acids, biogenic amines, aldehydes, free fatty acids, and esters [3, 23]. The number of Staphylococcus bacteria was correlated with the biosynthesis of the main organic acids in Baijiu [24]. Therefore, Staphylococcus has an important regulatory effect on the synthesis of four main esters (ethyl acetate, ethyl lactate, ethyl butyrate, and ethyl caproate), which greatly affects the quality of Baijiu. Few studies have focused on Kroppenstedtia, belonging to the family Thermoactinomycetaceae, which was isolated recently. Kroppenstedtia has been identified in several types of Daqu, namely, Baiyunbian high-temperature Daqu, light-flavor Daqu, and RSF Daqu, using high throughput sequencing [24,25,26,27]. Although its role in Baijiu manufacture is not clear, it is abundant at the beginning of fermentation but then decreased immediately, giving way to Lactobacillus [25]. Acetobacter, the prevalent bacteria during Baijiu production, greatly influences the flavor and taste of the final product, because it not only produces acetic acid, but also affords acetoin, a biosynthetic in Baijiu liquor [28]. Acetobacter can also select most of the acid-tolerant microbes owing to the high concentrations of acetic acid it produces [3]. In other words, Acetobacter can affect the microbial flora in the fermentation process of Baijiu, which will eventually affect the quality of the final product.
Overall structure and diversity of eukaryotic communities
After removing the low quality chimera reads and trimming of the PCR primers, a total of 950,891 high-quality reads, with an average read length of 442 bp, corresponding to 18S rDNA gene sequences, was obtained (Table 2). The rarefaction curves reached the saturation plateau, indicating that the eukaryotic communities were well captured in the current analysis. In contrast to the prokaryotic community, the diversity of the eukaryotic microbes was smaller. Unlike the prokaryotic community, the eukaryotic Chao 1 estimator, ACE, and observed OTUs first increased, then fluctuated during aging, indicating that the succession of the eukaryotic communities was different from that of the prokaryotic communities during the whole aging process. The microbial richness (indicated by Chao 1 and ACE) of the eukaryotic community was highest in samples A1 and A3, followed by A2, with the lowest richness in sample A0. The microbial diversity (Shannon) of A0 was significantly higher than that of A2, while those of A1 and A3 had similar intermediate values (Table 2). PCoA analysis and heatmap results showed that sample A2 formed a cluster, and samples A0, A1 and A3 another large cluster with A1 and A3 being clustered together (Additional file 2: Figs. S3 and S4).
At the phylum level, the predominant eukaryotic communities were Zygomycota and Ascomycota during the Daqu aging, which was consistent with previous reports [26]. In general, these two phyla constituted more than 99.0% of the total abundance during the entire aging process (Fig. 4a). However, their changes during aging showed opposite trends. In detail, Zygomycota was increased during the first two months of aging, and then decreased in the third month, while the pattern for Ascomycota was the opposite. These findings were similar to those in a previous report, where the abundance of Ascomycota and Zygomycota gradually approached each other at the end of aging, with some differences [14]. The same study showed that the population of Ascomycota was slightly higher than that of Zygomycota, perhaps due to the different manufacturing environments for these two Daqus, leading to a Baijiu that was different in style [14]. The eukaryotic community succession was investigated according to OTUs classified at the genus level (Fig. 4b). The results showed that Absidia and Unclassified were the dominant genera in all Daqu samples, with Trichocomaceae_norank and Rhizopus forming the subdominant genera. As aging proceeded, Absidia and Rhizopus initially increased for up to 2 months, and then declined at the end of aging, while Trichocomaceae_norank showed the opposite trend, becoming the predominant genus in mature Daqu. Interestingly, Thermomucor, which has only been reported in Daqu by Li and Qiu, was a subdominant genus at the beginning of aging, but had disappeared after 1 month [29]. This might have been due to the interaction between microbes in Daqu and the environment during aging. During aging, Syncephalastrum might be present in the environment because it was not present in Daqu at the beginning of aging and became almost a subdominant genus after aging. Therefore, environmental microbiota influenced the microbial succession during aging, meaning that they also played an important role in the maturation of Daqu during aging [25]. After aging, some rare genera, such as Rhizomucor and Saccharomyces, were observed.

Analysis of eukaryotic communities composition in four samples (A0, at the beginning of aging; A1, after 1 month of aging; A2, after 2 months of aging; A3, after 3 months of aging) using high-throughput sequencing. Results of taxonomy at the phylum level and genus level were shown in (a) and (b), respectively. The relative abundance defines sequence percentages in samples as depicted by colors in the bar chart. The top 20 phyla and genera detected in RSF Daqu are shown in (a) and (b), respectively. The rest of phyla and genera detected in all samples were classified as others
Molds and yeasts are mainly responsible for saccharification and alcoholic fermentation in the production of Baijiu. In the present study, four mold and one yeast genera were identified in Daqu, namely, Absidia, Rhizopus, Syncephalastrum, Rhizomucor, and Saccharomyces. Absidia and Rhizopus were the most frequent and important fungi found in Daqu [6]. Absidia, a main amylase producer, has a strong saccharification capability and contributes significantly to Baijiu fermentation [30]. Rhizopus also secretes large amounts of various hydrolyzing enzymes that decompose macromolecules and produce smaller metabolites and volatile compounds, such as ethanol, 2-methyl-1-butanol, and 3-methyl-1-butanol, which affect the flavor of Baijiu [6, 30, 31]. Syncephalastrum, first detected in Daqu, has been shown to secrete enzymes, such as amylase, glucosidase and pullulanase, that degrade starch [32]. Rhizomucor is also considered to be a functional microbe, with a high ability to produce saccharifying amylase [3, 9]. Therefore, Absidia, Rhizopus, Syncephalastrum, and Rhizomucor all contribute to saccharifying and liquefying activities in Daqu although their activity decreased as aging progressed [3, 6]. This might be due to several factors, such as: The activity of enzymes that degrade starch into sugar is different in these genera; the fluctuation in the number of these microorganisms is inconsistent during the whole aging process; and, in addition to these genera, there are other microorganisms, such as bacteria and yeasts, which exhibit saccharifying and liquefying activities. Saccharomyces is the most abundant yeast and the most efficient ethanol producer during the Baijiu fermentation stage, with the Baijiu produced being associated with high scores for harmony, and the highest values of floral aroma compared with other yeasts from sensory evaluation [33, 34]. Although the numbers of Saccharomyces decreased during aging, there were no significant changes in fermentation capacity in the Daqu samples. This might have been due to the balance between the abundance of Saccharomyces and other yeasts, belonging to Saccharomycetaceae, Saccharomycopsidaceae, or Saccharomycetales, which have a certain ability to produce ethanol.
Relationships between the physicochemical properties and microbial communities of Daqu
Microbial communities generally have significant effects on some physicochemical properties. In the present study, RDA and CCA were performed based on the prokaryotic and eukaryotic communities, and the physicochemical properties (Fig. 5). Overall, the two axes explained 97.17 and 86.39% of the total variance in bacterial (Fig. 5a, DCA = 4.10) and fungal (Fig. 5b, DCA = 1.15) community differentiation, respectively, suggesting remarkable correlations between the microbial communities and the physicochemical properties. Figure 5a shows that Pseudobutyrivibrio, Bifidobacterium, Subdoligranulum, Faecalibacterium, Bacteroides, Akkermansia, Streptococcus, Escherichia-Shigella, and Thermoactinomyces were positively correlated with sample A0, which was consistent with previous results (Fig. 3). These genera were positively correlated with the saccharifying and liquefying activities (Fig. 5a). In previous reports, most of these microorganisms can produce glucosidase and amylase [35,36,37,38,39,40,41,42,43]. Remarkable positive correlations were also determined between Kroppenstedtia, Acetobacter, and Staphylococcus and samples A2 and A3. These genera contributed to the high esterifying activity of both samples, possibly because they can yield lipase or esterase (Fig. 5a) [44, 45]. Another possible explanation was that these bacteria can produce organic acids, which are transformed into esters by lipase or esterase produced by other microbes or esterification with ethanol [24, 28]. The RDA results showed a significant positive correlation between Thermomucor, Rhizomucor, Saccharomycopsis, Unidentified_Eugregarinorida, and Rhizopus and the saccharifying and liquefying activities, which further confirmed that the above strains were the main producers of amylase or glucosidase [3, 6, 31]. RDA also revealed that Rasamsonia, Pichia, Syncephalastrum, Absidia, and Lichtheimia were positively correlated with esterifying activity. Some studies have found that these genera can produce lipase or esterase, causing the high esterifying activity [46,47,48,49].

Canonical correspondence analysis (CCA) of prokaryotic communities at genus level and physicochemical properties (a), and redundancy discriminate analysis (RDA) of eukaryotic communities at genus level and physicochemical properties (b) found in Daqu. Arrows represent different physicochemical properties; the symbols ■, ●, ▲, and ◆ represent samples A0 (at the beginning of aging), A1 (after 1 month of aging), A2 (after 2 months of aging), and A3 (after 3 months of aging), respectively. The tow axes explain 97.17 and 86.39% of the total variance in bacterial (DCA = 4.10) and fungal (DCA = 1.15) community differentiation, respectively
Correlations between microbial communities and flavor compounds
The heatmap revealed the correlations between the microbial communities and flavor compounds (Fig. 6). Ledol, a lipophilic aromatic compound, was positively correlated with Staphylococcus, Absidia, Lichtheimia, and Syncephalastrum. However, there have been no reports of these genera synthesizing or metabolizing the corresponding flavor substances. No report for p-toluic acid 2-ethylhexyl ester, and the analysis showed that this compound was positively correlated with eukaryotes, such as Saccharomycopsis, unidentified_Eugregarinorida, Wickerhamomyces, Meyerozyma, Saccharomyces, and Hyphopichia. In contrast, phthalic acid hept-4-yl isobutyl ester, a main compound in the flowers of musk melon (Cucumis melo L.) and roasted peanuts, was positively correlated with prokaryotes, such as Weissella, Mitochondria_norank, Lactobacillus, and Pediococcus [50, 51]. Higher fatty acid esters, such as 9,12-octadecadienoic acid ethyl ester, are colloidal substances that play a vital role in the stability and taste of Baijiu [52]. Figure 6 shows that Enterobacteriaceae_norank, and Kroppenstedtia were highly positively correlated with 9,12-octadecadienoic acid ethyl ester. However, there are no previous studies on this correlation and possible metabolism, which require further verification and study. Hexanal, associated with pleasant descriptors, such as herbal, fruity and flowery, was positively correlated with Staphylococcus, Absidia, Lichtheimia, and Syncephalastrum, which, except for Lichtheimia, were confirmed by previous results [53,54,55]. A previous report has shown that hexanal was oxidized from hexylamine by amine oxidases produced by the above genera [55]. Benzaldehyde was also positively correlated with Staphylococcus, Absidia, Lichtheimia and Syncephalastrum. However, except for the relationship between benzaldehyde and Staphylococcus, the relationships among others have not been reported [56]. Statistical analysis performed in the present study indicated a positive correlation between (E)-2-octenal/nonanal and Faecalibacterium, Lachnospiraceae_norank, Rhizopus, Gregarina, and Mucor. To our knowledge, only the relationship of (E)-2-octenal with Rhizopus and Mucor has been confirmed in previous studies, with the 2-alkenal reductase produced playing an important role in the synthesis of (E)-2-octenal [31, 57]. Faecalibacterium, Lachnospiraceae_norank, Rhizopus, Gregarina, and Mucor had an important influence on the compounds 2,2,4-trimethyl-1,3-pentanediol, 2-methyl-naphthalene, 1,7-dimethyl-naphthalene, 2-undecanone, and tetradecane. Their correlations and possible metabolism require further verification and study because no relevant studies have yet been reported. Pythium, Heterococcus, Luticola, and Spumella had positive loadings on 4-ethenyl-1,2-dimethoxy-benzene and (2-dodecen-1-yl) succinic anhydride from the heatmap. Alkanes pentadecane and hexadecane were positively correlated with unidentified_Mucorales, while 3-methyl-tetradecane was positively correlated with Saccharomycopsis, unidentified_Eugregarinorida, Wickerhamomyces, Meyerozyma, Saccharomyces, and Hyphopichia (Fig. 6). No studies have described these relationships previously.

Correlation between volatile compounds (V1-V34: different flavor compounds in Additional file 1 Table S1) and prokaryotic communities (a) and eukaryotic communities (b) found in Daqu. Scale bar colors denote the nature of the correlation, with 1 indicating a perfectly positive correlation (read) and − 1 indicating a perfectly negative correlation (blue) between them. “*” shows significant correlations (P < 0.05), and “NA” shows no correlations
Conclusions
This study has comprehensively demonstrated the changes in the physicochemical properties, flavor components, and microbial communities of Daqu during aging. The physicochemical properties of Daqu changed significantly during aging. The higher liquefying and saccharifying activities of Daqu at the beginning of aging, changed to a high esterifying activity, and moderate liquefying and saccharifying activities, at the end of aging. The aging process improved both the ester content and fragrance of Daqu. These changes were undoubtedly caused by changes in the microbial communities in Daqu, which were attributed to the external environment and interaction with external microorganisms during aging. Overall, the results showed that physicochemical properties, such as acidity, and sensory factors, such as flavor components and appearance, can provide a scientific basis to determine whether Daqu is mature. However, the microbial community is essential for understanding the aging process when making Daqu. This study has provided a deeper theoretical basis to explain the need for aging when making Daqu, and the means to design effective strategies to manipulate the preparation of Daqu to better control Baijiu production systems and to improve the quality of the final product.
Methods
Sampling
Four samples of Daqu from the same production set were taken at different times until it was judged to be mature by experienced workers: A0, at the beginning of aging, sampled on May 8, 2017 (non-aging Daqu); A1, after 1 month of aging, sampled on June 8, 2017; A2, after 2 months of aging, sampled on July 8, 2017; A3, after 3 months of aging, sampled on August 8, 2017 (end aging Daqu). The samples were then collected from the liquor (RSF) at Shandong Bandaojing Co. Ltd. (Shandong, China). The bricks of Daqu were freshly produced from wheat grains during spring 2017. To obtain reliable samples for analysis, each sample was randomly selected from upper, middle, lower and inside locations in the storage room (Fig. 1), then promptly ground to a powder, mixed well to form one sample, and collected by quartering [58]. The samples were transferred to sterile bags, sealed, and then stored at − 80 °C for further analysis.
Analysis of physicochemical properties
The physicochemical properties of the Daqu samples, namely, moisture, protein, and starch contents, acidity, pH, amino nitrogen (AN), reducing sugar (RS), and total ester (TE) levels, and saccharifying, liquefying, esterifying, and fermenting activities, were measured three times, as described in a previous report [14].
Analysis of volatile compounds
The contents of volatile compounds in the Daqu samples were determined using HS-SPME-GC-MS (TSQ 8000 Evo, Trace MS/GC; Thermo Fisher Scientific, Waltham, MA, USA) as previously described by Fan et al. [59]. Individual volatile compounds were identified by comparison with data from the NIST 05a spectra library (Gaithersburg, MD, USA), then quantified according to the amount of internal standard (2-octanol, Sigma-Aldrich Chemical Co., USA) added to the Daqu sample before extraction and expressed as mg per kg of Daqu. The analyses were performed in triplicate.
DNA extraction and quantitation
Daqu samples were prepared using a method described in a previous study [10]. A commercial PowerSoil DNA Isolation Kit (Mo Bio Laboratories Inc., Carlsbad, CA, USA) was used to extract the genomic DNA. After being quantified using spectrophotometry (ratio of optical densities at 260 and 280 nm) and electrophoresis in 0.6% (w/v) agarose gels, the genomic DNA was adjusted to equal concentrations for use as a template for PCR amplification.
PCR amplification and Illumina MiSeq sequencing
The V3-V4 regions on the 16S ribosomal RNA gene of the bacteria were amplified with forward primers 340F (5′-barcode-CCTACGGGNBGCASCAG-3′) and reverse primers 805R (5′-GACTACNVGGGTATCTAATCC-3′). The 18S ribosomal RNA gene of the fungi was amplified using forward primer 512F (5′-TATTCCAGCTCCAATAGCG-3′) and reverse primer 978R (5′-barcode-GACTACGATGGTATCTAATC-3′). The primer was modified by adding an error-correcting barcode, which was unique to each sample and served as a multiplexing marker. The PCR reactions and extraction of amplicons were performed as described in a previous report [59]. After quantification using the QuantiFluor-ST system (Promega Corp., Fitchburg, WI, USA), the purified amplicons were pooled in equimolar amounts then paired-end sequenced (2 × 250 bp) on an Illumina HiSeq2500 sequencing platform (Illumina Inc., San Diego, CA, USA) in accordance with the standard protocols. The raw reads have been deposited in the NCBI Sequence Read Archive (SRA) database (SRP161533).
Processing of sequencing data
The raw Illumina fastq files were demultiplexed, then quality-filtered using the QIIME (version 1.8) microbiome analysis software (quiime.org). Reads which could not be assembled were discarded, and chimeric sequences were identified and removed using UCHIME. The operational taxonomic units (OTUs) with ≥97% similarity were clustered using UPARSE (version 7.1, drive5.com/uparse/). The taxonomy of each 16S rRNA/18S rRNA gene sequence was obtained using RDP Classifier (rdp.cme.msu.edu/) against the Silva (SSU115) 16S rRNA and 18S rRNA databases with a confidence threshold of 70% (www.arb-silva.de).
The α-diversity, used to estimate the richness and diversity of OTUs, was calculated using QIIME to generate rarefaction curves, Good’s coverage, Chao 1, ACE, and Shannon and Simpson diversity indices. The β-diversity was evaluated using the UniFrac method. The weighted UniFrac distance matrices and OTU tables were calculated using QIIME to evaluate differences between the matrices and producers. The group_significance.py script of QIIME was run to compare the frequencies of OTUs across samples. Both the weighted and unweighted calculations were performed using principal coordinate analysis (PCoA). Redundancy analysis (RDA), canonical correspondence analysis (CCA), and heatmaps were performed using the vegan and pheatmap packages in R (vegan.r-forge.r-project.org/), to confirm the correlations between community structures and environmental variables, respectively.
Statistical analysis
Each treatment was replicated three times. All statistical analyses were performed using SPSS_16.0 (SPSS Inc. Chicago, IL. USA). The significance of differences between mean values was examined using Tukey’s test. Differences between means at P < 0.05 were considered significant.
Availability of data and materials
The data generated or analyzed during this study are included in this published article and its Additional file. The original sequencing data supporting the conclusions of this article is available in the National Center for Biotechnology Information Sequence Read Archive (SRA) database, accession no. SRP161533 and hyperlink to dataset in https://www.ncbi.nlm.nih.gov/sra/SRP161533.
Abbreviations
- AN:
-
Amino nitrogen
- CCA:
-
Canonical correspondence analysis
- HS-SPME-GC-MS:
-
Headspace solid-phase microextraction-gas chromatography-mass spectrometry
- HTS:
-
High-throughput sequencing
- LAB:
-
Lactic acid bacteria
- OTUs:
-
Operational taxonomic units
- PCoA:
-
Principal coordinate analysis
- RDA:
-
Redundancy analysis
- RS:
-
Reducing sugar
- RSF:
-
Roasted sesame-like flavor
- TE:
-
Total ester
References
Pang XN, Han BZ, Huang XN, Zhang X, Hou LF, Cao M, Gao LJ, Hu GH, Chen JY. Effect of the environment microbiota on the flavour of light-flavour Baijiu during spontaneous fermentation. Sci Rep. 2018; 8(3396): http://doi.org/https://doi.org/10.1038/s41598-018-21814-y.
Liu HL, Sun BG. Effect of fermentation processing on the flavor of baijiu. J Agr Food Chem. 2018;66(22):5425–32.
Wang XD, Ban SD, Qiu SY. Analysis of the mould microbiome and exogenous enzyme production in Moutai-flavor Daqu. J I Brewing. 2018;124(1):91–9.
Wang P, Wu Q, Jiang XJ, Wang ZQ, Tang JL, Xu Y. Bacillus licheniformis affects the microbial community and metabolic profile in the spontaneous fermentation of Daqu starter for Chinese liquor making. Int J Food Microbiol. 2017;250:59–67.
Ming HM, Zhou J, Zhu LL, Xu DF, Liu YH, Yao X. A comparative study of traditional/new daqu quality indicators system. Liquor-Making Sci Technol. 2015;4:32–6.
Li P, Aflakpui FWK, Yu H, Luo LX, Lin WT. Characterization of activity and microbial diversity of typical types of Daqu for traditional Chinese vinegar. Ann Microbiol. 2015;65(4):2019–27.
He GQ, Huang J, Zhou RQ, Wu CD, Jin Y. Effect of fortified daqu on the microbial community and flavor in Chinese strong-flavor liquor brewing process. Front Microbiol 2019; 10:56, https://doi.org/https://doi.org/10.3389/fmicb.2019.00056.
Li P, Lin WF, Liu X, Wang XW, Luo LX. Environmental factors affecting microbiota dynamics during traditional solid-state fermentation of Chinese daqu starter. Front Microbiol 2016; 7: https://doi.org/https://doi.org/10.3389/fmicb.2016.01237.
Zheng XW, Tabrizi MR, Nout MJR, Han BZ. Daqu - a traditional Chinese liquor fermentation starter. J I Brewing. 2011;117(1):82–90.
Zhang LQ, Wu CD, Ding XF, Zheng J, Zhou RQ. Characterization of microbial communities in Chinese liquor fermentation starters Daqu using nested PCR-DGGE. World J Microb Biot. 2014;30(12):3055–63.
Zheng XW, Yan Z, Nout MJR, Boekhout T, Han BZ, Zwietering MH, Smid EJ. Characterization of the microbial community in different types of Daqu samples as revealed by 16S rRNA and 26S rRNA gene clone libraries. World J Microb Biot. 2015;31(1):199–208.
Mo XL, Fan WL, Xu Y. Changes in volatile compounds of Chinese rice wine wheat Qu during fermentation and storage. J I Brewing. 2009;115(4):300–7.
Song RB, Shao ZL, Jun S. Change in physiochemical indexes and microbial quantity of nongxiang daqu during the storage process. Liquor-Making Sci Technol. 2017;1:59–61.
Fan GS, Fu ZL, Sun BG, Zhang YH, Wang XL, Xia YQ, Huang MQ, Li XT. Roles of aging in the production of light-flavored Daqu. J Biosci Bioeng. 2019;127(3):309–17.
Liu JJ, Chen JY, Fan Y, Huang XN, Han BZ. Biochemical characterisation and dominance of different hydrolases in different types of Daqu - a Chinese industrial fermentation starter. J Sci Food Agr. 2018;98(1):113–21.
Herrero A, Muro-Pastor AM, Flores E. Nitrogen control in cyanobacteria. J Bacteriol. 2001;183(2):411–25.
Yan ZK, Meng QY, Wang XZ, Du J. Analysis on physicochemical indexes and cultivation technology of traditional Xifeng Daqu sensory evaluation. Liquor Making. 2016;43(5):51–5.
Zhou P, Luo HB, Huang D, Deng B, Shen CP, Wu JF, Feng XY, Wang H. Influence of the different heat processing over the yeast on liquor-brewing. Food Sci Tech-Brazil. 2016;41(8):39–44.
Ding XF, Wu CD, Huang J, Zhou RQ. Characterization of interphase volatile compounds in Chinese Luzhou-flavor liquor fermentation cellar analyzed by head space-solid phase micro extraction coupled with gas chromatography mass spectrometry (HS-SPME/GC/MS). Lwt-Food Sci Technol. 2016;66:124–33.
Zhang CL, Ao ZH, Chui WQ, Shen CH, Tao WY, Zhang SY, Wang XJ. Changes in volatile compounds of Daqu during fermentation and storage by headspace solid phase microextraction combined with gas chromatography-mass spectrum. Food Ferment Ind. 2011;37(4):198–203.
Gou M, Wang HZ, Yuan HW, Zhang WX, Tang YQ, Kida KJ. Characterization of the microbial community in three types of fermentation starters used for Chinese liquor production. J I Brewing. 2015;121(4):620–7.
Wang L, Wang YY, Wang DQ, Xu J, Yang F, Liu G, Zhang DY, Feng Q, Xiao L, Xue WB, et al. Dynamic changes in the bacterial community in Moutai liquor fermentation process characterized by deep sequencing. J I Brewing. 2015;121(4):603–8.
Udomsil N, Rodtong S, Tanasupawat S, Yongsawatdigul J. Improvement of fish sauce quality by strain CMC5-3-1: a novel species of staphylococcus sp. J Food Sci. 2015;80(9):M2015–22.
Liu F, Zhou XH, Chen X, Chen J, Du GC, Fang F. Analysis of the microbial community of fermented grains and its correlation with organic acids biosynthesis during Chinese strong-aroma liquor manufacturing process. Acta Microbiol Sin 2018: https://doi.org/https://doi.org/10.13343/j.cnki.wsxb.20170616.
Wang XS, Du H, Zhang Y, Xu Y. Environmental microbiota drives microbial succession and metabolic profiles during Chinese liquor fermentation. Appl Environ Microb 2018; 84: https://doi.org/https://doi.org/10.1128/AEM.02369-17.
Hu Y, Dun Y, Li S, Fu B, Xiong X, Peng N, Liang Y, Zhao S. Changes in microbial community during fermentation of high-temperature Daqu used in the production of Chinese 'Baiyunbian' liquor. J I Brewing. 2017;123(4):594–9.
von Jan M, Riegger N, Poetter G, Schumann P, Verbarg S, Sproeer C, Rohde M, Lauer B, Labeda DP, Klenk H. Kroppenstedtia eburnea gen. Nov., Sp nov., a thermoactinomycete isolated by environmental screening, and emended description of the family Thermoactinomycetaceae Matsuo et al. 2006 emend. Yassin et al. 2009. Int J Syst Evol Micr. 2011;61(9):2304–10.
Gan X, Tang HL, Ye DD, Li P, Luo LX, Lin WF. Diversity and dynamics stability of bacterial community in traditional solid-state fermentation of Qishan vinegar. Ann Microbiol. 2017;67(10):703–13.
Li DN, Qiu SY. Overview of microbial community structure and function in Moutai-flavor Daqu. China Brewing. 2017;36(1):5–11.
Zheng XW, Yan Z, Han BZ, Zwietering MH, Samson RA, Boekhout T, Nout MJR. Complex microbiota of a Chinese "fen" liquor fermentation starter (fen-Daqu), revealed by culture-dependent and culture-independent methods. Food Microbiol. 2012;31(2):293–300.
Feng XM, Larsen TO, Schnurer J. Production of volatile compounds by Rhizopus oligosporus during soybean and barley tempeh fermentation. Int J Food Microbiol. 2007;113(2):133–41.
Ray RR. Beta-amylases from various fungal strains. A review Acta Microbiol Imm H. 2004;51(1–2):85–95.
Liu PL, Xiong XM, Wang S, Miao LH. Population dynamics and metabolite analysis of yeasts involved in a Chinese miscellaneous-flavor liquor fermentation. Ann Microbiol. 2017;67(8):553–65.
Cai HY, Zhang T, Zhang Q, Luo J, Cai CG, Mao JW. Microbial diversity and chemical analysis of the starters used in traditional Chinese sweet rice wine. Food Microbiol. 2018;73:319–26.
D'Elia JN, Salyers AA. Effect of regulatory protein levels on utilization of starch by Bacteroides thetaiotaomicron. J Bacteriol. 1996;178(24):7180–6.
Kamitori S, Kondo S, Okuyama K, Yokota T, Shimura Y, Tonozuka T, Sakano Y. Crystal structure of Thermoactinomyces vulgaris R-47 alpha-amylase II (TVAII) hydrolyzing cyclodextrins and pullulan at 2.6 angstrom resolution. J Mol Biol. 1999;287(5):907–21.
Kopecny J, Zorec M, Mrazek J, Kobayashi Y, Marinsek-Logar R. Butyrivibrio hungatei sp nov and Pseudobutyrivibrio xylanivorans sp nov., butyrate-producing bacteria from the rumen. Int J Syst Evol Micr. 2003;53(1):201–9.
Pokusaeva K, O'Connell-Motherway M, Zomer A, Fitzgerald GF, van Sinderen D. Characterization of two novel alpha-glucosidases from Bifidobacterium breve UCC2003. Appl Environ Microb. 2009;75(4):1135–43.
Rhim SL, Park MS, Ji GE. Expression and secretion of Bifidobacterium adolescentis amylase by Bifidobacterium longum. Biotechnol Lett. 2006;28(3):163–8.
Tan K, Tesar C, Wilton R, Keigher L, Babnigg G, Joachimiak A. Novel alpha-glucosidase from human gut microbiome: substrate specificities and their switch. FASEB J. 2010;24(10):3939–49.
Vacca-Smith AM, Venkitaraman AR, Quivey RGJ, Bowen WH. Interactions of streptococcal glucosyltransferases with alpha-amylase and starch on the surface of saliva-coated hydroxyapatite. Arch Oral Biol. 1996;41(3):291–8.
Yin YY, Liu YJ, Zhu WY, Mao SY. Effects of acarbose addition on ruminal bacterial microbiota, lipopolysaccharide levels and fermentation characteristics in vitro. Asian Austral J Anim. 2014;27(12):1726–35.
Hondoh H, Saburi W, Mori H, Okuyama M, Nakada T, Matsuura Y, Kimura A. Substrate recognition mechanism of alpha-1,6-glucosidic linkage hydrolyzing enzyme, dextran glucosidase from Streptococcus mutans. J Mol Biol. 2008;378(4):913–22.
Rosenstein G. Staphylococcal lipases: biochemical and molecular characterization. Biochimie. 2001;82(11):1005–14.
Horchani H, Aissa I, Ouertani S, Zarai Z, Gargouri Y, Sayari A. Staphylococcal lipases: biotechnological applications. J Mol Catal B-Enzym. 2012;76:125–32.
Chopra AK, Chander H. Factors affecting lipase production in Syncephalastrum racemosum. J Appl Bacteriol. 1983;54(2):163–9.
Lv M, Chen MB, Zhen D. Isolation of esterase-producing strain HSM from nong-flavor Daqu and study on its esterase-producing nutritional conditions. Liquor-making Sci Technol. 2014;1:12–5 20.
Zhou W, Liu XH, Ye L, Feng MQ, Zhou P, Shi XL. The biotransformation of astragalosides by a novel acetyl esterase from Absidia corymbifera AS2. Process Biochem. 2014;49(9):1464–71.
Wu XW, Zheng RC, Zheng YG. Overexpression of Talaromyces thermophilus lipase (TTL) and its application in biosynthesis of pregabalin intermediate. Chin J Bioproc Eng. 2016;14(5):37–44.
Manivannan P, Thangaraj S, Selvaraju P, Subbarayalu M. Spatial variation of volatile composition in musk melon (Cucumis melo L.) flowers and their cues as bee attractant. Int J Chem Stud. 2018;6(3):1362–70.
Jiao SS, Zhu DD, Deng Y, Zhao YY. Effects of hot air-assisted radio frequency heating on quality and shelf-life of roasted peanuts. Food Bioprocess Tech. 2016;9(2):308–19.
Zhou HY, Wang SM, Yu JX, Sun YL. A method for rapid determination of ethyl palmitate in Baijiu (liquor). Liquor-making Sci Technol. 2014;10:120–1.
Jin GY, Zhu Y, Xu Y. Mystery behind Chinese liquor fermentation. Trends Food Sci Tech. 2017;63:18–28.
Vergnais L, Masson F, Montel MC, Berdague JL, Talon R. Evaluation of solid-phase microextraction for analysis of volatile metabolites produced by staphylococci. J Agr Food Chem. 1998;46(1):228–34.
Hirano Y, Chonan K, Murayama K, Sakasegawa S, Matsumoto H, Sugimori D. Syncephalastrum racemosum amine oxidase with high catalytic efficiency toward ethanolamine and its application in ethanolamine determination. Appl Microbiol Biot. 2016;100(9):3999–4013.
Olesen PT, Meyer AS, Stahnke LH. Generation of flavour compounds in fermented sausages-the influence of curing ingredients, Staphylococcus starter culture and ripening time. Meat Sci. 2004;66(3):675–87.
Miyamura N, Matsui H, Tahara S, Mizutani J, Chiba S. Purification and some properties of 2-alkenal reductase of Mucor griseo-cyanus AHU6044. Agr Biol Chem. 1984;48(1):185–92.
Wang XS, Du H, Xu Y. Source tracking of prokaryotic communities in fermented grain of Chinese strong-flavor liquor. Int J Food Microbiol. 2017;244:27–35.
Fan GS, Sun BG, Fu ZL, Xia YQ, Huang MQ, Xu CY, Li XT. Analysis of physicochemical indices, volatile flavor components and microbial community of a light-flavor daqu. J Am Soc Brew Chem. 2018;76(3):209–18.
Acknowledgments
We thank Philip Creed and Simon Partridge, PhD for providing language help.
Funding
This research was supported by the National Natural Science Foundation of China (No. 31830069, 31701592 and 31671798), National Key R&D Program of China (2017YFD0400206). Funding bodies had no role in study design, collected data, analysis or writing.
Author information
Authors and Affiliations
Contributions
GF carried out the experiments, and wrote the manuscript. XL conceived and designed the study. ZF and PL carried out the lab work. CT and MR revised the manuscript. QW analyzed the data. The final manuscript is read and approved by all authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Table S1. Volatile compounds identified in RSF Daqu samples.
Additional file 2:
Figure S1. The clustering analysis was performed using UPGMA, which is a type of hierarchical clustering method based on unweighted UniFrac distance metrics, and showed the relationship of the prokaryotic communities in the four samples. Figure S2. PCoA analysis of the prokaryotic communities in the four samples. Figure S3. The clustering analysis was performed using UPGMA, which is a type of hierarchical clustering method based on unweighted UniFrac distance metrics, and showed the relationship of the eukaryotic communities in the four samples. Figure S4. PCoA analysis of the eukaryotic communities in the four samples.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fan, G., Fu, Z., Teng, C. et al. Effects of aging on the quality of roasted sesame-like flavor Daqu. BMC Microbiol 20, 67 (2020). https://doi.org/10.1186/s12866-020-01745-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-020-01745-3