
Abstract
Background
Previous investigations of phylogeny in Cervus recovered many clades without whole genomic support.
Methods
In this study, the genetic diversity and phylogeny of 5 species (21 subspecies/populations from C. unicolor, C. albirostris, C. nippon, C. elaphus and C. eldii) in the genus Cervus were analyzed using reduced-representation genome sequencing.
Results
A total of 197,543 SNPs were identified with an average sequencing depth of 16 x. A total of 21 SNP matrices for each subspecies/population and 1 matrix for individual analysis were constructed, respectively. Nucleotide diversity and heterozygosity analysis showed that all 21 subspecies/populations had different degrees of genetic diversity. C. eldii, C. unicolor and C. albirostris showed relatively high expected and observed heterozygosity, while observed heterozygosity in C. nippon was the lowest, indicating there was a certain degree of inbreeding rate in these subspecies/populations. Phylogenetic ML tree of all Cervus based on the 21 SNP matrices showed 5 robustly supported clades that clearly separate C. eldii, C. unicolor, C. albirostris, C. elaphus and C. nippon. Within C. elaphus clade, 4 subclades were well differentiated and statistically highly supported: C. elaphus (New Zealand), C. e. yarkandensis, C. c. canadensis and the other grouping the rest of C. canadensis from China. In the C. nippon clade, 2 well-distinct subclades corresponding to C. n. aplodontus and other C. nippon populations were separated. Phylogenetic reconstruction indicated that the first evolutionary event of the genus Cervus occurred approximately 7.4 millions of years ago. The split between C. elaphus and C. nippon could be estimated at around 3.6 millions of years ago. Phylogenetic ML tree of all samples based on individual SNP matrices, together with geographic distribution, have shown that there were 3 major subclades of C. elaphus and C. canadensis in China, namely C. e. yarkandensis (distributed in Tarim Basin), C. c. macneilli/C. c. kansuensis/C. c. alashanicus (distributed in middle west of China), and C. c. songaricus/C. c. sibiricus (distributed in northwest of China). Among them, C. e. yarkandensis was molecularly the most primitive subclade, with a differentiation dating back to 0.8–2.2 Myr ago. D statistical analysis showed that there was high probability of interspecific gene exchange between C. albirostris and C. eldii, C. albirostris and C. unicolor, C. nippon and C. unicolor, and there might be 2 migration events among 5 species in the genus Cervus.
Conclusions
Our results provided new insight to the genetic diversity and phylogeny of Cervus deer. In view of the current status of these populations, their conservation category will need to be reassessed.
Similar content being viewed by others


Background
The Cervidae phylogeny has proven difficult to resolve, due either to rapid radiation in Pecorans (superordinate clade of Cervide) in the Mid-Eocene or ambiguity and conflict between different analyses [1,2,3], especially phylogenetic relationships among formerly included in Cervus (Rusa, Rucervus, Przewalskium, Panolia and Cervus). The recent publication [4], showed that several species should be classified into the genus Cervus: Elaphurus davidianus, Przewalskium albirostris, Rucervus eldii, and all species of Rusa, and sometimes Panolia eldii was wrongly included in the genus Rucervus, the genetic analysis demonstrated the significant evolutionary and systematic distance between Panolia eldii and Rucervus duvaucelii. They were approved by some important publications [5, 6]. While some zoologists hold different opinions based on previous studies in the year 2001–2008 [7, 8]. At present, it is generally considered that there are 6 or 7 closely related taxa in the genus Cervus, namely C. timorensis, C. unicolor, C. albirostris, C. nippon, C. elaphus, C. canadensis and C. eldii. Past taxonomic studies, based on morphology [9], karyotypes [10, 11], serum proteins [12], mitochondrial DNA markers [13,14,15,16,17,18,19,20,21,22] and/or microsatellite DNA markers [23, 24] have led to partial consensus, reflecting the sparse amount of phylogenetic information available for some of these taxa, and disagreements between comparative anatomy and molecular systematics [1, 3].
Karyotype analysis showed that C. unicolor was the most primitive species in the genus Cervus, followed by C. albirostris and C. nippon, and then C. elaphus, C. eldii has close relationship with C. unicolor [11]. Based on the combined analysis of mitochondrial and nuclear genes, the genus Cervus was found polyphyletic, C. eldii form sister group to the clade plus all other species of Cervus (C. unicolor, C. albirostris, C. elaphus and C. nippon) [5]. Some researchers believed that C. elaphus and C. canadensis were derived from a single common ancestor, while C. elaphus form a sister group to C. canadensis and C. nippon, C. canadensis share more nucleotide similarities with C. nippon [19], while other researchers presumed that C. nippon is more primitive than C. elaphus and C. canadensis [13, 16, 17], eight subspecies of the C. elaphus probably originated from C. nippon in the middle Pleistocene [14, 25,26,27], however, C. nippon is allied with C. elaphus in nuclear gene analysis [5]. The Ag-No number and sites of C. nippon, C. elaphus and C. canadensis were same in karyotype analysis [11], the C-band and G-band were similar. Actually, the fawn of C. nippon, C. elaphus and C. canadensis were all spotted white, and there is no reproductive isolation between them. The above results imply that C. elaphus, C. canadensis and C. nippon could be very close-related species in the genus Cervus. Nonetheless, the classification proposed by all these studies are not in concordance with each other suggesting more work is needed to clarify these relationships.
The specific aims of our study were (1) to investigate the phylogeny of the genus Cervus, (2) to test the genetic diversity in the Cervus populations, and (3) to investigate interspecific gene exchange between C. unicolor, C. albirostris, C. nippon, C. elaphus and C. eldii.
To achieve these aims, SNP data from reduced-representation genome sequencing were used to provide phylogenetic relationships among 21 subspecies/populations derived from 5 species of genus Cervus, the divergence times, interspecific gene exchange and genetic diversity were estimated.
Methods
Ethics statement
All procedures concerning animals were organized to accord with the guidelines of care and use of experimental animals established by the Ministry of Agriculture of China, and all protocols were approved by the Institutional Animal Care and Use Committee of Institute of Special Animal and Plant Sciences, Chinese Academy of Agricultural Sciences, Changchun, China.
Samples
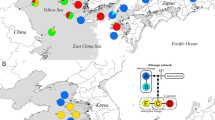
We sequenced a total of 195 samples, including 185 samples from 21 subspecies/populations, representing the 5 described species of Cervus (C. eldii, C. unicolor, C. albirostris, C. elaphus, C. nippon), and 10 samples from Rangifer (R. tarandus) as out-group. The information about species distribution, classification, source, and number was collected (Fig. 1 and Table 1). All samples, apart from C. n. aplodontus that were wild-caught, were raised in captivity, all of them are derived from wild-caught deer and have been maintained under closed flock breeding for a range of 5–50 generations. Chemical anesthesia was used during deer catching, Lumianning injection (070011777, Jilin Huamu Animal Health Products Co., Ltd., China), an anesthetic, was administered intramuscularly 1 ml per 100 kg body weight, peripheral vein blood of each sample were collected fresh and stored in − 20 °C until DNA extraction.

Map showing approximate sample collection sites. Numbers next to sites are equivalent to numbers in Table 1. Symbols for subspecies are identical to group symbols in Table 1. The lower right corner of the map is the geographic distribution of samples from New Zealand. Dotted elliptical circles represent three main subclades of C. elaphus and C. canadensis in China. The map was taken from maps package in R 3.2.0 software, we acknowledge its offer
Genomic DNA extraction
Genomic DNA extraction was carried out using whole blood genome DNA isolation kit (BioTeke Corporation, Beijing, China). The degradation and contamination of the genomic DNA was monitored on 1% agarose gels, DNA purity was checked using the NanoPhotometer® spectrophotometer (IMPLEN, CA, USA). DNA concentration was measured using Qubit® DNA Assay Kit in Qubit® 2.0 Flurometer (Life Technologies, CA, USA).
Library preparation and Illumina sequencing
We carried out a pre-design experiment, the restriction sites of Mse I, Nla III and Hae III in the reference genome assembly (mhl.quiver-7-1) were predicted, the result showed that the three enzyme approach is more favorable than one enzyme or two enzyme method in library construction for the reduced-representation genome sequencing. The average enzyme capture rate was 97.0% (Additional file 1: Table S1). So the three enzyme approach was selected in formal experiments. Genomic DNA was incubated at 37 °C with Mse I (New England Biolabs, NEB), T4 DNA ligase (NEB), ATP (NEB), and Mse I Y adapter N containing barcode. Restriction ligation reactions were heat-inactivated at 65 °C, and then digested for additional restriction enzyme Nla III (NEB) and Hae III (NEB) at 37 °C. The restriction ligation samples were purified with Agencourt AMPure XP (Beckman), then performed PCR reaction using purified samples, Phusion Master Mix (NEB) universal primer and index primer to add index, complete i5 and i7 sequence. The PCR productions were purified using Agencourt AMPure XP (Beckman) and pooled, then run out on a 2% agarose gel. Fragments with 400–425 bp (with indexes and adaptors) in size were isolated using a Gel Extraction Kit (Qiagen). These fragment products were then purified using Agencourt AMPure XP (Beckman), which was diluted for sequencing. Pair-end sequencing was performed upon the selected tags using an Illumina PE150 high throughput sequencing platform at the DNA sequencing laboratory (Tianjin Novogene Bioinformatics Technology Co., Ltd).
Read mapping and quality control
The sequences of each sample were sorted according to the barcodes. To make sure reads reliable and without artificial bias in the following analyses, raw data of fastq format was firstly processed through a series of quality control (QC) procedures in-house C scripts. QC standards as the following: (1) Removing reads with ≥10% unidentified nucleotides (N); (2) Removing reads with > 50% bases having phred quality < 5; (3) Removing reads with > 10 nt aligned to the adapter, allowing ≤10% mismatches. The assembly sika deer genome (mhl.quiver-7-1) was taken as reference genome (unpublished data). Comparison analysis of each sample was performed using BWA software (Parameter: mem -t 4 -k 32 -M -R).
SNP matrix construction
Population SNPs detection was performed with Stacks based on the clean data, the SNP datasets were filtered with the following criterion: maf = 0.01, mis = 0.7, dp = 2. For the population-based analysis, according to the clustering information (Table 1), the SNP information of each population is extracted from the filtered VCF file, and discarded those SNPs that may represent duplicated regions in the genome, finally the SNP matrix of each population is obtained. For the individual-based analysis, we constructed a SNP matrix with 197,543 SNP sites based on Stacks result.
Genetic diversity analysis
Nucleotide diversity (π) reflects population polymorphism, it was calculated using Arlequin software (http://cmpg.unibe.ch/software/arlequin35/). Expected heterozygosity (He) and Observed heterozygosity (Ho) were calculated according to the formula provided by Nei [28].
Phylogenetic analysis
Phylogenetic analyses were conducted using maximum likelihood (ML) on inter-population SNP matrix and individual SNP matrix, respectively. All ML analyses were conducted using the on line version of RAxML program [29] with the general time-reversible and gamma model (GTR + G model) of sequence evolution. The topology was evaluated with 1000 bootstrap replicates. A Yule speciation process was implemented as the tree prior and 1 X 106 steps were used in the Markov chain Monte Carlo (MCMC) iterations with sampling every 1000 iterations. Divergence times were estimated using MCMCTree 4.9e program (http://abacus.gene.ucl.ac.uk/software/paml.html), divergence time for R. tarandus and C. n. aplodontus 13.60–13.88 millions of years (http://www.timetree.org/home) were used as time correction point, the results were visualized using FigTree software.
Gene exchange analysis
Interspecific gene exchange of C. eldii, C. unicolor, C. albirostris, C. elaphus and C. nippon were analyzed by D-statistics using AdmixTools software. D-statistics is a method included in AdmixTools to analyze the admixture of ancestral population. It infers the distances of genetic relationships between populations by determining gene exchange or gene infiltration between populations. This method usually takes four populations W, X, Y and Z as analysis objects, among them, Z is an exogenous group and Y is an ancestral group. If the analysis showed that there were gene flows between W and Y, X and Z, the BABA model is fitted, so the relationship between W and Y is closer; If there is gene flow between W and Z, X and Y, the ABBA model is fitted, so the relationship between X and Y is closer. In this study, for SNP locus i, the allele frequencies of different populations were w’, x’, y’, z’, respectively. Firstly, BABA was defined, which meant that the alleles of W and Y were the same, the alleles of X and Z were the same, and the alleles of W and X were different. Similarly, ABBA was defined, then gene exchange analysis was carried out accordingly. Population differentiation and mixing were estimated by TreeMix based on genome-wide allele frequency, 2–5 gradients were set respectively, combined with D statistical results, calculated the number of migration events among 5 species [30].
Results
Reduced-representation genome sequencing
The sequencing of 195 individuals resulted in a total of 84,231,376,992 bp raw de-multiplexed reads with an average 431,955,779 bp reads per individual. After strict filtration, high quality clean data were obtained. All the sequencing data have been statistical analyzed, including output, error rate, Q20, Q30, GC content (Additional file 2: Table S2). The raw sequencing reads were deposited in NCBI SRA (Accession numbers: PRJNA355630). We successfully mapped 97.51% of reads per sample to the reference genome. The average estimated site coverage per sample was 16.21 x. The average 1 x genome coverage rate was 3.00% (Additional file 3: Table S3).
SNP matrices
Under inter-population combined analysis, we generated a total of 21 SNP matrices for the phylogenetic analyses. The average number of SNPs among 21 subspecies/populations was 120,030 (Fig. 2), C. nippon (dongda) had the largest number of SNPs (150,082 SNPs), C. eldii had the least number of SNPs (75,530 SNPs) in Cervus, followed by C. n. aplodontus (95,583 SNPs). In all populations, the number of SNPs in R. tarandus was the least (69,103 SNPs). Individual analysis produced a matrix containing 197,543 SNPs.

Number of SNPs among 21 subspecies/populations. Population SNPs detection was obtained by Stacks based on the clean data. The average number of SNPs among 21 subspecies/populations was 120,030, C. nippon (dongda) had the largest number of SNPs (150,082 SNPs), C. eldii had the least number of SNPs (75,530 SNPs) in Cervus, followed by C. n. aplodontus (95,583 SNPs)
Genetic diversity analysis
Nucleotide diversity (π) ranged from 0.009 to 0.022 among 21 subspecies/populations, with C. nippon (aodong) having the lowest (0.009) and C. c. alashanicus having the highest (0.022) π average (Table 2). Furthermore, we found that He was higher than other subspecies/populations in C. c. alashanicus and C. eldii (0.573 and 0.542, respectively). Subspecies/populations in C. elaphus (New Zealand) and C. c. xanthopygus showed relatively low average He (0.373 and 0.388, respectively) compared to other species of Cervus, and Ho was higher than other subspecies/populations in C. eldii and C. albirostris (0.632 and 0.396, respectively), while subspecies/populations in C. nippon (aodong) and C. nippon (xingkaihu) showed relatively low average Ho (0.103 and 0.106, respectively). Generally, all 21 subspecies/populations showed different degrees of genetic diversity. C. eldii, C. unicolor and C. albirostris showed relatively high He and Ho, while Ho in C. nippon was the lowest, indicating there was a certain degree of inbreeding rate in these subspecies/populations.
Phylogeny of Cervus
Phylogenetic ML tree of all Cervus clades based on the 21 SNP matrices showed that 5 clades that clearly separate C. eldii, C. unicolor, C. albirostris, C. elaphus and C. nippon from 21 subspecies/populations with the highest bootstrap support (Fig. 3). Within C. elaphus clade, 4 subclades were well differentiated and statistically highly supported: C. elaphus (New Zealand), C. e. yarkandensis, C. c. canadensis and the other grouping the rest of C. canadensis from China. In the C. nippon clade, 2 well-distinct subclades corresponding to C. n. aplodontus and other C. nippon populations were separated. Our phylogenetic reconstruction indicated that the first evolutionary event of the genus Cervus occurred approximately 7.4 millions of years ago. The divergence from a common ancestor of C. unicolor and C. eldii would have occurred at 2.7–6.1 millions of years (Myr) ago, so C. unicolor and C. eldii might be the most primitive Cervus deer. C. albirostris was the closest to them with divergence time of 3.3–5.8 Myr ago. The split between C. elaphus and C. nippon could be estimated at around 3.6 millions of years ago, the results were consist with previous report [6]. C. elaphus, C. canadensis and C. nippon might have the same ancestor. It is surprising because C. elaphus (New Zealand) and C. e. yarkandensis were sisters to C. canadensis while usually C. canadensis turn out to be more closely related to C. nippon than to C. elaphus.

Phylogenetic ML tree and divergence time analysis of all Cervus clades based on the 21 SNP matrices. Phylogenetic analyses were conducted using maximum likelihood (ML) on inter-population SNP matrix, divergence times were estimated using MCMCTree 4.9e program, divergence time for R. tarandus and C. n. aplodontus 13.60–13.88 millions of years was used as time correction point. Phylogenetic ML tree of all Cervus clades based on the 21 SNP matrices showed that 5 clades that clearly separate C. eldii, C. unicolor, C. albirostris, C. elaphus and C. nippon with the highest bootstrap support. Our phylogenetic reconstruction indicated that the first evolutionary event of the genus Cervus occurred approximately 7.4 millions of years ago
In the C. elaphus clade, the divergence between C. elaphus (New Zealand) and the other would have occurred at 1.1–3.0 Myr ago, it was accordant with fossil record [31]. The separation of C. e. yarkandensis and C. c. canadensis took place more recently, nearly 0.8–2.2 and 0.4–1.4 Myr ago. Whereas the bootstrap support of divergence was not high among other C. canadensis populations. In the C. nippon clade, there was a distinct divergence between C. n. aplodontus and other C. nippon populations, and the divergence time between them was 1.1–3.0 Myr. The divergence of other C. nippon populations was not obvious, so C. n. aplodontus might be the most primitive subspecies of C. nippon in our study.
Three main subclades of C. elaphus and C. canadensis in China
The C. elaphus and C. canadensis were geographically widespread in history. At present, it was considered that there were 7 or 8 subspecies of C. elaphus and C. canadensis lived in mainland China, and they were very restricted in distribution. All the subspecies except C. c. wallichii were collected and analyzed in this study.
Phylogenetic ML tree of all Cervus species based on the individual SNP matrices was basically the same as inter-population combined analysis, but some of the samples fell within their respective groups as anticipated (Fig. 4), such as C. c. songaricus and C. c. sibiricus, C. c. macneilli and C. c. kansuensis (these populations do have overlapping ranges), C. c. xanthopygus was split to 2 populations, one of which has close relationships with C. c. sibiricus and C. c. songaricus, and the other was grouped with C. c. alashanicus. Similarly, the C. nippon populations except C. n. aplodontus could not be well-separated. These results were probably caused by no geographical isolation between these local populations.

Phylogenetic ML tree of all Cervus species based on the individual SNP matrices. Phylogenetic analyses were conducted using maximum likelihood (ML) on individual SNP matrix. One colour for each group: R. tarandus, OrangeRed; C. eldii, LightSlateGray; C. unicolor, Green; C. albirostris, HotPink; C. elaphus (New Zealand), DarkMagenta; C. e. yarkandensis, Yellow; C. c. songaricus, LightCoral; C. c. sibiricus, LightPink; C. c. xanthopygus, Indigo; C. c. macneilli, DodgerBlue; C. c. kansuensis, RoyalBlue; C. c. alashanicus, Crimson; C. c. canadensis, Orchid; C. nippon (shuangyang), SpringGreen; C. nippon (tonghua), Olive; C. nippon (dongda), Blue; C. nippon (dongfeng), MidnightBlue; C. nippon (siping), GreenYellow; C. nippon (aodong), PaleVioletRed; C. nippon (ussuri), Goldenrod; C. nippon (xingkaihu), DarkSlateGray; C. n. aplodontus, CadetBlue. Phylogenetic ML tree of all Cervus species based on the individual SNP matrices was basically the same as inter-population combined analysis, but some of the samples fell within their respective groups as anticipated, such as C. c. songaricus and C. c. sibiricus, C. c. macneilli and C. c. kansuensis (these populations do have overlapping ranges), C. c. xanthopygus was split to 2 populations, one of which has close relationships with C. c. sibiricus and C. c. songaricus, and the other was grouped with C. c. alashanicus. Similarly, the C. nippon populations except C. n. aplodontus could not be well-separated. The phylogenetic ML tree of all C. elaphus and C. canadensis based on the individual SNP matrices, together with geographic distribution, have showed that, there were three major subclades in China, namely C. e. yarkandensis (distributed in Tarim Basin, with 92% bootstrap support), C. c. macneilli/C. c. kansuensis/C. c. alashanicus (distributed in middle west of China, with 89% bootstrap support), and C. c. songaricus/C. c. sibiricus (distributed in northwest and northeast of China, with 89% bootstrap support)
The phylogenetic ML tree of all C. elaphus and C. canadensis samples based on the individual SNP matrices, together with geographic distribution, have showed that, there were three major subclades in China, namely C. e. yarkandensis (distributed in Tarim Basin, with 92% bootstrap support), C. c. macneilli/C. c. kansuensis/C. c. alashanicus (distributed in middle west of China, with 89% bootstrap support), and C. c. songaricus/C. c. sibiricus (distributed in northwest and northeast of China, with 89% bootstrap support), whereas there might be introgression in the current C. c. xanthopygus population (Figs. 1 and 4). Among them, C. e. yarkandensis was molecularly the most primitive subclade, with a differentiation dating back to 0.8–2.2 Myr ago.
Interspecific gene exchange analysis
Based on D statistical results, p-value was transformed to Z value, if the absolute value of Z is greater than 2 (namely P-value< 0.05), the higher the probability of interspecific genetic exchange events is. The result was showed in Table 3. Group I, II, III conformed to BABA model, indicating C. albirostris and C. eldii, C. albirostris and C. unicolor had more probability of interspecific genetic exchange; Group IV, V conformed to ABBA model, indicating C. unicolor and C. albirostris, C. unicolor and C. nippon had more probability of interspecific genetic exchange. Population differentiation and mixing could be explained by the estimation of migration events, which included both geographic migration and evolutionary process, and they might all be marked by the genetic evidences. In this study, migration events estimated by TreeMix was shown in Fig. 5, there might be 2 migration events among 5 species in the genus Cervus: both migrations started from a common ancestor of C. eldii and C. unicolor, some of the ancestors migrated to C. albirostris, and another part migrated to the common ancestor of C. nippon and C. albirostris.

Migration events between species of the genus Cervus estimated by TreeMix. Population differentiation and mixing were estimated by TreeMix based on genome-wide allele frequency, 2–5 gradients were set respectively, combined with D statistical results, we found that there might be 2 migration events among 5 species in the genus Cervus. The direction of migration events are shown by arrows, and intensity is represented by color depth. Both migrations started from a common ancestor of C. eldii and C. unicolor, some of the ancestors migrated to C. albirostris, and another part migrated to the common ancestor of C. nippon and C. albirostris
Discussion
Phylogeny and genetic diversity analysis using whole genome SNPs in the genus Cervus
In many studies, molecular markers were used to verify the accuracy of genetic resources [32,33,34,35], whereas species in the genus Cervus had close relationships, all the species could hybridize with each other. Little genetic markers could be used to disentangle phylogenetic relationships among the closely related species. A series of methods were used for these analysis, such as using more data to reconstruct phylogeny [36], or developing a gene-by-gene method [37], however, the amount of markers is still limited in these methods, and most markers were not suitable for inferring the species phylogenies [38]. Recently, the improvements of next generation sequencing based genotyping methods have made the classification and characterization of genetic resources more feasible. Large numbers of SNP markers could be generated and used to infer phylogenetic relationships among very closely related species. Based on these strategies, reduced-representation genome sequencing (RRGS) [39] was developed, such as reduced representation shotgun sequencing (RRS) [40], restriction site associated DNA sequencing (RAD) [41,42,43,44], genotyping by sequencing (GBS) [45] have been successfully used to infer phylogenetic relationships [46]. Among these methods, GBS is relatively simple and quick for phylogenetic studies [39]. The GBS protocol was first developed using one restriction enzyme [47] and then modified by using two restriction enzymes [48]. The two enzyme GBS approach could generate uniform complexity reduction and avoid sequencing of repetitive regions. GBS has been successfully applied in crops to resolve phylogenetic relationship and genetic diversity [49,50,51]. Therefore, reduced-representation genome sequencing has great potential for phylogenetic inference based on large data sets.
We report here the first genome-wide study on 21 subspecies/populations in the genus Cervus. The sequencing depth is enough for SNP screening and analysis [52]. All the subspecies/populations showed a certain degree of genetic diversity,whereas in C. nippon, C. elaphus and C. canadensis, the genetic varation was relatively low, this might be caused by the small population size and inbreeding effect. The result was accordant with other genetic diversity study of C. nippon and C. elaphus [53]. More importantly, the mapping of 5 species in the genus Cervus to the sika deer genome resulted in a high mapping rate, providing a useful data set for the study of genetic diversity and phylogeny of Cervus deer. In comparison to previous studies, which used mitochondrial DNA [4, 13, 14, 54] or microsatellite DNA [23, 24], the genome-wide data set is remarkable for its amount and quality of data [2], and the genomic results were close to the theoretical limit of uncertainty for the ages of mammalian ordinal and supraordinal clades [55].
Phylogeny of closely related C. elaphus, C. canadensis and C. nippon complex
It is usually considered that C. elaphus and C. canadensis are not conspecific [7], they constitute three well-supported groups that are often ranked at species level: European and West-Asian red deer form a group, Central Asian red deer form a second group, and East Asian and North American wapiti form a third group [56, 57]. Our study do follow these opinions, in the C. elaphus clade, the divergence between C. elaphus (New Zealand) and the other occurred firstly, form one group; then the separation of C. e. yarkandensis, form a second group; and C. c. canadensis and other C. canadensis populations form a third group.
Interestingly, the phylogenetic position of the C. e. yarkandensis and C. elaphus (New Zealand) were surprising in this study, because they were sisters to the C. canadensis while usually C. canadensis turn out to be more closely related to C. nippon than to C. elaphus [56, 58]. C. elaphus (New Zealand) in this study are in all likelihood descendants of European red deer, while the New Zealand sample might also be derived from C. c. canadensis, but in this study, C. elaphus (New Zealand) were not clustered with C. c. canadensis, so our New Zealand samples could be the descendants of C. elaphus. The estimated divergence of this artificial population from the indigenous Eurasian Cervus has a little value and does not show a real chronology of evolutionary processes. Apparently, the obtained time of divergence at Pliocene or Early Pleistocene time is an overestimation and may be related to the founder effect, artificial selection and other genetic phenomena usual in the case of an artificial introduced population. Since the present data are genomic and not just mtDNA or single nuclear loci, this is potentially very interesting as it would restore C. elaphus and C. canadensis monophyly to the exclusion of C. nippon. In the following studies, European red deer samples (from Europe proper, covering the different phylogeographic lineages there) would be included to see whether this pattern holds.
Subclades of C. elaphus and C. canadensis in China
Most of studies agreed with that the clade of C. elaphus and C. canadensis was divided to western and eastern subclades, the western subclade comprised the C. e. yarkandensis and the European populations, the eastern subclade consisted of C. c. songaricus, C. c. sibiricus and populations from other Asian areas (Alashan, Gansu, Tibet, Mongolia, and northeastern China), and C. c. canadensis [15, 18]. It was revealed that a boundary separating the western subclade from the eastern subclade occurs between Tarim Basin and Tienshan Mountains in China, C. e. yarkandensis was more closer to European red deer, meanwhile, C. c. canadensis were genetically closer to Asian populations in north China and Mongolia, supporting that C. elaphus might immigrated from northeastern Eurasia to North America through the glacier-induced land-bridge (Beringia) which had formed between the two continents after Late Pleistocene [57], this was accordant with our phylogenetic tree result. Based on our analysis of good quality and non-redundant SNPs, we suspected that C. e. yarkandensis might be a subspecies of European red deer, C. c. canadensis might originated from C. elaphus or C. e. yarkandensis, so they probably migrated from these regions. Other C. canadensis in China were clearly separated from C. e. yarkandensis, it might be interpreted by the independent evolution of C. e. yarkandensis in a relatively closed natural environment after they separated from other C. elaphus populations [14, 59].
The genetic analysis based on mitochondrial DNA cytb showed that, C. c. kansuensis and C. c. macneilli had the same genotypes [60, 61], we also found that samples from these populations were clustered as one group, it was indicated that these samples should be classified as C. c. kansuensis, the populations in Qinghai should not be considered as a subspecies. Other studies based on mitochondrial DNA D-loop regions found that there were gene exchanges between C. c. sibiricus, C. c. songaricus and C. c. kansuensis, probably caused by hybridization between populations [62], in the group we are studying also provided further evidence that there had been putative intra-subspecific crosses of C. c. songaricus and C. c. sibiricus, C. c. xanthopygus and other C. canadensis, however, difficult specimens in the genus may often be characterized as “hybrids” without adequate evidence of their hybrid status [63].
Interspecific gene exchange of the genus Cervus
The genus Cervus had wide range of distribution. Every evolutionary type, from primitive to modern, were preserved during the evolution. Among them, C. albirostris is unique to China, it has similar karyotype to C. unicolor and C. eldii [64], the characteristics of their skulls are almost the same. Besides, their antlers also show continuous changes. In this study, interspecific gene exchange of the genus Cervus showed that C. albirostris had gene exchange with C. unicolor and C. eldii. It is consistent with previous research findings. C. elaphus and C. nippon are closely related, under natural conditions, they can hybridize with each other. But in this study, we didn’t find the evidence of gene exchange between the two species, we speculated that the similarity of gene frequencies between them is not necessarily due to the fusion effect of gene exchange, but to the same selection pressure. Gene exchange between species of the genus Cervus is a very interesting event, and it is still worth exploring in many different ways.
Implications for conservation
Along with increasingly extensive and intensive human activities, the influence on Cervus deer is increasing gradually. Many subspecies in the genus Cervus are extinct in the wild (e.g. C. n. taiouanus, C. n. grassianus and C. n. mandarinus) [65]. Several species are listed as “Endangered” and “Vulnerable” by the IUCN (e.g. C. eldii, C. albirostris, C. unicolor), however, C. elaphus, C. canadensis and C. nippon were classified as “Least Concern” species, most of their subspecies are on the brink of extinction. As to C. canadensis in China, no distinction between C. c. songaricus and C. c. sibiricus, C. c. macneilli and C. c. kansuensis was evident from phylogenetic ML tree based on the individual SNP matrices. C. c. xanthopygus was split to 2 populations, one of which has close relationships with C. c. sibiricus and C. c. songaricus, and the other was grouped with C. c. alashanicus. Similarly, the C. nippon populations except C. n. aplodontus could not be well-separated. The conservation status of these populations will need to be reassessed, urgent protection measures were required to prevent introgression or extinction of these populations.
Conclusions
The high-performance and low-cost of reduced-representation genome sequencing provides numerous high quality SNPs in the genus Cervus, enabled us to undertake phylogenetic analyses to answer the initial questions: (1) C. unicolor and C. eldii might be the most primitive Cervus deer. C. albirostris was the most closest to them with divergence time of 3.3–5.8 Myr ago. The split between C. elaphus and C. nippon could be estimated at around 3.6 millions of years ago. C. elaphus, C. canadensis and C. nippon might have the same ancestor. There were three major C. elaphus and C. canadensis subclades in China, within them, C. e. yarkandensis was molecularly the most primitive subclade, with a differentiation dating back to 0.8–2.2 Myr ago. (2) All 21 subspecies/populations had different degrees of genetic diversity. C. eldii, C. unicolor and C. albirostris showed relatively high expected and observed heterozygosity, while observed heterozygosity in C. nippon was the lowest, indicating there was a certain degree of inbreeding rate in these subspecies/populations. (3) There was high probability of interspecific gene exchange between C. albirostris and C. eldii, C. albirostris and C. unicolor, C. nippon and C. unicolor, and there might be 2 migration events among 5 species in the genus Cervus.
Our results provided new insight to the genetic diversity and phylogeny of Cervus deer. A revision of the current results based on comparison of phenotypic and molecular data is desirable for future research. In view of the current status of these populations, their conservation category will need to be reassessed.
Abbreviations
- C. c. :
-
Cervus canadensis
- C. e. :
-
Cervus elaphus
- C. n. :
-
Cervus nippon
- C. :
-
Cervus
- ddRADseq:
-
Double digestion RADseq
- GBS:
-
Genotyping by sequencing
- QC:
-
Quality control
- R. :
-
Rangifer
- RADseq:
-
Restriction site associated DNA sequencing
- RRGS:
-
Reduced-representation genome sequencing
- RRS:
-
Reduced representation shotgun
- SNPs:
-
Single nucleotide polymorphisms
References
Hernandez Fernandez M, Vrba ES. A complete estimate of the phylogenetic relationships in Ruminantia: a dated species-level supertree of the extant ruminants. Biol Rev Camb Philos Soc. 2005;80(2):269–302.
Decker JE, Pires JC, Conant GC, McKay SD, Heaton MP, Chen K, Cooper A, Vilkki J, Seabury CM, Caetano AR, et al. Resolving the evolution of extant and extinct ruminants with high-throughput phylogenomics. Proc Natl Acad Sci U S A. 2009;106(44):18644–9.
Heckeberg NS, Erpenbeck D, Worheide G, Rossner GE. Systematic relationships of five newly sequenced cervid species. PeerJ. 2016;4:e2307.
Hassanin A, Delsuc F, Ropiquet A, Hammer C, Jansen van Vuuren B, Matthee C, Ruiz-Garcia M, Catzeflis F, Areskoug V, Nguyen TT, et al. Pattern and timing of diversification of Cetartiodactyla (Mammalia, Laurasiatheria), as revealed by a comprehensive analysis of mitochondrial genomes. C R Biol. 2012;335(1):32–50.
Gilbert C, Ropiquet A, Hassanin A. Mitochondrial and nuclear phylogenies of Cervidae (Mammalia, Ruminantia): systematics, morphology, and biogeography. Mol Phylogenet Evol. 2006;40(1):101–17.
Pitra C, Fickel J, Meijaard E, Groves PC. Evolution and phylogeny of old world deer. Mol Phylogenet Evol. 2004;33(3):880–95.
Wilson DE, Mittermeier RA. Handbook of the Mammals of the World. 2011;2. Barcelona: Lynx Edicions.
Groves CP, Grubb P. Ungulate Taxonomy. Baltimore: Johns Hopkins University Press; 2011.
Flerov K. Morphology and ecology of Cervidae during its evolution. Journal of Paleontology Translation. 1957;1(2):2–16.
Neitzel H. Chromosome evolution in der family der Hirsche (Cervidae). Bongo, Berlin. 1979;3:27–38.
Wang Z, Du R. Evolution of karyotype of the genus Cervus. Acta Genet Sin. 1982;9(11):24–31.
Wang Z, Jia F, Massoud M. An electrophoretic analysis of the serum protein in Cervidae-SDS-PAGE. Acta Theriologica Sinica. 1988;8(1):13–20.
Cronin MA. Mitochondrial-DNA phylogeny of deer (Cervidae). J Mammal. 1991;72(3):553–66.
Li M, Wang X, Sheng H. Mitochondrial DNA divergence and phylogeny of four species of deer of the genus Cervus. Acta Zool Sin. 1999;45(1):99–105.
Randi E, Mucci N, Claro-Hergueta F, Bonnet A, Douzery EJP. A mitochondrial DNA control region phylogeny of the Cervinae: speciation in Cervus and implications for conservation. Anim Conserv. 2001;4:1–11.
Emerson BC, Tate ML. Genetic analysis of evolutionary relationships among deer (subfamily Cervinae). J Hered. 1993;84(4):266–73.
Tamate HB, Tsuchiya T. Mitochondrial DNA polymorphism in subspecies of the Japanese sika deer, Cervus nippon. The Journal of heredity. 1995;86(3):211–5.
Ludt CJ, Schroeder W, Rottmann O, Kuehn R. Mitochondrial DNA phylogeography of red deer (Cervus elaphus). Mol Phylogenet Evol. 2004;31(3):1064–83.
Polziehn RO, Strobeck C. A phylogenetic comparison of red deer and wapiti using mitochondrial DNA. Mol Phylogenet Evol. 2002;22(3):342–56.
Li M, Wang X, Sheng H. Origin and genetic differentiation study for four subspecies of wapiti. Zool Res. 1998;19(3):177–83.
Liu X, Wang Y, Liu Z. Phylogenetic relationships of cervinae based on sequence of mitochondrial cytochrome b gene. Zool Res. 2003;24(1):27–33.
Stankovic A, Doan K, Mackiewicz P, Ridush B, Baca M, Gromadka R, Socha P, Weglenski P, Nadachowski A, Stefaniak K. First ancient DNA sequences of the Late Pleistocene red deer (Cervus elaphus) from the Crimea, Ukraine. Quat Int. 2011;245(2):262–7.
Polziehn RO, Hamr J, Mallory FF, Strobeck C. Microsatellite analysis of north American wapiti (Cervus elaphus) populations. Mol Ecol. 2000;9(10):1561–76.
Xu Y, Pan Z, Xu Z, Yang S, Jin Y, Bai S. Status of microsatellites as genetic markers in cervids. J For Res. 2001;12(1):55–8.
Geist V. The relation of social evolution and dispersal in ungulates during the Pleistocene, with emphasis on the old world deer and the genus bison. Quat Res. 1971;1(3):285–315.
Ohtaishi N, Gao Y. A review of the distribution of all species of deer (Tragulidae, Moschidae and Cervidae) in China. Mammal Rev. 1990;20(2–3):125–44.
Kuwayama R, Ozawa T. Phylogenetic relationships among European Red Deer, wapiti, and sika deer inferred from mitochondrial DNA sequences. Molecular Phylogenetics & Evolution. 2000;15(1):115–23.
Nei M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics. 1978;89(3):583–90.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3.
Pickrell JK, Pritchard JK. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8(11):e1002967.
Meiri M, Kosintsev P, Conroy K, Meiri S, Barnes I, Lister A. Subspecies dynamics in space and time: a study of the red deer complex using ancient and modern DNA and morphology. J Biogeogr. 2018;45(2):367–80.
van de Wouw M, van Treuren R, van Hintum T. Authenticity of old cultivars in Genebank collections: a case study on lettuce. Crop Sci. 2011;51(2):736–46.
Samec D, Bogovic M, Vincek D, Martincic J, Salopek-Sondi B. Assessing the authenticity of the white cabbage (Brassica oleracea var. capitata f. alba) cv. 'Varazdinski' by molecular and phytochemical markers. Food Res Int. 2014;60:266–72.
Dossett M, Bassil NV, Finn CE. SSR fingerprinting of black raspberry cultivars shows discrepancies in identification. Acta Hortic. 2012;(946):49–53.
Kostamo K, Toljamo A, Antonius K, Kokko H, Karenlampi SO. Morphological and molecular identification to secure cultivar maintenance and management of self-sterile Rubus arcticus. Ann Bot-London. 2013;111(4):713–21.
Kluge AG. A concern for evidence and a phylogenetic hypothesis of relationships among Epicrates (Boidae, Serpentes). Syst Biol. 1989;38(1):7–25.
Rannala B, Yang ZH. Phylogenetic inference using whole genomes. Annu Rev Genom Hum G. 2008;9:217–31.
Eaton DAR, Ree RH. Inferring phylogeny and introgression using RADseq data: an example from flowering plants (Pedicularis: Orobanchaceae). Syst Biol. 2013;62(5):689–706.
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet. 2011;12(7):499–510.
Altshuler D, Pollara VJ, Cowles CR, Van Etten WJ, Baldwin J, Linton L, Lander ES. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature. 2000;407(6803):513–6.
Amores A, Catchen J, Ferrara A, Fontenot Q, Postlethwait JH. Genome evolution and meiotic maps by massively parallel DNA sequencing: spotted gar, an outgroup for the teleost genome duplication. Genetics. 2011;188(4):799–808.
Emerson KJ, Merz CR, Catchen JM, Hohenlohe PA, Cresko WA, Bradshaw WE, Holzapfel CM. Resolving postglacial phylogeography using high-throughput sequencing. Proc Natl Acad Sci U S A. 2010;107(37):16196–200.
Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, Cresko WA. Population genomics of parallel adaptation in Threespine stickleback using sequenced RAD tags. PLoS Genet. 2010;6(2):e1000862.
Hohenlohe PA, Amish SJ, Catchen JM, Allendorf FW, Luikart G. Next-generation RAD sequencing identifies thousands of SNPs for assessing hybridization between rainbow and westslope cutthroat trout. Mol Ecol Resour. 2011;11:117–22.
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6(5):e19379.
Hipp AL, Eaton DA, Cavender-Bares J, Fitzek E, Nipper R, Manos PS. A framework phylogeny of the American oak clade based on sequenced RAD data. PLoS One. 2014;9(4):e93975.
Sonah H, Bastien M, Iquira E, Tardivel A, Legare G, Boyle B, Normandeau E, Laroche J, Larose S, Jean M, et al. An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. PLoS One. 2013;8(1):e54603.
Poland JA, Brown PJ, Sorrells ME, Jannink JL. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS One. 2012;7(2):e32253.
Labate JA, Robertson LD, Strickler SR, Mueller LA. Genetic structure of the four wild tomato species in the Solanum peruvianum s.l. species complex. Genome. 2014;57(3):169–80.
Escudero M, Eaton DAR, Hahn M, Hipp AL. Genotyping-by-sequencing as a tool to infer phylogeny and ancestral hybridization: a case study in Carex (Cyperaceae). Mol Phylogenet Evol. 2014;79:359–67.
Girma G, Hyma KE, Asiedu R, Mitchell SE, Gedil M, Spillane C. Next-generation sequencing based genotyping, cytometry and phenotyping for understanding diversity and evolution of Guinea yams. Theor Appl Genet. 2014;127(8):1783–94.
De Donato M, Peters SO, Mitchell SE, Hussain T, Imumorin IG. Genotyping-by-sequencing (GBS): a novel, efficient and cost-effective genotyping method for cattle using next-generation sequencing. PLoS One. 2013;8(5):e62137.
Ba HX, Jia BY, Wang GW, Yang YF, Kedem G, Li CY. Genome-wide SNP discovery and analysis of genetic diversity in farmed sika deer (Cervus nippon) in Northeast China using double-digest restriction site-associated DNA sequencing. G3-Genes Genom Genet. 2017;7(9):3169–76.
Zhang WQ, Zhang MH. Phylogeny and evolution of Cervidae based on complete mitochondrial genomes. Genetics and molecular research : GMR. 2012;11(1):628–35.
dos Reis M, Inoue J, Hasegawa M, Asher RJ, Donoghue PCJ, Yang Z. Phylogenomic datasets provide both precision and accuracy in estimating the timescale of placental mammal phylogeny. Proceedings Biological sciences. 2012;279(1742):3491–500.
Lorenzini R, Garofalo L. Insights into the evolutionary history of Cervus (Cervidae, tribe Cervini) based on Bayesian analysis of mitochondrial marker sequences, with first indications for a new species. J. Zool. Syst. Evol. Res. 2015;53(4):340–9.
Mahmut H, Masuda R, Onuma M, Takahashi M, Nagata J, Suzuki M, Ohtaishi N. Molecular phylogeography of the red deer (Cervus elaphus) populations in Xinjiang of China: comparison with other Asian, European, and north American populations. Zool Sci. 2002;19(4):485–95.
Doan K, Mackiewicz P, Sandoval-Castellanos E, Stefaniak K, Ridush B, Dalén L, Węgleński P, Stankovic A: The history of Crimean red deer population and Cervus phylogeography in Eurasia. Zool J Linnean Soc. 2017;183:208–25.
Mahmut H, Ganzorig S, Onuma M, Masuda R, Suzuki M, Ohtaishi N. A preliminary study of the genetic diversity of Xinjiang Tarim red deer (Cervus elaphus yarkandensis) using the microsatellite DNA method. Jpn J Vet Res. 2001;49(3):231–7.
Zhang L: Genetic diversity and phylogeny evolution of white-lipped deer and five subspecies of wapiti using mitochondrial CytB complete sequence. Gansu Agricultural University 2008.
Lei T: Analysis genetic diversity of Gansu red deer and genetic relationship with Qinghai red deer using microsatellite DNA. Gansu Agricultural University 2009.
Li Q, Deng Z, Ren Z, Xiong J, Wang L, Wang H. Genetic diversity, origin and evolution of wapiti (red deer) in Xinjiang. China Herbivore Science. 2013;4:8–11.
Cayouette J, Catling PM. Hybridization in the genus Carex with special reference to North America. Bot Rev. 1992;58(58):351–438.
Neitzel H: Chromosome Evolution of Cervidae: Karyotypic and Molecular Aspects; 1987.
Hu P, Liu H, Xing X. Characteristics of germplasm resources and methods of conservation and utilization of domestic sika deer in China. China Animal Husbandry and Veterinary Medicine. 2015;42(10):2732–8.
Acknowledgements
The authors gratefully acknowledge Xingyao Hua, Ilia Sheremetev and Binyu Jing for their assistance in the sample collection.
Funding
This work was supported by the Agricultural Science and Technology Innovation Program of China (CAAS-ASTIP-201X-ISAPS) and the Special Economic Animals Sharing Platform in China. The funders had no role in study design, data collection, and analysis, decision to publish, or the preparation of the manuscript.
Availability of data and materials
The raw data are available from the SRA (http://www.ncbi.nlm.nih.gov/sra/) data repository under the submission number of PRJNA355630.
Author information
Authors and Affiliations
Contributions
PH conceived of the study, designed and oversaw all experiments; analyzed sequencing data, prepared figures and drafted the manuscript. YS and JX prepared DNA samples, helped to analyze respective data and prepared input data for analysis. TW, YL, HL and MR carried out data assays and evolution analysis. WS, BC, SC and XC performed SNP analysis and helped to draft the manuscript. FY, HT and XX conceived of the study, interpreted the data, oversaw the research and finalized the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All procedures concerning animals were organized to accord with the guidelines of care and use of experimental animals established by the Ministry of Agriculture of China, and all protocols were approved by the Institutional Animal Care and Use Committee of Institute of Special Animal and Plant Sciences, Chinese Academy of Agricultural Sciences, Changchun, China.
The IUCN Red List species involved in this study include Cervus eldii, Cervus unicolor and Cervus albirostris, they are artificially bred for species conservation purposes in China, that is in compliance with the provisions of Chapter 25 of the law of the People’s Republic of China on wildlife protection. The deer samples collected in this study were approved by the Department of Wildlife Protection and Nature Reserve Management of the State Forestry Administration and the local government.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Information on enzyme digestion results. (XLSX 21 kb)
Additional file 2:
Table S2. Statistics of output, error rate, Q20, Q30, GC content of all the sequencing data. (XLSX 24 kb)
Additional file 3:
Table S3. Summary of mapping rate and expected site coverage. (XLSX 29 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hu, P., Shao, Y., Xu, J. et al. Genome-wide study on genetic diversity and phylogeny of five species in the genus Cervus. BMC Genomics 20, 384 (2019). https://doi.org/10.1186/s12864-019-5785-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5785-z