
Abstract
Background
Maize is one of the primary crops of genetic manipulation, which provides an excellent means of promoting stress resistance and increasing yield. However, the differences in induction and regeneration capacity of embryonic callus (EC) among various genotypes result in genotypic dependence in genetic transformation.
Results
In this study, embryonic calli of two maize inbred lines with strong redifferentiation capacity and two lines with weak redifferentiation capability were separately subjected to transcriptome sequencing analysis during the early redifferentiation stages (stage I, 1–3 d; stage II, 4–6 d; stage III, 7–9 d) along with their corresponding controls. A total of ~ 654.72 million cDNA clean reads were yielded, and 62.64%~ 69.21% clean reads were mapped to the reference genome for each library. In comparison with the control, the numbers of differentially expressed genes (DEGs) for the four inbred lines identified in the three stages ranged from 1694 to 7193. By analyzing the common and specific DEGs of the four materials, we found that there were 321 upregulated genes and 386 downregulated genes identified in the high-regeneration lines (141 and DH40), whereas 611 upregulated genes and 500 downregulated genes were specifically expressed in the low-regeneration lines (ZYDH381–1 and DH3732). Analysis of the DEG expression patterns indicated a sharp change at stage I in both the high- and low-regeneration lines, which suggested that stage I constitutes a crucial period for EC regeneration. Notably, the specific common DEGs of 141 and DH40 were mainly associated with photosynthesis, porphyrin and chlorophyll metabolism, ribosomes, and plant hormone signal transduction. In contrast, the DEGs in ZYDH381–1 and DH3732 were mainly related to taurine and hypotaurine metabolism, nitrogen metabolism, fatty acid elongation, starch and sucrose metabolism, phenylpropanoid biosynthesis, and plant circadian rhythm. More importantly, WOX genes, which have an ancestral role in embryo development in seed plants and promote the regeneration of transformed calli, were specifically upregulated in the two high-regeneration lines.
Conclusions
Our research contributes to the elucidation of molecular regulation during early redifferentiation in the maize embryonic callus.
Similar content being viewed by others


Background
Maize (Zea mays L.) is a primary global crop supplying the food, feed, and industrial materials industries. Genetic transformation is presently widely used to improve yield and stress resistance and for gene function validation in maize, which largely depend on callus induction and regeneration from maize immature embryos [1,2,3]. Armstrong et al. [1] classified maize calli into three types, namely, I-, II -, and III-type calli, based on the callus characteristics. Among these types, only the II-type callus, known as embryonic callus, has cell totipotency and the ability to regenerate into whole plants and is therefore widely applied to genetic transformation in maize. Previous studies revealed that the genotype is an important factor that restricts the regeneration of the maize embryonic callus [4,5,6,7, 85]. Research on quantitative trait locus (QTL) mapping revealed that the regenerative capability of the embryonic callus is controlled by multiple genes in maize [8, 86].
Several functional genes have been shown to play important roles in callus regeneration in plants. The root stem cell regulators PLETHORA1 (PLT1) and PLETHORA2 (PLT2) must be activated by PLETHORA3 (PLT3), PLETHORA5 (PLT5), and PLETHORA7 (PLT7) to establish competent shoot regeneration progenitor cells [11, 12, 14]. A CDK (cyclin-dependent kinase) inhibitor (inhibitor of cyclin-dependent kinase, ICK) has been reported to improve the regenerative capacity of embryonic callus in Arabidopsis [20]. Meanwhile, the expression of WOX5 (WUSCHEL-related homeobox 5) in the quiescent center (QC) is considered a marker of Arabidopsis root stem cell niche [10]. Whereas, WUSCHEL (WUS), a marker of shoot apical meristem (SAM) identity, together with CLAVATA1/3 (CLV1/3), maintains the SAM stem cell niche through a feedback pathway in Arabidopsis [15]. WUS also influences shoot stem cell induction activity in the roots [16] and the conversion of root apical meristems (RAMs) to SAMs depending on the exogenous plant growth hormones applied in vitro [17]. In addition, as an AP2/ERF transcription factor, WIND1 (WOUND INDUCED DEDIFFERENTIATION 1) was proven to upregulate the expression of ESR1 (ENHANCER OF SHOOT REGEENRATION 1) gene that encodes another AP2/ERF transcription factor, promoting Arabidopsis shoot regeneration [9, 13]. SOMATIC EMBRYOGENESIS RECEPTOR KINASE 1 (SERK1), which is involved in the acquisition of embryogenic competence in plant tissue culture, is strongly expressed during the early stages of somatic embryogenesis in Arabidopsis [18, 19]. The downregulation of multiple CDK inhibitor ICK/KRP genes additively enhances both the shoot and root regeneration abilities of root-derived callus in Arabidopsis, indicating that CDK activity is a major factor for in vitro organogenesis [20]. Nishimura et al. reported that a ferredoxin-nitrite reductase (NiR) was responsible for rice regeneration ability [83]. In a recent study, WOX2 (WUSCHEL-related homeobox 2) and BBM (Baby Boom) genes were together introduced into maize by genetic transformation, resulting in the increased number of resistant seedlings regenerated from the transformed immature embryos [79]. In our latest study, 40 candidate genes were identified as being associated with the regenerative capacity of embryonic callus in maize, with regulators in cell fate determination, auxin transport, seed germination, or embryo development [85]. The present study was aimed at revealing the regulatory mechanisms associated with the early redifferentiation of embryonic callus by using the transcriptome data of four maize inbred lines with different regeneration capacities.
Results
Phenotypic evaluation of the four inbred lines
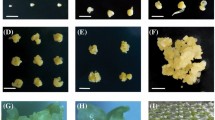
The EC regeneration capacities of the four lines were investigated in our previous study [85]. The CDR (callus differentiating rate) and CPN (callus plantlet number) of inbred lines 141 and DH40 were much higher than DH3732 and ZYDH381–1 (Fig. 1a) [85]. For the high-regeneration materials (141 and DH40), some small adventitious buds grew from the callus at 3 d, a mass of adventitious buds were generated at 6 d, and little plantlets formed at 9 d. For the low-regeneration materials (DH3732 and ZYDH381–1), only some calli became green after 6 d, and no adventitious bud formation was observed during the whole process (Fig. 1b). Based on the morphological features of 141 and DH40, the early redifferentiation of EC was divided into three stages: stage I (1–3 d), stage II (4–6 d), and stage III (7–9 d).

Phenotypic evaluation of the four inbred lines. a Regeneration ability of the EC of the four inbred lines; b The growth status of the EC of maize inbred lines 141 and DH3732 at 0 d, 3 d, 6 d, and 9 d
Transcriptome sequencing of maize EC
In total, the transcriptome sequencing of 48 libraries produced ~ 654.72 million clean reads after filtering low-quality reads, adaptor-polluted reads, and high unknown base (N) reads. The number of clean reads ranged from 13,342,096 to 14,067,230 (average = 13,640,022) among the samples. The results of the base composition and quality analysis showed that the clean reads had a good base composition (the T curve was in accordance with the A curve, and the G curve was in accordance with the C curve), and the percentage of low-quality reads was lower than 20% (Additional file 1: Figure S1). The B73 genome was used as the reference for read mapping, and HISAT was used to map the reads against the reference. Finally, only 60.73 to 67.38% of the remaining reads could be uniquely mapped onto the reference genome sequence (Table 1).
Gene expression levels were calculated using FPKM and were estimated using Cufflinks [24,25,26]. A correlation value between biological replicates for each of the stages was calculated according to the FPKM result. The Pearson’s correlations were mostly higher than 0.90 (Additional file 2: Figure S2), indicating good repeatability of the sequencing data.
Reliability validation of DEG expression via qRT-PCR
Ten DEGs involved in different biological processes (photosynthesis, plant hormone signal transduction, and protein phosphorylation) were randomly selected for qRT-PCR to validate the reliability of the transcriptome sequencing data. The results showed that the Pearson’s correlation coefficients between the data generated from the two platforms were all higher than 0.9 (R2 > 0.90) for the three stages (Fig. 2), indicating that the RNA-Seq data were reliable.

Correlation of the differential expression ratio between qRT-PCR and RNA-Seq in the three stages
Specific and common DEGs in the lines with different regeneration capacities
To gain insight into the regulatory network of embryonic callus redifferentiation, we analyzed the similarities and differences in the DEGs among the four maize inbred lines. For each line, the common DEGs among all the stages were much higher than the specific DEGs detected by each of the stages (Fig. 3), indicating that only a small number of the DEGs were involved in the early redifferentiation capability of EC. We further focused on the common DEGs shared by the high-regeneration lines (141 and DH40), which showed differential expression patterns in each of the low-regeneration lines (ZYDH381–1 and DH3732) during the process of redifferentiation (Fig. 4). These DEGs were accordingly called specific common DEGs in the high-regeneration lines, which were positively correlated with EC regeneration. Conversely, the specific common DEGs in the low-regeneration lines probably exerted inhibitory effects on EC regeneration. Detailed information on these specific common DEGs is shown in Additional file 3: Tables S3 and S4. In total, 385 (149 up and 236 down), 436 (199 up and 237 down), and 318 (126 up and 192 down) specific common DEGs were detected at stage I, stage II, and stage III, respectively, amounting to 707 DEGs in the three stages. In contrast, in the low-regeneration lines, a total of 1111 specific common DEGs were identified (Fig. 4). Specifically, 418 (174 up and 244 down), 576 (324 up and 252 down), and 787 (443 up and 344 down) DEGs were expressed at stage I, stage II, and stage III, respectively. According to the adjusted P-value, Zm00001d019518 (Photosystem I reaction center subunit IV A), Zm00001d042178 (Photosystem II reaction center psb28 protein), and Zm00001d039687 (Photosystem I reaction center subunit XI chloroplastic) were the most significant DEGs in 141 and DH40 (Table 2). Conversely, Zm00001d007049 (cysteine proteinases superfamily protein), Zm00001d044122 (dihydroflavonol-4-reductase), and Zm00001d024281 (polyamine oxidase 1) were the most significant DEGs in ZYDH381–1 and DH3732 (Table 3).

Venn diagram of the DEGs for each inbred line

Venn diagram of the DEGs between each inbred line
Expression patterns of specific common DEGs during redifferentiation
To assess the expression patterns of these specific common DEGs during early redifferentiation, we conducted a K-means approach using ExpressCluster software, as described in Ge et al. [84]. The expression patterns of the up- or downregulated specific common DEGs were classified into five clusters (Fig. 5). Interestingly, the majority of the clusters in both the high- and low-regeneration lines indicated sharp changes in DEGs at stage I, but more moderate changes at stage II and III, which suggested that stage I played a key role in the regeneration of EC. Somatic embryogenesis, which controls the future morphogenesis of plantlets, mainly occurred at stage I, and thus we supposed that these DEGs were particularly important for embryoid formation. Specifically, the upregulated DEGs in cluster 5 for the high-regeneration lines exhibited a greater change than the other clusters (Additional file 4: Table S5), and the majority of these DEGs were related to photosynthesis, such as Zm00001d019518 (photosystem I subunit IV) and Zm00001d042178 (photosystem II 13 kDa protein). Furthermore, the downregulated DEGs in cluster 3 for the high-regeneration lines showed identical and significant declines in stage I (Additional file 4: Table S6), and these DEGs were involved in the cellular protein modification process (Zm00001d042551, integrin-linked kinase), primary metabolic process (Zm00001d008952, endoglucanase; Zm00001d014244, alpha-L-fucosidase), and defense response (Zm00001d034461, indole-3-glycerol-phosphate lyase). For the DEGs in the low-regeneration lines, the upregulated DEGs in cluster 1 continuously increased during the three stages (Additional file 4: Table S7), and most of these genes were related to the metabolic process, such as Zm00001d044122 (dihydroflavonol-4-reductase) and Zm00001d018161 (ferredoxin-nitrite reductase). These downregulated specific common DEGs of cluster 3 displayed consistent expression trends in both the lines, with a sharp reduction in stage I and a slight increase in stage II and III (Additional file 4: Table S8), which included Zm00001d017913 (somatic embryogenesis receptor kinase 1), Zm00001d048647 (transcription factor MYB108-like), and Zm00001d032376 (disease resistance protein RPM1). These DEGs may account for the low regeneration capacity in ZYDH381–1 and DH3732.

The expression patterns of specific common genes in high (low) regeneration lines
GO analysis for specific common DEGs
To obtain functional annotations of the genes involved in EC regeneration capacity, we carried out GO analysis for the specific common DEGs. Both the upregulated and downregulated genes of the specific common DEGs of 141 and DH40 were categorized into three functions: Biological Process (BP), Cellular Component (CC), and Molecular Function (MF). As shown in Additional file 5: Figure S3, many terms of plant growth and development were significantly enriched in these biological processes, and the significantly enriched entries (FDR ≤ 0.05 or P-value ≤0.05) are displayed in Additional file 6: Tables S9–S11. Among the upregulated genes, 265 were assigned with functional annotations involved in 332 terms for BP, 65 terms for CC, and 13 terms for MF. BP included photosynthesis, glyceraldehyde-3-phosphate metabolic process, isopentenyl diphosphate biosynthetic process, cellular aldehyde metabolic process, phospholipid biosynthetic process, biosynthetic process and metabolic process of pigment, shoot and system form morphogenesis, the process of embryonic development, and seed development. CC involved chloroplast and chloroplast part, plasmids and plasmid part, and NAD(P)H dehydrogenase complex. Meanwhile, MF included the structural constituent of ribosome, transcription factor activity and core RNA polymerase binding, and chlorophyll binding. In the downregulated genes, 247 were annotated with functions, including seven terms in BP (regulation of jasmonic acid-mediated signaling pathway, response to wounding, regulation of defense response, regulation of response to stress, regulation of signaling and signal transduction, and regulation of cell communication), two in CC (integral component of membrane, intrinsic component of membrane), and four in MF (transcription co-repressor activity, transcription cofactor activity, transcription factor activity and transcription factor binding, and protein binding).
The specific common DEGs of ZYDH381–1 and DH3732 with 611 upregulated genes and 500 downregulated genes were also divided into three functional categories of BP, CC, and MF (Additional file 7: Figure S4), and the significant GO terms of enrichment (FDR ≤ 0.05) are shown in Additional file 8: Tables S12–S14. For the upregulated genes, a total of 458 DEGs were assigned functional annotations, including 13 terms in BP (nucleosome assembly and nucleosome organization, protein-DNA complex assembly and protein-DNA complex subunit organization, DNA conformation change and DNA packaging, oxidation–reduction process, monocarboxylic acid biosynthetic process, and fatty acid biosynthetic process), 16 in CC (protein-DNA complex, nucleosome, chromosomal part, chromosome, and cell wall), and 14 in MF (protein heterodimerization activity, protein dimerization activity, beta-glucosidase activity, galactosidase activity, and beta-galactosidase activity). As for the downregulated genes, 314 DEGs had functional annotations with 47 GO terms in BP (regulation of nucleobase-containing compound metabolic process, regulation of RNA metabolic process, regulation of nitrogen compound metabolic process, regulation of macromolecule biosynthetic process, auxin-activated signaling pathway, oligosaccharide biosynthetic process, the metabolic process and biosynthetic process of strigolactone, and cellular response to light stimulus), one in CC (nucleus), and six in MF (DNA binding, nucleic acid binding transcription factor activity, polysaccharide binding, and photoreceptor activity).
Pathways enriched by specific common DEGs
To further understand the functional role of the DEGs in the process of EC regeneration, KEGG pathway analysis of the specific common DEGs of 141 and DH40 was conducted (Additional file 9: Table S15). In stage I, 66 upregulated DEGs and 85 downregulated DEGs with functional annotations were involved in 40 and 58 metabolic pathways, respectively. In stage II, 91 upregulated DEGs and 87 downregulated DEGs with functional annotations were included in 61 and 51 metabolic pathways, respectively. In stage III, 56 upregulated DEGs and 69 downregulated DEGs with functional annotations affected 44 and 51 metabolic pathways, respectively. Photosynthesis, ribosome, and porphyrin and chlorophyll metabolism were three of the most significantly enriched upregulated pathways and were shared by the three stages of EC regeneration, indicating the conserved and important roles of these three metabolic pathways in the process of EC regeneration. Moreover, plant hormone signal transduction was the most significantly enriched pathway for the downregulated DEGs shared by the three stages of EC regeneration, suggesting that the decreased expression of the genes in this pathway positively influences EC regeneration. The significantly enriched pathways for the specific common DEGs of 141 and DH40 in stage I, II, and III samples are listed in Table 4.
Pathways enriched in the specific common DEGs of ZYDH381–1 and DH3732 are indicated in Additional file 10: Table S16. In stage I, 69 upregulated DEGs and 79 downregulated DEGs were involved in 71 and 62 metabolic pathways, respectively. In stage II, 118 upregulated DEGs and 100 downregulated DEGs were included in 78 and 76 metabolic pathways, respectively. In stage III samples, 178 upregulated DEGs and 112 downregulated affected 90 and 71 metabolic pathways, respectively. For upregulated DEGs, taurine and hypotaurine metabolism was the only significantly enriched pathway that was shared by the three stages of EC regeneration, and nitrogen metabolism was the only enriched pathway shared by stages I and II. In addition, 10 significant enrichment pathways (fatty acid elongation, starch and sucrose metabolism, phenylpropanoid biosynthesis, glycosaminoglycan degradation, sphingolipid metabolism, galactose metabolism, glycosphingolipid biosynthesis-ganglio series, phenylalanine metabolism, other glycan degradation, and stilbenoid, diarylheptanoid, and gingerol biosynthesis) were shared by stages II and III. Among these, numerous DEGs were involved in the pathway of phenylpropanoid biosynthesis, which is the most significant metabolic pathway enriched in stage III. For the downregulated DEGs, seven pathways (plant circadian rhythm, carbon metabolism, valine, leucine and isoleucine degradation, plant hormone signal transduction, glycolysis/gluconeogenesis, propanoate metabolism, and cyanoamino acid metabolism) were identified in the three stages of EC regeneration, among which the plant circadian rhythm was the most significant pathway in stages I and II and the second most significant pathway in stage III, whereas plant hormone signal transduction was the most significant pathway in stage III. These significant pathways common to the three stages might have a repressive effect on EC regeneration (Table 5).
Specific common DEGs involved in EC regeneration
The early redifferentiation of EC is a process of somatic embryogenesis and adventitious shoot regeneration. By comparing the DEGs of the four inbred lines, we found that a number of genes were involved in processes of photosynthesis, hormone signaling transduction, cell cycle, embryo and meristem initiation, circadian rhythm of plant, and phenylpropanoid biosynthesis. Based on the functional annotations, we identified some significantgenes closely related to callus regeneration (listed in Additional file 11: Table S17).
Photosynthesis mechanism: Photosynthesis is essential for the survival and development of plantlets. According to the functional annotations of the specific common DEGs of 141 and DH40, 14 upregulated genes were involved in the pathway of photosynthesis (Fig. 6a and Additional file 12: Figure S5). In addition, 12 genes were involved in porphyrin and chlorophyll metabolism (Fig. 6a and Additional file 13: Figure S6). Moreover, three DEGs including Zm00001d021906, Zm00001d018157, and Zm00001d006587, were involved in the pathway of photosynthetic antenna proteins, all of which were only upregulated in stage I (Fig. 6a and Additional file 14: Figure S7). We speculated that photosynthesis could effectively promote the regeneration of embryogenic callus in the two high-regeneration lines.

Relative expression patterns of the DEGs involved in tissue regeneration. a The relative expression pattern of DEGs involved in photosynthesis mechanism; b The relative expression pattern of DEGs involved in plant circadian rhythm; c The relative expression pattern of DEGs involved in plant hormone signal transduction mechanisms; d The relative expression pattern of DEGs involved in phenylpropanoid biosynthesis; e The relative expression pattern of DEGs related to cell cycling and WUSCHEL homeobox protein
Plant circadian rhythm: A total of 17 specifically downregulated DEGs in both ZYDH381–1 and DH3732 were associated with plant circadian rhythm (Fig. 6b). Of these DEGs, seven were annotated as Dof zinc finger protein DOF5.5 (CDF1), and two DEGs (Zm00001d003477 and Zm00001d016915) were annotated as cryptochrome 1 (CRY1). Especially CRY1 and ELF3, which respond to blue light and therefore affect the photomorphogenesis of the plant, the other DEGs respond to red light and regulate plant cell elongation and flowering (Additional file 15: Figure S8).
Plant hormone signal transduction mechanisms: Plant hormones play an important role in EC regeneration [53,54,55,56,57,58,59,60, 80]. Based on the specific common DEGs of 141 and DH40, a total of 29 downregulated genes were involved in hormone signal transduction (Fig. 6c). Of these, nine genes were annotated as jasmonate ZIM domain-containing protein (JAZ), and four genes were annotated as gibberellin receptor GID1 (GID1). Some specifically upregulated genes in 141 and DH40 were also related to the hormone transduction signal (Fig. 6c), including ABA receptor PYR/PYL family (PYL) (Zm00001d016105) and GID1 (Zm00001d007908, Zm00001d022320). Notably, JAZ and MYC2, as important factors of jasmonic acid signal transduction, were found to play critical roles in senescence and the plant stress response [88] (Additional file 16: Figure S9). Furthermore, GID1 and PIF4, which participate in the signal transduction pathway of gibberellin, have important functions in stem growth and the induction of seed germination (Additional file 16: Figure S9). For the specific common DEGs of ZYDH381–1 and DH3732, a total of 30 downregulated genes were related to plant hormone signal transduction (Fig. 6c). Among them, 11 genes were annotated as auxin-responsive protein IAA (IAA). SAUR (Zm00001d017397, Zm00001d018200), auxin responsive GH3 gene family (GH3) (Zm00001d010697), and ARF (Zm00001d014690) were identified in the downregulated genes of the specific common DEGs of ZYDH381–1 and DH3732. These genes are all involved in auxin signal transduction, which is important for cell enlargement and plant growth (Additional file 17: Figure S10). Additionally, EIN3 and EBF play roles in ethylene signal transduction, which is related to senescence and fruit ripening (Additional file 17: Figure S10). Ten specifically upregulated genes in ZYDH381–1 and DH3732 were also involved in ethylene signal transduction. In summary, these DEGs participate in the signal transduction pathways of several hormones, including auxin, cytokinin (CTK), gibberellin (GA), jasmonic acid (JA), and brassinosteroid (BR), indicating that the cross-talking of these hormones plays important roles in EC regeneration.
Phenylpropanoid biosynthesis: A total of 24 upregulated DEGs were related to the metabolic pathway of phenylpropanoid biosynthesis, specifically in ZYDH381–1 and DH3732 (Fig. 6d and Additional file 18: Figure S11). Of these, beta-glucosidase is a glycoside hydrolase enzyme that participates in various cell functions, such as the catalysis of disaccharide and oligosaccharide bonds, catalysis of cell wall components, and the activation of phytohormones [27,28,29,30,31]. POD protects tissues and cells from oxidative damage by catalyzing the reduction of H2O2, and its accumulation accelerates the browning of the callus [28]. PAL catalyzes the nonoxidative elimination of ammonia from L-Phe to yield trans-cinnamate in the first step of the phenylpropanoid pathway, which plays an essential role in plant development and the stress response [29,30,31]. In the two lines with poor EC regeneration, the DEGs involved in phenylpropanoid biosynthesis were all increased at the same stage, which suggested that phenylpropanoid biosynthesis negatively influenced EC redifferentiation by accelerating callus browning.
The DEGs related to cell cycling: For the specific common DEGs of 141 and DH40, one upregulated gene and two downregulated genes were related to cell cycle regulation (Fig. 6e), including cell division protease FtsH (Zm00001d036371) and cyclin-dependent kinase 12/13 (Zm00001d022041, Zm00001d042285). In addition, 10 upregulated genes and four downregulated genes were assigned to this category for specific common DEGs of ZYDH381–1 and DH3732 (Fig. 6e). Of these, cyclin-dependent kinase 12/13 (CDK12_13) belongs to CDKs, which are the core regulators of the cell cycle and are assumed to control cell differentiation and proliferation in response to phytohormone signals [32, 33]. Furthermore, cyclin A (CCNA) and cyclin B (CCNB) are both members of the cyclin family, which regulates CDK activity [34]. Cell division protease FtsH (FtsH) is a part of ATP-dependent proteases, which function in protein quality control and regulation [35].
Moreover, Zm00001d035535, which encodes a putative WUSCHEL homeobox protein (WOX), was specifically upregulated in 141 and DH40 at all the three stages, whereas Zm00001d042821, annotated as WUSCHEL-related homeobox 9-like (WOX9), was significantly downregulated in ZYDH381–1 and DH3732 in stage III (Fig. 6e). WOX family proteins have been proved to regulate embryo development and callus regeneration [78, 82]. Therefore, the cross-talk of these DEGs related to the cell cycle and WOX could positively influence embryonic callus regeneration.
Cluster analysis of the relative expressions of these DEGs (Additional file 19: Figure S12) showed that inbred lines 141 and DH40 were clustered into one group, while ZYDH381–1 and DH3732 as well as the three stages of each lines were separately clustered together. Furthermore, stage II and III were categorized into one class, whereas stage I was alone. These results indicated that the expression levels of these DEGs between the high- and low-regeneration lines were significantly distinct. The responses of these DEGs were rapid, being largest in the first stage and smaller in the following stages.
Discussion
Effect of light on callus regeneration
Light, which is closely related to the gain of energy and metabolic processes, is usually an important factor affecting growth, organogenesis, and the formation of plant products, including both primary and secondary metabolites [36]. A number of reports have mentioned that plant regeneration from a callus requires light and that plant regeneration derived from a callus is effectively increased in light compared to the dark [36,37,38,39,40,41]. Rikiishi et al. reported that light probably controls shoot regeneration from calli by modifying cytokinin levels and/or responses, and blue light signals act in the photoinhibition of shoot regeneration in immature barley embryo culture [38]. Other researchers have shown that light intensity and photoperiod exert a significant influence on shoot regeneration from a callus [36, 39]. However, there are few reports related to the specific molecular mechanism of the effect of light on the regeneration of embryo-derived EC in maize. In this study, we found that many specifically upregulated DEGs in the high-regeneration lines were related to photosynthesis. Furthermore, plant circadian rhythm was the most significant pathway enriched with downregulated DEGs in the low-regeneration lines. It is clear that photosynthesis and plant circadian rhythm both require light (Additional file 12: Figure S5 and Additional file 15: Figure S8). We speculated that light might regulate callus regeneration mainly by responding to photosynthesis and plant circadian rhythm.
Photosynthesis is the basic energy conversion process on Earth, facilitating the utilization of the energy from sunlight by living organisms [42]. In this study, photosynthesis, porphyrin and chlorophyll metabolism, and photosynthetic antenna proteins were the three significantly upregulated enriched pathways in the high-regeneration lines. Photosynthetic antenna proteins, which are specialized pigment–protein complexes, allow for the capture of energy from sunlight, thereby participating in the initial step of photosynthesis [43]. Chlorophyll, a compound of magnesium porphyrin that absorbs energy from light, is one of the most important pigments related to photosynthesis [44]. The DEGs involved in the pathway of photosynthesis were associated with photosystem I (PS I), photosystem II (PS II), photosynthetic electron transport, and F-type ATPase, all of which play significant roles in the process of photosynthesis. Sergio et al. showed that adventitious root formation in leafy cuttings of hazelnut (Corylus avellana L.) was affected by leaf photosynthesis, which provides the carbohydrate supply for this intensive metabolic process [45]. Some previous studies reported that the process of somatic embryogenesis and embryo germination demonstrated photosynthetic capacity [46,47,48]. The present study also indicated that EC gradually developed the photosynthetic apparatus and photosynthetic capacity for further autotrophy in this regeneration process, considering that the relative expression levels of most DEGs related to photosynthesis were gradually increased (Fig. 6a and Additional file 12: Figure S5).
Plant circadian rhythms are associated with the synchrony of the plant with the light cycle of its surrounding environment, providing the plant with information on the season, thereby informing flowering and pollinator attraction [49, 50]. In the pathway of plant circadian rhythm, Dof zinc finger proteins, phytochrome A and B (PHYA, PHYB), cryptochrome 1 (CRY1) were associated with downregulated expression in the low regeneration lines. Of these, Dof proteins, as transcription factors, are a subfamily of zinc finger proteins particular to the plant kingdom that are essential in the regulation of many plant growth and development processes, such as seed germination and the expression of some genes associated with photosynthesis [51, 52]. Moreover, PHYA is the main phytochrome in seedlings grown in the dark but rapidly degrades in light to produce CRY1 [49, 50]. Therefore, the downregulated expression of the DEGs related to plant circadian rhythms might be detrimental to the redifferentiation of EC (Fig. 6b and Additional file 15: Figure S8).
Effects of plant hormones on callus regeneration
A number of DEGs were related to plant hormone signal transduction in both the high-and low-regeneration lines. Interestingly, in 141 and DH40, the specific common DEGs involved in hormone signal transduction were mainly associated with the signal transduction of JA, GA, BR, and ABA, whereas the majority of those in ZYDH381–1 and DH3732 were related to the signal transduction of auxin, CTK, BR, and GA (Additional file 16: Figure S9 and Additional file 17: Figure S10). JA plays an important role in plant growth and development, the stress response, and secondary metabolism processes, and JAZ is a plant-specific negative regulator of JA responsive genes, containing the conserved domain of ZIM and the Jas domain [53, 54]. In tissue culturing of the garlic bulb, the efficiency of shoot regeneration was improved by the addition of 1–10 μM JA in B5 basic medium [55]. In our study, JAZ was significantly downregulated in 141 and DH40. Given its negative regulation of downstream JA responsive genes, we speculated that the downregulated expression of these genes prompted the expression of downstream responsive genes and therefore motivated the regeneration of the callus. GA regulates plant growth by influencing stem growth and inducing germination [56], and the receptor protein GID1, DELLA protein, and PIF are involved in the signal transduction pathway of GA. In the tissue culture of immature barley embryo, the accumulation of ABA inhibited the shoot regeneration of the callus derived from immature barley embryos [57]. The downregulated DEGs related to the transduction of GA and ABA in our study might play a role in the regeneration of EC derived from immature maize embryos. Previous studies indicated that the balance of auxin and CTK is key to controlling the dedifferentiation and differentiation of plant cells, and either shoots or roots can be regenerated from a callus by adjusting the auxin–cytokinin ratios of the induction medium [58,59,60]. For example, a study on de novo shoot regeneration in Arabidopsis thaliana revealed that ARF3 directly bound to the promoter of ATP/ADP ISOPENTENYLTRANSFERASE5 (AtIPT5) and negatively regulated AtIPT5 expression, thereafter causing the abnormal biosynthesis of CTK and ultimately resulting in organ regeneration disruption [60]. Our study indicated that most of the DEGs related to auxin and CTK transduction were increased in 141 and DH40 but decreased in the poor regeneration lines (Fig. 6c), suggesting that the transduction of auxin and CTK is critical for callus regeneration. BR is a naturally produced class of plant steroid hormones and is typically involved in cell elongation, cell division, and differentiation throughout the plant life cycle and regulates many developmental processes, from seed germination to flowering and senescence [61, 62]. Research on cotton regeneration via somatic embryogenesis showed that BR had a stimulatory effect on the maturation of somatic embryos, but negatively affected callus growth [63, 64]. In this study, the DEGs associated with the transduction of BR were downregulated in 141 and DH40 but displayed inverse trends in the other two lines, which indicated that the transduction of BR might be negatively correlated with EC redifferentiation. In combination, these findings suggest that the regeneration process of embryogenic callus depends on the coordination of various hormones.
Phenylpropanoid biosynthesis in callus regeneration
In our previous studies, the low-regenerating embryonic calli turned brown more easily, which is associated with cell disorganization and eventual cell death. Phenylpropanoid biosynthesis is induced by several stresses [65, 81], and we found it had significantly enriched pathways with specific common DEGs in ZYDH381–1 and DH3732, mainly including POD, PAL, and β-glucosidase. Notably, all of the DEGs were upregulated in ZYDH381–1 and DH3732 but downregulated or unchanged in 141 and DH40. Some studies have indicated that callus browning is a typical feature and a major obstacle in callus culturing and is related to polyphenol oxidase (PPO) and POD enzymatic activities [66,67,68]. A study on the plant regeneration of pine species via somatic organogenesis showed that cell death was correlated with elevated levels of peroxides, and the addition of antioxidants inhibited callus browning by reducing the accumulation of peroxidase [28]. PAL activity is correlated with xylogenesis and nodule induction in bean callus culture [69]. β-glucosidase activity is associated with cell lignification, and the production of lignin compounds in the callus of pine species was considered to be the result of stress reactions [70]. In the present study, the upregulated expression of genes related to phenylpropanoid biosynthesis was probably due to the response to environmental stress during EC regeneration, which might cause embryonic callus browning and hinder embryonic callus regeneration.
The role of other regulators involved in callus regeneration
Some genes involved in cell cycle regulation and embryogenesis were differentially expressed, including FtsH, CDK, CCNA, CCNB, and WOX. Most of the cell cycle-related genes were upregulated in ZYDH381–1 and DH3732 but downregulated or unchanged in 141 and DH40, except embryogenesis-related genes (WOX), which were upregulated in 141 and DH40 but downregulated in ZYDH381–1 and DH3732. Among them, FtsH is the main thylakoid membrane protease found in organisms that performs oxygenic photosynthesis, and its malfunction causes cell division defects and growth arrest [35, 71]. Cell cycle regulation involves the differential expression of some cell-cycle genes during all phases of plant development. CDKs are a type of core cell cycle regulator that are regulated by the presence of cyclins [33, 72, 73]. We discovered that most of the cell cycle-related DEGs (FtsH, CDK, CCNA, CCNB) were significantly upregulated in the two poor regeneration materials but exhibited opposite trends in the two high-regeneration materials. Accordingly, we suggested that the increased expression of these DEGs facilitated the maintenance of the condition of EC and hindered the redifferentiation of EC. The WOX family (with 15 members) is a class of transcription factors that specifically exists in plants, including WUS and WOX1-WOX14 in Arabidopsis, and plays an important role in the stem cell maintenance of SAM and RAM, the development of lateral organs, the formation of floral organs, and embryo development [74,75,76,77]. Previous studies showed that WOX played an ancestral role in embryo development in seed plants, and the connection among polar auxin transport (PAT), PIN-FORMED (PIN), and WOX in the regulation of embryo patterning in seed plants was strengthened by the study of Palovaara et al. [78]. Lowe et al. also obtained plantlets with increased resistance from transgenic callus by overexpressing the maize BBM and WUSCHEL2 (WUS2) genes [79]. In this study, the WOX genes were upregulated in the two high-regeneration capacity materials but downregulated in the other lines, which verified the positive regulatory role of WOX genes in callus regeneration.
Conclusions
In maize, the obvious differences in induction and regeneration capabilities of EC among various genotypes result in genotype dependence in genetic transformation. In this study, transcriptome analysis of the EC of the four maize inbred lines showed that the specific common DEGs of the high-regeneration lines were mainly associated with photosynthesis, porphyrin and chlorophyll metabolism, ribosomes, and plant hormone signal transduction, while those of the low regeneration lines were mainly related to taurine and hypotaurine metabolism, nitrogen metabolism, fatty acid elongation, starch and sucrose metabolism, phenylpropanoid biosynthesis, and plant circadian rhythm. More importantly, WOX genes that have an ancestral role in embryo development in seed plants and promote the regeneration of transformed calli were specifically upregulated in the two high-regeneration lines. Our research provides new insight into molecular regulation during the early redifferentiation of a maize embryonic callus.
Methods
Samples and RNA isolation
In our previous study, we obtained two high-regeneration lines (141, DH40) and two low-regeneration lines (ZYDH381–1 and DH3732) from a natural population of 144 maize inbred lines [85, 89]. Furthermore, 141 and DH3732 both belong to NSS group, whereas DH40 and ZYDH381–1 were classified into Tropical group and SS group, respectively [90]. In the present study, the immature embryos of 1.0–1.5 mm in length were collected from the four lines at 12 d after self-pollination and then cultured in modified N6 medium supplemented with 2,4-D under aseptic and aphotic conditions at 27 °C for 25 d to induce embryonic callus (EC). The embryo-derived EC was subsequently transferred to new N6 medium and cultured for 20 d to subculture the EC, which was then cultured in MS medium at 27 °C in 12 h light/d for 9 d for redifferentiation. No replacement of fresh media was conducted during each of the culture phases. The specific components of the medium were listed in Additional file 20: Table S1. Total RNA was isolated from a pool of three calli for each sample at 0–9 d during redifferentiation by using Trizol Reagent (Invitrogen). RNA from 1 to 3 d samples after redifferentiation (stage I sample, green callus forming) were mixed in equal proportions, as were RNA from 4 to 6 d (stage II sample, embryoid and less adventitious bud emerged) and 7–9 d (stage III sample, mass adventitious bud forming), with the 0 d RNA being used as the control (CK). The four RNA samples of each line were then separately submitted to transcriptome analysis using Illumina sequencing, with three biological replicates.
Illumina sequencing and data analysis
Clean reads were obtained by filtering low-quality reads, adaptor-polluted reads, and reads with a high content of unknown base (N) reads. Then, the clean reads were mapped to the reference genome and genes of maize available at RefGen_V4 ( http://www.gramene.org/) using Bowtie2. Gene expression levels were calculated with RSEM. The Fragments Per Kilobase of transcript per Million mapped reads (FPKM) method was used to estimate transcript expression levels in all the samples. DESeq2 was used to detect differentially expressed genes (DEGs) between each selected sample pair. The DESeq2 parameters included fold change (FC) ≥ 2.00 and adjusted P-value ≤0.05. Hierarchical clustering of the DEGs was performed using the pheatmap package in R. The DEGs were clustered according to their expression levels using ExpressCluster software to investigate their expression patterns. Gene Ontology (GO) and pathway annotations and enrichment analyses were based on the Gene Ontology Database (www.geneontology.org) [21] and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway (www.genome.jp/kegg) [22], respectively. The calculated P-values was subjected to Bonferroni correction [23], using the corrected P-value ≤0.05 as a threshold.
DEG statistics
In this study, DESeq2 was used to identify the DEGs between each comparison with a threshold of false discovery rate (FDR)-adjusted P-value ≤0.05 and FC ≥ 2 (|Log2FC| ≥ 1) [22]. A large number of DEGs were detected at the three stages of EC regeneration for each inbred line in comparison to the control (Fig. 7a). At stage I, there were 1116, 1727, 1059, and 3714 upregulated DEGs and 1578, 2304, 1265, and 3479 downregulated DEGs for lines 141, DH40, ZYDH381–1, and DH3732, respectively. For Stage II, there were 1322, 2007, 1182, and 3784 upregulated DEGs and 1367, 2277, 1171, and 2876 downregulated DEGs in 141, DH40, ZYDH381–1, and DH3732, respectively. In contrast, in Stage III, 1108, 1563, 1498, and 3779 DEGs showed increased expression for 141, DH40, ZYDH381–1, and DH3732, respectively, and 1043, 1668, 1112, and 2999 displayed decreased expression.

Numbers and transcription levels of DEGs of the four maize inbred lines. a Number of DEGs for each maize. inbred line. b Relative expression levels of the DEGs at the three stages of EC redifferentiation for each maize inbred line. Each box plot shows the distribution of the relative transcription level [log2 (fold-change)] of the DEGs. The red line indicates a one-fold change relative to the transcription level of the control samples
The expression level ranges of the DEGs are displayed in Fig. 7b. In line 141, the relative expressions (compared to CK) of these DEGs ranged from − 5.864 to 5.863, − 6.516 to 6.366, and − 6.479 to 6.404 at stage I, stage II, and stage III, respectively. For DH40, the FC of the DEGs ranged from − 6.449 to 6.488, − 7.613 to 8.679, and − 6.257 to 8.477 in the three stages, respectively. The expression of the DEGs in ZYDH381–1 ranged from − 5.214 to 5.914, − 6.726 to 6.144, and − 5.130 to 6.573 at stage I, stage II, and stage III, respectively. In DH3732, these DEGs were more sensitive to EC regeneration than the other lines, with FC values ranging from − 10.440 to 8.431, − 10.246 to 8.402, and − 10.397 to 9.122 at these stages, respectively.
Quantitative real-time PCR validation
To validate the DEGs obtained from the Illumina sequencing, 10 genes (Zm00001d041327, Zm00001d033049, Zm00001d018178, Zm00001d047789, Zm00001d019518, Zm00001d018157, Zm00001d008230, Zm00001d022041, Zm00001d049387, Zm00001d014723) were randomly selected to conduct quantitative real-time PCR analysis using an Applied Biosystems 7500 Real-Time PCR System. GADPH (Zm00001d049641) was used as the endogenous control. cDNA synthesis was carried out using 1 μg of total RNA. The corresponding primers were designed using the Primer 3 online tool and are listed in Additional file 21: Table S2. The amplification program was performed according to the standard protocol of the Applied Biosystems 7500 Real-Time PCR System: 40 cycles of 98 °C for 2 min, 98 °C for 2 s, and 59 °C for 10 s, followed by a thermal denaturing step to generate the melting curves for amplification specificity verification. All reactions were run in triplicate, including non-template controls. Statistical analysis was performed using the 2-△△CT method [87].
Abbreviations
- ABA:
-
Abscisic acid
- ARF:
-
Auxin response factor
- BP:
-
Biological Process
- BR:
-
Brassinosteroid
- BRI:
-
Protein brassinosteroid insensitive 1
- CC:
-
Cellular Component
- CCNA:
-
Cyclin A
- CCNB:
-
Cyclin B
- CDK:
-
Cyclin-dependent kinase
- CDR:
-
Callus differentiating rate
- CK:
-
Control
- CPN:
-
Callus plantlet number
- CTK:
-
Cytokinin
- DEGs:
-
Differentially expressed genes
- EBF:
-
EIN3-binding F-box protein
- EC:
-
Embryonic callus
- EIN3:
-
Ethylene-insensitive protein 3
- FPKM:
-
Million mapped reads
- FtsH:
-
Cell division protease FtsH
- GA:
-
Gibberellin
- GID1:
-
Gibberellin receptor GID1
- GO:
-
Gene Ontology
- JA:
-
Jasmonic acid
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MF:
-
Molecular Function
- MYC2:
-
Transcription factor MYC2
- PAL:
-
Phenylalanine ammonia-lyase
- PIF4:
-
Phytochrome-interacting factor 4
- POD:
-
Peroxidase
- RAMs:
-
Root apical meristems
- SAM:
-
Shoot apical meristem
- SAUR:
-
SAUR family protein
- WOX:
-
WUSCHEL homeobox protein
- WUS:
-
WUSCHEL
References
Armstrong C, Green C. Establishment and maintenance of friable embryogenic maize callus and theinvolvement of L-proline. Planta. 1985;164(2):207–14.
Shen Y, Jiang Z, Yao X, Zhang Z, Lin H, Zhao M, Liu H, Peng H, Li S, Pan G. Genome expression profile analysis of the immature maize embryo during dedifferentiation. PLoS One. 2012;7(3):e0032237.
Bronsema FB, Van Oostveen WJ, Van Lammeren AA, et al. Comparative analysis of callus formation and regeneration on cultured immature maize embryos of the inbred lines A188 and A632. Plant Cell Tissue Organ Cult. 1997;50(1):57-65.
Prioli LM, Silva WJD, Söndahl MR. Tissue, Cell, and Protoplast Culture of Maize ( Zea mays, L.). Progress in Plant Cellular and Molecular Biology: Proceedings of the VIIth International Congress on Plant Tissue and Cell Culture, Amsterdam, The Netherlands, 24–29 June 1990[M]. Springer Science & Business Media. 2012;1990:38-43.
Tomes DT, Smith OS. The effect of parental genotype on initiation of embryogenic callus from elite maize (Zea mays L.) germplasm. Theor Appl Genet. 1985;70(5):505–9.
Armstrong CL, Green CE, Phillips RL, et al. Genetic control of plant regeneration from maize tissue cultures. Maize Genet Coop Newsl. 1985;59:92–3.
Armstrong CL, Romero-Severson J, Hodges TK. Improved tissue culture response of an elite maize inbred through backcross breeding and identifieation of chromosomal regions important for regeneration by RFLP analysis. Theor APPI Genet. 1992;84(5–6):755–62.
Krakowsky MD, Lee M, Garay L, et al. Quantitative trait loci for callus initiation and totipotency in maize ( Zea mays, L.). Theor Appl Genet. 2006;113(5):821–30.
Iwase A, Mita K, Nonaka S, et al. WIND1-based acquisition of regeneration competency in Arabidopsis and rapeseed. J Plant Res. 2015;128(3):389–97.
Sarkar AK, Luijten M, Miyashima S, et al. Conserved factors regulate signalling in Arabidopsis thaliana shoot and root stem cell organizers. Nature. 2007;446(7137):811–4.
Aida M, Beis D, Heidstra R, et al. The PLETHORA genes mediate patterning of the Arabidopsis root stem cell niche. Cell. 2004;119(1):109–20.
Galinha C, Hofhuis H, Luijten M, et al. PLETHORA proteins as dose-dependent master regulators of Arabidopsis root development. Nature. 2007;449(7165):1053–7.
Iwase A, Harashima H, Ikeuchi M, et al. WIND1 promotes shoot regeneration through transcriptional activation of ENHANCER OF SHOOT REGENERATION1 in Arabidopsis. Plant Cell. 2017;29(1):54–69.
Kareem A, Durgaprasad K, Sugimoto K, et al. PLETHORA genes control regeneration by a two-step mechanism. Current Biology Cb. 2015;25(8):1017–30.
Schoof H, Lenhard M, Haecker A, et al. The stem cell population of Arabidopsis shoot meristems in maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell. 2000;100(6):635–44.
Gallois JL, Nora FR, Mizukami Y, et al. WUSCHEL induces shoot stem cell activity and developmental plasticity in the root meristem. Genes Dev. 2004;18(4):375–80.
Chatfield SP, Capron R, Severino A, et al. Incipient stem cell niche conversion in tissue culture: using a systems approach to probe early events in WUSCHEL-dependent conversion of lateral root primordia into shoot meristems. Plant J. 2013;73(5):798–813.
Hecht V, Viellecalzada JP, Hartog MV, et al. The Arabidopsis SOMATIC EMBRYOGENESIS RECEPTOR KINASE 1 gene is expressed in developing ovules and embryos and enhances embryogenic competence in culture. Plant Physiol. 2001;127(3):803–16.
Zhang Y, Peng L, Wu Y, et al. Analysis of global gene expression profiles to identify differentially expressed genes critical for embryo development in Brassica rapa. Plant Mol Biol. 2014;86(4):425–42.
Cheng Y, Liu H, Cao L, et al. Down-regulation of multiple CDK inhibitor ICK/KRP genes promotes cell proliferation, callus induction and plant regeneration in Arabidopsis. Front Plant Sci. 2015;6:825.
Consortium T G O. The gene ontology project in 2008. Nucleic Acids Res. 2008;36(Database issue):D440–4.
Kanehisa M. The KEGG database. Novartis Found Symp. 2002;247(247):91.
Kanehisa M, Araki M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–4.
Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc. 2012;7(3):562.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106.
Pertea G. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5.
Khazaei M, Maaliamiri R, Talei AR, et al. Differential transcript accumulation of dhydrin and beta-glucosidase genes to cold-induced oxidative stress in chickpea. J Agric Sci Technol. 2015;17(3):725–34.
Tang W, Harris LC, Outhavong V, et al. Antioxidants enhance in vitro plant regeneration by inhibiting the accumulation of peroxidase in Virginia pine (Pinus virginiana mill.). Plant Cell Rep. 2004;22(12):871–7.
Jones DH. Phenylalanine ammonia-lyase: regulation of its induction, and its role in plant development. Phytochemistry. 1984;23(7):1349–59.
Rösler J, Schmid J. Maize phenylalanine ammonia-lyase has tyrosine ammonia-lyase activity. Plant Physiol. 1997;113(1):175–9.
Ritter H, Schulz GE. Structural basis for the entrance into the Phenylpropanoid metabolism catalyzed by phenylalanine Ammonia-Lyase. Plant Cell. 2004;16(12):3426.
Yamaguchi M, Kato H, Yoshida S, et al. Control of in vitro organogenesis by cyclin-dependent kinase activities in plants. Proc Natl Acad Sci U S A. 2003;100(13):8019.
Dante RA, Larkins BA, Sabelli PA. Cell cycle control and seed development. Front Plant Sci. 2014;5(1):493.
Wang G, Kong H, Sun Y, et al. Genome-wide analysis of the cyclin family in Arabidopsis and comparative phylogenetic analysis of plant cyclin-like proteins. Plant Physiol. 2004;135(2):1084.
Bieniossek C, Schalch T, Bumann M, et al. The molecular architecture of the metalloprotease FtsH. Proc Natl Acad Sci U S A. 2006;103(9):3066–71.
Liu C, Moon K, Honda H, et al. Enhanced regeneration of rice (Oryza sativa L.) embryogenic callus by light irradiation in growth phase. J Biosci Bioeng. 2001;91(3):319.
Moon HK, Stomp AM. Effects of medium components and light on callus induction, growth, and frond regeneration in Lemna gibba, (duckweed). Vitro Cell Dev Biol Plant. 1997;33(1):20–5.
Rikiishi K, Matsuura T, Maekawa M, et al. Light control of shoot regeneration in callus cultures derived from barley (Hordeum vulgare L.) immature embryos. Breed Sci. 2008;58(2):129–35.
Farhadi N, Panahandeh J, Azar AM, et al. Effects of explant type, growth regulators and light intensity on callus induction and plant regeneration in four ecotypes of Persian shallot ( Allium hirtifolium ). Sci Hortic. 2017;218:80–6.
Siddique AB, Islam SS. Effect of light and dark on callus induction and regeneration in tobacco (Nicotiana tabacum L.). Bangladesh J Bot. 2015;44:643–51.
Din ARJM, Ahmad FI, Wagiran A, et al. Improvement of efficient in vitro regeneration potential of mature callus induced from Malaysian upland rice seed (Oryza sativa cv. Panderas). Saudi J Biol Sci. 2016;23(1):S69–77.
Vass I, Cser K, Cheregi O, et al. Molecular mechanisms of light stress of photosynthesis. Ann N Y Acad Sci. 2007;1113(1):114–22.
Fassioli F, Dinshaw R, Arpin PC, et al. Photosynthetic light harvesting: excitons and coherence. J R Soc Interface. 2014;11(92):–20130901.
Woodward RB, Ayer WA, Beaton JM, et al. The total synthesis of chlorophyll. Tetrahedron. 1990;46(22):7599–659.
Sergio T, Alberto P, Stefano P, et al. Influence of light and shoot development stage on leaf photosynthesis and carbohydrate status during the adventitious root formation in cuttings of Corylus avellanaL. Front Plant Sci. 2015;6(943):1–14.
Sato-Nara K, Demura T, Fukuda H. Expression of photosynthesis-related genes and their regulation by light during somatic embryogenesis in Daucus carota. Planta. 2004;219(1):23–31.
Borisjuk L, Rolletschek H, Walenta S, et al. Energy status and its control on embryogenesis of legumes: ATP distribution within Vicia faba embryos is developmentally regulated and correlated with photosynthetic capacity. Plant J Cell Mol Biol. 2003;36(3):318–29.
Ge X, Zhang C, Wang Q, et al. iTRAQ protein profile differential analysis between somatic globular and cotyledonary embryos reveals stress, hormone, and respiration involved in increasing plantlet regeneration of Gossypium hirsutum L. J Proteome Res. 2014;14(1):268–278. 8.
Webb AA. The physiology of circadian rhythms in plants. New Phytologist. 2003;160(2):281-303.
Mcclung CR. Plant circadian rhythms. Plant Cell. 2006;18(4):792–803.
Gualberti G, Papi M, Bellucci L, et al. Mutations in the Dof zinc finger genes DAG2 and DAG1 influence with opposite effects the germination of Arabidopsis seeds. Plant Cell. 2002;14(6):1253–63.
Yanagisawa S, Sheen J. Involvement of maize Dof zinc finger proteins in tissue-specific and light-regulated gene expression. Plant Cell. 1998;10(1):75.
Chini A, Fonseca S, Fernandez G, et al. The JAZ family of repressors is the missing link in jasmonate signalling. Nature. 2007;448(7154):666–71.
Chico JM, Chini A, Fonseca S, et al. JAZ repressors set the rhythm in jasmonate signaling. Curr Opin Plant Biol. 2008;11(5):486.
Ravnikar M, Žel J, Plaper I, et al. Jasmonic acid stimulates shoot and bulb formation of garlic in vitro. J Plant Growth Regul. 1993;12(2):73.
Sakamoto T, Miura K, Itoh H, et al. An overview of gibberellin metabolism enzyme genes and their related mutants in Rice. Plant Physiol. 2004;134(4):1642.
Rikiishi K, Matsuura T, Ikeda Y, et al. Light inhibition of shoot regeneration is regulated by endogenous abscisic acid level in Calli derived from immature barley embryos. PLoS One. 2015;10(12):e0145242.
Su YH, Liu YB, Bai B, et al. Establishment of embryonic shoot-root axis is involved in auxin and cytokinin response during Arabidopsis somatic embryogenesis. Front Plant Sci. 2015;5:792.
Fehér A, Pasternak TP, Dudits D. Transition of somatic plant cells to an embryogenic state. Plant Cell Tiss Org Cult. 2003;74(3):201–28.
Cheng ZJ, Wang L, Sun W, et al. Pattern of auxin and cytokinin responses for shoot meristem induction results from the regulation of cytokinin biosynthesis by AUXIN RESPONSE FACTOR3. Plant Physiol. 2013;161(1):240–51.
Gou X, Yin H, He K, et al. Genetic evidence for an indispensable role of somatic embryogenesis receptor kinases in brassinosteroid signaling. PLoS Genet. 2012;8(1):e1002452.
Müssig C. Brassinosteroid-promoted growth. Plant Biol. 2010;7(2):110–7.
Aydin Y, Talas-Ogras T, Ipekçi-Altas Z, et al. Effects of brassinosteroid on cotton regeneration via somatic embryogenesis. Biologia. 2006;61(3):289–93.
Aydin Y, Ogras T, Altınkut A, İsmailoğlu I, Arican E, Gozukirmizi N. Cytohistological Studies During Cotton Somatic Embryogenesis With Brassinosteroid Application. IUFS Journal of Biology Research Articles. J Biol. 2010;33:33–9.
Dixon R, Paiva NL. Stress-induced Phenylpropanoid metabolism. Plant Cell. 1995;7(7):1085–97.
Yang H, Guo XL, Ran L, et al. Changes in morphology and biochemical indices in browning callus derived from Jatropha curcas hypocotyls. Plant Cell Tiss Org Cult. 2009;98(1):11–7.
Khosroushahi AY, Naderi-Manesh H, Simonsen HT. Effect of antioxidants and carbohydrates in callus cultures of Taxus brevifolia: evaluation of Browning, callus growth. Total Phenolics Paclitaxel Prod Bioimpacts. 2011;1(1):37.
Jittayasothorn Y, Lu J, Xu X, et al. A simple and highly efficient protocol for somatic embryogenesis and plant regeneration from proembryonic mass suspension culture in'Autumn Royal Seedless'. VITIS-J Grapevine Res. 2015;46(1):45.
Haddon L, Northcote DH. Correlation of the induction of various enzymes concerned with Phenylpropanoid and lignin synthesis during differentiation of bean callus (Phaseolus vulgaris L.). Planta. 1976;128(3):255–62.
Laukkanen H, Rautiainen L, Taulavuori E, et al. Changes in cellular structures and enzymatic activities during browning of scots pine callus derived from mature buds. Tree Physiol. 2000;20(7):467–75.
Malnoë A, Wang F, Girardbascou J, et al. Thylakoid FtsH protease contributes to photosystem II and cytochrome b6f remodeling in Chlamydomonas reinhardtii under stress conditions. Plant Cell. 2014;26(1):373.
Cho JW, Sun CP, Shin EA, et al. Cyclin D1 and p22 ack1, play opposite roles in plant growth and development. Biochem Biophys Res Commun. 2004;324(1):52–7.
Doerner PW. Cell cycle regulation in plants. Plant Physiol. 1994;106(3):823–7.
Yadav RK, Reddy GV. WUSCHEL protein movement and stem cell homeostasis. Plant Signal Behav. 2012;7(5):592–4.
Stahl Y, Wink RH, Ingram GC, et al. A signaling module controlling the stem cell niche in Arabidopsis root meristems. Curr Biol. 2009;19(11):909.
Shimizu R, Ji J, Kelsey E, et al. Tissue specificity and evolution of meristematic WOX3 function. Plant Physiol. 2009;149(2):841–50.
Ueda M, Zhang Z, Laux T. Transcriptional activation of Arabidopsis axis patterning genes WOX8/9 links zygote polarity to embryo development. Dev Cell. 2011;20(2):264–70.
Palovaara J, Hallberg H, Stasolla C, et al. Comparative expression pattern analysis of WUSCHEL-related homeobox 2 (WOX2) and WOX8/9 in developing seeds and somatic embryos of the gymnosperm Picea abies. New Phytol. 2010;188(1):122–35.
Lowe K, Wu E, Wang N, et al. Morphogenic Regulators Baby boom and Wuschel Improve Monocot Transformation. Plant Cell. 2016;28(9). https://doi.org/10.1105/tpc.16.00124.
Lee K, Park OS, Seo PJ. RNA-Seq analysis of the Arabidopsis transcriptome in pluripotent Calli. Mol Cells. 2016;39(6):484–94.
Wang X, Yang Z, Wang M, et al. The BRANCHING ENZYME1, gene, encoding a glycoside hydrolase family 13 protein, is required for in vitro plant regeneration in Arabidopsis. Plant Cell Tiss Org Cult. 2014;117(2):279–91.
Wang P, Han X, Ye Z, et al. Genome-wide high-resolution mapping of DNA methylation identifies epigenetic variation across embryo and endosperm in maize ( Zea may ). BMC Genomics. 2015;16(1):1–14.
Nishimura A, Ashikari M, Lin S, et al. Isolation of a rice regeneration quantitative trait loci gene and its application to transformation systems. Proc Natl Acad Sci U S A. 2005;102(33):11940–4.
Ge F, Luo X, Huang X, et al. Genome-wide analysis of transcription factors involved in maize embryonic callus formation. Physiol Plant. 2016;158(4):452–62.
Ma L, Liu M, Yan Y, et al. Genetic dissection of maize embryonic callus regenerative capacity using multi-locus genome-wide association studies. Front Plant Sci. 2018;9:561.
Pan G, Zhang Z, Wei X, et al. QTL analysis of maize ( Zea mays L.) embryo culturing capacity. Zuo Wu Xue Bao. 2006;32:7–13.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods. 2001;25(4):402–8.
Hu Y, Jiang Y, Han X, et al. Jasmonate regulates leaf senescence and tolerance to cold stress: crosstalk with other phytohormones. J Exp Bot. 2017;68(6):1361–9.
Zhang X, Long Y, Ge F, et al. A genetic study of the regeneration capacity of embryonic callus from the maize immature embryo culture. Hereditas. 2017;39:143–55.
Zhang X, Zhang H, Li LJ, et al. Characterizing the population structure and genetic diversity of maize breeding germplasm in Southwest China using genome-wide SNP markers. BMC Genomics. 2016;17:697.
Acknowledgments
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Funding
This work was supported by the Natural Science Foundation of China (31871637) and the Young Scientists Fund of Sichuan Province (2016JQ0008) and the Key Fund of the Department of Education of Sichuan Province (15ZA0004).
Availability of data and materials
The reference genome and genes of maize are available from RefGen_V4 (http://www.gramene.org/). Gene Ontology (GO) and pathway annotations and enrichment analyses were based on the Gene Ontology Database (www.geneontology.org) [21] and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway (www.genome.jp/kegg) [22], respectively.
Author information
Authors and Affiliations
Contributions
YS and GP designed the experiments. XZ, YW, YY, HP, YL, and YZ conducted the experiment and performed the analysis. XZ, YW, YS, ZJ, PL, CZ, HWP, and GP drafted the manuscript. All the authors critically revised and approval the final version of this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Base composition and quality of clean data. (JPG 1466 kb)
Additional file 2:
Figure S2. CorrelationHeatmap of AllSamples. (PDF 17 kb)
Additional file 3:
Table S3. The detailed information about the specific common DEGs of 141 and DH40; Table S4. The detailed information about the specific common DEGs of DH3732 and ZYDH381–1.). (XLSX 874 kb)
Additional file 4:
Table S5. Up specific common DEGs of the two high regeneration lines in 5 clusters; Table S6. Down specific common DEGs of the two high regeneration lines in 5 clusters; Table S7. Up specific common DEGs of the two low regeneration lines in 5 clusters; Table S8. Down specific common DEGs of the two low regeneration lines in 5 clusters.). (XLSX 195 kb)
Additional file 5:
Figure S3. GO analysis of specific common DEGs of 141 and DH40 (A. up-regulated gene; B. down-regulated gene) (JPG 1422 kb)
Additional file 6:
Table S9. List of GO analysis (BP) for the specific common DEGs of 141 and DH40 (All GO terms shown were significant at FDR ≤ 0.05); Table S10. List of GO analysis (CC) for the specific common DEGs of 141 and DH40 (GO terms shown were significant at FDR ≤ 0.05 for up-regulated genes, and P-value ≤0.05 for down-regulated genes); Table S11. List of GO analysis (MF) for the specific common DEGs of 141 and DH40 (All GO terms shown were significant at FDR ≤ 0.05).). (DOCX 45 kb)
Additional file 7:
Figure S4. GO analysis of specific common DEGs of ZYDH381–1 and DH3732 (A. up-regulated gene; B. down-regulated gene) (JPG 2215 kb)
Additional file 8:
Table S12. List of GO analysis (BP) for the specific common DEGs of DH3732 and ZYDH381–1 (All GO terms shown were significant at FDR ≤ 0.05); Table S13. List of GO analysis (CC) for the specific common DEGs of DH3732 and ZYDH381–1 (All GO terms shown were significant at FDR ≤ 0.05); Table S14. List of GO analysis (MF) for the specific common DEGs of DH3732 and ZYDH381–1 (All GO terms shown were significant at FDR ≤ 0.05).). (DOCX 20 kb)
Additional file 9:
Table S15. List of enriched pathways for the specific common DEGs of 141 and DH40 at three stages (stage I II and III). (XLSX 32 kb)
Additional file 10:
Table S16. List of enriched pathways for specific common DEGs of ZYDH381–1 and DH3732 at three stages (stage I II and III). (XLSX 44 kb)
Additional file 11:
Table S17. List of DEGs related to embryonic callus regeneration. (DOCX 20 kb)
Additional file 12:
Figure S5. KEGG Pathway Map of Photosynthesis for the specific common DEGs of 141 and DH40. (PNG 43 kb)
Additional file 13:
Figure S6. KEGG Pathway Map of Protein and Chlorophyll Metabolism. (PNG 42 kb)
Additional file 14:
Figure S7. KEGG Pathway Map of Photosynthesis-Antenna Protein. (PNG 50 kb)
Additional file 15:
Figure S8. KEGG Pathway Map of Circadian Rhythm-Plant. (PNG 13 kb)
Additional file 16:
Figure S9. KEGG Pathway Map of Plant Hormone Signal Transduction for the specific common DEGs of 141 and DH40. (PNG 445 kb)
Additional file 17:
Figure S10. KEGG Pathway Map of Plant Hormone Signal Transduction for the specific common DEGs of ZYDH381–1 and DH3732. (PNG 152 kb)
Additional file 18:
Figure S11. KEGG Pathway Map of Phenylpropanoid Biosynthesis. (PNG 21 kb)
Additional file 19:
Figure S12. Expression clustering of specific common DEGs involved in EC regeneration. (JPG 4769 kb)
Additional file 20:
Table S1. Plant culture medium formula. (DOCX 14 kb)
Additional file 21:
Table S2. Primers of real-time qRT-PCR assay used in this study. (DOCX 15 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Zhang, X., Wang, Y., Yan, Y. et al. Transcriptome sequencing analysis of maize embryonic callus during early redifferentiation. BMC Genomics 20, 159 (2019). https://doi.org/10.1186/s12864-019-5506-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5506-7