
Abstract
Background
Lipopolysaccharide (LPS) from Gram-negative bacteria cause innate immune responses in animals and plants. The molecules involved in LPS signaling in animals are well studied, whereas those in plants are not yet as well documented. Recently, we identified Arabidopsis AtLBR-2, which binds to LPS from Pseudomonas aeruginosa (pLPS) directly and regulates pLPS-induced defense responses, such as pathogenesis-related 1 (PR1) expression and reactive oxygen species (ROS) production. In this study, we investigated the pLPS-induced transcriptomic changes in wild-type (WT) and the atlbr-2 mutant Arabidopsis plants using RNA-Seq technology.
Results
RNA-Seq data analysis revealed that pLPS treatment significantly altered the expression of 2139 genes, with 605 up-regulated and 1534 down-regulated genes in WT. Gene ontology (GO) analysis on these genes showed that GO terms, “response to bacterium”, “response to salicylic acid (SA) stimulus”, and “response to abscisic acid (ABA) stimulus” were enriched amongst only in up-regulated genes, as compared to the genes that were down-regulated. Comparative analysis of differentially expressed genes between WT and the atlbr-2 mutant revealed that 65 genes were up-regulated in WT but not in the atlbr-2 after pLPS treatment. Furthermore, GO analysis on these 65 genes demonstrated their importance for the enrichment of several defense-related GO terms, including “response to bacterium”, “response to SA stimulus”, and “response to ABA stimulus”. We also found reduced levels of pLPS-induced conjugated SA glucoside (SAG) accumulation in atlbr-2 mutants, and no differences were observed in the gene expression levels in SA-treated WT and the atlbr-2 mutants.
Conclusion
These 65 AtLBR-2-dependent up-regulated genes appear to be important for the enrichment of some defense-related GO terms. Moreover, AtLBR-2 might be a key molecule that is indispensable for the up-regulation of defense-related genes and for SA signaling pathway, which is involved in defense against pathogens containing LPS.
Similar content being viewed by others
Background
The endotoxin lipopolysaccharide (LPS), a major component of the outer membranes of Gram-negative bacteria, is one of the most studied pathogen-associated molecular patterns (PAMPs). The perception of LPS triggers various defense responses in plants and animals [1]. In plants, LPS-induced defense responses have been well studied; these include LPS-induced generation of reactive oxygen species (ROS) and nitrogen oxide (NO), salicylic acid (SA) accumulation, expression of pathogenesis-related (PR) genes, and stomatal closure [2,3,4,5]. SA, in particular, is an important signal molecule in plant defense. The accumulation of SA is involved in local defenses as well as in systemic acquired resistance (SAR) [6].
The LPS recognition mechanism has been well studied in animals. In mammals, LPS-binding protein (LBP) and bactericidal/permeability-increasing protein (BPI) play important roles in the regulation of immune responses against LPS [7]. Although both the proteins directly bind to LPS, BPI inhibits whereas LBP enhances the binding of LPS to Toll-like receptor 4, a mammalian LPS receptor. Recently, we identified two Arabidopsis LBP/BPI-related proteins, AtLBR-1 and AtLBR-2 [8]. When we incubated recombinant forms of both AtLBR-1 and AtLBR-2 with Pseudomonas aeruginosa LPS (pLPS) separately, they exhibited the capability to bind to it directly; atlbr mutants showed deficiencies in pLPS-induced PR1 gene expression and ROS generation. We predicted that AtLBR-2 would be more important than AtLBR-1 in the induction of defense responses to LPS because the binding affinity of AtLBR-2 for LPS appeared higher than that of AtLBR-1, and AtLBR-2 is located in the apoplastic region.
In the present study, we investigated the importance of AtLBR-2 in the dynamic changes in Arabidopsis transcriptome in response to LPS treatment. To achieve this goal, we performed a transcriptome analysis using high-throughput mRNA sequencing (RNA-Seq). RNA-Seq analysis using WT and the atlbr-2-1 identified 65 AtLBR-2-dependent genes that were up-regulated after LPS treatment. These 65 genes appear to be important for the enrichment of some defense-related gene ontology (GO) terms. Our findings highlight the indispensable role of AtLBR-2 in defense signaling mechanism against LPS.
Results
Transcriptomic analysis of P. aeruginosa LPS-responsive genes in WT Arabidopsis
To examine and compare the LPS-induced transcriptional changes between wild-type (WT) and the atlbr-2-1, we treated them with LPS from P. aeruginosa (pLPS); total RNA was extracted and RNA-Seq analysis was performed.
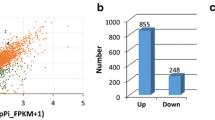
Firstly, we analyzed the pLPS-responsive genes in the WT. The RNA-Seq data obtained from untreated WT were compared with that of pLPS-treated WT. We observed that the transcript levels of 2139 genes changed significantly in pLPS-treated WT. Of these, 605 genes were identified as up-regulated genes in pLPS-treated WT (Fig. 1a). Moreover, 1534 genes were identified as down-regulated genes in pLPS-treated WT (Fig. 1b). We performed gene ontology (GO) analysis of these genes using the functional annotation chart of DAVID. The biological process (BP) GO classification of the 605 up-regulated genes identified 33 GO terms (P < 0.01, Fig. 2, blue line) (Additional file 1: Table S1), including not only defense-related GO terms, but also several metabolic processes-related terms. This finding corresponded with the results reported from transcriptional analysis on Arabidopsis seedlings treated with LPS from Burkholderia cepacia [9]. Defense-related GO terms included, “response to bacterium”, “response to SA stimulus”, “response to abscisic acid (ABA) stimulus”, “response to jasmonic acid stimulus”, “response to ROS”, and “response to wounding”. In contrast, 1534 down-regulated genes were classified via 43 GO terms (P < 0.01) (Additional file 1: Table S2). Interestingly, defense-related GO terms, other than “response to bacterium”, “response to SA stimulus”, “response to ABA stimulus”, were also common in these 43 GO terms. These findings suggested that up-regulation, but not down-regulation, of genes related to bacterial responses may be a characteristic of normal pLPS-induced gene expression. It can also be inferred that SA- and ABA-related pathways may be important for the up-regulation, but not down-regulation, of genes after pLPS treatment.

The number of differentially expressed genes in pLPS-treated WT plants. Each RNA-Seq data set obtained from untreated WT, pLPS-treated atlbr-2-1, and untreated atlbr-2-1 plants, were compared with that obtained from pLPS-treated WT plants. The numbers in the parentheses indicate the number of genes identified as up-regulated (a) or down-regulated (b) in the pLPS-treated WT plants. The numbers of genes, which were up- or down-regulated only in pLPS-treated WT but not in the other three conditions, are indicated in bold type

GO classification of pLPS-responsive up-regulated genes. BP GO terms obtained from the GO analysis of 605 pLPS-induced up-regulated genes in WT are shown with the blue line. The same analysis conducted with 540 genes, which excluded 65 AtLBR-2-dependent up-regulated genes from the 605 genes, are shown with the red line. The red font highlights no enriched GO terms in the 540 genes. An arrow indicates the GO term identified only in 540 genes. Scale of y axis shows the percentage of genes that are annotated for each biological process. P < 0.01
Furthermore, cellular component (CC) GO analysis showed that 23.0% of the 605 up-regulated genes were categorized as “endomembrane system”; also, 22.9% and 11.9% of the 1534 down-regulated genes were categorized as “endomembrane system” and “intrinsic to membrane”, respectively (Additional file 1: Table S3). These results indicated that genes activated in the membrane-related region were most affected by the pLPS treatment.
Identification of AtLBR-2-dependent up- or down-regulated genes
To elucidate the importance of AtLBR-2 in pLPS-induced transcriptional responses, we identified the genes that were up-regulated in an AtLBR-2-dependent manner after 24 h of pLPS treatment. We compared each of the three RNA-Seq data with that of pLPS-treated WT (Fig. 1). Furthermore, we studied the genes that were up- or down-regulated only in the pLPS-treated WT plants and not in the other 3 data sets; these were then identified as AtLBR-2-dependent up- or down-regulated genes. A total of 65 candidate genes were identified to be AtLBR-2-dependent up-regulated genes (Fig. 1a, Table 1). We focused on these 65 genes and analyzed them further; only two genes, “unfertilized embryo sac 11 (UNE11: AT4G00080)” and the gene for an “uncharacterized protein (AT3G20340)”, were identified to be AtLBR-2-dependent down-regulated genes (Fig. 1b).
AtLBR-2 is indispensable for pLPS-induced defense-related GO terms
To determine the importance of the 65 AtLBR-2-dependent up-regulated genes in the GO classification of 605 up-regulated genes, we performed GO analysis for 540 genes, excluding the above-mentioned 65 genes from the 605 up-regulated genes (P < 0.01, Fig. 2, red line) (Additional file 1: Table S1). Comparing the results of the GO analysis revealed that 540 genes showed no enrichment for defense-related GO terms, including responses to bacterium, SA stimulus, ABA stimulus, wounding, and drug. These results highlight the importance of 65 genes in defense-related GO terms, and demonstrate that AtLBR-2 might be indispensable for the expression of pLPS-induced defense-related genes.
Characterization of 65 AtLBR-2-dependent up-regulated genes
The details of the 65 pLPS-induced AtLBR-2-dependent up-regulated genes are shown in Table 1. We expected that PR1 would be one of the 65 AtLBR-2-dependent up-regulated genes, because we previously reported that atlbr mutants showed deficiencies in the expression of pLPS-induced PR1 (AT2G14610) [8]. Consistent with our prediction, as shown in Table 1, PR1 was the most differentially-expressed gene among the AtLBR-2-dependent up-regulated genes. Furthermore, most of the 65 genes have been reported to be associated with plant–pathogen interaction and with SA-regulated responses.
These 65 genes were also annotated to each of the three GO categories; CC, molecular function (MF), and BP. These genes were assigned the CC GO terms, “endomembrane system” (27.1%), “membrane” (17.1%), and “membrane-bound organelle” (11.4%, Table 2). Thus, approximately 50% of these genes were categorized as membrane-related CC GO terms, highlighting the inter-relationship between the proteins encoded by these genes and AtLBR-2 [8]. Furthermore, the MF GO terms related to catalytic (44.1%), binding (35.3%), and transporter (11.8%) activities were mostly enriched. They were also predicted to participate in 25 BP GO terms, mainly in the “response to stress” (12.0%), “cellular metabolic process” (10.6%), “response to biotic stimulus” (9.2%), and in “response to other organisms” (9.1%). Thus, interestingly, approximately 50% of these genes were involved in response to stress and stimuli. To further define the functions of these 65 genes, the enriched pathways were identified by KOBAS (Table 3). The pathway analysis also revealed that the 65 genes were involved in defense-related pathways, including “camalexin biosynthesis”, “glutathione-mediated detoxification II”, and “plant–pathogen interaction”.
atlbr-2-1 mutants showed defect in up-regulation of six pLPS-induced genes
To confirm the RNA-Seq results of AtLBR-2-dependent up-regulated genes, we assigned and tested several genes involved in plant–pathogen interaction by conducting quantitative RT-PCR (qRT-PCR) using appropriate primers (Additional file 1: Table S4; Fig. 3). In all of the tested 6 genes, putative cytochrome P450 (CYP71A13), late upregulated in response to Hyaloperonospora parasitica (LURP1), plant natriuretic peptide A (PNP-A), avrRpt2-induced gene 1 (AIG1), glutathione S-transferase 7 (GSTF7), and pleiotropic drug resistance 12 (PDR12), we could detect the significant differences between WT and the atlbr-2-1 at 24 h of pLPS treatments, indicating that AtLBR-2-dependent up-regulated genes identified by RNA-Seq were also confirmed by qRT-PCR. However, the expression levels of these 6 genes were not completely abolished in pLPS-treated atlbr-2-1 mutants. These results suggested the possibility that AtLBR-1, the paralog of AtLBR-2, may compensate for the absence of the AtLBR-2 gene. Therefore, we conducted qRT-PCR analysis for these 6 genes with the atlbr-1 mutant seedlings by the same method that was used on the atlbr-2-1 mutant (Additional file 1: Figure S1) [8]. Similar to the results shown in Fig. 3, we detected significant differences in all tested genes in pLPS-treated WT and atlbr-1 mutants, suggesting that AtLBR-1 might also play an important role in some AtLBR-2-dependent up-regulated genes. Furthermore, to exclude the possibility of contamination by other bacterial components, we purified the commercial pLPS. Seedlings were treated with purified pLPS and were analyzed by qRT-PCR (Additional file 1: Figure S2). Similar to the results shown in Fig. 3, we could detect the significant differences between WT and atlbr-2-1 in all of the genes tested at 24 h of purified pLPS treatments, confirming that this phenomenon was not caused by contamination.

qRT-PCR analysis of six AtLBR-2-dependent up-regulated genes. Among the 65 pLPS-induced AtLBR-2-dependent up-regulated genes, 6 defense-related genes (CYP71A13, LURP1, PNP-A, AIG1, GSTF7, and PDR12) were randomly selected and analyzed by qRT-PCR in WT and atlbr-2-1 plants treated with pLPS. The mean expression values were calculated from the results of three independent experiments. Means ± standard errors are presented. Significant differences among the means were determined by two-way ANOVA followed by post hoc Bonferroni test compared to WT plants; *P < 0.05, **P < 0.01, ***P < 0.001
Although we did not observe read counts from the region downstream of the T-DNA insertion site of AtLBR-2 (Additional file 1: Figure S3), the possibility of slight expression of AtLBR-2 is not excluded because the T-DNA insertion site of the atlbr-2-1 is located just next to the stop codon of the gene. Thereafter, we conducted qRT-PCR using the cDNA obtained from another pLPS-treated T-DNA insertion line, atlbr-2-2 [8]. Similar to that in the atlbr-2-1, we could detect the significant differences between pLPS-treated WT and the atlbr-2-2 (Additional file 1: Figure S4).
Validation of RNA-Seq data by qRT -PCR
To validate RNA-Seq results, we conducted the expression analysis by qRT-PCR for randomly selected pLPS-responsive genes, including AtLBR-2-dependent or -independent up- or down-regulated genes. Figure 4 shows a comparison between the results from qRT-PCR and RNA-Seq analysis. For all 20 tested genes, transcript levels determined by qRT-PCR analysis were similar to those detected using RNA-Seq, indicating the reliability of the RNA-Seq data.

Comparison of RNA-Seq results with those of qRT-PCR. The Log2FC values for transcript levels observed in RNA-Seq data (white bar) of randomly selected 20 genes, including AtLBR-2-dependent -independent up- or down-regulated genes, were compared to the results obtained from qRT-PCR (gray bar). AtLBR-2-dependent up- or down-regulated genes are indicated with asterisk: CYP71A13, LURP1, PNP-A, AIG1, GSTF7, PDR12, UNE11, and AT3G20340. AtLBR-2-independent up- or down-regulated genes are indicated without asterisk: HVA22B (AT5G62490), HVA22 homologue B; PDF1.3 (AT2G26010), plant defensin 1.3; CLE21 (AT5G64800), clavata3/ESR-related 21; HAI2 (AT1G07430), highly ABA-induced 2; LEA4–5 (AT5G06760), late embryogenesis abundant 4–5; HR2 (AT3G50460), homolog of RPW8 2; ATH2 (AT3G47740), A. thaliana ABC2 homolog 2; MES13 (AT1G26360), methyl esterase 13; M17 (AT2G41260), late embryogenesis abundant gene; PR12 (AT1G75830); PP2-A6 (AT5G45080), phloem protein 2-A6; PROPEP3 (AT5G64905), elicitor peptide 3 precursor
A proposed pathway: AtLBR-2-mediated SAG accumulation and following SA-related gene expression
In the WT, pLPS treatment induced differential expression of many genes related to SA, a potent inducer of pathogen-induced defense responses (Additional file 1: Table S5). Among these, 14 genes were identified as AtLBR-2-dependent up-regulated genes. In addition, 65 AtLBR-2-dependent up-regulated genes were responsible for the enrichment of GO term “response to SA stimulus” (Fig. 2). These RNA-Seq data analyses suggested the association of AtLBR-2 with pLPS-induced SA signaling. Therefore, first, we investigated whether pLPS-induced SA accumulation levels were altered in atlbr-2-1 plants. Treatment of both WT and the atlbr-2-1 with pLPS did not result in significant changes in the content of free SA (Fig. 5a, left panel). In contrast, the accumulation levels of conjugated SA glucoside (SAG) between WT and the atlbr-2-1 showed significant differences at 8 h after pLPS treatment (Fig. 5a, right panel). These results indicated that AtLBR-2 has an important role in pLPS-induced SAG accumulation. Furthermore, to confirm the relationship between AtLBR-2-mediated SAG accumulation and SA-related gene expression, we investigated the expression levels of 3 AtLBR-2-dependent up-regulated genes, PR1, LURP1, and PDR12, in SA-treated WT plants and atlbr-2-1 mutants by qRT-PCR. These 3 genes are known as SA-related genes, and atlbr-2-1 mutants showed significant differences in the expression of these genes after pLPS treatment ([8] and Fig. 3). As shown in Fig. 5b, we observed the up-regulation of all tested genes in the SA-treated atlbr-2-1 to be at the same level, as or more, than that of WT plants. These results suggested that AtLBR-2 may act upstream of the SA signaling (SAG accumulation) pathway induced by pLPS treatment.

pLPS-induced free SA and SAG accumulation and SA-induced gene expression. (a) Quantification of the total levels of free SA and SAG were measured by HPLC using samples extracted from Arabidopsis treated with pLPS for the indicated time. (b) Among the 65 pLPS-induced AtLBR-2-dependent up-regulated genes, the expression of the 3 SA-related genes (PR1, LURP1, and PDR12) were analyzed by qRT-PCR in WT and the atlbr-2-1 mutants plants treated with SA. The mean values were calculated from the results of three independent experiments. Means ± standard errors are presented. Significant differences among the means were determined by two-way ANOVA followed by post hoc Bonferroni test compared to WT plants; *P < 0.05, **P < 0.01. FW, fresh weight
Discussion
Our primary interest in this study was in understanding the role of AtLBR-2 in LPS-induced plant defense responses. In a previous study, we concluded that apoplast-localized AtLBR-2 might play an important role in binding and transferring LPS to the LPS receptor. However, the functional properties of AtLBR-2 have not been characterized in detail. Therefore, in this study, we analyzed, for the first time, the effect of AtLBR-2 on transcriptional changes involved in pLPS treatment using RNA-Seq technology. By our RNA-Seq analysis, we identified that pLPS-induced up-regulated genes were associated with defense-related, as well as metabolism-related processes, which is consistent with a previous study that used LPS from B. cepacia [9]. Furthermore, we found a strong association between AtLBR-2 and SA by our RNA-Seq analysis and, in fact, we demonstrated the reduced level of pLPS-induced SAG accumulation in the atlbr-2-1, and the SA-induced normal gene expression in atlbr-2-1 mutants. Our data revealed the importance of AtLBR-2 in SA-mediating signaling pathway in response to LPS or when triggered by LPS.
AtLBR-2 and SA signaling
In this study, we identified 65 AtLBR-2-dependent genes that were up-regulated after pLPS treatment. The pathway analysis of these 65 genes revealed a significant enrichment of defense-related pathways. In fact, 44 of these genes encode proteins related to defense responses, and 14 genes, including PR1, LURP1, tyrosine aminotransferase 3 (TAT3), azelaic acid induced 1 (AZI1), peptide transporter 3 (PTR3), homolog of RPW8 4 (HR4), cysteine-rich receptor-like kinase (RLK) 5 (CRK5), elicitor-activated gene 3–2 (ELI3–2), flg22-induced RLK 1 (FRK1), glutathione S-transferase 6 (GSTF6), cell wall-associated kinase 1 (WAK1), PDR12, AT2G25510, and AT5G03350, are known to be induced by SA (Additional file 1: Table S5). The SA signaling is mediated by at least two mechanisms, one requiring the NPR1 (NON-EXPRESSOR OF PR1) and the second, which is independent of NPR1 [6]. PR1 is well known as a marker for NPR1-dependent SA-induced pathway. In addition, Blanco et al. identified some NPR1-dependent SA-induced genes, including HR4, WAK1, and AT5G03350 by microarray analysis in SA-treated WT and npr1–1 plants [10]. Furthermore, they also identified NPR1-independent SA-induced genes, including TAT3 and AT2G25510. These facts, along with our findings in this study, suggested the possibility that AtLBR-2 plays an important role in both NPR1-dependent and -independent signaling pathways triggered by LPS. Furthermore, we demonstrated that LPS-induced SAG accumulation was dependent on AtLBR-2. The LPS-induced accumulation of SAG, but not of SA, has been observed in previous studies, and might cause the transient production of SA and the stable accumulation of SAG [11]. In addition, the SA-treated atlbr-2-1 showed up-regulation of SA-related genes at levels similar to or more than those exhibited by WT plants. These results as well as our previous findings support the hypothesis that AtLBR-2 binds to LPS directly in the apoplastic region of Arabidopsis and, subsequently, induces the accumulation of SA (or SAG), leading to the activation of both NPR1-dependent and -independent signaling pathways and the gene expression that follows them [8].
Interestingly, NPR1 expression appeared to be up-regulated after inoculation with P. syringae pv. tomato (Pst) DC3000 or upon SA treatment [12, 13]; however, no difference was observed between the untreated and pLPS-treated WT plants in the present study (FDR < 0.01). The WRKY transcription factors, including WRKY18, WRKY38, and WRKY53, reported to be the targets of NPR1 during SAR [14], appear to be down-regulated in the pLPS-treated WT plants (Additional file 1: Table S5). Shah suggested the existence of a negative feedback loop involving NPR1, which regulates the accumulation of SA [6]. Moreover, in the present study, we showed that SAG concentration returned to the basal levels after 24 h of pLPS treatment. Therefore, based on these observations, we speculated that long-term treatment of pLPS (24 h) might induce SA negative feedback loop involving NPR1 suppression and result in a decline in SA (SAG) to basal levels.
AtLBR-2 and camalexin
The 65 AtLBR-2-dependent up-regulated genes included CYP71A13 and phytoalexin deficient 3 (PAD3). These genes encode cytochrome P450 enzymes, which contribute to the enrichment of the “camalexin biosynthesis” pathway. Camalexin is an indole alkaloid phytoalexin produced by Arabidopsis that is thought to be important for resistance to necrotrophic fungal pathogens. A previous study revealed the LPS-induced camalexin production in plants [15]. CYP71A13 catalyzes the conversion of indole acetaldoxime to indole-3-acetonitrile, an intermediate in the camalexin biosynthesis [16]. PAD3 catalyzes the conversion of dihydrocamalexic acid to camalexin, which is the last step of camalexin biosynthesis [17]. Interestingly, Nafisi et al. demonstrated that the expression levels of CYP71A13 and PAD3 were coregulated in response to infection by P. syringae [16]. Furthermore, SA is required for camalexin synthesis, which is mediated by an NPR1-independent pathway [18, 19]. These findings from previous research and the RNA-Seq analysis performed in this study led to the hypothesis that the binding and transfer of LPS to LPS receptor by AtLBR-2 and further SA (SAG) induction might be necessary to activate efficient LPS-induced camalexin biosynthesis. More research is needed to better understand the relationship between AtLBR-2 and camalexin biosynthesis in Arabidopsis.
AtLBR-2 and ATPase
Four genes (AT1G43910, AT3G28540, AT3G28580, and AT3G63380) encoding proteins that function as an ATPase were also among the 65 AtLBR-2-dependent up-regulated genes. Relationship between the four genes and their defense responses have been reported previously. AT1G43910 and AT3G28540 were consistently higher in sni1, a transcription repressor of NPR1, when compared with the WT plants [20]. AT3G28580 has been used as singlet oxygen (1O2)-responsive gene [21, 22]. 1O2 is a singular ROS that can be produced by phytotoxins during plant–pathogen interactions [23]. In addition, Frei dit Frey showed that AT3G63380 (auto-inhibited Ca2+-ATPase 12) cannot interact with FLS2, the bacterial flagellin peptide flg22 receptor, but might contribute to the control of cytosolic Ca2+ levels during flg22 responses [24]. Furthermore, a previous report described the relationship between the plasma membrane ATPase of plants and PAMPs-induced rapid extracellular alkalinization [25]. These reports lead us to speculate that AtLBR-2 might be related to LPS-induced ATPase-related responses, e.g. extracellular alkalinization and Ca2+ influx/efflux.
AtLBR-2 and protein kinases
For understanding the LPS recognition mechanism(s), it is essential to investigate the LPS receptor that binds to LPS and initiates a signaling cascade inside the cell via its kinase activity. The well known PAMPs, including flg22, bacterial elongation factor Tu (EF-Tu) peptide elf18, and fungal cell wall component chitin, are recognized by pattern recognition receptors, flagellin-sensitive 2 (FLS2), EF-Tu receptor (EFR), and chitin elicitor receptor kinase 1 (CERK1), respectively [26]. Interestingly, the genes induced or repressed by PAMPs are clearly correlated. The flg22 treatment induced the expression not only of EFR and CERK1, in addition to that of FLS2 [27]. In this study, we identified seven AtLBR-2-dependent up-regulated genes, which encode protein kinases (CRK5, CRK38, FRK1, WAK1, AT1G51820, AT1G67520, and AT4G17660). The bulb-type lectin S-domain-1 RLK LORE (AT1G61380), which is required for sensing of LPS from Pseudomonas and Xanthomonas species [28], was not, but lectin protein kinase family protein (AT1G67520) was included in the list. AT1G67520 is known as G-type lectin RLK and is similar to LORE; however, it lacks the transmembrane region [29]. Interestingly, both flg22 and elf26 treatments induced LORE and AT1G67520 [27]. These results suggest that AT1G67520 might be involved in recognition of LPS or other related PAMPs. The LPS-responsive S-domain RLK (Nt-Sd-RLK) was also identified in Nicotiana tabacum [30]. Signal transduction via RLKs seems to be required for the activation of LPS-induced plant defense responses. We also focused on CRK5, which is potential target gene of WRKY transcription factors. Chen et al. reported that CRK5 expression was up-regulated by SA treatment and constitutive over-expression of CRK5 led to increased resistance to Pst DC3000, which was associated with rapidly induced expression of PR1 after pathogen infection [31]. Moreover, we also focused on leucine-rich repeat (LRR) protein kinase family protein encoded by AT1G51820, because FLS2, EFR, and their co-receptor brassinosteroid insensitive 1 (BRI1)-associated receptor kinase 1 (BAK1) belong to the LRR-RLK family. Interestingly, AT1G51820 expression was up-regulated after infection with oomycete downy mildew pathogen [32]. These studies suggest that AtLBR-2-related protein kinases might be involved in the perception of PAMP. We speculate on possible LPS perception systems via unknown protein kinases other than LORE or those, which cooperate with LORE.
Conclusion
In summary, we have reported here the first analysis of the genome-wide effect of AtLBR-2 on pLPS-induced gene expression. The transcriptome analyses performed in this study identify the 65 AtLBR-2-dependent up-regulated genes, and reveal the indispensable role of AtLBR-2 in the up-regulation of pLPS-induced genes associated with defense responses. Further experiments also suggested the existence of an SA-mediated LPS signaling system via AtLBR-2. Thus, we suggest that 65 AtLBR-2-related proteins might be the key candidate molecules in LPS-induced defense mechanisms in plants.
Methods
Arabidopsis growth conditions and pLPS or SA treatment
Arabidopsis ecotype Col-0 was used as the wild-type (WT) plants in this study. All mutants were in the Col background. After 5 d of growth on MS agar plates at 22–24 °C under a 16 h light/8 h dark cycle, the seedlings were transferred to two separate liquid MS medium supplemented with 100 μg/ml LPS from P. aeruginosa serotype 10 (pLPS; Sigma-Aldrich Japan, Tokyo, Japan) and 200 μM SA (Wako, Osaka, Japan) that were then kept under continuous light conditions for 24 h and 6 h, respectively. Untreated seedlings were used as controls (0 h). The experiments were performed in three biological replicates.
RNA sample preparation and high-throughput sequencing
After the treatment, the seedlings were powdered in liquid nitrogen and RNA was extracted using the plant total RNA extraction Miniprep system (Viogene, Taipei, Taiwan) according to the manufacturer’s instructions. The quality of total RNA obtained from pLPS-treated or untreated seedlings was evaluated using the value of RNA Integrity Number (RIN) in the Agilent Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA). The total RNA concentration was measured using a Qubit RNA assay kit (Thermo Fisher Scientific, Waltham, MA, USA). The mRNAs were purified from 100 ng total RNAs by NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs, Ipswich, MA, USA), according to the manufacturer’s instructions. The sequencing libraries were prepared by NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA, USA), according to the manufacturer’s instructions. The quality of the libraries was assessed by a microchip electrophoresis system (MCE-202 MultiNA; Shimadzu, Kyoto, Japan) and their quantities were measured by Qubit dsDNA BR assay kit (Thermo Fisher Scientific, Waltham, MA, USA). After equimolar amounts of the libraries were pooled, they were used for paired-end read sequencing (2 × 101 bp) on Illumina Hiseq 4000 (Illumina, San Diego, CA, USA).
Read mapping and transcript assembly
After the quality evaluation and removal of adapter-containing reads, more than 88% left and right reads could be mapped to the Arabidopsis TAIR 10 genomes using TopHat software (http://www.ccb.jhu.edu/software/tophat/) (Additional file 1: Table S6) [33]. The concordant pair alignment rate was more than 86%. The transcripts were assembled and fragments per kilobase of transcript per million fragments mapped (FPKM) were estimated using Cufflinks software (http://cole-trapnell-lab.github.io/cufflinks/) (Additional file 2: Table S7) [33].
Identification of AtLBR-2-dependent up- or down-regulated genes
The differential expression between pLPS-treated WT and the three other data sets (untreated WT, pLPS-treated atlbr-2-1, and untreated atlbr-2-1) were calculated in R packages, edgeR (https://bioconductor.org/packages/release/bioc/html/edgeR.html) [34] and TCC (http://bioconductor.org/packages/release/bioc/html/TCC.html) [35] using the read counts of each data set, which were calculated by HTSeq-count (https://htseq.readthedocs.io/en/release_0.9.1/index.html) [36]. When compared to RNA-Seq data obtained from pLPS-treated WT, genes with negative Log fold change (Log2FC) value changes (FDR; false discovery rate < 0.01, Log2FC < −1.35) were identified as up-regulated genes in pLPS-treated WT. In contrast, genes with positive Log2FC value changes (FDR < 0.01, Log2FC > 1.35) were identified as down-regulated genes in pLPS-treated WT. Commonly up-regulated or down-regulated genes were investigated and 65 and 2 genes were identified as AtLBR-2-dependent up- and down-regulated genes, respectively.
Gene ontology (GO) enrichment analysis
The GO enrichment analysis for pLPS-induced up- or down-regulated genes in the WT plants was performed using functional annotation charts of DAVID Bioinformatics Resources 6.7 (https://david-d.ncifcrf.gov/) [37]. The distributions of GO terms at levels 1 or 2 for CC, MF, and BP for the 65 AtLBR-2-dependent up-regulated genes were analyzed using the VirtualPlant 1.3 online tool (http://virtualplant.bio.nyu.edu/cgi-bin/vpweb/) [38].
Pathway analysis
The pathway analysis of the 65 AtLBR-2-dependent up-regulated genes was performed via KOBAS 3.0 (http://kobas.cbi.pku.edu.cn/), which uses the BioCyc (https://biocyc.org/), KEGG (http://www.genome.jp/kegg/), and PANTHER (http://pantherdb.org/) databases [39, 40]. Only pathways with P-value <0.01 were listed.
Real-time PCR (qRT-PCR) analysis
After the pLPS or SA treatment and total RNA isolation, as above, reverse transcription was performed using 1 μg of total RNA [8]. qRT-PCR was run on a PikoReal real-time PCR system (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s recommendations using the following conditions: 1 min at 95 °C, and 40 cycles of 5 s at 95 °C and 30 s at 60 °C. Because the FPKM values of β-tubulin4 (AT5G44340) were not affected by pLPS treatment, it was used as a non-responsive reference gene (Additional file 1: Fig. S5). The sequences of gene-specific primers are mentioned in Table S4 (Additional file 1). Each experiment was repeated at least three times.
Free SA and SAG measurement
Two-week-old Arabidopsis seedlings grown on MS agar plates were transferred to liquid MS medium and treated with 100 μg/ml pLPS for the indicated time points. After the treatment, 80 mg of whole plants were harvested. Both free SA and SAG extraction was performed, based on a previously-reported method [41, 42]. Free SA and SAG were quantified by reverse-phase HPLC on a C18 column (YMC, Kyoto, Japan) and monitored by UV-detection at 240 nm. The column was eluted with 50 to 90% acetonitrile gradient in water containing 0.1% trifluoroacetic acid.
Abbreviations
- ABA:
-
abscisic acid
- AtLBR-2:
-
Arabidopsis LBP/BPI-related 2
- BP:
-
biological process
- BPI:
-
bactericidal/permeability-increasing protein
- CC:
-
cellular component
- FDR:
-
false discovery rate
- GO:
-
gene ontology
- LBP:
-
LPS-binding protein
- Log2FC:
-
log fold change
- LPS:
-
lipopolysaccharide
- MF:
-
molecular function
- NPR1 :
-
non-expressor of PR1
- PAMP:
-
pathogen-associated molecular pattern
- pLPS:
-
commercial LPS from Pseudomonas aeruginosa
- PR1 :
-
pathogenesis-related 1
- RNA-Seq:
-
mRNA sequencing
- SA:
-
salicylic acid
- SAG:
-
conjugated SA glucoside
References
Ranf S. Immune sensing of lipopolysaccharide in plants and animals: same but different. PLoS Pathog. 2016;12:e1005596.
Desaki Y, Miya A, Venkatesh B, Tsuyumu S, Yamane H, Kaku H, et al. Bacterial lipopolysaccharides induce defense responses associated with programmed cell death in rice cells. Plant Cell Physiol. 2006;47:1530–40.
Zeidler D, Zähringer U, Gerber I, Dubery I, Hartung T, Bors W, et al. Innate immunity in Arabidopsis Thaliana: lipopolysaccharides activate nitric oxide synthase (NOS) and induce defense genes. Proc Natl Acad Sci. 2004;101:15811–6.
Mishina TE, Zeier J. Pathogen-associated molecular pattern recognition rather than development of tissue necrosis contributes to bacterial induction of systemic acquired resistance in Arabidopsis. Plant J. 2007;50:500–13.
Melotto M, Underwood W, Koczan J, Nomura K, He SY. Plant stomata function in innate immunity against bacterial invasion. Cell. 2006;126:969–80.
Shah J. The salicylic acid loop in plant defense. Curr Opin Plant Biol. 2003;6:365–71.
Weiss J. Bactericidal/permeability-increasing protein (BPI) and lipopolysaccharide-binding protein (LBP): structure, function and regulation in host defence against gram-negative bacteria. Biochem Soc Trans. 2003;31:785–90.
Iizasa S, Iizasa E, Matsuzaki S, Tanaka H, Kodama Y, Watanabe K, et al. Arabidopsis LBP/BPI related-1 and -2 bind to LPS directly and regulate PR1 expression. Sci Rep. 2016;6:27527.
Madala NE, Molinaro A, Dubery IA. Distinct carbohydrate and lipid-based molecular patterns within lipopolysaccharides from Burkholderia cepacia contribute to defense-associated differential gene expression in Arabidopsis Thaliana. Innate Immun. 2012;18:140–54.
Blanco F, Salinas P, Cecchini NM, Jordana X, Hummelen PV, Alvarez ME, et al. Early genomic responses to salicylic acid in Arabidopsis. Plant Mol Biol. 2009;70:79–102.
Zeidler D, Dubery IA, Schmitt-Kopplin P, Von Rad U, Durner J. Lipopolysaccharide mobility in leaf tissue of Arabidopsis Thaliana. Mol Plant Pathol. 2010;11:747–55.
Lim CW, Luan S, Lee SCA. Prominent role for RCAR3-mediated ABA signaling in response to pseudomonas syringae pv. Tomato DC3000 infection in Arabidopsis. Plant Cell Physiol. 2014;55:1691–703.
Datta R, Sinha R, Chattopadhyay S. Changes in leaf proteome profile of Arabidopsis Thaliana in response to salicylic acid. J Biosci. 2013;38:317–28.
Ishihama N, Yoshioka H. Post-translational regulation of WRKY transcription factors in plant immunity. Curr Opin Plant Biol. 2012;15:431–7.
Beets CA, Huang J-C, Madala NE, Dubery I. Activation of camalexin biosynthesis in Arabidopsis Thaliana in response to perception of bacterial lipopolysaccharides: a gene-to-metabolite study. Planta. 2012;236:261–72.
Nafisi M, Goregaoker S, Botanga CJ, Glawischnig E, Olsen CE, Halkier BA, et al. Arabidopsis cytochrome P450 monooxygenase 71A13 catalyzes the conversion of indole-3-acetaldoxime in camalexin synthesis. Plant Cell. 2007;19:2039–52.
Schuhegger R, Nafisi M, Mansourova M, Petersen BL, Olsen CE, Svatoš A, et al. CYP71B15 (PAD3) catalyzes the final step in camalexin biosynthesis. Plant Physiol. 2006;141:1248–54.
Zhao J, Last RL. Coordinate regulation of the tryptophan biosynthetic pathway and indolic phytoalexin accumulation in Arabidopsis. Plant Cell. 1996;8:2235–44.
Glazebrook J, Rogers EE, Ausubel FM. Isolation of Arabidopsis mutants with enhanced disease susceptibility by direct screening. Genetics. 1996;143:973–82.
Mosher RA, Durrant WE, Wang D, Song J, Dong X. A comprehensive structure–function analysis of Arabidopsis SNI1 defines essential regions and transcriptional repressor activity. Plant Cell. 2006;18:1750–65.
Kim C, Meskauskiene R, Zhang S, Lee KP, Ashok ML, Blajecka K, et al. Chloroplasts of Arabidopsis are the source and a primary target of a plant-specific programmed cell death signaling pathway. Plant Cell. 2012;24:3026–39.
Šimková K, Moreau F, Pawlak P, Vriet C, Baruah A, Alexandre C, et al. Integration of stress-related and reactive oxygen species-mediated signals by topoisomerase VI in Arabidopsis Thaliana. Proc Natl Acad Sci. 2012;109:16360–5.
Triantaphylidès C, Havaux M. Singlet oxygen in plants: production, detoxification and signaling. Trends Plant Sci. 2009;14:219–28.
Dit FNF, Mbengue M, Kwaaitaal M, Nitsch L, Altenbach D, Häweker H, et al. Plasma membrane calcium ATPases are important components of receptor-mediated signaling in plant immune responses and development. Plant Physiol. 2012;159:798–809.
Elmore JM, Coaker G. The role of the plasma membrane H+-ATPase in plant–microbe interactions. Mol Plant. 2011;4:416–27.
Segonzac C, Zipfel C. Activation of plant pattern-recognition receptors by bacteria. Curr Opin Microbiol. 2011;14:54–61.
Zipfel C, Kunze G, Chinchilla D, Caniard A, Jones JDG, Boller T, et al. Perception of the bacterial PAMP EF-Tu by the receptor EFR restricts agrobacterium-mediated transformation. Cell. 2006;125:749–60.
Ranf S, Gisch N, Schäffer M, Illig T, Westphal L, Knirel YA, et al. A lectin S-domain receptor kinase mediates lipopolysaccharide sensing in Arabidopsis Thaliana. Nat Immunol. 2015;16:426–33.
Vaid N, Pandey PK, Tuteja N. Genome-wide analysis of lectin receptor-like kinase family from Arabidopsis and rice. Plant Mol Biol. 2012;80:365–88.
Sanabria NM, van Heerden H, Dubery IA. Molecular characterisation and regulation of a Nicotiana Tabacum S-domain receptor-like kinase gene induced during an early rapid response to lipopolysaccharides. Gene. 2012;501:39–48.
Chen F, D’Auria JC, Tholl D, Ross JR, Gershenzon J, Noel JP, et al. An Arabidopsis Thaliana gene for methylsalicylate biosynthesis, identified by a biochemical genomics approach, has a role in defense. Plant J. 2003;36:577–88.
Hok S, Danchin EGJ, Allasia V, Panabières F, Attard A, Keller H. An Arabidopsis (malectin-like) leucine-rich repeat receptor-like kinase contributes to downy mildew disease. Plant Cell Environ. 2011;34:1944–57.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc. 2012;7:562–78.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40.
Sun J, Nishiyama T, Shimizu K, Kadota KTCC. An R package for comparing tag count data with robust normalization strategies. BMC Bioinformatics. 2013;14:219.
Anders S, Pyl PT, Huber W. HTSeq—a python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4:44–57.
Katari MS, Nowicki SD, Aceituno FF, Nero D, Kelfer J, Thompson LP, et al. VirtualPlant: a software platform to support systems biology research. Plant Physiol. 2010;152:500–15.
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39(suppl 2):W316–22.
Wu J, Mao X, Cai T, Luo J, Wei LKOBAS. Server: a web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006;34(suppl 2):W720–4.
Li X, Zhang Y, Clarke JD, Li Y, Dong X. Identification and cloning of a negative regulator of systemic acquired resistance, SNI1, through a screen for suppressors of npr1-1. Cell. 1999;98:329–39.
Bowling SA, Guo A, Cao H, Gordon AS, Klessig DF, Dong X. A mutation in Arabidopsis that leads to constitutive expression of systemic acquired resistance. Plant Cell. 1994;6:1845–57.
Uknes S, Mauch-Mani B, Moyer M, Potter S, Williams S, Dincher S, et al. Acquired resistance in Arabidopsis. Plant Cell. 1992;4:645–56.
Bricchi I, Bertea CM, Occhipinti A, Paponov IA, Maffei ME. Dynamics of membrane potential variation and gene expression induced by Spodoptera Littoralis, Myzus Persicae, and pseudomonas syringae in Arabidopsis. PLoS One. 2012;7:e46673.
Morán-Diez E, Rubio B, Domínguez S, Hermosa R, Monte E, Nicolás C. Transcriptomic response of Arabidopsis Thaliana after 24h incubation with the biocontrol fungus Trichoderma harzianum. J Plant Physiol. 2012;169:614–20.
Borges AA, Dobon A, Expósito-Rodríguez M, Jiménez-Arias D, Borges-Pérez A, Casañas-Sánchez V, et al. Molecular analysis of menadione-induced resistance against biotic stress in Arabidopsis. Plant Biotechnol J. 2009;7:744–62.
Van LLC, Rep M, CMJ P. Significance of inducible defense-related rroteins in infected plants. Annu Rev Phytopathol. 2006;44:135–62.
Kus JV, Zaton K, Sarkar R, Cameron RK. Age-related resistance in Arabidopsis is a developmentally regulated defense response to pseudomonas syringae. Plant Cell Online. 2002;14:479–90.
Bourdais G, Burdiak P, Gauthier A, Nitsch L, Salojärvi J, Rayapuram C, et al. Large-scale phenomics identifies primary and fine-tuning roles for CRKs in responses related to oxidative stress. PLoS Genet. 2015;11:e1005373.
Breitenbach HH, Wenig M, Wittek F, Jordá L, Maldonado-Alconada AM, Sarioglu H, et al. Contrasting roles of the APOPLASTIC aspartyl protease APOPLASTIC, ENHANCED DISEASE SUSCEPTIBILITY1-DEPENDENT1 and LEGUME LECTIN-LIKE PROTEIN1 in Arabidopsis systemic acquired resistance. Plant Physiol. 2014;165:791–809.
Jakab G, Manrique A, Zimmerli L, Métraux J-P, Mauch-Mani B. Molecular characterization of a novel lipase-like pathogen-inducible gene family of Arabidopsis. Plant Physiol. 2003;132:2230–9.
Jung HW, Tschaplinski TJ, Wang L, Glazebrook J, Greenberg JT. Priming in systemic plant immunity. Science. 2009;324:89–91.
Reuber TL, Ausubel FM. Isolation of Arabidopsis genes that differentiate between resistance responses mediated by the RPS2 and RPM1 disease resistance genes. Plant Cell Online. 1996;8:241–9.
Mészáros T, Helfer A, Hatzimasoura E, Magyar Z, Serazetdinova L, Rios G, et al. The Arabidopsis MAP kinase kinase MKK1 participates in defence responses to the bacterial elicitor flagellin. Plant J. 2006;48:485–98.
Denoux C, Galletti R, Mammarella N, Gopalan S, Werck D, De Lorenzo G, et al. Activation of defense response pathways by OGs and Flg22 elicitors in Arabidopsis seedlings. Mol Plant. 2008;1:423–45.
Du Z, Xu D, Li L, Shi Y, Schläppi M, Xu Z-Q. Inhibitory effects of Arabidopsis EARLI1 against Botrytis Cinerea and Bradysia Difformis. Plant Cell Tissue Organ Cult PCTOC. 2012;110:435–43.
Karim S, Holmström K-O, Mandal A, Dahl P, Hohmann S, Brader G, et al. AtPTR3, a wound-induced peptide transporter needed for defence against virulent bacterial pathogens in Arabidopsis. Planta. 2006;225:1431–45.
Asano T, Kimura M, Nishiuchi T. The defense response in Arabidopsis Thaliana against fusarium sporotrichioides. Proteome Sci. 2012;10:61.
Chen K, Fan B, Du L, Chen Z. Activation of hypersensitive cell death by pathogen-induced receptor-like protein kinases from Arabidopsis. Plant Mol Biol. 2004;56:271–83.
Quirino BF, Normanly J, Amasino RM. Diverse range of gene activity during Arabidopsis Thaliana leaf senescence includes pathogen-independent induction of defense-related genes. Plant Mol Biol. 1999;40:267–78.
Yi SY, Shirasu K, Moon JS, Lee S-G, Kwon S-Y. The activated SA and JA signaling pathways have an influence on flg22-triggered oxidative burst and callose deposition. PLoS One. 2014;9:e88951.
Gruner K, Griebel T, Návarová H, Attaran E, Zeier J. Reprogramming of plants during systemic acquired resistance. Front Plant Sci. 2013;4:252.
Lieberherr D, Wagner U, Dubuis P-H, Métraux J-P, Mauch F. The rapid induction of glutathione S-transferases AtGSTF2 and AtGSTF6 by avirulent pseudomonas syringae is the result of combined salicylic acid and ethylene signaling. Plant Cell Physiol. 2003;44:750–7.
Tsuda K, Sato M, Glazebrook J, Cohen JD, Katagiri F. Interplay between MAMP-triggered and SA-mediated defense responses. Plant J. 2008;53:763–75.
Blanco F, Garretón V, Frey N, Dominguez C, Pérez-Acle T, der Straeten DV, et al. Identification of NPR1-dependent and independent genes early induced by salicylic acid treatment in Arabidopsis. Plant Mol Biol. 2005;59:927–44.
Siemens J, Keller I, Sarx J, Kunz S, Schuller A, Nagel W, et al. Transcriptome analysis of Arabidopsis clubroots indicate a key role for cytokinins in disease development. Mol Plant-Microbe Interact. 2006;19:480–94.
Gechev TS, Gadjev IZ, Hille J. An extensive microarray analysis of AAL-toxin-induced cell death in Arabidopsis Thaliana brings new insights into the complexity of programmed cell death in plants. Cell Mol Life Sci CMLS. 2004;61:1185–97.
Bethke G, Grundman RE, Sreekanta S, Truman W, Katagiri F, Glazebrook J, Arabidopsis PECTINMETHYLESTERASE. Contribute to immunity against pseudomonas syringae. Plant Physiol. 2014;164:1093–107.
Ferrari S, Galletti R, Denoux C, Lorenzo GD, Ausubel FM, Dewdney J. Resistance to Botrytis Cinerea induced in Arabidopsis by elicitors is independent of salicylic acid, ethylene, or jasmonate signaling but requires PHYTOALEXIN DEFICIENT3. Plant Physiol. 2007;144:367–79.
Campbell EJ, Schenk PM, Kazan K, Penninckx IAMA, Anderson JP, Maclean DJ, et al. Pathogen-responsive expression of a putative ATP-binding cassette transporter gene conferring resistance to the diterpenoid sclareol is regulated by multiple defense signaling pathways in Arabidopsis. Plant Physiol. 2003;133:1272–84.
Acknowledgements
We are grateful to Dr. S. Mitsutake and Dr. T. Ueda of Saga University for their support and helpful suggestions. We also want to thank our research assistant, Ms. A. Sakai, for her tireless assistance.
Funding
This work was supported by Research grant for advanced research, United Graduate School of Agricultural Sciences, Kagoshima University.
Availability of data and materials
The RNA-Seq raw data (Additional file 1: Table S6) presented in this paper were downloaded from the DNA Data Bank of Japan (DDBJ) (ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/DRA005/DRA005496).
Author information
Authors and Affiliations
Contributions
SI, EI, and YN designed the study and wrote the manuscript; SI, EI, KW, and YN performed the bioinformatic analysis and analyzed the data; SI and EI performed the experiments. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:Table S1-S6.
and Figure S1-S5. (PDF 579 kb)
Additional file 2: Table S7.
The FPKM of all transcripts in each data sets. (XLSX 4687 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Iizasa, S., Iizasa, E., Watanabe, K. et al. Transcriptome analysis reveals key roles of AtLBR-2 in LPS-induced defense responses in plants. BMC Genomics 18, 995 (2017). https://doi.org/10.1186/s12864-017-4372-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-4372-4