
Abstract
Background
The current chassis organisms or various types of cell factories have considerable advantages and disadvantages. Therefore, it is necessary to develop various chassis for an efficient production of different bioproducts from renewable resources. In this context, synthetic biology offers unique potentialities to produce value-added products of interests. Microbial genome reduction and modification are important strategies for constructing cellular chassis and cell factories. Many genome-reduced strains from Escherichia coli, Bacillus subtilis, Corynebacterium glutamicum and Streptomyces, have been widely used for the production of amino acids, organic acids, and some enzymes. Some Pseudomonas strains could serve as good candidates for ideal chassis cells since they grow fast and can produce many valuable metabolites with low nutritional requirements and strong environmental adaptability. Pseudomonas chlororaphis GP72 is a non-pathogenic plant growth-promoting rhizobacterium that possesses capacities of tolerating various environmental stresses and synthesizing many kinds of bioactive compounds with high yield. These include phenazine-1-carboxylic acid (PCA) and 2-hydroxyphenazine (2-OH-PHZ), which exhibit strong bacteriostatic and antifungal activity toward some microbial pathogens.
Results
We depleted 685 kb (10.3% of the genomic sequence) from the chromosome of P. chlororaphis GP72(rpeA-) by a markerless deletion method, which included five secondary metabolic gene clusters and 17 strain-specific regions (525 non-essential genes). Then we characterized the 22 multiple-deletion series (MDS) strains. Growth characteristics, production of phenazines and morphologies were changed greatly in mutants with large-fragment deletions. Some of the genome-reduced P. chlororaphis mutants exhibited more productivity than the parental strain GP72(rpeA-). For example, strain MDS22 had 4.4 times higher production of 2-OH-PHZ (99.1 mg/L) than strain GP72(rpeA-), and the specific 2-OH-PHZ production rate (mmol/g/h) increased 11.5-fold. Also and MDS10 had the highest phenazine production (852.0 mg/L) among all the studied strains with a relatively high specific total phenazine production rate (0.0056 g/g/h).
Conclusions
In conclusion, P. chlororaphis strains with reduced genome performed better in production of secondary metabolites than the parent strain. The newly developed mutants can be used for the further genetic manipulation to construct chassis cells with the less complex metabolic network, better regulation and more efficient productivity for diverse biotechnological applications.
Similar content being viewed by others

Background
The exploration of minimal bacterial gene set has become a research hotspot since the smallest known bacterial genome of any free-living organism was fully sequenced, which is about 470 protein-coding genes in a 580 kb Mycoplasma genitalium genome [1]. Until now, great progresses have been made in certain model strains under particular growth conditions, e.g., 303 essential genes in Escherichia coli [2], 261 (including 2 functional RNAs genes) in Bacillus subtilis [3], 335 in P. aeruginosa species [4], and 14.7% of the Streptococcus agalactiae genes are essential and critical [5]. Herein, the Database of Essential Genes (DEG) provides comprehensive information on essential genes of each strain [6], which could guide further research on other strains or species. Essential genes play important roles in understanding the last universal common ancestor [7], determining minimal gene set for cellular life [8], identifying therapeutic candidates [9], and designing chassis cells from the perspective of synthetic biology [10].
The synthetic biology principle facilitates the construction of microbial cell factories or chassis cells for biotechnological applications [11]. An ideal bacterial chassis used in biotechnology should have enough genetic information to maintain the basic biological functions, such as self-maintenance, robust cell growth and strong stress resistance, and simplified genome to make effective use of the intracellular energy [12]. The microbial genome reduction and modification are important strategies for constructing cellular chassis, such as E. coli strains ∆16 [13] and MGF-01 [14], B. subtilis strain MG1M [15], C. glutamicum strains [16], S. avermitilis SUKA17 [17] and S. coelicolor M145 [18]. Great achievements have been made in these model organisms due to their well-characterized background or easily genetic manipulation [19].
New chassis cells from different microbial genotypes should also be needed to produce complex molecules and valuable secondary metabolites, and sometimes they have to tolerate substantial environmental stresses [20]. A recent study shed light that several species of the genus Pseudomonas could serve as good candidates for chassis cells [21]. Pseudomonads are endowed with extensive metabolic and physiological diversity and can tolerate various stresses, which make them ideal resources of bacterial chassis [21]. P. putida with 4.3% eliminated genome was proved to be an enhanced host to express heterologous genes (GFP-LuxCDABE reporter system) [22]. This largely owns to Pseudomonas remarkable biodegradation capabilities accompanied by high tolerance to organic solvents. However, a biological chassis from Pseudomonas for the production of secondary metabolites has not been reported yet.
P. chlororaphis strains without virulence factors are well known plant growth-promoting rhizobacteria and widely used in agriculture and production of bio-pesticides [23]. P. chlororaphis GP72 was isolated from green pepper rhizosphere [24]. Previous studies indicated that this strain could withstand both endogenous and exogenous stresses, and synthesize different type of phenazines, including PCA, 2-hydroxy-phenazine-1-carboxylic acid (2-OH-PCA) and 2-OH-PHZ [25, 26]. Phenazines are a group of over 100 natural and more than 6000 synthetic nitrogen-containing heterocyclic compounds [27]. Strains of Pseudomonas and Streptomyces are main phenazine producers, and formers are used to produce simple phenazine compounds, while latters tend to generate more diverse and complex molecules [28]. Phenazine metabolites make enormous contributions to agriculture due to their broad spectrum antibiotic properties [29], in industrial application owing to the electron transfer ability [30], and in health area because of the anti-cancer activity [31]. In P. chlororaphis GP72, the production of 2-OH-PHZ has been greatly improved through genetic engineering [32,33,34], indicating that strain GP72 has noteworthy potential for enhanced production of secondary metabolites. Herein, we aimed to implement a comparative genomic approach together with genome mining strategy, to design and construct valuable genome-reduced Pseudomonas mutants.
Results
Computational prediction of deletion targets and set of essential genes
Maximal deletion regions on the chromosome of P. chlororaphis were selected using comparative genomics between P. chlororaphis GP72 and P. stutzeri A1501. As P. stutzeri strains are nitrogen-fixing root-associated bacteria harboring relative small genomes among pseudomonads [35], and strain A1501, a model organism of this species [36], is phylogenetically distant from strain GP72 [25]. Conserved genes between them are likely to be essential [37], while strain-specific ones are nonessential. Previous studies indicated that strain GP72 has six secondary metabolite gene clusters which cannot be found in the genome of strain P. stutzeri A1501 [25, 26]. So the five clusters, except the phenazine operon which can be used for testing the production capacity of secondary metabolites in mutants, can be eliminated firstly. Based on the in silico subtractive hybridization analysis via mGenomeSubtractor online software [38], there were 714 conserved genes and 3351 strain-specific ones in the genome of strain GP72. Therefore, regions with strain-specific genes longer than 15 kb were selected as candidate deletion regions, which provide a framework for the design of deletion targets.
Chassis cells must be efficient and profitable, which contain essential genes to sustain bacterial vitality, and set of favorable and selected genes for industrial use [39]. So those selected candidate regions were checked for the presence of predicted essential genes. The predicted essential gene was selected when the corresponding protein was conserved at least in eight of 15 essential gene data sets using the basic local alignment search tool (BLAST) under the threshold of 35% identity [40]. These sets of essential genes were from 15 g-negative bacteria [6], including P. aeruginosa UCBPP-PA14, E. coli MG1655 II, Burkholderia pseudomallei K96243, Acinetobacter baylyi ADP1, Shewanella oneidensis MR-1, Salmonella enterica subsp. enterica serovar Typhimurium SL1344, Haemophilus influenzae Rd. KW20, Francisella novicida U112, Caulobacter crescentus, Sphingomonas wittichii RW1, Vibrio cholerae N16961, Porphyromonas gingivalis ATCC 33277, Bacteroides fragilis 638R, Campylobacter jejuni NCTC 11168 and Helicobacter pylori G27. Two hundred fifteen genes were predicted to be essential out of the total 6176 genes in the genome of strain GP72 (Additional file 1). In each essential gene set, the number of homologous genes that can be blasted in GP72 is shown in Additional file 2.
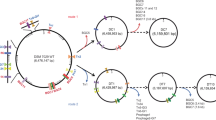
Given the above alignments, 22 strain-specific genomic regions without essential genes were picked as deletion targets (Fig. 1). Based on the gene annotation, we hypothesized that the selected deletion regions could not affect the basic biochemistry and physiology quality of engineered strains.

An overview of the reduced genome of P. chlororaphis MDS22. Deletions of MD1 through MD22 were illustrated on the circular map of the GP72 genome. The green and purple circles represent the genomes of GP72 and the largest deletion mutant (MDS22), respectively
Construction of deletion strains
The selected regions were markerless deleted through homologous recombination via genetically modified plasmids, which were constructed using pK18 mobsacB. We sequentially deleted 22 multiple-deletion (MD) regions (MD1 to MD22) (Table 1), 575 genes in total (Additional file 3). And MDS22 lacked 685,750 base pairs (bp), accounting for 10.3% of the parent genome (6,663,241 bp). Deletion endpoint was designed in noncoding regions next to the deleted gene, or in some cases, it was in the deletion region, which may not affect the expression of remaining genes.
The deletion strategy we implemented was that genes responsible for biosynthesis of secondary metabolites were deleted as the first choice, including MD1–5. MDS1 was created by deleting the achromobactin operon synthesizing a temperature-regulated secondary siderophore [41]. Then the MDS2 was constructed based on MDS1 by removing MD2. Genes in MD2 are responsible for a P. fluorescens insecticidal toxin which exhibits oral insecticidal activity [42]. Another siderophore operon pyoverdine (Pvd, excluding pvdL, pvdS and pvdY) was deleted to design MDS3. MDS4 was constructed by deleting hydrogen cyanide gene cluster which cannot be found in the genome of A1501 but existed in many other pseudomonads [25]. The pyrrolnitrin operon [43] encoding the chlorinated phenylpyrrole compound based on L-tryptophan was also removed, resulting in the fifth mutant MDS5.
Afterwards, unnecessary metabolisms or transport genes of the strain when inoculated in the rich medium were selected for deletion (MD6–18). Those include genes related to amino acid metabolism or transport, carbohydrate metabolism or transport, inorganic ion transport, and signal transduction system. In MD6, four of the 21 genes may be related to threonine transport and metabolism based on the cluster of orthologous group (COG) prediction and nine of them have not been functionally predicted. The threonine dehydratase (TdcB) may play roles in threonine or serine degradation, but it is only expressed during anaerobic growth in the absence of glucose [44]. The osmoprotectant glycine betaine could be absorbed from the environment [45]. So its biosynthetic genes (MOK_00378–00406) in MD7 were removed. According to the gene annotation and metabolic pathway analysis, genes MOK_00955–00968 in MD8 might be involved in the biosynthesis of terpenoids. The MOK_00957 encoding phytoene dehydrogenase was conserved only in a few Pseudomonas strains which mainly belonged to the species of P. chlororaphis. Its amino acid sequence showed 89% identities with coverage of 99% to that found in an uncultured proteobacterium QS1, and showed 37% identities with more than 94% coverage to that found in Streptomyces. But their capacity of synthesizing terpenoids has not been experimental verified. And the last non-ribosomal peptide synthase gene (pvdL) together with some strain-specific genes existed in MD9 also deleted. The pea genes encoding quinol hemoprotein amine dehydrogenase and related functional proteins in response to primary amines to provide the host with carbon and energy source [46], were removed together with the nearby genes in MD12. The operon rmd-gmd-wbpW-wzm-wzt-wbpX-wbpY-wbpZ in MD13 can be found in both pathogen and nonpathogenic strains, which has been proved to be responsible for the assembly of rhamnan polysaccharide according to the phenotype of P. aeruginosa mutants (wbpX, wbpY and wbpZ) [47]. And genes related to amino acid transport, carbohydrate metabolism, inorganic ion transport, or signal transduction system were removed in MD10, MD11 and MD14–18.
Finally, strain-specific genes without functional prediction and those encoding hypothetical proteins were deleted, including MD19–22. In MD21, there was a czc operon encoding a cation-proton antiporter, which is responsible for cobalt, zinc, and cadmium resistances [48].
Characterization of deletion strains
Growth features of MDS strains and the parent strain GP72(rpeA-) were first compared in fermentation condition. The growth curves can be divided into three groups based on features of the maximum cell population: Group 1 (including GP72(rpeA-) and MDS1–8) (Fig. 2a), Group 2 (including MDS9–18) (Fig. 2b) and Group 3 (including MDS19–22) (Fig. 2c). Strain MDS7 belonging to Group 1 with a 153.6 kb reduced genome exhibited robust cell growth. Strains in Group 2 had a slightly reduced growth, while mutants in Group 3, such as MDS19 showed a dramatically slower growth patterns than that of the parent strain.

Growth curves of parent strain and genome-reduced mutants. Growth trends of all 23 strains were divided into three groups. a Group 1: GP72(rpeA-) and MDS1–8. b Group 2: MDS9–18. c Group 3: MDS19–22. Strains were grown in KMB liquid medium adding 40 mg/L Km at 28 °C and 180 rpm. The values are presented as the means of three replicates, and the error bars indicate the standard deviation (SD)
Here we observed colony morphologies of GP72(rpeA-) and its derivate mutants. Dietrich et al. [49] reported that the colony morphology could be modulated by phenazine production in P. aeruginosa strains, for the overproducer remained smooth after 6 days, while the phenazine null strain started to wrinkle earlier than the wild type. Phenotypes of representative strains with different levels of phenazine production are portrayed in Fig. 3a. The colonies of GP72(rpeA-), MDS10 and MDS15 remained smooth, but low-producing strain MDS22 started to wrinkle on the second day and severely wrinkled in the 6th day. The rugose colonial morphology also occurred in MDS19–21.

Phenotypic characterization of the parent strain GP72(rpeA-) and the genome-reduced mutants (MDS10 and MDS22). a Colony morphology assays were performed on 10 g/L tryptone broth plates (1% agar) containing Congo Red (40 μg/ml) and Coomassie Brilliant Blue (20 μg/ml). Strains were grown in LB liquid medium to late exponential phase, and then 10 μl were spotted onto plates and incubated at 28 °C for 6 days [49]. b Flagellar swimming assays were performed by using semi solid agarose plates. The swimming status was photographed after overnight incubation at 28 °C [51]. Three replicates were used for each sample and experiments repeated for twice
Similarly, motility is one of the physiological characteristics of microorganism and plays important roles in pseudomonads, such as access to nutrient effectively, avoidance of toxic substances, and dispersal in the optimal environment [50, 51]. Mutants MDS1–18 and strain GP72(rpeA-) moved rapidly across the agar surface and covered the entire surface after 24 h, and there was no significant difference among them, whereas strains MDS19–22 showed diminished or absent swimming abilities.
Ability of producing secondary metabolites in GP72(rpeA-) and its derivate mutants
Three phenazine derivatives: PCA, 2-OH-PCA, and 2-OH-PHZ were analyzed in P. chlororaphis strains and results are displayed in Fig. 4. The 2-OH-PHZ is derived from 2-OH-PCA without the involvement of any enzyme [52], and PCA is converted to 2-OH-PCA via an aromatic monooxygenase encoded by phzO [33]. After 24 h of cultivation, the production of 2-OH-PHZ of MDS10 and MDS22 were 98.3 and 99.1 mg/L, respectively, both of which were 4.4 times higher than that of GP72(rpeA-) (22.2 mg/L) (p < 0.01). The significantly improved production was first occurred in MDS8, which was 5.6 times higher than that of the parent strain (p < 0.01). Notably, MDS10 had the highest production of PCA and total phenazines, 660.4 mg/L and 852.0 mg/L, respectively. Unfortunately, the PCA production showed varying degrees of decline in certain mutants, like MDS15 and MDS19.

Production of phenazines by the parent strain and genome-reduced mutants. Production of PCA, 2-OH-PCA, and 2-OH-PHZ by the parent strain GP72(rpeA-) and its mutants after 24 h of fermentation in KMB medium at 28 °C and 180 rpm. The phenazines were extracted and measured by HPLC at a wavelength of 254 nm. The bar indicates the group mean and the error bar indicates SD from triplicate experiments
Table 2 summarized the fermentation characteristics of the three representative strains GP72(rpeA-), MDS10 and MDS22 during the fermentation stage of 14 to 24 h. Compared with the parental strain, the specific growth rates of the mutants decreased, while the specific PCA production rates increased 3.3-fold (p < 0.01) and 1.2-fold (p < 0.05) in MDS10 and MDS22, respectively, and the specific total phenazine production rates changed similarly. Notably, the specific production rates of intermediate 2-OH-PCA in MDS10 and MDS22 were lower than that in the parental strain GP72(rpeA-) (p < 0.01), but the specific production rates of the final product 2-OH-PHZ significantly increased in mutants (p < 0.01), especially in MDS22 (11.5-fold).
Biolog phenotype microarray analysis of selected deletion strains
The three strains GP72(rpeA-), MDS10 and MDS22 were analyzed for differences in biosynthetic pathways using Biolog phenotype microarrays [53]. Biolog PM5 plate containing different nutrient supplements was used, and the kinetic curve displayed the growth phenotype (Fig. 5). Strains MDS10 and GP72(rpeA-) had highly comparable kinetic curves, while strain MDS22 showed decreased substrate utilization capability. MDS10 and the parent strain showed identical growth characteristics in the negative well (minimal medium F-0 GN base without adding any supplement) or in the F8 well (IF-0 GN base with glucose). When supplied with substrates, such as L-serine, L-threonine, L-tryptophan, L-citrulline, cytosine, cytidine, 2′-deoxy cytidine and p-amino-benzoic acid, the growth of MDS10 has improved as compared to the parental strain GP72(rpeA-).

Metabolism of strains GP72(rpeA-), MDS10 and MDS22 on Biolog PM5 plates. Suspensions were added to each well of the PM5 microplates at a volume of 150 μl and incubated at 28 °C for 72 h [86]. Red and blue curves denote growth trends of GP72(rpeA-) and MDS10, respectively, while the black one shows information of MDS22, which indicates a significant defect in substance utilization. Wells A1 and A2 are negative control and positive control, respectively. The information of other 94 wells is listed in Additional file 6
Discussion
Previous studied showed that numbers of essential genes vary among different genera, species, strains, even same strain in different studies. This difference might result from mutant complementation, different cultural conditions [54] and different study strategies, like different transposons [4]. Therefore, we chose the overlapping set of conserved genes as candidate essentials in the genome of GP72, which showed a certain degree of consistency with the minimal bacterial gene set comprising 206 protein-coding genes proposed by Gil et al. [55]. The biggest difference is the transport genes, for they only have a PTS for glucose and pitA to provide the phosphate. Because the authors recognized it is hard to define specific transporters depending on the nutrients available in the environment or the cell membrane characteristics [55]. The data set of essential genes in this study provides frameworks for further research, such as optimizing the metabolic network or constructing chassis cells. And the exactly essential genes in GP72(rpeA-) under certain conditions should be verified by targeted gene deletions [2], saturation transposon mutagenesis [5] or RNA interference [56].
Here we used homologous recombination strategy to implement large-scale genome reduction to develop series sequential deletion mutants. The method we used was suitable to construct large-scale deletions. The largest deleted region covered more than 87 kb. The 5.977-Mb genome of MDS22 is smaller than any genome of P. chlororaphis strains in the available genomic data (6.66 to 7.30 Mb) [23]. Other large fragment deletion strategies were also widely used in Pseudomonas, such as Flp/FRT, Cre/loxP and I-SceI site-specific recombination systems [57,58,59]. While Flp/FRT and Cre/loxP systems cannot generate the true scarless mutant, leaving one copy of frt or loxP site in the genome, and the foreign sequences may interfere with gene expression or subsequent deletions.
In this study, we aimed to construct Pseudomonas mutants with less energy consumption and higher productivity from the perspective of synthetic biology and its applications. So genes that were unessential when bacteria in nutrient-rich medium were selected for the deletion, The previous study showed that the achromobactin operon is expressed under iron deficient conditions [41] and pyoverdine biosynthesis genes are unessential in P. aeruginosa [4]. Although pyoverdine is a high-affinity siderophore under the iron starvation condition, which is beneficial for the growth of strain; but on the other hand, its biosynthesis and excretion also require much energy and amino acids [60]. And Pseudomonas could also get iron via other mechanisms, such as the strain P. stutzeri A1501 [36].
Deletions of multiple futile metabolic pathways may be useful to save cellular energy and resources and thereby improve the growth of MDS7. The reduced growth rate might be due to the deletion of genomic loci MOK_00502, MOK_00503 (encoding PvdL and PvdS, respectively), MOK_00505 (encoding amino acid ABC transporter substrate-binding protein) and MOK_00506–00507 (encoding a repressor AtuR and a protein AtuA related to acyclic terpene utilization) in MD9, and MOK_03695 (related to transcriptional regulator) and MOK_03714–03716 (encoding outer membrane receptor protein, Fe2+-dicitrate sensor and RNA polymerase sigma factor, respectively) in MD19. PvdL is the core enzyme and necessary for the production of pyoverdine [61]. The deletion of MD3 may not affect the synthesis of siderophore, for the strain can use isoenzymes on the genome. But when MD9 containing pvdL was deleted, MDS9 may have defect in iron acquisition, resulting in slower growth trend than that of MDS3. As a pvdL mutant derived from P. fluorescens, SBW25 displays reduced cell density when compared to the wild-type in iron depleted medium [62]. Besides, the cumulative effect of consecutive deletions may play an important role in the growth of MDS19. E. coli ∆16 lacked up to 29.7% of the parental chromosome grew more slowly than the parent strain MG1655 [13], whereas the mutant reduced the genome size by 7% exhibited normal growth [63].
Besides the reduction of phenazine production, the rugose colonies may be due to the overproduction of an exopolysaccharide [64] or an extracellular cellulose-like material [65], or the metabolism of carbohydrates [66]. The swimming character mainly depends on flagella [50], but the deletion in MD19 did not contain any biosynthetic genes of flagella. We speculate that the defects may occur at the functional level, such as defects in chemotaxis and the cytoplasmic signal transduction system [51, 67], which may be under the control of GntR family (MOK_03695) and TetR family (MOK_03712) transcriptional regulator. Overall, phenotypic differences between the two types of strains: rough colonies with defective flagellar motility and smooth colonies with normal flagellar activity were performed by the results found in P. aeruginosa 57RP [68]. The exact reasons for these different phenotypic traits need further research.
Comprehensive observation of the growth and phenazines production in representative strains: GP72(rpeA-), MDS10 and MDS22, we found mutants grew slower than the parent strain, but the production capacities of phenazines, including initial product and the final one, were improved greatly. So keeping the specific growth rate at a certain level may facilitate the productivity of secondary metabolites. The higher 2-OH-PHZ production firstly occurred in mutant MDS8, which is produced abiotically from 2-OH-PCA [52]. The deletion of MD8 contained genes encoding quorum sensing system QsyI/R (MOK_00967 and MOK_00968) which may affect the production of phenazines. QsyR is a LuxR family transcriptional regulator, and its amino acid sequence was conserved among P. chlororaphis. The sequence in strain GP72(rpeA-) showed 54% identity to that found in P syringae. In P syringae, it coordinates regulation of pathogenesis-related genes, but the mechanism has not been illustrated [69]. It also showed 31% identity to the amino acid sequence of qscR found in P. aeruginosa. QscR is a negative regulator that could repress the transcription of phenazines [70]. The increased total phenazines production may be from the deletion of pvdL in MD9. The pvdL gene encodes a non-ribosomal peptide synthetase involved in incorporating L-glutamate, D-tyrosine and L-2,4-diaminobutanoate in a precursor peptide chain [71]. The elimination of this gene may save cellular resources and energy, and facilitate the synthesis of phenazines.
The MDS15 as the progenitor of following low-producing strains lacked genes of apr gene cluster encoding alkaline protease (AprA), its inhibitor (AprI) and secretion system (AprDEF). These genes showed high identities to those found in other pseudomonads. Previous studies showed that endoproteases including alkaline protease were involved in the regulation of numerous cellular processes by degrading specific proteins [72], while AprI is a highly potent inhibitor to protect periplasmic proteins from the AprA proteolysis [73]. The protease secretion mechanisms among different species, such as Pseudomonas and Erwinia, are quite similar [74]. Another protease namely serine protease Lon has been proved to be a powerful negative regulator, and the mutant can greatly improve the pyoluteorin production [75]. So the relationship between AprA and phenazine production is unclear and needs further study. Besides, it also lacked genes of dmpA (MOK_02632), which catalyzes peptide to release the N-terminal D and L amino acids. However, in MDS19 and other strains, the reduced concentrations may result from the interruption of the transport system.
In PM assays, we found that the substrates could make the growth characteristics difference between mutant MDS10 and the parental strain. Previous studies showed that some of those substrates also influence the production of phenazines [76]. For example, phenazine-1-carboxamide production was improved by adding individual amino acids threonine and tryptophan in P. chlororaphis PCL1391 [76]. And p-amino-benzoic acid was used to suppress phenazine production during triparental matings in P. aureofaciens 30–84 [77]. Further work is needed to check the importance of these substrates in the performance of Pseudomonas chlororaphis mutants.
Conclusions
The genome reduction strategy in this study is useful to construct Pseudomonas and other microbial cell factories for the production of valuable metabolites. P. chlororaphis strain GP72(repA-) showed the feasibility of constructing genome-reduced Pseudomonas strains. In the next step, the genome-scale metabolic reconstruction as well as regulatory network analysis will give more information to construct a desirable chassis cell with system simplicity and practicability for biotechnological application.
Methods
Bacterial strains, plasmid and culture conditions
For constructing genomic deletion strains, we used P. chlororaphis GP72(rpeA-) as the parent strain, which derives from inactivation of rpeA from strain GP72 and shows stable and high 2-OH-PHZ production [33]. E. coli DH5ɑ (TransGen Biotech, Beijing, China) was used to construct series deletion strains, and E. coli S17–1 (λpir) served as the donor strain to transform plasmids to P. chlororaphis in conjugations [78]. The plasmid pK18 mobsacB used to harbor homologous fragments with the target sequences deriving from pK18 is a broad-host-range vector containing genetic loci sacB and Kmr (accession No. FJ437239) [79]. Bacterial strains and the plasmids used in this study are summarized in Table 3. All studied Pseudomonas strains were grown in Luria-Bertani (LB) medium (tryptone 10.0 g, yeast extract 5.0 g, NaCl 10.0 g/L) or King’s medium B (KMB) (glycerol 20 g, tryptone 20 g, MgSO4 0.732 g, K2HPO4 0.514 g/L) at 28 °C and 180 rpm, while E. coli strains were cultured in LB medium at 37 °C and 220 rpm. If required, antibiotics in the medium were used at the following concentrations: 40 mg/L kanamycin (Km), 100 mg/L gentamicin (Gm), 24 mg/L isopropyl-β-D-1-thiogalactopyranoside (IPTG), 40 mg/L 5-bromo-4-chloro-3-indolyl-beta-D-galactopyranoside (X-gal), and 15% (w/v) sucrose.
Selection of deletion targets
For comparative genomic analysis, we used the genome sequence of P. chlororaphis GP72 (accession No. NZ_AHAY01000000) [80]. All predicted protein sequences and genome annotations were obtained from the integrated microbial genomes system [81]. BLASTP programs were conducted between protein sequences of strain GP72 and P. stutzeri A1501 (accession No. CP000304) [36] using the mGenomeSubtractor (http://202.120.12.134/mGS2/) [38]. The strain-specific genes were identified under a homology value cutoff of 0.42 at E-value < 10−5 [38]. The regions with continuous strain-specific genes were selected as elimination candidates.
For assessing these selected genes, we identified essential genes existing in strain GP72. There are 33 prokaryotic organisms in total listed on the database of essential genes (DEG) version 10.5 [6], including 23 g-negative bacteria. Since there are multiple bacterial records for one species and significant overlap among sets of essential genes derived from gram-negative bacteria [82, 83], we chose the newest data set for each species in this study. For example, the recent set of P. aeruginosa essential genes is more accurate based on combining gene information from both PAO1 and PA14 insertion libraries [4]. Besides, previous studies show that predicting essential genes via homology searches are significantly affected by phylogenetically unrelated organisms [84]. Therefore, 15 sets of essential genes were selected among gram-negative bacteria and used to create local databases. The local BLASTP programs were carried out on NCBI BLASTP+ version 2.2.30+. The homology searches were launched between protein coding sequences of GP72 and each local database to identify essential genes in P. chlororaphis. These were carried out under an E-value cut-off of 10−10 and 35% identity threshold [40]. Finally, five known secondary metabolite clusters, and 17 strain-specific regions larger than 15 kb and containing more than 10 unnecessary continuous genes were selected for deletion [14].
Construction of multiple deletion strains
For DNA manipulations, plasmid DNA and genomic DNA were isolated from E. coli using a spin plasmid preparation kit and P. chlororaphis using a genomic DNA isolation kit, respectively, according to the manufacturer’s instructions (TransGen Biotech, Beijing, China). All the enzymes were purchased from TaKaRa (Dalian, China) except for Taq polymerase (TransGen Biotech, Beijing, China). All primers used in this study were synthesized at Shanghai Sunny Biotechnology Co, Ltd. (Shanghai, China) and are listed in Additional file 4. DNA products were gel purified using AxyPrep DNA gel extraction kit (Corning, Shanghai, China). DNA sequencing was conducted at BGI Tech Solutions Co., Ltd. (Shenzhen, China) and sequences were analyzed on the NCBI website.
Gene deletions in P. chlororaphis strain were conducted using a markerless deletion method with pK18mobsacB as described previously [85]. The method was slightly modified based on the specific strain (Additional file 5). Firstly, the upstream and downstream homologous arms ranging from 400 to 800 bp were amplified from the parent strain genomic DNA using two pairs of primers (F1 and R1) and (F2 and R2), respectively. Two primers of R1 and F2 had an 18–20 bp homology region between them. So purified polymerase chain reaction (PCR) fragments were joined by an overlap PCR using primers of F1 and R2, both of which contained different restriction sites at their 5′ ends. The purified DNA product was then digested and cloned into the plasmid pK18 mobsacB. Secondly, the ligation and purified controls were transformed into chemically competent DH5ɑ cells using standard protocol followed by blue/white screening on LB plates containing Km X-gal and IPTG. The selected white colonies (KmR) were verified by PCR amplification and DNA sequencing. Thirdly, the deletion construct was transformed into E. coli S17–1 (λpir), resulting in the S17–1 (λpir) harboring the construct (KmRGmS) and then conjugated it into P. chlororaphis to get single crossover merodiploid transconjugants, which were selected on LB plates with antibiotics Km and Gm. At each step, the positive clones were confirmed by PCR amplification using primers of F1 and R2. Finally, merodiploid (KmRGmR) were transferred to LB agar plates with 15% (w/v) sucrose to counter-select the integration, which resulted in the loss of the sacB gene. SucRGmRKmS colonies were selected and screened by PCR using primers specific to sequences in the remaining genome (F1 and R2) and deleted fragment (NF and NR). Then the positive clones were also confirmed by spotting them on LB plates only containing Km, on which clones cannot grow. The subsequent deletion mutants were constructed based on the same strategy as mentioned above.
Fermentation processing
A single colony was selected and grown in a test tube containing 5 mL KMB liquid medium supplemented with 40 mg/L Km at 28 °C overnight. Then the seed culture was transferred to 50 mL KMB medium in a 250 mL baffled flask at an OD600 of 0.02. The fermentation process was carried at 28 °C and 180 rpm during 4 days. Cultures were collected for the measurement of OD600, biomass, and production of phenazine compounds. There are triplicate experiments for each fermentation test, and experiments were repeated twice. The result value is expressed as the mean ± SD. Statistical significances of the differences between groups were examined by Student’s t test, using paired data.
Quantification of phenazine compounds
Samples were prepared following the previous procedure [33]. The fermentation broth was adjusted to pH 2.0 using 6 M HCl and extracted with three volumes of ethyl acetate by vigorous shaking. The organic layer was then mixed with 1/10 volume of distilled water and shaken vigorously. Finally, the organic phase containing phenazine compounds was evaporated under vacuum pressure. The phenazine compounds were dissolved in methanol for further analysis. The analysis was carried out by using the Agilent 1260 Infinity high-performance liquid chromatography (HPLC) series with a C-18 reverse phase column and ultraviolet light detector (Agilent Technologies, USA). The mobile phase changed during the detection process methanol: ammonium acetate =20:80 (v/v) in the first 5 min, then changing to 50:50 in 5 to 25 min and 20:80 again in the last 5 min (25–30 min). The retention times for PCA, 2-OH-PCA, and 2-OH-PHZ were approximately 9.1 min, 13.3 min and 21.7 min, respectively.
Morphology assays
Colony morphology assays were performed as previously described by using 10 g/L tryptone broth plates (1% agar) with the addition of Congo Red (40 μg/ml) and Coomassie Brilliant Blue (20 μg/ml) [49]. Strains were grown in LB medium to late exponential phase, and then 10 μl were spotted onto plates and incubated for 6 days. Flagellar swimming assays were done by stabbing tryptone swim plates (1% tryptone, 0.5% NaCl, 0.3% agar) as described previously [51]. These plates were inoculated with bacteria from an overnight culture on LB agar plates using a sterile toothpick. The swimming status was photographed after overnight. All plates were wrapped with saran to prevent dehydration and cultured at 28 °C.
Biolog phenotype microarray
The biosynthetic pathways of strain P. chlororaphis GP72(rpeA-), MDS10 and MDS22 were compared using phenotype microarray (PM) technology in plate PM5 containing 94 different nutrients (BIOLOG Inc. CA, USA). All procedures were performed as described by Bochner [86]. The tested strains were grown overnight at 28 °C using Biolog Universal Growth agar (BIOLOG Inc. CA, USA) plates. The single colony was swabbed from the plate and suspended using IF-0 GN base inoculating fluid (BIOLOG Inc. CA, USA) to a density of 85% transmittance in the Biolog turbidimeter. Suspensions were added to each well of the PM5 microplates at a volume of 150 μl and incubated at 28 °C for 72 h. Plates were placed in the OmniLog instrument, and cell growth was recorded by the respiration-dependent color change of tetrazolium violet every 15 min. Finally, the readouts were analyzed based on the different kinetic curves reflecting the phenotypic changes. The added nutrient for each well is listed in Additional file 6. The PM assay was performed twice.
Abbreviations
- 2-OH-PCA:
-
2-hydroxyphenazine-1-carboxylic acid
- 2-OH-PHZ:
-
2-hydroxyphenazine
- BLAST:
-
Basic local alignment search tool
- bp:
-
Base pairs
- COG:
-
Cluster of orthologous group
- DEG:
-
Database of essential genes
- Gm:
-
Gentamicin
- HPLC:
-
High-performance liquid chromatography
- IPTG:
-
Isopropyl-β-D-1-thiogalactopyranoside
- Km:
-
Kanamycin
- KMB:
-
King’s medium B
- LB:
-
Luria-Bertani
- MD:
-
Multiple-deletion
- MDS:
-
Multiple-deletion series
- PCA:
-
Phenazine-1-carboxylic acid
- PCR:
-
Polymerase chain reaction
- PM:
-
Phenotype microarray
- Pvd:
-
Pyoverdine
- SD:
-
Standard deviation
- X-gal:
-
5-bromo-4-chloro-3-indolyl-beta-D-galactopyranoside
References
Fraser CM, Gocayne JD, White O, Adams MD, Clayton RA, Fleischmann RD, et al. The minimal gene complement of Mycoplasma genitalium. Science. 1995;270(5235):397–403.
Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006–0008.
Commichau FM, Pietack N, Stulke J. Essential genes in Bacillus subtilis: a re-evaluation after ten years. Mol BioSyst. 2013;9(6):1068–75.
Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, Wu G, et al. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci U S A. 2006;103(8):2833–8.
Hooven TA, Catomeris AJ, Akabas LH, Randis TM, Maskell DJ, Peters SE, et al. The essential genome of Streptococcus agalactiae. BMC Genomics. 2016;17(1):1–12.
Luo H, Lin Y, Gao F, Zhang CT, Zhang R. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 2014;42(Database issue):D574–80.
Koonin EV. Comparative genomics, minimal gene-sets and the last universal common ancestor. Nat Rev Microbiol. 2003;1(2):127–36.
Gerdes S, Edwards R, Kubal M, Fonstein M, Stevens R, Osterman A. Essential genes on metabolic maps. Curr Opin Biotechnol. 2006;17(5):448–56.
Butt AM, Nasrullah I, Tahir S, Tong Y. Comparative genomics analysis of Mycobacterium ulcerans for the identification of putative essential genes and therapeutic candidates. PLoS One. 2012;7(8):e43080.
Seo SW, Yang J, Min BE, Jang S, Lim JH, Lim HG, et al. Synthetic biology: tools to design microbes for the production of chemicals and fuels. Biotechnol Adv. 2013;31(6):811–7.
Sauer M, Mattanovich D. Construction of microbial cell factories for industrial bioprocesses. J Chem Technol Biotechnol. 2012;87(4):445–50.
Foley PL, Shuler ML. Considerations for the design and construction of a synthetic platform cell for biotechnological applications. Biotechnol Bioeng. 2010;105(1):26–36.
Hashimoto M, Ichimura T, Mizoguchi H, Tanaka K, Fujimitsu K, Keyamura K, et al. Cell size and nucleoid organization of engineered Escherichia coli cells with a reduced genome. Mol Microbiol. 2005;55(1):137–49.
Mizoguchi H, Sawano Y, Kato J, Mori H. Superpositioning of deletions promotes growth of Escherichia coli with a reduced genome. DNA Res. 2008;15(5):277–84.
Ara K, Ozaki K, Nakamura K, Yamane K, Sekiguchi J, Ogasawara N. Bacillus Minimum genome factory: effective utilization of microbial genome information. Biotechnol Appl Biochem. 2007;46(Pt 3):169–78.
Unthan S, Baumgart M, Radek A, Herbst M, Siebert D, Bruhl N, et al. Chassis organism from Corynebacterium glutamicum--a top-down approach to identify and delete irrelevant gene clusters. Biotechnol J. 2015;10(2):290–301.
Komatsu M, Uchiyama T, Omura S, Cane DE, Ikeda H. Genome-minimized Streptomyces host for the heterologous expression of secondary metabolism. Proc Natl Acad Sci U S A. 2010;107(6):2646–51.
Gomez-Escribano JP, Bibb MJ. Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb Biotechnol. 2011;4(2):207–15.
Juhas M, Reuss DR, Zhu B, Commichau FM. Bacillus subtilis and Escherichia coli essential genes and minimal cell factories after one decade of genome engineering. Microbiology. 2014;160(Pt 11):2341–51.
Vickers CE, Blank LM, Kromer JO. Grand challenge commentary: chassis cells for industrial biochemical production. Nat Chem Biol. 2010;6(12):875–7.
Nikel PI, Martinez-Garcia E, de Lorenzo V. Biotechnological domestication of pseudomonads using synthetic biology. Nat Rev Microbiol. 2014;12(5):368–79.
Martinez-Garcia E, Nikel PI, Aparicio T, de Lorenzo V. Pseudomonas 2.0: Genetic upgrading of P. putida KT2440 as an enhanced host for heterologous gene expression. Microb Cell Factories. 2014;13:159.
Chen YW, Shen XM, Peng HS, Hu HB, Wang W, Zhang XH. Comparative genomic analysis and phenazine production of Pseudomonas chlororaphis, a plant growth-promoting rhizobacterium. Genom Data. 2015;4:33–42.
Liu HM, Dong D, Peng HS, Zhang XH, Xu YQ. Genetic diversity of phenazine- and pyoluteorin-producing pseudomonads isolated from green pepper rhizosphere. Arch Microbiol. 2006;185(2):91–8.
Shen XM, Hu HB, Peng HS, Wang W, Zhang XH. Comparative genomic analysis of four representative plant growth-promoting rhizobacteria in Pseudomonas. BMC Genomics. 2013;14:271.
Liu HM, He YW, Jiang H, Peng HS, Huang XQ, Zhang XH, et al. Characterization of a phenazine-producing strain Pseudomonas chlororaphis GP72 with broad-spectrum antifungal activity from green pepper rhizosphere. Curr Microbiol. 2007;54(4):302–6.
Pierson LS 3rd, Pierson EA. Metabolism and function of phenazines in bacteria: impacts on the behavior of bacteria in the environment and biotechnological processes. Appl Microbiol Biotechnol. 2010;86(6):1659–70.
Laursen JB, Nielsen J. Phenazine natural products: biosynthesis, synthetic analogues, and biological activity. Chem Rev. 2004;104(3):1663–86.
Mavrodi DV, Blankenfeldt W, Thomashow LS. Phenazine compounds in fluorescent Pseudomonas spp. biosynthesis and regulation. Annu Rev Phytopathol. 2006;44:417–45.
Rabaey K, Boon N, Hofte M, Verstraete W. Microbial phenazine production enhances electron transfer in biofuel cells. Environ Sci Technol. 2005;39(9):3401–8.
Cimmino A, Evidente A, Mathieu V, Andolfi A, Lefranc F, Kornienko A, et al. Phenazines and cancer. Nat Prod Rep. 2012;29(4):487–501.
Liu HM, Yan A, Zhang XH, Xu YQ. Phenazine-1-carboxylic acid biosynthesis in Pseudomonas chlororaphis GP72 is positively regulated by the sigma factor RpoN. World J Microbiol Biotechnol. 2008;24(9):1961–6.
Huang L, Chen MM, Wang W, Hu HB, Peng HS, Xu YQ, et al. Enhanced production of 2-hydroxyphenazine in Pseudomonas chlororaphis GP72. Appl Microbiol Biotechnol. 2011;89(1):169–77.
Liu KQ, Hu HB, Wang W, Zhang XH. Genetic engineering of Pseudomonas chlororaphis GP72 for the enhanced production of 2-hydroxyphenazine. Microb Cell Factories. 2016;15(1):131.
Silby MW, Winstanley C, Godfrey SA, Levy SB, Jackson RW. Pseudomonas genomes: diverse and adaptable. FEMS Microbiol Rev. 2011;35(4):652–80.
Yan YL, Yang J, Dou YT, Chen M, Ping SZ, Peng JP, et al. Nitrogen fixation island and rhizosphere competence traits in the genome of root-associated Pseudomonas stutzeri A1501. Proc Natl Acad Sci U S A. 2008;105(21):7564–9.
Mushegian AR, Koonin EV. A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc Natl Acad Sci U S A. 1996;93(19):10268–73.
Shao YC, He XY, Harrison EM, Tai C, Ou HY, Rajakumar K, et al. mGenomeSubtractor: a web-based tool for parallel in silico subtractive hybridization analysis of multiple bacterial genomes. Nucleic Acids Res. 2010;38:W194–200.
Xavier JC, Patil KR, Rocha I. Systems biology perspectives on minimal and simpler cells. Microbiol Mol Biol Rev. 2014;78(3):487–509.
Duffield M, Cooper I, McAlister E, Bayliss M, Ford D, Oyston P. Predicting conserved essential genes in bacteria: in silico identification of putative drug targets. Mol BioSyst. 2010;6(12):2482–9.
Smith CL, Weiss BL, Aksoy S, Runyen-Janecky LJ. Characterization of the achromobactin iron acquisition operon in Sodalis glossinidius. Appl Environ Microbiol. 2013;79(9):2872–81.
Castagnola A, Stock SP. Common virulence factors and tissue targets of entomopathogenic bacteria for biological control of lepidopteran pests. Insects. 2014;5(1):139–66.
Gross H, Loper JE. Genomics of secondary metabolite production by Pseudomonas spp. Nat Prod Rep. 2009;26(11):1408–46.
Umbarger HE, Brown B. Threonine deamination in Escherichia coli. II. Evidence for two L-threonine deaminases. J Bacteriol. 1957;73(1):105–12.
Csonka LN. Physiological and genetic responses of bacteria to osmotic stress. Microbiol Rev. 1989;53(1):121–47.
Nakai T, Deguchi T, Frebort I, Tanizawa K, Okajima T. Identification of genes essential for the biogenesis of quinohemoprotein amine dehydrogenase. Biochemistry. 2014;53(5):895–907.
Rocchetta HL, Burrows LL, Pacan JC, Lam JS. Three rhamnosyltransferases responsible for assembly of the A-band D-rhamnan polysaccharide in Pseudomonas aeruginosa: a fourth transferase, WbpL, is required for the initiation of both A-band and B-band lipopolysaccharide synthesis. Mol Microbiol. 1998;28(6):1103–19.
Nies DH. The cobalt, zinc, and cadmium efflux system CzcABC from Alcaligenes eutrophus functions as a cation-proton antiporter in Escherichia coli. J Bacteriol. 1995;177(10):2707–12.
Dietrich LE, Teal TK, Price-Whelan A, Newman DK. Redox-active antibiotics control gene expression and community behavior in divergent bacteria. Science. 2008;321(5893):1203–6.
Jarrell KF, McBride MJ. The surprisingly diverse ways that prokaryotes move. Nat Rev Microbiol. 2008;6(6):466–76.
Rashid MH, Kornberg A. Inorganic polyphosphate is needed for swimming, swarming, and twitching motilities of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2000;97(9):4885–90.
Chen MM, Hx C, Peng HS, Hu HB, Wang W, Zhang XH. Reaction kinetics for the biocatalytic conversion of phenazine-1-carboxylic acid to 2-hydroxyphenazine. PLoS One. 2014;9(6):e98537.
Bochner BR. New technologies to assess genotype-phenotype relationships. Nat Rev Genet. 2003;4(4):309–14.
Glass JI, Assad-Garcia N, Alperovich N, Yooseph S, Lewis MR, Maruf M, et al. Essential genes of a minimal bacterium. Proc Natl Acad Sci U S A. 2006;103(2):425–30.
Gil R, Silva FJ, Pereto J, Moya A. Determination of the core of a minimal bacterial gene set. Microbiol Mol Biol Rev. 2004;68(3):518–37. table of contents
Ji Y, Zhang B, Van SF, Horn WP, Woodnutt G, et al. Identification of critical staphylococcal genes using conditional phenotypes generated by antisense RNA. Science. 2001;293(5538):2266–9.
Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene. 1998;212(1):77–86.
Quenee L, Lamotte D, Polack B. Combined sacB-based negative selection and cre-lox antibiotic marker recycling for efficient gene deletion in Pseudomonas aeruginosa. BioTechniques. 2005;38(1):63–7.
Martínezgarcía E, De LV. Engineering multiple genomic deletions in gram-negative bacteria: analysis of the multi-resistant antibiotic profile of Pseudomonas putida KT2440. Environ Microbiol. 2011;13(10):2702–16.
Ravel J, Cornelis P. Genomics of pyoverdine-mediated iron uptake in pseudomonads. Trends Microbiol. 2003;11(5):195–200.
Mossialos D, Ochsner U, Baysse C, Chablain P, Pirnay JP, Koedam N, et al. Identification of new, conserved, non-ribosomal peptide synthetases from fluorescent pseudomonads involved in the biosynthesis of the siderophore pyoverdine. Mol Microbiol. 2002;45(6):1673–85.
Moon CD, Zhang XX, Matthijs S, Schafer M, Budzikiewicz H, Rainey PB. Genomic, genetic and structural analysis of pyoverdine-mediated iron acquisition in the plant growth-promoting bacterium Pseudomonas fluorescens SBW25. BMC Microbiol. 2008;8:7.
Yu BJ, Sung BH, Koob MD, Lee CH, Lee JH, Lee WS, et al. Minimization of the Escherichia coli genome using a Tn5-targeted Cre/loxP excision system. Nat Biotechnol. 2002;20(10):1018–23.
Yildiz FH, Schoolnik GK. Vibrio cholerae O1 el tor: identification of a gene cluster required for the rugose colony type, exopolysaccharide production, chlorine resistance, and biofilm formation. Proc Natl Acad Sci U S A. 1999;96(7):4028–33.
Spiers AJ, Kahn SG, Bohannon J, Travisano M, Rainey PB. Adaptive divergence in experimental populations of Pseudomonas fluorescens. I. Genetic and phenotypic bases of wrinkly spreader fitness. Genetics. 2002;161(1):33–46.
Friedman L, Kolter R. Genes involved in matrix formation in Pseudomonas aeruginosa PA14 biofilms. Mol Microbiol. 2004;51(3):675–90.
Dasgupta N, Wolfgang MC, Goodman AL, Arora SK, Jyot J, Lory S, et al. A four-tiered transcriptional regulatory circuit controls flagellar biogenesis in Pseudomonas aeruginosa. Mol Microbiol. 2003;50(3):809–24.
Deziel E, Comeau Y, Villemur R. Initiation of biofilm formation by Pseudomonas aeruginosa 57RP correlates with emergence of hyperpiliated and highly adherent phenotypic variants deficient in swimming, swarming, and twitching motilities. J Bacteriol. 2001;183(4):1195–204.
Choi YH, Lee JS, Yun S, Baik HS. A LuxR-type transcriptional regulator, PsyR, coordinates regulation of pathogenesis-related genes in Pseudomonas syringae pv. tabaci. J Life Sci. 2015;25(2):136–50.
Chugani SA, Whiteley M, Lee KM, D'Argenio D, Manoil C, Greenberg EP. QscR, a modulator of quorum-sensing signal synthesis and virulence in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2001;98(5):2752–7.
Visca P, Imperi F, Lamont IL. Pyoverdine siderophores: from biogenesis to biosignificance. Trends Microbiol. 2007;15(1):22–30.
Pel MJ, van Dijken AJ, Bardoel BW, Seidl MF, van der Ent S, van Strijp JA, et al. Pseudomonas syringae evades host immunity by degrading flagellin monomers with alkaline protease AprA. Mol Plant-Microbe Interact. 2014;27(7):603–10.
Bardoel BW, van Kessel KP, van Strijp JA, Milder FJ. Inhibition of Pseudomonas aeruginosa virulence: characterization of the AprA-AprI interface and species selectivity. J Mol Biol. 2012;415(3):573–83.
Guzzo J, Duong F, Wandersman C, Murgier M, Lazdunski A. The secretion genes of Pseudomonas aeruginosa alkaline protease are functionally related to those of Erwinia chrysanthemi proteases and Escherichia coli Alpha-haemolysin. Mol Microbiol. 1991;5(2):447–53.
Takaya A, Tabuchi F, Tsuchiya H, Isogai E, Yamamoto T. Negative regulation of quorum-sensing systems in Pseudomonas aeruginosa by ATP-dependent Lon protease. J Bacteriol. 2008;190(12):4181–8.
Van Rij ET, Wesselink M, Chin-A-Woeng TFC, Bloemberg GV, Lugtenberg BJJ. Influence of environmental conditions on the production of phenazine-1-carboxamide by Pseudomonas chlororaphis PCL1391. Mol Plant-Microbe Interact. 2004;17:557–66.
Pierson LS 3rd, Keppenne VD, Wood DW. Phenazine antibiotic biosynthesis in Pseudomonas aureofaciens 30-84 is regulated by PhzR in response to cell density. J Bacteriol. 1994;176(13):3966–74.
Hoffmann A, Thimm T, Droge M, Moore ER, Munch JC, Tebbe CC. Intergeneric transfer of conjugative and mobilizable plasmids harbored by Escherichia coli in the gut of the soil microarthropod Folsomia candida (Collembola). Appl Environ Microbiol. 1998;64(7):2652–9.
Schafer A, Tauch A, Jager W, Kalinowski J, Thierbach G, Puhler A. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 1994;145(1):69–73.
Shen XM, Chen MM, Hu HB, Wang W, Peng HS, Xu P, et al. Genome sequence of Pseudomonas chlororaphis GP72, a root-colonizing biocontrol strain. J Bacteriol. 2012;194(5):1269–70.
Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y, et al. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012;40(Database issue):D115–22.
Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, et al. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2003;100(24):14339–44.
Deng J, Deng L, Su S, Zhang M, Lin X, Wei L, et al. Investigating the predictability of essential genes across distantly related organisms using an integrative approach. Nucleic Acids Res. 2011;39(3):795–807.
Juhas M, Eberl L, Glass JI. Essence of life: essential genes of minimal genomes. Trends Cell Biol. 2011;21(10):562–8.
Kvitko BH, Collmer A. Construction of Pseudomonas syringae pv. tomato DC3000 mutant and polymutant strains. Methods Mol Biol. 2011;712:109–28.
Bochner BR, Gadzinski P, Panomitros E. Phenotype microarrays for high-throughput phenotypic testing and assay of gene function. Genome Res. 2001;11(7):1246–55.
Acknowledgements
We thank Prof. Yawen He for valuable technical assistance and thank GENEWIZ Company for the oligonucleotide synthesis and sequencing service. We thank Dr. Ruilian Yao and Dr. Muhammad Bilal for advice on the manuscript. We gratefully acknowledge the thoughtful comments and suggestions from the editor and three anonymous reviewers of the journal.
Funding
This work was supported by the National Natural Science Foundation of China (No. 31670033 and No. 31270084), the National Key Basic Research Program of China (No. 2012CB721005), and the National High Technology Research and Development Program of China (No. 2012AA022107).
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and its Additional files].
Author information
Authors and Affiliations
Contributions
XHZ, XMS, XQH, and HBH conceived, coordinated, and designed the study. WW, XHZ, and XMS were responsible for sequencing. XMS, XHZ, and ZW performed experiments and data analyses. XHZ and XMS drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Predicted essential genes of P. chlororaphis strain. (XLSX 29 kb)
Additional file 2:
Homology search of P. chlororaphis essential genes. Essential genes in the genome of P. chlororaphis were predicted using homology searches based on local BLASTP program. (TIFF 169 kb)
Additional file 3:
Deleted genes in each MD region. (XLSX 51 kb)
Additional file 4:
Primers used in this study. (XLSX 14 kb)
Additional file 5:
Flowchart of the markerless deletion method based on pK18mobsacB. (TIFF 110 kb)
Additional file 6:
Nutrient added in each well of the Biolog PM5 plate. (XLSX 12 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Shen, X., Wang, Z., Huang, X. et al. Developing genome-reduced Pseudomonas chlororaphis strains for the production of secondary metabolites. BMC Genomics 18, 715 (2017). https://doi.org/10.1186/s12864-017-4127-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-4127-2