
Abstract
Background
Knowledge about the distribution of the genetic variation of marine species is fundamental to address species conservation and management strategies, especially in scenarios with mass mortalities. In the Mediterranean Sea, Petrosia ficiformis is one of the species most affected by temperature-related diseases. Our study aimed to assess its genetic structure, connectivity, and bottleneck signatures to understand its evolutionary history and to provide information to help design conservation strategies of sessile marine invertebrates.
Results
We genotyped 280 individuals from 19 locations across the entire distribution range of P. ficiformis in the Atlanto-Mediterranean region at 10 microsatellite loci. High levels of inbreeding were detected in most locations (especially in the Macaronesia and the Western Mediterranean) and bottleneck signatures were only detected in Mediterranean populations, although not coinciding entirely with those with reported die-offs. We detected strong significant population differentiation, with the Atlantic populations being the most genetically isolated, and show that six clusters explained the genetic structure along the distribution range of this sponge. Although we detected a pattern of isolation by distance in P. ficiformis when all locations were analyzed together, stratified Mantel tests revealed that other factors could be playing a more prominent role than isolation by distance. Indeed, we detected a strong effect of oceanographic barriers impeding the gene flow among certain areas, the strongest one being the Almeria-Oran front, hampering gene flow between the Atlantic Ocean and the Mediterranean Sea. Finally, migration and genetic diversity distribution analyses suggest a Mediterranean origin for the species.
Conclusions
In our study Petrosia ficiformis showed extreme levels of inbreeding and population differentiation, which could all be linked to the poor swimming abilities of the larva. However, the observed moderate migration patterns are highly difficult to reconcile with such poor larval dispersal, and suggest that, although unlikely, dispersal may also be achieved in the gamete phase. Overall, because of the high genetic diversity in the Eastern Mediterranean and frequent mass mortalities in the Western Mediterranean, we suggest that conservation efforts should be carried out specifically in those areas of the Mediterranean to safeguard the genetic diversity of the species.
Similar content being viewed by others

Background
The Mediterranean basin is one of the most diverse places on Earth in terms of animal and plant diversity, yet one of the most threatened (e.g., [1, 2]. Most of these threats are human-related and comprise pollution, direct and indirect mechanical damage and extraction of species, which result in habitat degradation, fragmentation, and loss, and alterations to the native biodiversity, among others [1, 2]. According to Coll and collaborators [2], several areas in the Mediterranean, such as the North Adriatic Sea, the Spanish coasts, and the Gulf of Lion in France, are known to have high diversity in terms of marine invertebrates (at least those with commercial value). These diversity hotspots are also the most threatened, which presents a dramatic scenario for the conservation of invertebrates in this area of the planet.
Most notably, climate-related natural disasters are considered one of the acutest threats for invertebrates in the Mediterranean Sea, with the Eastern Mediterranean being the most affected region [2,3,4]. Although the conservation emphasis is often on commercially important species or those considered as threatened in the Red List [5], a wide variety of sessile invertebrates are strongly affected by temperature anomalies, pollutants or its combined effect [3, 6]. In particular, sponges of the order Dictyoceratida have been considered one of the most affected invertebrates by warming oceans [3, 7,8,9]. Other than dictyoceratids, the emblematic haplosclerid sponge Petrosia (Petrosia) ficiformis (Poiret, 1789) has been recurrently reported to be affected by necrotic processes that decimated their populations in several areas of the Mediterranean [3, 10]. In all cases, mortalities were linked to dramatic increase in seawater temperature and heavier rainfall in the area [10]. The necrotic white tissue observed in Ligurian and Spanish populations of P. ficiformis was attributed to the loss of cyanobacterial symbionts and a heat shock destabilizing the adhesion and cytoskeletal organization of the exopinacoderm [10]. A similar situation was described when corals bleach due to the loss of zooxanthellae [11].
Petrosia ficiformis was originally described from what was then called the Berber coast (now Morocco, Algeria, Tunisia and Libya). It is a common and ubiquitous shallow water species [12], distributed along the entire Mediterranean Sea and the Macaronesia (Azores, Madeira, and Canary Islands) [13]. Petrosia ficiformis is a massive or lobate wine-colored sponge that grows slowly (approximately 1% monthly increase in volume) [14]. Besides being a frequent component of benthic assemblages in the Atlanto-Meditterranean shallow waters (10–50 m depth) and a structural species, it has economic value because of its ability to produce bioactive products [12]. Petrosia ficiformis is an oviparous species, which exhibits a special type of ciliated crawling stereoblastula [15] with very limited dispersal potential. These features, slow growth and restricted dispersal, make P. ficiformis a highly vulnerable species with a poor ability to recovery during disease episodes.
The shallow waters of the Atlanto-Mediterranean region experience a range of climatic conditions, including subtropical, temperate and subarctic conditions, leading to a high diversity of marine invertebrates generally, and of sponges in particular [16]. This area has experienced major geological events such as the Messinian Salinity Crisis (MSC), approximately 5 Myr BP, with a massive drop in available coastal habitat [17]. In addition, sea surface temperature and sea level drastically decreased during the Last Glacial Maximum (LGM, 30–19 Myr BP). All these geological events have profoundly shaped the marine fauna diversity patterns [16, 18, 19], and in particular, the present-day sponge fauna in the Mediterranean Sea is suggested to be the result of a recolonization from Atlantic refugia (Ibero-Moroccan and West African areas) after the MSC [20, 21]. More recently, during the interglacial periods of the Pleistocene, the warmer Mediterranean Sea received colder shallow Atlantic surface water, and the reverse occurred during glacial periods [17, 18]. In this sense, the Alboran Sea is considered to be an important area containing information on the past and recent faunistic interchange between the Atlantic and the Mediterranean Sea [18, 19]. According to Patarnello and collaborators [22], two plausible scenarios can be drawn for species spanning their distributions in the Mediterranean Sea and the Atlantic Ocean: 1) “complete genetic separation between Atlantic–Mediterranean populations since the early Pliocene”, and 2) “complete absence of population differentiation, usually following late Pleistocene recolonization”.
Population genetic analyses of marine Atlanto-Mediterranean invertebrate species have been performed for some taxa, including echinoderms [23,24,25,26,27,28], molluscs (e.g., [29, 30]), arthropods (e.g., [31, 32]), and sponges (e.g., [33,34,35,36]). In general, for most marine invertebrates, migration of Atlantic populations into the Mediterranean is lower than between the Eastern and Western Mediterranean basins mostly due to the presence of the Almeria-Oran front (e.g. [26, 30]). Also, higher local population differentiation is observed for sessile invertebrates with short larval duration phases, while the genetic structure of mobile invertebrates with long larval duration phases seems to be largely affected by a strongest effect of oceanographic fronts [37]. However, the genetic structure and connectivity patterns of sponges, covering both the Eastern and Western Mediterranean sub-basins and the Northeastern Atlantic, have been studied in few species, mostly with mitochondrial markers with low variability, providing poor resolution to infer detailed patterns [38, 39] or are based on limited sampling in the Atlantic region [34, 35]. Interestingly, the phylogeographic patterns inferred for Atlanto-Mediterreanean sponges appear to be the result of contrasting past demographic events. While Crambe crambe (Schmidt, 1862) is a Mediterranean species that later colonized Macaronesia via human transport [38], Phorbas fictitius (Bowerbank, 1866) originated in the Macaronesian archipelagos, colonizing the mainland using currents that flow towards the Iberian Peninsula [39].
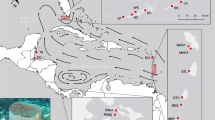
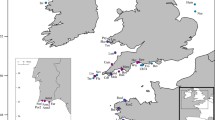
The main goal of this study was to evaluate the distribution of the genetic diversity and assess population differentiation and connectivity of P. ficiformis in its current distribution range (Fig. 1 and Table 1), including areas where massive mortalities have been reported, using highly informative microsatellite markers. In addition, our aim was to infer the phylogeographic history of the species to understand recent demographic events and establish its potential geographical origin.

Sampling sites spanning the entire distribution range of Petrosia ficiformis. Maps were obtained and modified from Google maps (Map data: Google, DigitalGlobe, 2018)
Results
Genetic diversity in Petrosia ficiformis
The average number of alleles per site was 37.05, ranging from 24 in FLO, LIG, NAP, SLO, SCRO, JECRO, CRE, and ISR to 81 in BLA (Table 2, Additional files 1, 2). However, the number of effective alleles (Table 2) was highest in ISR (4.276) and lowest in FLO (1.532), similar to what we observed for the allelic richness following rarefaction (Table 2, Additional file 1). The allele frequency per locus and population is shown in Additional file 2. Only four individuals were identified as potential clones with GENODIVE: two in CAR and two in ULL, although one of the two potential clones observed in CAR was not genotyped for one microsatellite marker (30PETRO).
Genetic diversity was highest in the Eastern Mediterranean site of ISR (He = 0.738) and the Western Mediterranean sites of BLA (He = 0.718), LIG (He = 0.706), and NAP (He = 0.682) (Table 2, Fig. 2, and Additional file 1). In the Atlantic, CAN showed the highest genetic diversity (He = 0.618), while the rest of sites showed values below 0.500 (Table 2, Fig. 2, and Additional file 1). The lowest genetic diversity value was detected in FLO (He = 0.341) (Table 2, Fig. 2, and Additional file 1). The average genetic diversity (He) for the 19 populations was 0.578. Within the Mediterranean populations, observed heterozygosity values (Ho) were higher in ISR, SLO, NAP and BLA, while in the Atlantic populations, CAN showed the highest Ho (Table 2 and Additional file 1). Significant differences in He values were detected in the comparisons including Atlantic populations (Atlantic vs. Western Mediterranean: p-value = 0.00482; Atlantic vs. Eastern-Mediterranean: p-value = 0.0014) but not in the comparisons between Western and Eastern-Mediterranean (p-value = 0.1133).

Correlation between genetic diversity (He) of Petrosia ficiformis and geographical location. The sea surface temperature (SST) anomalies during the last glacial maximum (30,000–19,000 years ago) is plotted onto the graph. Data for SST was obtained from [79]
The inbreeding coefficients (FIS) were positive in 11 out of the 19 sites (SMI, FLO, MAD, CAN, CART, BLA, MRS, NIZ, NAP, LIG, and JECRO), LIG being the site with the highest value (FIS = 0.311; Table 2, Additional file 1), indicating that individuals in these sites were more related than expected if random mating occurs. The closest value to perfect HWE (FIS = 0) was detected in SCRO (FIS = − 0.001; Table 2, Additional file 1), indicating that individuals within this site were less related than expected, and other populations (ULL, ISR, CRE, SLO, ESC, FEL, and CAR) also showed negative (then effectively 0) values for FIS. The populations of CAR, CART, FEL, ESC, NAP, SLO, SCRO, CRE and ISR were in HWE (Table 2, Additional file 1). The microsatellite markers that contributed the most to any HWE deviation in the rest of populations were 1PETRO, 17PETRO and 19PETRO (Table 2, Additional file 1), but excluding these three markers did not change the result, and therefore we included them in all subsequent analyses.
Population differentiation in Petrosia ficiformis
The optimal number of clusters identified using delta K (Additional file 3A) for P. ficiformis was six, then eight, and finally ten (Fig. 3, Additional file 3A–B). In the optimal scenario considering K = 6, the following clusters were obtained (Fig. 3a–b): Cluster 1 (purple): grouping the populations in Azores (SMI, FLO); Cluster 2 (crimson): grouping the other populations in Southern Macaronesia (MAD, CAN) and the south of Spain (CAR); Cluster 3 (green): containing ULL and half of the population of BLA, as well as some individuals from the populations CART, FEL, LIG, and SCRO; Cluster 4 (blue): containing the rest of Western Mediterranean populations (half of the population of BLA, most FEL, some individuals in ESC, MRS, NIZ, half LIG, and some individuals in NAP); Cluster 5 (pink): comprised of some individuals in CART, most ESC individuals, half of the NAP population, and the Eastern Mediterranean population of ISR; and finally Cluster 6 (orange): the Adriatic populations (SLO, SCRO, JECRO) and the Eastern Mediterranean population of CRE. The second optimal clustering identified 8 clusters (Fig. 3c–d), detecting substructure in Cluster 2 (Fig. 3c), with the Southern Macaronesia populations and CAR appearing in two different clusters (clusters 2 and 7) and a new cluster (cluster 8) grouping individuals from BLA, ESC, MRS, and LIG (Fig. 3c–d). The intriguing cluster 5 from K = 6 and K = 8, grouping individuals from quite disparate populations (ESC, NAP and ISR), was not detected in the scenario with K = 10, showing separation of those populations in two different clusters (Additional file 3B). Using a multivariate approach, the population structure and individual assignment to a given population assessed with adegenet revealed that 6 to 10 clusters would define the most probable structure (Additional file 3C), the 10-cluster hypothesis having the lowest BIC (Additional file 3D). In the 10-cluster scenario (Additional file 3D), most individual assignments were similar to those obtained by STRUCTURE (Fig. 3c-d and Additional file 3D), except for those of CAN, CAR, and SLO, which were assigned here to separate clusters (Additional file 3D).

Individual genotype assignment of Petrosia ficiformis to clusters (K) as inferred by STRUCTURE for all studied sites with a–b. K = 6 and c–d. K = 10. In b and d, pie frequency charts depict the percentage of individuals assigned to each of the 5 clusters from b and c for all sites. Colors are assigned as in b and d except grey which shows the percentage of individuals for which the assignation was not clear (Q-value cutoff 60). Red lines indicate barriers detected by BARRIER ranked a to f in order of importance. Maps were modified from Wikipedia under a Creative Commons license (User Canuckguy)
Population differentiation was also assessed using FST pairwise comparisons, revealing significant differences among all population pairs (p < 0.008), except for the population pairs CRE and SCRO (Fig. 4a and Additional file 4). The highest FST values (> 0.400) were observed between Atlantic sites and Western and Eastern Mediterranean sites: first between SMI and the following sites: (1) MAD, ULL, SCRO, and CRE; (2) between FLO and ULL, MRS, SCRO, CRE, and (3) between MRS and MAD (Fig. 4a and Additional file 4). The lowest value of FST (0.02233) was observed between SCRO and CRE (Additional file 4). The global FST for all populations was 0.2348 and the average values were around 0.15 (Fig. 4b).

a. FST comparisons between 19 populations of Petrosia ficiformis. All comparisons showed significant p-values except for the comparison between CRE and SCRO. Actual values for each comparison can be found in Additional file 2. b. Average values (and standard errors) of FST for each sampling site across Petrosia ficiformis distribution
To test whether the population differentiation observed with the FST pairwise analyses was due to an isolation by distance (IBD), a Mantel test was performed for the whole dataset, and approximately 47% of the variation observed could be attributable to IBD (r2 = 0.466, p = 0.03). However, due to the limitations of the Mantel test [40, 41], we also performed stratified Mantel tests within each of the clusters separated by genetic breaks identified by BARRIER (see below), finding no significant IBD in any of them (Clusters tested: a: CAN, MAD, CAR: r2 = 0.615, p = 0.310; b: Western Mediterranean 3: r2 = − 0.168, p = 0.307; c: Eastern Mediterranean: r2 = 0.245, p = 0.173).
BARRIER located six a priori selected barriers in order of importance: a, separating the Azores sites; b, between SMI and FLO; c, separating the Atlantic sites from the Mediterranean ones; d, located approximately at the Almeria-Oran front and separating CAR and CART from the rest of the Mediterranean sites; e, located approximately at the Strait of Sicily front separating the Western and Eastern Mediterranean sites; and finally f, isolating the Spanish sites (BLA, FEL, ULL, and ESC) from the rest of the Mediterranean sites (Fig. 3b, d).
The DAPC analyses for all the sampling sites and only the Mediterranean sites (Fig. 5a–b) rendered different patterns. When the matrix containing all the sampling sites was analysed, the Azores populations (SMI and FLO) were clearly separated from the rest considering both axes (Fig. 5a) and the Y-axis also separated the populations of the Eastern Mediterranean + Southern Macaronesia (MAD, CAN) and those in the Western Mediterranean and ISR (Fig. 5a). When only the Mediterranean sites were analysed (Fig. 5b), a clear separation of the Eastern and Western Mediterranean sites was observed, with only the CAR population (named Pre-Alboran site because it is located before the Almeria-Oran Front) separated from the rest of Mediterranean sites. It is important to note that in these two cases, the ISR site was closer to the Western Mediterranean sites than to the Eastern sites (Fig. 5a–b).

Population differentiation in Petrosia ficiformis. a. Subdivision of all sites according to DAPC analysis. b. Subdivision of the Mediterranean sites according to DAPC analysis
All the AMOVA analyses (A. Atlantic vs. Mediterranean; B. Atlantic vs. Western Mediterranean vs. Eastern Mediterranean; and C. Azores vs. Southern Macaronesia vs. Western Mediterranean vs. Eastern Mediterranean) were significant among groups, populations and individuals (Table 3). However, the analysis that rendered the highest value for the percentage of variance explained among the groups used (13.2%) was that using the groups Azores, Southern Macaronesia, Western Mediterranean, and Eastern Mediterranean (Table 3).
Migration patterns in Petrosia ficiformis
We identified only 7 individuals as last generation migrants: one from FEL into CAR, another one from FEL into CART, one from MRS into BLA, one from ULL into FEL, two from NIZ into ESC and LIG, and one from NAP into LIG, with mean distances among these populations ranging from 43 to 632 km. The migration patterns obtained with GENODIVE showed that Atlantic sites did not contain any migrant among their individuals (Fig. 6a). In the Mediterranean, the different sites showed a percentage of migrants between 3.7% (from FEL in CAR) and 28% (from CRE in SCRO), except for SLO, where we did not observe any (Fig. 6a). Interestingly, when analyzing the directionality of recent migration events among areas, the main gene flow appeared between the Adriatic Sea and the Eastern Mediterranean basin in both directions (Fig. 6b), and then from the Eastern Mediterranean into the Western Mediterranean and vice versa (Fig. 6b). This analysis also indicates that there were low levels of migration from the Atlantic populations of MAD and CAN (MAC) into the Western Mediterranean (Fig. 6b), and almost negligible migration from the Azores to the Western Mediterranean (Fig. 6b). When the migration patterns were analysed in more detail using pairwise comparisons among all populations (Fig. 6c and Additional file 5), it was clear that the gene flow was relatively high among the Macaronesian islands (SMI, FLO, MAD, and CAN) and the Southern Spain populations (CAR and CART) and also with CRE, and moderate with the rest of the Mediterranean, but no gene flow was detected from the Mediterranean into the Atlantic populations. Within the Mediterranean, we detected high migration rates among the populations in the Western Mediterranean and also with the Aegean Sea, and from the Adriatic into ESC and NAP (Fig. 6c and Additional file 5). The highest effective population sizes, assessed from theta, were found for some populations from the Atlantic, Western Mediterranean and the Adriatic: FLO, MAD, CAN, CAR, CART, BLA, ULL, SLO and JECRO (Table 2).

Migration patterns in Petrosia ficiformis. a. Number of last generation migrants indicated in percentage of the population for each sampling site. b. Migration estimates among the four areas considered AZO (SMI, FLO), CAN, MAD, WES (CAR, CART, BLA, ULL, FEL, ESC, MRS, NIZ, LIG, and NAP), and EAS (SLO, SCRO, JECRO, CRE, and ISR). Only values over 0.1 are shown in the graph. c. Heatmap of coalescent migration estimations. The scale refers to migration rates between 0 and 250 (actual data can be seen in Additional file 5)
Demographic events in Petrosia ficiformis
Several populations showed signs of a recent reduction of their population size using the Wilcoxon rank tests under two different models: with SMM, the populations of CAR, CART, BLA, MRS, NIZ, SCRO, JECRO, and ISR appeared to have a bottleneck, but bottlenecks in only MRS, NIZ, and SLO were detected under the TPM model (Table 2). In any case, our results should be taken with caution since our sample numbers were sometimes too low to robustly detect bottleneck events.
Discussion
Negligible clonality in populations of Petrosia ficiformis
Sponges are commonly considered clonal organisms since they are capable of both sexual and asexual reproduction, the latter including gemmulation, fission, and budding [42]. The larvae of many clonal organisms, not only sponges but also of bryozoans and cnidarians, are usually short-lived small propagules that remain within close proximity of their progenitors [43]. Therefore, other dispersal mechanisms maybe responsible for the large distribution ranges observed in many clonal organisms, including rafting and passive dispersal of newly settlers by oceanic currents [43]. However, even though that might be true for some sponges (e.g., [44]), Petrosia ficiformis seems to not rely on any dispersal mechanism via asexual reproduction. In our analysis, only two out of 280 individuals (0.7%) showed potential clonality signatures. This almost negligible clonality contrasts with the high values obtained for other sponges [45] using similar sampling strategies and geographic separation of locations, but could be easily explained by their different biological traits. Petrosia ficiformis is a massive sponge, with no asexual reproduction documented so far [15]. In contrast, both C. crambe and Cliona spp. are encrusting sponges known to undergo frequent fission processes [46, 47], which explains their high rates of clonality. In our case, it seems that the connectivity and gene flow between populations of P. ficiformis is mostly accomplished by sexual reproductive strategies since no asexual reproduction or any other dispersal strategy (e.g., rafting) have ever been reported for P. ficiformis.
Contrasting patterns in inter- and intrapopulation structure along the distribution range of Petrosia ficiformis
The average genetic diversity (He) of P. ficiformis (0.578), falls into the range (0.4–0.9) observed using microsatellite markers in other Mediterranean sponges, including poecilosclerids, dictyoceratids, and scopalinids [33,34,35,36, 48], as well as in clionaids, petrosiids and calcareous sponges in other latitudes (e.g., [45, 49,50,51]). Interestingly, relatively similar (although slightly higher) values of genetic diversity (He) to those of P. ficiformis are usually reported for other Mediterranean sessile (e.g., ~ 0.8 in Corallium rubrum, [52]; ~ 0.7 in Paramuricea clavata, [53]) and vagile organisms (e.g., ~ 0.6 in the bluefin tuna, [54]; ~ 0.75 in the sea bass, [55]; ~ 0.8 in Tripterygion delaisi, [56], ~ 0.8 in Paracentrotus lividus, [57]; ~ 0.7 in Echinaster sepositus, [26]).
The departures from HWE in 10 out of the 19 populations of P. ficiformis analysed (Table 2, Additional file 1) can be explained by several factors. In the Macaronesian archipelagos (except in CAN), the Western Mediterranean populations of BLA, MRS, NIZ, and LIG, and the Eastern Mediterranean population of JECRO, heterozygote deficiencies (and therefore inbreeding) seemed to be responsible. For many other organisms, the presence of null alleles, Wahlund effect [58], and the production of high numbers of sperm cells in free-spawners are also responsible for HW disequilibrium patterns [59]. For P. ficiformis, the incidence of null alleles was negligible in all populations (only one microsatellite showed null alleles), and no substructure attributable to Wahlund effect was detected in the population differentiation analyses. Other factors related to the sexual reproductive strategy of P. ficiformis might be playing a significant role in the departures for HWE observed here. Most sponge larvae have very limited dispersal capabilities, despite being free-swimming larvae [60,61,62], but P. ficiformis falls into the extreme end of the spectrum. In theory, the average distance of dispersal should be greater in oviparous species than in viviparous species, because it inevitably includes external fertilization [43]. However, even though P. ficiformis is an oviparous species, its larva has very peculiar features that restrict its dispersal capabilities compared to other sponge larvae. Instead of swimming in the water, they crawl for short periods (up to 2 weeks) until settlement [15]. Therefore, its dispersal capabilities are strongly limited by the slow crawling of the larvae. In addition, P. ficiformis is a gonochoristic species, and it devotes almost all its internal tissues to massive oocyte and sperm production [15]. But although large numbers of gametes are released in the water that could potentially disperse farther, the incrementing dilution factor favours fertilization within few meters in experiments performed in the field for marine invertebrates. Indeed, fertilization rates drop to less than 5% when gametes are dispersed to 1 m in sea urchins [63], and to 0% at 7 m in hydroids [64], although some species could still fertilize within 10 m [65]. In fact, using genetic markers in corals, the distance that sperm dispersed and mantained fertilization success was always less than 20 m [66]. However, a long lifespan (16–26 h) for the sperm of the ascidian Botryllus schlosseri [67] has been reported, and a similar phenomenon may exist in P. ficiformis. In all, inbreeding and population self-recruitment seem the most plausible causes of homozygote excess in P. ficiformis.
Only two populations that were not in HWE showed excess of heterozygotes (CAN and ULL). Whereas in studies with single nucleotide polymorphisms, gain of heterozygosis in populations not in HWE were mainly caused by genotyping errors [68], biological strategies such as selection against homozygotes, or extreme longevity of individuals were suggested for excess of heterozygotes in a sympatric Mediterranean sponge [33].
Molecular signatures of demographic contractions in Petrosia ficiformis
Garrabou and collaborators documented die-offs of Petrosia ficiformis populations due to a disease outbreak in 2003 in the Provence Coast and the Gulf of Genoa [3], necrotic events in the Gulf of Genoa have also been reported in previous years [10]. In our study, the populations of MRS and NIZ had signatures of bottlenecks under different models (Table 2). But although BLA also presented a bottleneck, no similar die-offs were documented for the Catalan coast. However, during our periodic surveys conducted in 2004–2007 to establish the reproductive patterns of P. ficiformis in the area, we recurrently observed necrotic processes after egg spawning in February–March (Riesgo personal observations). Even though we did not record dead sponges, such necrotic processes could have led to die offs in other years in these populations.
In addition, although the sampling sites of SLO, SCRO, JECRO, CRE, and ISR also showed signatures of bottleneck in one test, massive population die-offs were never reported for those areas. Interestingly, while JECRO showed heterozygosity deficiency, SLO, SCRO, and ISR were in HWE (Table 2). In this case, the results of the bottleneck analysis should be taken with caution given the small sample sizes analysed, which could explain the incongruence between levels of allelic richness and heterozygosity in these populations.
Population differentiation and connectivity in Petrosia ficiformis
We found strong population structure in our dataset both at small (i.e. significant FST between nearby populations), medium (i.e. significant differences between populations within basins) and large (i.e. significant population differentiation and AMOVAs among basins) geographical scales. The global and average Fst values for P. ficiformis were in the upper extreme of the range for sponges [69]. In P. ficiformis, population differentiation was even detected between populations located less than 50 km apart, similar to what was reported for I. fasciculata [36] although the differentiation was much more conspicuous in P. ficiformis.
Both population clustering approaches employed here, situated the most probable number of clusters (or K) between 6 and 10, rendering very similar results. While in the sympatric sponge Ircinia fasciculata [36] two clusters mostly explained the distribution of the genetic diversity structure observed in the Mediterranean Sea, for P. ficiformis we found a much more structured scenario. This could be attributed to a combination of both the limited dispersal potential of P. ficiformis (discussed above) and the stronger effect of oceanographic barriers to gene flow among the populations analysed here. Although in P. ficiformis an IBD pattern was detected initially in this and previous studies [70], after performing stratified Mantel tests for each of the clustering groups, IBD was not evident. Similarly, many sponge species have been reported as not showing IBD [36].
Indeed, instead of IBD, it seems that oceanographic fronts play a more prominent role in the spatial distribution of neutral genetic diversity of marine organisms around the globe (e.g., [71, 72]), but especially in sessile organisms [69]. In the Mediterranean, the most prominent barriers to gene flow and well-known for their isolating effects of the marine fauna are: the Almeria-Oran front, the Sicily Channel and the Otranto Strait [22, 37, 73]. In our study, the two first barriers overlapped with genetic discontinuities. However, the main barrier to gene flow was not found in the Mediterranean, but between the Azores populations and the continental ones (Fig. 3b, d), possibly driven by depth (> 4000 m) or distance (> 2000 km), with lack of suitable habitat for P. ficiformis in intermediate waters. Water circulation between the continent and the Azores archipelago [74] does not favour migration between them [75], and deep water is likely a barrier to the shallow-dwelling P. ficiformis [71, 76], potentially separating Azores from the rest of the Macaronesia (Madeira and the Canary Islands). Within the Mediterranean Sea, we detected the usual barriers to gene flow (Almeria-Oran front and Otranto Channel) reported in many other marine organisms [22, 37, 73], but interestingly, we found a genetic discontinuity in the Gulf of Lion, separating the populations in the Catalan coast from those in the French coast (Fig. 3b, d). In this area, freshwater from the Rhône river and the strong cooling induced by the Mistral south of the Rhône delta generate a layer of cold and less saline water during winter and spring [77] that could act like a barrier for sponge larval dispersal.
In our study, the closest populations geographically (BLA, ULL, FEL y ESC) did not appear in the same genetic cluster, and showed diverging proportions of individuals of 3 (when k = 6) or 4 (when k = 8) clusters. Some degree of larval retention could play a role in the case of ULL. That site is located in a submarine pinnacle that goes down to 65 m and is separated few km from the coast. It was identified as a highly-isolated population in K = 10, which could partially explain the low allelic richness of the site. The rest of microscale patterns of differentiation observed here are difficult to explain. In other sessile organisms with low dispersal abilities such as gorgonians, microscale differentiation has been explained mostly by a higher degree of self-recruitment [78]. In our case, also local gyres or connectivity with other non-sampled populations located in the Balearic Islands, Corsica and Sardinia, could be behind such genetic differentiation patterns.
Colonization patterns and migration in Petrosia ficiformis
Interestingly, the total number of alleles, regardless of the estimate used, and the genetic diversity (He) were always highest in the Mediterranean populations and lowest in those of the Macaronesian region (Table 2, Fig. 2 and Additional file 1), suggesting a Mediterranean origin for the species and subsequent colonization of Atlantic sites. A similar pattern was observed for the dictyoceratid Spongia officinalis [34] and the poecilosclerid Crambe crambe [38], where a colonization of the Macaronesia from the Mediterranean was also suggested. Using other molecular markers, the origin of P. ficiformis was also hypothesized to be Mediterranean, the source population most likely being located in the Eastern Mediterranean, which suffered a less dramatic seawater temperature drop (Fig. 2) during the LGM [70] (3 °C below current values [79]) and would likely have offered refuge as previously suitable habitat disappeared [22]. The affinity of P. ficiformis for warm waters is further supported by the fact that its reproductive season is triggered by the temperature rising in summer, and release of gametes with the abrupt temperature drop that characterizes the arrival of winter [36, 80].
The most recent calibrated phylogenetic tree for demosponges places the appearance of Petrosia ficiformis most likely during a period between the Cretaceous (approx. 100 Mya) and the Paleogene (23 Mya) [81], when the Mediterranean was still considered to be the Tethys Sea and harboured a highly-diverse warm-water biota [82]. Therefore, the appearance of P. ficiformis likely pre-dated the Messinian Salinity Crisis (MSC) during the Miocene (dated on 5.6 Mya, [17]), when several Mediterranean evaporitic basins could have acted as refugia for P. ficiformis [83]. Even though true marine conditions have been reported for some areas during the MSC [83], most areas show evidence of extensive desiccation and formation of hypersaline and freshwater sub-basins [84], but there is still controversy about the surviving fauna [85,86,87]. Alternatively, during the MSC, P. ficiformis could have survived in marine caves [82] where it is particularly abundant today. These scenarios are impossible to test with our dataset since the genomic traces of such events would not be detected with microsatellites (fast evolving markers), but certainly the higher levels of genetic diversity in the Eastern Mediterranean and the warm-water affinity of P. ficiformis point to this direction. The alternate possibility of P. ficiformis being an Atlantic species introduced in the Mediterranean after the Zanclean flood of the Mediterranean Sea, approximately 5.33 Mya [17], is not supported by the current lower genetic diversity and relative isolation of the Atlantic populations.
During the Pliocene, circulation patterns similar to the ones we currently find were established, preventing gene flow from the Mediterranean towards the Atlantic [17]. This was supported by our migration results showing null migration rates in that direction. On the other hand, our results suggest moderate gene flow between the populations of the Atlantic Ocean and those in the Alboran Sea, located closer to the Strait of Gibraltar, in both directions (Fig. 6), and moderate to low gene flow from the Atlantic towards the main Mediterranean basin (Fig. 6), except from the high values obtained from some Macaronesian populations into the Aegean Sea (Fig. 6c). The circulation patterns in the Mediterranean would have promoted connectivity from the Atlantic populations, along the southward coasts, and into the Aegean Sea (CRE), which could explain the high migration values detected. Currently, the strongest barrier to gene flow between the Atlantic Ocean and the Mediterranean is the Almeria-Oran front (see reviews of [22, 37]), an oceanographic barrier that has been shown to impair the connectivity of many benthic invertebrates (e.g., [25, 26, 88]), including sponges [35, 36]. Indeed, besides the genetic discontinuities found in the Macaronesian populations suggesting the presence of barriers to gene flow in the area, the Almeria-Oran front seems to be a prominent factor driving the current gene flow among the populations of P. ficiformis (Fig. 6c), preventing gene flow from the Atlantic into the Mediterranean. Moderate levels of migration were detected among the Mediterranean populations, with higher migration rates between the Adriatic and Eastern Mediterranean populations (Fig. 6c). In fact, all last generation migrants detected were exchanged among Mediterranean populations, although surprisingly among the Western ones, in a range from 40 to 630 km. This sort of direct migration is difficult to explain given the low dispersal abilities of the larva of P. ficiformis, and more research is definitely needed to understand the importance of gamete dispersal in the connectivity of this particular species. In this sense, a long gamete lifespan in P. ficiformis, as described in other organisms [67], could explain some of the dispersal necessary to cover such distances.
Conclusions
Our study provides a detailed account of the distribution of genetic diversity across the geographic natural range of P. ficiformis, including information on how that diversity is connected and maintained during massive die-offs. In summary, it seems that the Eastern Mediterranean (Crete and Israel) harbours the greatest diversity, and we suggest the possibility that those could act as a reservoir in case population decimation affects other areas in the Mediterranean. However, that area shows higher levels of connectivity with the Adriatic Sea, and therefore in light of our results efforts should be made to protect populations of P. ficiformis not only in already existent Marine Protected Areas –MPAs– in the Eastern Mediterranean (or to create new MPAs to preserve them), but also in the Adriatic and Western Mediterranean in order to provide westward “gene flow corridors” for the species. In addition, the Atlantic populations deserve special attention, since they show the highest degree of reproductive isolation and if impacted, they will not receive recruits from closely related areas, which could end in their disappearance.
There are ongoing efforts to assess the impact of climate change on mass mortalities in the Mediterranean by modelling the current mass mortality risk associated with thermal stress of benthic coastal ecosystems [89]. However, those studies have only used one model species, Paramuricea clavata, for which genetic diversity and connectivity data are available (e.g., [53, 90]), and it is necessary to use more species in the models to obtain a more comprehensive assessment of the risks and impacts. Therefore, the information we provide in the present study is very much needed to design sound management and conservation strategies to mitigate the short term effects of global warming in the Mediterranean.
Methods
A total of 280 sponges were sampled by the authors between 2008 and 2013 using SCUBA diving in 19 sites spanning the entire distribution range for P. ficiformis: the Atlanto-Mediterranean region that includes the Macaronesian islands of Azores, Madeira and the Canary Islands, and the entire Mediterranean Sea except for the Southern Mediterranean coastline (Fig. 1 and Table 1). Samples from some localities were collected by colleagues from their focal study areas (see acknowledgements). Variation in geographical distance among sampling sites ranged from 43 to 9115 km for the whole studied area. All sponges were collected at depths ranging from 15 to 35 m. Even though Petrosia ficiformis is a relatively common sponge in the Atlanto-Mediterranean, in some localities it was impossible to find more than 12 specimens. Tissue pieces of approximately 2 cm3 were excised with scissors/knives and preserved in 96% ethanol, replaced three times with fresh ethanol, and stored at − 20 °C. DNA was extracted with the DNeasy Blood & Tissue™ kit (Qiagen) following the manufacturer’s indications with minor modifications on the lysis time (performed overnight) and the elution step (performed twice using 50 μl of buffer EB each time).
Microsatellite amplification and analysis
All 280 individuals were genotyped at 10 unlinked microsatellite loci (1PETRO, 4PETRO, 7PETRO, 11PETRO, 15PETRO, 17PETRO, 18PETRO, 19PETRO, 25PETRO, and 30PETRO) previously described [91], using the PCR conditions described therein. The sizes of the fluorescently labeled PCR products were estimated using an internal size marker (GeneScanTM 500 LIZ) on an ABI Prism™ 7700 Sequencer (Applied Biosystems) and analysed with Peak Scanner v1.0™ (Applied Biosystems). All populations (and therefore individuals) were randomly located in the 12 plates genotyped.
Genetic diversity analyses in Petrosia ficiformis
GenAlEx 6.5 [92] was used for the estimations of the observed (Ho) and expected (He) heterozygosity, and the fixation index (FIS). Differences between He values among basins (Atlantic Ocean: FLO, SMI, MAD, CAN; Western Mediterranean: CAR, CART, BLA, ULL, FEL, MRS, NIZ, LIG, NAP, and Eastern Mediterranean: SLO, SCRO, JECRO, CRE, and ISR) were calculated using two-tailed T tests in Statplus 6 (AnalystSoft). In addition, Genepop on the web version 4.0.10 [93, 94] was used to obtain values for departure from Hardy Weinberg equilibrium (HWE) by locus and population using a probability test with level of significance and the following Markov chain parameters: 5000 dememorization steps, 1000 batches, and 5000 iterations per batch. Other measures of genetic diversity, such as the total number of alleles per locus and population, and number of effective alleles (=1/∑π2, where ∑π2 is the sum of the squared population allele frequencies) were calculated with GenAlEx and GENODIVE version 2.0b23 [95]. In order to correct for differences in sample sizes, the rarefaction method implemented in FSTAT 2.9.3.2 [96] was used to obtain the allelic richness at each locus. Clonality was assessed using GENODIVE and ignoring missing alleles.
Population structure and differentiation in Petrosia ficiformis
Several methods to assess population structure and differentiation in P. ficiformis were used: two based on clustering approaches (STRUCTURE and a Discriminant Analysis of Principal Components, DAPC) and four distance-based methods: FST estimations, Isolation by Distance (IBD), BARRIER, and Analyses of the Molecular Variance (AMOVA).
The assignment of individuals to each population was performed using a Bayesian clustering approach in STRUCTURE 2.3.4 [97], that calculates population allele frequencies and then assigns individuals to populations probabilistically, always based on the estimates of Hardy Weinberg equilibrium (HWE) and/or linkage equilibrium. The specific parameters used were admixture, since although we had no previous knowledge on the origin of the populations studied, we assumed that a proportion of individuals can have recent ancestor coming from multiple populations, no locprior, since no additional sample-characteristic data was available, and correlated allele frequencies, because we had no previous knowledge on the correlation levels across populations [98]. The program was run with a burn-in time of 100,000 repetitions and 100,000 iterations (MCMC), setting the putative K (predicted number of genetic units) from 2 to 20 (one cluster more than the number of sampling sites considered in the analysis) and twenty replicated runs. The estimation of log probabilities of data Pr(X | K) for each value of K was evaluated by calculating ∆K, which accounts for the rate of change in the log probability of data between successive K values, currently considered a more reliable predictor of the true number of populations [99]. Convergence was assessed with the alpha parameter. Calculations and evaluation of ∆K were performed with STRUCTURE HARVESTER [100]. We then used CLUMPAK web server [101] to find the major and minor best alignment of the results across the range of K values by averaging the probabilities of each K cluster. Graphs were visualized in CLUMPAK [101] and the major mode solution was selected.
For further assessment of population differentiation, the multivariate DAPC method was applied using the adegenet 2.1.1 package [102] implemented in R 2.14 [103]. DAPC defines clusters using the algorithm k-means on transformed data with Principal Component Analysis (PCA), which is then run sequentially with increasing values of k. The resulting clustering solutions are compared using Bayesian Information Criterion (BIC), with the optimal cluster solution corresponding to the lowest value of BIC. Before performing the analysis, the optimal number of PCs to be retained was explored by a cross-validation method as implemented in the same package.
Population differentiation between pairwise sampling sites was estimated with the FST statistic using an infinite allele model (IAM) in the software Arlequin 3.5 [104]. Significance of FST values was analysed with 20,000 permutations and corrected using the B-Y [105] False Discovery Rate (FDR) approach as described in [106]. In addition, the frequency of null alleles was estimated with Microchecker 2.2.3 [107]. Only the microsatellite 17PETRO contained null alleles, and we corrected allele frequencies and FST values after it, using the ENA method [108] described in FreeNA [109]. Global FST and average FST per population following [110] with UPGMA clustering of populations were obtained using the R packages adegenet 2.1.1 [102] and hierfstat v0.04–22 [111].
To determine whether genetic differentiation was driven by geographical distance creating a pattern of IBD, linearized pairwise FST estimates (FST /1- FST) were correlated against log-transformed geographical distances between samples [112] using a Mantel test with all the sites together and stratified Mantel tests using the clusters separated by oceanographic barriers obtained below in GENODIVE version 2.0b23 [95]. Geographical distances were estimated as the minimum linear distance between pairs of locations by sea. Furthermore, to localize the occurrence of genetic breaks in the population structure of P. ficiformis (i.e., oceanographic fronts), pairwise FST values and coordinates for sampling sites were implemented in the software BARRIER v2.2 [113]. BARRIER links the matrix of geographical coordinates with their corresponding distance matrix (FST), and applies the Monmonier’s maximum distance algorithm to identify ‘barriers’ to gene flow among sites, namely the zones where differences between pairs of sites are the largest.
Finally, an Analysis of Molecular Variance (AMOVA) was performed to determine the hierarchical distribution of genetic variation in GENODIVE version 2.0b23 [95]. To reveal the source of variation for the genetic differentiation, we a priori defined several groupings: 1) two groups: Atlantic (SMI, FLO, CAN, MAD) vs. Mediterranean (CART, CAR, BLA, FEL, ULL, ESC, MRS, NIZ, LIG, NAP, SLO, SCRO, JECRO, CRE, ISR) populations; 2) three groups: Atlantic (SMI, FLO, CAN, MAD), Western Mediterranean (CART, CAR, BLA, FEL, ULL, ESC, MRS, NIZ, LIG, NAP), and Eastern Mediterranean (SLO, SCRO, JECRO, CRE, ISR) populations; and 3) four groups: Azores (SMI, FLO), Madeira and Canary Islands (CAN, MAD), Western Mediterranean (CART, CAR, BLA, FEL, ULL, ESC, MRS, NIZ, LIG, NAP), and Eastern Mediterranean (SLO, SCRO, JECRO, CRE, ISR) populations. The significance of the AMOVAs was calculated with 10,000 permutations of the original data.
Demographic events and migration patterns in Petrosia ficiformis
Given the documented episodes of mass mortalities for P. ficiformis [3, 10] in the Mediterranean Sea, we tested for recent effective population size reductions (bottlenecks) based on allele data frequencies using the software BOTTLENECK [114]. The software operates under the assumption that “populations that have gone through a recent reduction of their effective population size show a reduction of the allelic diversity and heterozygosity, even though the number of alleles are reduced faster than the heterozygosity” [114]. We used the “Wilcoxon sign-rank test” [115] which can be used when more than 5 (but less than 20) loci are included. We performed the analyses with two models of mutation: two phase model (TPM) and the stepwise mutation model (SMM), using default values (a proportion of SMM in the TPM = 0.000 and a variance of the geometric distribution for TPM = 0.36), since they are the recommended models for microsatellites.
In addition, a population assignment analysis was performed calculating the likelihood ratio thresholds for all 19 populations based on the Monte Carlo test with an alpha of 0.002 and 1000 replicated datasets using GENODIVE version 2.0b23 [95]. This method assigns or excludes reference populations as possible origins of individuals on the basis of multilocus genotypes. Furthermore, the detection of last generation migrants was performed based on the calculations of the likelihood of an individual belonging to a given population, which was then done replacing the zero frequencies by a random 0.005 frequency in 4000 permutations using GENODIVE. Migration estimates among areas were obtained with diveRsity package (https://diversityinlife.weebly.com/) in R [103], which uses the method described in [116] to plot the relative migration levels between population samples from microsatellite allele frequency data. We only considered relative migration values over 0.1 obtained using the Gst statistic in a bootstrapped analysis (100 replicates). Since the method is still in experimental stages, results should be interpreted with caution. The sampling sites were pooled into the following groups: AZO (SMI, FLO), MAC (MAD, CAN), WES (CART, CAR, BLA, FEL, ULL, ESC, MRS, NIZ, LIG, NAP), ADR (SLO, SCRO, JECRO), EAS (CRE, ISR). To assess long-term migration rates among populations, a coalescent approach using Bayesian implementations in MIGRATE-n was used to obtain both migration rate and theta (θ) [117]: we selected the Brownian model, more appropriate due to the high variability of the microsatellite loci, and then ran 4 replicated chains with an increment of 1500 and 15,000 recorded steps with a 10% burn-in after an initial test run to obtain the correct set of priors for the migration and theta parameters (total visited parameters 1500,000). Convergence was assessed using the effective sample size threshold of 200 and the when the Gelman’s convergence criterion was close to 1. Migration rates were only taken into account when their 95% CIs did not overlap zero.
Abbreviations
- AMOVA:
-
Analysis of Molecular Variance
- BIC:
-
Bayesian Inference Criterion
- BP:
-
Before Present
- CI:
-
Confidence Intervals
- FDR:
-
False Discovery Rate
- HWE:
-
Hardy-Weinberg Equilibrium
- IAM:
-
Infinite allele model
- IBD:
-
Isolation By Distance
- LGM:
-
Last Glacial Maxima
- MPA:
-
Marine Protected Area
- Myr:
-
Million Years
- PCA:
-
Principal Component Analysis
- SMM:
-
Stepwise Mutation Model
- TPM:
-
Two-Phase Model
References
Cuttelod A, García N, Malak DA, Temple HJ, Katariya V. The Mediterranean: a biodiversity hotspot under threat, Wildlife in a Changing World–an analysis of the 2008 IUCN Red List of Threatened Species; 2009. p. 89.
Coll M, Piroddi C, Albouy C, Ben Rais Lasram F, Cheung WW, Christensen V, Karpouzi VS, Guilhaumon F, Mouillot D, Paleczny M. The Mediterranean Sea under siege: spatial overlap between marine biodiversity, cumulative threats and marine reserves. Glob Ecol Biogeogr. 2012;21(4):465–80.
Garrabou J, Coma R, Bensoussan N, Bally M, Chevaldonné P, Cigliano M, Diaz D, Harmelin J-G, Gambi M, Kersting D. Mass mortality in northwestern Mediterranean rocky benthic communities: effects of the 2003 heat wave. Glob Chang Biol. 2009;15(5):1090–103.
Pérez T, Vacelet J. Effect of climatic and anthropogenic disturbances on sponge fisheries. In: The Mediterranean Sea. Netherlands: Springer; 2014. p. 577–87.
Santangelo G, Abbiati M. Red coral: conservation and management of an over-exploited Mediterranean species. Aquat Conserv Mar Freshwat Ecosyst. 2001;11(4):253–9.
Cebrian E, Agell G, Marti R, Uriz M. Response of the Mediterranean sponge Chondrosia reniformis Nardo to copper pollution. Environ Pollut. 2006;141(3):452–8.
Maldonado M, Sánchez-Tocino L, Navarro C. Recurrent disease outbreaks in corneous demosponges of the genus Ircinia: epidemic incidence and defense mechanisms. Mar Biol. 2010;157(7):1577–90.
Cebrian E, Uriz MJ, Garrabou J, Ballesteros E. Sponge mass mortalities in a warming Mediterranean Sea: are cyanobacteria-harboring species worse off? PLoS One. 2011;6(6):e20211.
Stabili L, Cardone F, Alifano P, Tredici SM, Piraino S, Corriero G, Gaino E. Epidemic mortality of the sponge Ircinia variabilis (Schmidt, 1862) associated to proliferation of a Vibrio bacterium. Microb Ecol. 2012;64(3):802–13.
Cerrano C, Magnino G, Sarà A, Bavestrello G, Gaino E. Necrosis in a population of Petrosia ficiformis (Porifera, Demospongiae) in relation with environmental stress. Ital J Zool. 2001;68(2):131–6.
Gates RD, Baghdasarian G, Muscatine L. Temperature stress causes host cell detachment in symbiotic cnidarians: implications for coral bleaching. Biol Bull. 1992;182(3):324–32.
Voultsiadou E, Vafidis D, Antoniadou C. Sponges of economical interest in the eastern Mediterranean: an assessment of diversity and population density. J Nat Hist. 2008;42(5–8):529–43.
Van Soest R, Boury-Esnault N, Hooper J, Kd R, De Voogd N, Alvarez de Glasby B, Hajdu E, Pisera A, Manconi R, Schoenberg C. World Porifera Database; 2017.
Ferretti C, Vacca S, De Ciucis C, Marengo B, Duckworth AR, Manconi R, Pronzato R, Domenicotti C. Growth dynamics and bioactivity variation of the Mediterranean demosponges Agelas oroides (Agelasida, Agelasidae) and Petrosia ficiformis (Haplosclerida, Petrosiidae). Mar Ecol. 2009;30(3):327–36.
Maldonado M, Riesgo A. Gametogenesis, embryogenesis, and larval features of the oviparous sponge Petrosia ficiformis (Haplosclerida, Demospongiae). Mar Biol. 2009;156(10):2181–97.
Xavier JR, Van Soest RW. Diversity patterns and zoogeography of the Northeast Atlantic and Mediterranean shallow-water sponge fauna. Hydrobiologia. 2012;687(1):107–25.
Garcia-Castellanos D, Estrada F, Jiménez-Munt I, Gorini C, Fernández M, Vergés J, De Vicente R. Catastrophic flood of the Mediterranean after the Messinian salinity crisis. Nature. 2009;462(7274):778.
Maldonado M, Uriz M-J. Biotic affinities in a transitional zone between the Atlantic and the Mediterranean: a biogeographical approach based on sponges. J Biogeogr. 1995;22(1):89–110.
Bianchi CN, Morri C. Marine biodiversity of the Mediterranean Sea: situation, problems and prospects for future research. Mar Pollut Bull. 2000;40(5):367–76.
Klausewitz W. New aspects of the paleogeography of the Mediterranean ichtyofauna. Ichthyologia (Yugosl). 1973;5:79–86.
Por F. Tethys returns to the Mediterranean: success and limits of tropical re-colonization. In: Environment, biodiversity and conservation in the Middle East Proceedings of the first middle eastern biodiversity congress, Aqaba, Jordan; 2008. p. 20–3.
Patarnello T, Volckaert FA, Castilho R. Pillars of Hercules: is the Atlantic–Mediterranean transition a phylogeographical break? Mol Ecol. 2007;16(21):4426–44.
Wangensteen OS, Turon X, Pérez-Portela R, Palacín C. Natural or naturalized? Phylogeography suggests that the abundant sea urchin Arbacia lixula is a recent colonizer of the Mediterranean. PLoS One. 2012;7(9):e45067.
Pérez-Portela R, Almada V, Turon X. Cryptic speciation and genetic structure of widely distributed brittle stars (Ophiuroidea) in Europe. Zool Scr. 2013;42(2):151–69.
TTaboada S, Pérez-Portela R. Contrasted phylogeographic patterns on mitochondrial DNA of shallow and deep brittle stars across the Atlantic-Mediterranean area. Sci Rep. 2016;6:32425.
Garcia-Cisneros A, Palacín C, Khadra YB, Pérez-Portela R. Low genetic diversity and recent demographic expansion in the red starfish Echinaster sepositus (Retzius 1816). Sci Rep. 2016;6:33269.
Borrero-Pérez G, González-Wangüemert M, Marcos C, Pérez-Ruzafa A. Phylogeography of the Atlanto-Mediterranean Sea cucumber Holothuria (Holothuria) mammata: the combined effects of historical processes and current oceanographical pattern. Mol Ecol. 2011;20(9):1964–75.
Zulliger DE, Tanner S, Ruch M, Ribi G. Genetic structure of the high dispersal Atlanto-Mediterreanean Sea star Astropecten aranciacus revealed by mitochondrial DNA sequences and microsatellite loci. Mar Biol. 2009;156(4):597–610.
Pérez-Losada M, Nolte MJ, Crandall KA, Shaw PW. Testing hypotheses of population structuring in the Northeast Atlantic Ocean and Mediterranean Sea using the common cuttlefish Sepia officinalis. Mol Ecol. 2007;16(13):2667–79.
Fernández R, Lemer S, McIntyre E, Giribet G. Comparative phylogeography and population genetic structure of three widespread mollusc species in the Mediterranean and near Atlantic. Mar Ecol. 2015;36(3):701–15.
Stamatis C, Triantafyllidis A, Moutou K, Mamuris Z. Mitochondrial DNA variation in Northeast Atlantic and Mediterranean populations of Norway lobster, Nephrops norvegicus. Mol Ecol. 2004;13(6):1377–90.
Triantafyllidis A, Apostolidis A, Katsares V, Kelly E, Mercer J, Hughes M, Jørstad K, Tsolou A, Hynes R, Triantaphyllidis C. Mitochondrial DNA variation in the European lobster (Homarus gammarus) throughout the range. Mar Biol. 2005;146(2):223–35.
Blanquer A, Uriz MJ. Population genetics at three spatial scales of a rare sponge living in fragmented habitats. BMC Evol Biol. 2010;10(1):13.
Dailianis T, Tsigenopoulos C, Dounas C, Voultsiadou E. Genetic diversity of the imperilled bath sponge Spongia officinalis Linnaeus, 1759 across the Mediterranean Sea: patterns of population differentiation and implications for taxonomy and conservation. Mol Ecol. 2011;20(18):3757–72.
Pérez-Portela R, Noyer C, Becerro MA. Genetic structure and diversity of the endangered bath sponge Spongia lamella. Aquat Conserv Mar Freshwat Ecosyst. 2015;25(3):365–79.
Riesgo A, Pérez-Portela R, Pita L, Blasco G, Erwin P, López-Legentil S. Population structure and connectivity in the Mediterranean sponge Ircinia fasciculata are affected by mass mortalities and hybridization. Heredity. 2016;117(6):427.
Pascual M, Rives B, Schunter C, Macpherson E. Impact of life history traits on gene flow: a multispecies systematic review across oceanographic barriers in the Mediterranean Sea. PLoS One. 2017;12(5):e0176419.
Duran S, Giribet G, Turon X. Phylogeographical history of the sponge Crambe crambe (Porifera, Poecilosclerida): range expansion and recent invasion of the Macaronesian islands from the Mediterranean Sea. Mol Ecol. 2004;13(1):109–22.
Xavier JR, Soest R, Breeuwer JA, Martins AM, Menken SB. Phylogeography, genetic diversity and structure of the poecilosclerid sponge Phorbas fictitius at oceanic islands. Contrib Zool. 2010;79(3):119–29.
Meirmans PG. Seven common mistakes in population genetics and how to avoid them. Mol Ecol. 2015;24(13):3223–31.
Meirmans PG. The trouble with isolation by distance. Mol Ecol. 2012;21(12):2839–46.
Simpson TL. The cell biology of sponges. New York: Springer Verlag; 1984.
Jackson J. Modes of dispersal of clonal benthic invertebrates: consequences for species' distributions and genetic structure of local populations. Bull Mar Sci. 1986;39(2):588–606.
Maldonado M, Uriz MJ. Sexual propagation by sponge fragments. Nature. 1999;398(6727):476.
Chaves-Fonnegra A, Feldheim KA, Secord J, Lopez JV. Population structure and dispersal of the coral-excavating sponge Cliona delitrix. Mol Ecol. 2015;24(7):1447–66.
Turon X, Tarjuelo I, Uriz M. Growth dynamics and mortality of the encrusting sponge Crambe crambe (Poecilosclerida) in contrasting habitats: correlation with population structure and investment in defence. Funct Ecol. 1998;12(4):631–9.
Carballo JL, Vega C, Cruz-Barraza JA, Yanez B, Nava H, Avila E, Wilson M. Short-and long-term patterns of sponge diversity on a rocky tropical coast: evidence of large-scale structuring factors. Mar Ecol. 2008;29(2):216–36.
Duran S, Pascual M, Estoup A, Turon X. Strong population structure in the marine sponge Crambe crambe (Poecilosclerida) as revealed by microsatellite markers. Mol Ecol. 2004;13(3):511–22.
DeBiasse M, Richards VP, Shivji MS. Genetic assessment of connectivity in the common reef sponge, Callyspongia vaginalis (Demospongiae: Haplosclerida) reveals high population structure along the Florida reef tract. Coral Reefs. 2010;29(1):47–55.
Guardiola M, Frotscher J, Uriz M-J. High genetic diversity, phenotypic plasticity, and invasive potential of a recently introduced calcareous sponge, fast spreading across the Atlanto-Mediterranean basin. Mar Biol. 2016;163(5):123.
Giles EC, Saenz-Agudelo P, Hussey NE, Ravasi T, Berumen ML. Exploring seascape genetics and kinship in the reef sponge Stylissa carteri in the Red Sea. Ecol Evol. 2015;5(13):2487–502.
Ledoux JB, Mokhtar-Jamaï K, Roby C, Féral JP, Garrabou J, Aurelle D. Genetic survey of shallow populations of the Mediterranean red coral [Corallium rubrum (Linnaeus, 1758)]: new insights into evolutionary processes shaping nuclear diversity and implications for conservation. Mol Ecol. 2010;19(4):675–90.
Pérez-Portela R, Cerro-Gálvez E, Taboada S, Tidu C, Campillo-Campbell C, Mora J, Riesgo A. Lonely populations in the deep: genetic structure of red gorgonians at the heads of submarine canyons in the North-Western Mediterranean Sea. Coral Reefs. 2016;35(3):1013–26.
Carlsson J, McDOWELL J, DÍAZ-JAIMES P, Carlsson JE, Boles SB, Gold JR, Graves JE. Microsatellite and mitochondrial DNA analyses of Atlantic bluefin tuna (Thunnus thynnus thynnus) population structure in the Mediterranean Sea. Mol Ecol. 2004;13(11):3345–56.
Bahri-Sfar L, Lemaire C, Hassine OKB, Bonhomme F. Fragmentation of sea bass populations in the western and eastern Mediterranean as revealed by microsatellite polymorphism. Proc R Soc Lond B Biol Sci. 2000;267(1446):929–35.
Carreras-Carbonell J, Macpherson E, Pascual M. High self-recruitment levels in a Mediterranean littoral fish population revealed by microsatellite markers. Mar Biol. 2007;151(2):719–27.
Calderón I, Palacín C, Turon X. Microsatellite markers reveal shallow genetic differentiation between cohorts of the common sea urchin Paracentrotus lividus (Lamarck) in Northwest Mediterranean. Mol Ecol. 2009;18(14):3036–49.
Freeland JR, Kirk H, Petersen S. Molecular genetics in ecology. Mol Ecol Second Edition. 2011:1–34.
Addison J, Hart M. Spawning, copulation and inbreeding coefficients in marine invertebrates. Biol Lett. 2005;1(4):450–3.
Uriz MJ, Turon X, Becerro MA. Morphology and ultrastructure of the swimming larvae of Crambe crambe (Demospongiae, Poecilosclerida). Invertebr Biol. 2001;120(4):295–307.
Mariani S, Uriz M-J, Turon X, Alcoverro T. Dispersal strategies in sponge larvae: integrating the life history of larvae and the hydrologic component. Oecologia. 2006;149(1):174–84.
Maldonado M. The ecology of the sponge larva. Can J Zool. 2006;84(2):175–94.
Pennington JT. The ecology of fertilization of echinoid eggs: the consequences of sperm dilution, adult aggregation, and synchronous spawning. Biol Bull. 1985;169(2):417–30.
Yund PO. An in situ measurement of sperm dispersal in a colonial marine hydroid. J Exp Zool. 1990;253(1):102–6.
Benzie J, Black K, Moran P, Dixon P. Small-scale dispersion of eggs and sperm of the crown-of-thorns starfish (Acanthaster planci) in a shallow coral reef habitat. Biol Bull. 1994;186(2):153–67.
Warner PA, Willis BL, van Oppen MJ. Sperm dispersal distances estimated by parentage analysis in a brooding scleractinian coral. Mol Ecol. 2016;25(6):1398–415.
Johnson SL, Yund PO. Remarkable longevity of dilute sperm in a free-spawning colonial ascidian. Biol Bull. 2004;206(3):144–51.
Chen B, Cole JW, Grond-Ginsbach C. Departure from hardy Weinberg equilibrium and genotyping error. Front Genet. 2017;8:167.
Pérez-Portela R, Riesgo A. Population genomics of early-splitting lineages of metazoans; 2018.
Xavier JR. Chapter 6: Phylogeography of the haplosclerid sponge Petrosia ficiformis (Porifera, Petrosiidae) reveals a deep-water affinity of some shalow-water populations. Amsterdam: University of Amsterdam; 2009.
Galarza JA, Carreras-Carbonell J, Macpherson E, Pascual M, Roques S, Turner GF, Rico C. The influence of oceanographic fronts and early-life-history traits on connectivity among littoral fish species. Proc Natl Acad Sci. 2009;106(5):1473–8.
Cowen RK, Sponaugle S. Larval dispersal and marine population connectivity; 2009.
Villamor A, Costantini F, Abbiati M. Genetic structuring across marine biogeographic boundaries in rocky shore invertebrates. PLoS One. 2014;9(7):e101135.
Worthington LV. On the North Atlantic circulation. Johns Hopkins Oceanographic Studies. 1976;6:100p.
Shaw P, Pierce G, Boyle P. Subtle population structuring within a highly vagile marine invertebrate, the veined squid Loligo forbesi, demonstrated with microsatellite DNA markers. Mol Ecol. 1999;8(3):407–17.
Shaw P, Arkhipkin A, Al-Khairulla H. Genetic structuring of Patagonian toothfish populations in the Southwest Atlantic Ocean: the effect of the Antarctic polar front and deep-water troughs as barriers to genetic exchange. Mol Ecol. 2004;13(11):3293–303.
Estournel C, Durrieu de Madron X, Marsaleix P, Auclair F, Julliand C, Vehil R. Observation and modeling of the winter coastal oceanic circulation in the Gulf of Lion under wind conditions influenced by the continental orography (FETCH experiment). J Geophys Res. 2003;108(C3):8059.
Costantini F, Gori A, Lopez-González P, Bramanti L, Rossi S, Gili J-M, Abbiati M. Limited genetic connectivity between gorgonian morphotypes along a depth gradient. PLoS One. 2016;11(8):e0160678.
Hayes A, Kucera M, Kallel N, Sbaffi L, Rohling EJ. Glacial Mediterranean Sea surface temperatures based on planktonic foraminiferal assemblages. Quat Sci Rev. 2005;24(7):999–1016.
Scalera Liaci L, Sciscioli M, Matarrese A. Raffronto tra il comportamento sessuale di alcune Ceractinomorpha. Riv Biol. 1973;66:135–62.
Schuster A, Vargas S, Knapp I, Pomponi SA, Toonen RJ, Erpenbeck D, Woerheide G. Divergence times in demosponges (Porifera): first insights from new mitogenomes and the inclusion of fossils in a birth-death clock model. BMC Evol Biol. 2018;18(1):114.
Bianchi CN, Morri C, Chiantore M, Montefalcone M, Parravicini V, Rovere A. Mediterranean Sea biodiversity between the legacy from the past and a future of change. Life in the Mediterranean Sea: a look at habitat changes; 2012. p. 1–55.
Rouchy JM, Caruso A. The Messinian salinity crisis in the Mediterranean basin: a reassessment of the data and an integrated scenario. Sediment Geol. 2006;188:35–67.
Krijgsman W, Hilgen F, Raffi I, Sierro FJ, Wilson D. Chronology, causes and progression of the Messinian salinity crisis. Nature. 1999;400(6745):652.
Penzo E, Gandolfi G, Bargelloni L, Colombo L, Patarnello T. Messinian salinity crisis and the origin of freshwater lifestyle in western Mediterranean gobies. Mol Biol Evol. 1998;15(11):1472–80.
Hrbek T, Meyer A. Closing of the Tethys Sea and the phylogeny of Eurasian killifishes (Cyprinodontiformes: Cyprinodontidae). J Evol Biol. 2003;16(1):17–36.
Huyse T, Van Houdt J, Volckaert FA. Paleoclimatic history and vicariant speciation in the “sand goby” group (Gobiidae, Teleostei). Mol Phylogenet Evol. 2004;32(1):324–36.
Pérez-Portela R, Turon X. Cryptic divergence and strong population structure in the colonial invertebrate Pycnoclavella communis (Ascidiacea) inferred from molecular data. Zoology. 2008;111(2):163–78.
Pairaud IL, Bensoussan N, Garreau P, Faure V, Garrabou J. Impacts of climate change on coastal benthic ecosystems: assessing the current risk of mortality outbreaks associated with thermal stress in NW Mediterranean coastal areas. Ocean Dyn. 2014;64(1):103–15.
Mokhtar-Jamaï K, Pascual M, LEDOUX JB, Coma R, FÉRAL JP, Garrabou J, Aurelle D. From global to local genetic structuring in the red gorgonian Paramuricea clavata: the interplay between oceanographic conditions and limited larval dispersal. Mol Ecol. 2011;20(16):3291–305.
Taboada S, Riesgo A, Blasco G, Solà J, Xavier JR, López-Legentil S. Development of 10 microsatellite markers for the Atlanto-Mediterranean sponge Petrosia ficiformis. Conserv Genet Resour. 2015;7(4):895–7.
Peakall R, Smouse PE. GENALEX 6: genetic analysis in excel. Population genetic software for teaching and research. Mol Ecol Resour. 2006;6(1):288–95.
Raymond M, Rousset F. An exact test for population differentiation. Evolution. 1995;49(6):1280–3.
Raymond M, Rousset F. GENEPOP (Version 1.2): Population genetics software for exact tests and ecumenicalism. J Hered. 1995;83:239.
Meirmans PG, Van Tienderen PH. GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes. 2004;4(4):792–4.
Goudet J. FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered. 1995;86(6):485–6.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–59.
Porras-Hurtado L, Ruiz Y, Santos C, Phillips C, Carracedo Á, Lareu M. An overview of STRUCTURE: applications, parameter settings, and supporting software. Front Genet. 2013;4:98.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14(8):2611–20.
Earl DA. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4(2):359–61.
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I. Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour. 2015;15(5):1179–91.
Jombart T. Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24(11):1403–5.
R Development Core Team (2008). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, http://www.R-project.org.
Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol Ecol Resour. 2010;10(3):564–7.
Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Stat. 2001;29(4):1165–88.
Narum SR. Beyond Bonferroni: less conservative analyses for conservation genetics. Conserv Genet. 2006;7(5):783–7.
Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Resour. 2004;4(3):535–8.
Weir B. Genetic analysis II. Sunderland: Sinauer Associates; 1996.
Chapuis M-P, Estoup A. Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol. 2006;24(3):621–31.
Nei M. Molecular evolutionary genetics. New York: Columbia university press; 1987.
Goudet J. Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol Ecol Resour. 2005;5(1):184–6.
Rousset F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics. 1997;145(4):1219–28.
Manni F, Guerard E, Heyer E. Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier’s algorithm. Hum Biol. 2004;76(2):173–90.
Cornuet JM, Luikart G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics. 1996;144(4):2001–14.
Luikart G, Cornuet JM. Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv Biol. 1998;12(1):228–37.
Sundqvist L, Keenan K, Zackrisson M, Prodöhl P, Kleinhans D. Directional genetic differentiation and relative migration. Ecol Evol. 2016;6(11):3461–75.
Beerli P, Felsenstein J. Maximum-likelihood estimation of migration rates and effective population numbers in two populations using a coalescent approach. Genetics. 1999;152(2):763–73.
Acknowledgements
The authors wish to warmly thank Maurizio Pansini, Thanos Dailianis, Thierry Perez, Peter Wirtz, and Tatjana Bakran-Petricioli (to JRX) Laura Steindler and Eduardo Serrano, Juan Moles Joan Giménez, and Carlos Angulo-Preckler (to AR) for sending specimens from their research locations. We are also indebted to the two reviewers and the Associate Editor Prof. Sally Leys who greatly improved our manuscript. AR and ST also wish to thank Martín Taboada Riesgo for the support along the study.
Funding
This research was funded by the Spanish Government project MARSYMBIOMICS CTM2013–43287-P, the Catalan Government grant 2014SGR-336 for Consolidated Research Groups, the European project SponGES (Grant Agreement No. 679849), and internal funds of the NHM to AR (SDF14032). AR and RPP were funded by Juan de la Cierva fellowships of the Spanish Government during half the study. The funding bodies did not have any role in the design of the study and collection, analysis, interpretation of data or in writing the manuscript.
Availability of data and materials
The complete matrix of genotypes can be accessed in PANGAEA under https://doi.org/10.1594/PANGAEA.882098. The coordinates and locations for each population are listed in Table 1.
Author information
Authors and Affiliations
Contributions
AR designed and performed the research, analysed data and wrote the paper. ST participated in population genotyping, data analysis and manuscript writing. RPP contributed to the data analysis and manuscript writing. PM contributed to population genotyping and revised the manuscript draft. JRX contributed to the sample collection and manuscript writing. GB participated in population genotyping. SLL contributed funding and participated in the research design and manuscript writing. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Descriptors of genetic diversity for all 19 locations of Petrosia ficiformis. Abbreviations: Ns, number of genotyped individuals; A, number of alleles; Ae, Number of effective alleles; rA, number of alleles after rarefaction; Ho, observed heterozygosity; He, expected heterozygosity; FIS, inbreeding coefficient; HWE, deviation from Hardy-Weinberg equilibrium: ns, not significant; * = p < 0.05; ** = p < 0.01; *** = p < 0.001. Population codes as in Table 1. (XLSX 58 kb)
Additional file 2:
Allele frequencies per locus and population for the entire study. (PDF 1776 kb)
Additional file 3:
A. Graph depicting delta K and likelihood of K obtained from STRUCTURE. B. Individual genotype assignment to clusters (K) as inferred by STRUCTURE for all studied sites with K = 10. C. Number of clusters obtained by adegenet for Petrosia ficiformis. Using Bayesian Information Criterion, the optimal clusters correspond to the lowest values, here shown in red circles. D. Individual assignment to each of the 10 clusters inferred using BIC. (PDF 331 kb)
Additional file 4:
FST comparisons between 19 populations of Petrosia ficiformis. Bold numbers reflect significant p-values after correction (p < 0.008). (XLSX 54 kb)
Additional file 5:
Migration rates (median and 95% CIs) obtained with MIGRATE-N for Petrosia ficiformis. Values with no overlapping CIs are shown in bold and green shading. (XLSX 19 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Riesgo, A., Taboada, S., Pérez-Portela, R. et al. Genetic diversity, connectivity and gene flow along the distribution of the emblematic Atlanto-Mediterranean sponge Petrosia ficiformis (Haplosclerida, Demospongiae). BMC Evol Biol 19, 24 (2019). https://doi.org/10.1186/s12862-018-1343-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-018-1343-6