
Abstract
Background
Recurrent (RP) and chronic pancreatitis (CP) may complicate Cystic Fibrosis (CF). It is still unknown if mutations in genes involved in the intrapancreatic activation of trypsin (IPAT) or in the pancreatic secretion pathway (PSP) may enhance the risk for RP/CP in patients with CF.
Methods
We enrolled: 48 patients affected by CF complicated by RP/CP and, as controls 35 patients with CF without pancreatitis and 80 unrelated healthy subjects. We tested a panel of 8 genes involved in the IPAT, i.e. PRSS1, PRSS2, SPINK1, CTRC, CASR, CFTR, CTSB and KRT8 and 23 additional genes implicated in the PSP.
Results
We found 14/48 patients (29.2%) with mutations in genes involved in IPAT in the group of CF patients with RP/CP, while mutations in such genes were found in 2/35 (5.7%) patients with CF without pancreatitis and in 3/80 (3.8%) healthy subjects (p < 0.001). Thus, we found mutations in 12 genes of the PSP in 11/48 (22.9%) patients with CF and RP/CP. Overall, 19/48 (39.6%) patients with CF and RP/CP showed one or more mutations in the genes involved in the IPAT and in the PSP while such figure was 4/35 (11.4%) for patients with CF without pancreatitis and 11/80 (13.7%) for healthy controls (p < 0.001).
Conclusions
The trans-heterozygous association between CFTR mutations in genes involved in the pathways of pancreatic enzyme activation and the pancreatic secretion may be risk factors for the development of recurrent or chronic pancreatitis in patients with CF.
Similar content being viewed by others

Background
Cystic Fibrosis (CF) is the most common inherited autosomal recessive disease in Caucasians. It is caused by defects in the CF transmembrane conductance regulator (CFTR) gene, which encodes a cAMP-regulated chloride channel. Defects in the CFTR protein cause abnormal chloride transport across the apical membranes of epithelial cells in the airways, pancreas, intestine, vas deferens, and sweat glands leading to progressive lung disease, pancreatic dysfunction, male infertility, and elevated sweat electrolytes, respectively (Castellani & Assael, 2017). About 85% of individuals affected by CF suffer from pancreatic insufficiency (PI), in most cases since the birth. However, 15% of the patients retain pancreatic sufficiency (PS) that permits adequate digestion (Walkowiak et al., 2008). About 85% of individuals affected by CF suffer from pancreatic insufficiency (PI), in most cases since the birth. However, 15% of the patients retain pancreatic sufficiency (PS) that permits adequate digestion (Walkowiak et al., 2008).
Recurrent pancreatitis (RP) and chronic pancreatitis (CP) may complicate CF. It was firstly mentioned by Shwachman et al. in 1975 (Shwachman et al., 1975). They reported, in a period of 20 years in 2000 patients with CF, 10 cases of pancreatitis (0.5%), all with PS. Currently, a frequency of recurrent/chronic pancreatitis between 17% (Durno et al., 2002) and 22% (Ooi et al., 2011) is estimated in patients with CF.
The dysfunction of the CFTR protein has a role in the pathogenesis of pancreatitis because it causes the impaired secretory function of pancreatic duct cells and the altered flow of digestive pro-enzymes into the duodenum triggering recurrent episodes of pancreatitis that in some patients may evolve to chronic pancreatitis (Walkowiak et al., 2008; Lew et al., 2017). This complication is more frequent in patients with CF and PS (that frequently have at least one class IV-V CFTR mutation), in which pancreatic acinar islets still produce pancreatic enzymes that may be prematurely activated within the pancreas. Recurrent/chronic pancreatitis has been observed also independently by the development of CF. In fact, Bishop et al. showed a frequency up to 30% of RP/CP in subjects carrying only one CFTR mutation in the absence of any sign of CF (Bishop et al., 2005). However, only a small percentage of patients with CFTR mutations or with CF experience RP or CP, suggesting that other risk factors must be involved (Walkowiak et al., 2008).
In fact, in addition to CFTR, patients with idiopathic recurrent or chronic pancreatitis have been investigated for other genes related to the premature intra-pancreatic activation of trypsin pathway. The first gene related to pancreatitis was the cationic trypsinogen gene protease serine 1 (PRSS1) in 1996: a gain of function missense mutation i.e., the R122H, was identified as a risk factor for CP (Whitcomb et al., 1996). In the following years, loss-of-function variants in the pancreatic secretory trypsin inhibitor (SPINK1) (Chen et al., 2000), calcium-sensing receptor (CASR) (Felderbauer et al., 2003) and chymotrypsinogen C (CTRC) (Szmola & Sahin-Toth, 2007) genes, firmly established the pivotal role of prematurely activated trypsin within the pancreas in the etiology of pancreatitis. Moreover, our group demonstrated that mutations in several dozens of genes bearing to six different pancreatic pathways represent risk factors for recurrent/chronic pancreatitis (Sofia et al., 2016) reinforcing the concept that trans-heterozygous mutations in different genes are involved in the pathogenesis of idiopathic pancreatitis.
Interestingly, we described trans-heterozygosity for mutations in different genes also in a patient with CF and RP that was compound heterozygous for the [delta]F508 and G91G CFTR mutations and had a pathogenic mutation in the CTRC gene (Tomaiuolo et al., 2015).
Thus, to better define the role of trans-heterozygosity for mutations in different genes as a risk factor for RP/CP in patients with CF, in this study we investigated a cohort of CF patients with RP/CP in comparison to patients with CF without pancreatitis and to healthy subjects to compare the frequency of mutations in a panel of genes related to the intra-pancreatic activation of trypsin (IPAT) and a group of other genes related to pancreatic secretion pathways (PSP) previously reported to contribute to to the pathogenesis of pancreatitis (Sofia et al., 2016).
Methods
Patients
The informed consent was obtained from all patients or from the parents or guardians of minors. The study was approved by the Ethical Committee (Scientific Board of “Bambino Gesù” Children’s Hospital, IRCCS, Rome, Italy) and was conducted in accordance with the Helsinki Declaration.
We enrolled 48 unselected patients affected by CF complicated by RP or CP recruited through a multicentric study involving 9 Italian CF centres. The main data (i.e., age at diagnosis of CF, age at diagnosis of RP/CP, CFTR genotype and pancreatic status) are reported in Table 2. As control populations, we studied 35 unselected patients with CF without symptoms or history of pancreatitis (see Table 3 for the data of age at diagnosis of CF, CFTR genotype and pancreatic status) and 80 unrelated, adult healthy subjects of the same ethnic group of the patients with CF (i.e., Italian from at least two generations) whose DNA samples and anonymized clinical data (in particular absence of CF and of any pancreatic disorder) were available, in the biological bank of our Institution.
The diagnosis of CF was done according to the international criteria (Farrell et al., 2017). Pancreatic sufficiency in patients with CF was defined on the basis of two values of faecal pancreatic elastase > 200 mg/g measured in subjects free from acute gastrointestinal events (Walkowiak et al., 2016) or on the basis of normal 72-h fecal fat balance (Walkowiak et al., 2008). Recurrent pancreatitis was diagnosed in patients that had at least two episodes of acute pancreatitis (at a distance of at least six months after the resolution of the previous episode) each one with abdominal pain (once excluded other causes) in association with the increase of serum lipase (at least 2X the upper reference limit) and/or imaging evidence (e.g., pancreatic edema, hemorrhage or necrosis) (Morinville et al., 2012; Kumar et al., 2016). Chronic pancreatitis was diagnosed according to the M-ANNHEIM criteria (Schneider et al., 2007) in patients in which instrumental analysis revealed calcifications or characteristic ductal changes. All the patients with CP had a positive anamnesis for episodes of recurrent pancreatitis.
Next-generation targeted sequencing of pancreatic genes
Targeted resequencing was performed using a uniquely customized design TruSeq Custom Amplicon Low Input technology (Illumina, San Diego, CA) with the MiSeq sequencing platform (Illumina). This technology is a fully integrated DNA-to-data solution, including online probe design and ordering through the Illumina website, sequencing assay, automated data analysis, and offline software for reviewing results. Online probe design was performed by entering target genomic regions into Design Studio software (Illumina). We designed a panel of eight genes included in IPAT genes (Sofia et al., 2016; Chen & Férec, 2009; Mahurkar et al., 2006; Cavestro et al., 2003): CFTR (NM_000492.3), SPINK1 (NM_003122.3), PRSS1 (NM_002769.4), protease, serine 2 (PRSS2) (NM_002770.2), CTRC (NM_007272.2), CASR (NM_001178065.1), cathepsin B (CTSB) (NM_147780.2) and keratin 8 (KRT8) (NM_002273). The sequence of these genes was obtained consulting the University of California, Santa Cruz, Genome Browser Home (https://genome.ucsc.edu/cgi-bin/hgGateway, last accessed October 2015) with a coverage of 100%. MiSeq system provides fully integrated on-instrument data analysis software. Each single variant reported in the vCard output file was evaluated for the coverage and the Q score and visualized via Integrative Genomics Viewer (Thorvaldosdottir et al., 2013; Robinson et al., 2011). All mutations identified by MiSeq Reporter were validated by Sanger sequencing using standard protocols.
In the second step, we selected the genes encoding proteins related to the pancreatic activation of zymogens (Sofia et al., 2016). Such genes were selected among the genes annotated in the “Pancreatic Secretion Pathway” (map04972), available in the KEGG database (Kanehisa et al., 2014). The 23 genes selected were classified into four groups according to the activity of the encoded protein or their role in the pathogenesis of pancreatitis: (i) genes encoding proteins involved in pancreatic secretion and ion homeostasis (PPY, F2RL1, TMPRSS15, SCL4A2, SLC4A4, SLC26A3, CPB1, CLPS) (Berni Canani et al., 2010; Sharma et al., 2005; Stevens et al., 2004; Multigner et al., 1985); (ii) genes encoding proteins involved in calcium (Ca2+) signalling and zymogen granules exocytosis (PRKCD, ITPR3, GP2, TRPC3, STIM1, ATP2C2, TRPV1, TRPV5, TRPV6, PIK3CG (Jin et al., 2015; Williams, 2008; Ramnath et al., 2010; Lupia et al., 2004); (iii) genes encoding proteins involved in autophagy (HSP90AA1, LAMP2, MAP1LC3B) (Willemer et al., 1989; Gukovskaya & Gukovsky, 2012; Fortunato & Kroemer, 2009) and (iv) autoimmune pancreatitis-related genes (CA4, ABCF1) (Ohmuraya & Yamamura, 2008). To search mutations in such genes, we used the targeted resequencing performed by a uniquely customized design: TruSeq Custom Amplicon Low Input Kit (Illumina) with the MiSeq sequencing platform (Illumina). The probe design (locus-specific oligos) was carried out by entering the target genomic regions into Design Studio software (Illumina). The design was performed over a cumulative target region of 99.328 bp and generated a panel of 677 amplicons with a coverage of 100% of the cumulative region. Library preparation and sequencing runs have been performed according to the manufacturer’s procedure. Only the PRSS2 gene was analyzed by Sanger sequencing because its genomic sequence was updated in the University of California Santa Cruz (UCSC) genome database after the design of the resequencing panel.
Data and bioinformatic analysis
The MiSeq Reporter software, a data analysis software included in the MiSeq system, performs secondary analysis on the base calls and quality score (Qscore) generated by the Real-Time Analysis software during the sequencing run and provides a list of all detected variants compared with the reference genome (Homo sapiens, hg19, build 37.2). Each single variant reported in the output file was evaluated for the coverage and the Qscore and visualized via the Integrative Genome Viewer (Thorvaldosdottir et al., 2013). Based on the guidelines of the American College of Medical Genetics and Genomics (Rehm et al., 2013), all regions that had been sequenced with a sequencing depth < 30 were considered not suitable for the analysis. Furthermore, we established a minimum threshold in Qscore of 30 (base call accuracy of 99.9%). All identified variants were analyzed with bioinformatic softwares evaluating the impact of change in amino-acidic structure on protein functionality with several parameters, and we filtered all variants to retain those alterations with a high disease-causing potential. We used four tools based on different parameters: PolyPhen-2 (Adzhubei et al., 2010), Align-GVGD (Mathe et al., 2006; Hicks et al., 2011), DNA SIFT (Ng & Henikoff, 2001) and MutationTaster (Schwarz et al., 2010). To facilitate the analysis of the potential splicing mutations, we used Human Splicing Finder to predict the effects of mutations on splicing signals or motifs in any human sequence (Desmet et al., 2009). Sanger sequencing using standard protocols validated the variants that have been predicted as “damaging” by at least three tools. For each of these mutations we assessed the frequency in the general population reported by the ExAC (Exome Aggregation Consortium) tool.
Results
All individuals from the three groups, i.e., patients with CF and RP/CP (n = 48), patients with CF and without pancreatitis (n = 35) and healthy subjects (n = 80) were investigated for mutations in the 8 genes encoding proteins involved in IPAT and in the 23 genes encoding proteins involved in the PSP (Table 1). All the 48 patients with CF and RP/CP (Table 2) and the 35 with CF without pancreatitis (Table 3) had a pathological sweat test (i.e., > 60 mEq/L) with the exception of a patient with CF without pancreatitis that had a value of 53 mEq/L, and all patients from both the groups had two CFTR mutations with the exception of a patient with CF and RP in which only one mutation was known. Among the 48 patients with CF and RP/CP we found 39 patients (81.2%) with PS and 9 patients with PI (18.8%); these figures were 13/33 (30.4%) and 20 (69.6%), respectively, among the patients with CF without pancreatitis (p < 0.001).
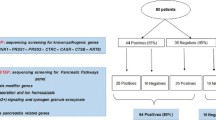
As shown in Table 1 and Fig. 1a, in the group of patients with CF and RP/CP we found 14/48 patients (29.2%) with mutations in IPAT genes, while mutations in such genes were found in 2/35 (5.7%) patients with CF without pancreatitis (Table 1 and Fig. 1b) and in 3/80 (3.8%) healthy subjects (Table 1 and Fig. 1c) (chi square: 20.4, p < 0.001).

Flowchart of the results of molecular analysis in 48 patients affected by CF and recurrent/chronic pancreatitis (a), in 35 patients with CF and without pancreatitis (b) and in 80 healthy subjects (c)
Going to the type of mutations in IPAT genes in the patients with CF and RP/CP: 2 were heterozygous for a splicing mutation in PRSS1 in cis, 4 patients were heterozygous for CTRC mutations, 3 patients were heterozygous for PRSS2 mutations, 2 patients were heterozygous for CASR mutations and 2 patients were heterozygous for KRT8 mutations; 1 patient had a splicing mutation in PRSS1 and a missense mutation in KRT8 (Table 2). All these mutations were absent in our controls and were found with a frequency < 1% in the general population as annotated in the ExAC tool (Additional file 1: Table S1). While, the 2 patients with CF without pancreatitis had, in IPAT genes, a heterozygous PRSS1/PRSS2 hybrid mutation and a heterozygous missense mutation of CTRC, respectively (Table 3). Finally, the three healthy subjects had all a heterozygous missense mutation in the KRT8, CASR and SPINK1 genes, respectively (Table 4).
Thus, all individuals from the three groups were studied for mutations in the 23 genes encoding proteins of PSP. In the group of patients with CF and RP/CP (Fig. 1a, Table 1, Table 2), the analysis revealed mutations in 11/48 (22.9%) patients. While, as shown in Fig. 1b and c and in Tables 3 and 4, mutations in such genes were found in 3/35 (8.5%) patients with CF without pancreatitis and in 8/80 (10%) healthy subjects (chi square: 4.39; p = 0.11).
Going to the type of mutations, of the 11 patients with CF and RP/CP (Table 2) 7 patients showed heterozygous mutations, 1 displayed a homozygous mutation and 3 patients were trans-heterozygous for mutations in more than one gene. Finally (Table 2 and Additional file 1: Table S1), in this group of patients, we found 13 missense mutations in 12 genes encoding proteins of PSP: (i) SLC4A2, TMPRSS15, SLC26A3 and SLC4A4 genes encoding proteins involved in pancreatic secretion and ion homeostasis; (ii) TRPV1, TRPV5, TRPV6, PIK3CG, PRKCD and ATP2C2 genes encoding proteins involved in calcium (Ca2+) signalling and zymogen granules exocytosis and (iii) MAP1LC3B, LAMP2 genes encoding proteins involved in autophagy. One nonsense homozygous mutation was found in TRPV1. All these mutations were not present in the 35 CF patients without pancreatitis and in the 80 healthy subjects (Table 2 and Additional file 1: Table S1).
Among the 35 patients with CF without pancreatitis (Table 3), one patient was trans-heterozygous for mutations in the genes of both panels and 2 patients had mutations only in the genes of the PSP. In the cohort of healthy controls (Table 4), we found 7 individuals with variants in at least one gene of the PSP.
All the mutations found in patients and controls had a frequency < 1% in the general population (data not shown).
Finally, 19/48 (39.6%) patients with CF and RP/CP had mutations in at least one gene of the IPAT or PSP pathway. While, this is true for 4/35 (11.4%) patients with CF without CP and for 11/80 (13.7%) healthy subjects, chi square: 14.5, p < 0.001 (Fig. 1 and Table 1). Additional file 1: Table S1 reports a summary of all gene mutations found in the three groups of subjects studied.
Discussion
Our study confirms that the occurrence of RP/CP is more frequent in patients with CF and PS (Walkowiak et al., 2008) and demonstrates that patients with CF and RP/CP have a significantly higher frequency of mutations in genes encoding proteins that may promote the auto-activation of pancreatic proenzymes or regulate pancreatic secretion. The small number of cases precluded clinical comparison between patients bearing mutations and those wild-type for all genes tested. Thus, the trans-heterozygosity for mutations in the CFTR and in other genes represents a risk factor for pancreatitis even in patients with CF, as we recently demonstrated for patients with idiopathic RP/CP (Sofia et al., 2016).
Among the genes encoding proteins involved in the premature activation of trypsin, we found mutations in 14 patients with CF and RP/CP in PRSS1, PRSS2, CTRC, CASR and KRT8 genes. Eight of such mutations were known as pathogenic, while for other 6 mutations, three bioinformatic tools predicted a pathogenic effect and the ExAC tool reported the absence or the very low frequency in the general population. Going in detail, we found two mutations in PRSS1: the first is the [c.592-11C > T;c.592-8C > T] complex allele was found in two siblings. Keiles et al. (Keiles & Kammesheidt, 2006) described the same complex allele in an 18-years old woman with pancreatitis; she also carried the T908 N CFTR mutation. Furthermore, the PRSS1 splicing mutation c.592-24C > T previously described in two siblings with CP (Singhi et al., 2014) was found in a patient with the CFTR genotype [delta]F508/2789 + 5G > A. The patient carried also a KRT8 mutation.
Thus, we found three mutations in PRSS2. The T230I and K98X mutations are novel. The T230I was reported as pathogenic by the three bioinformatic tools; the K98X is a nonsense mutation causing an early stop codon. The third mutation in PRSS2 gene, i.e., the G191R, had been analysed by Witt et al. (Witt et al., 2006). They demonstrated that the recombinant G191R protein showed a complete loss of trypsin activity owing to the introduction of a new tryptic cleavage site rendering the enzyme hypersensitive to autocatalytic proteolysis. Furthermore, we found three missense mutations in CTRC. The K172E was identified in two patients. Masson et al. described the K172E mutation in a patient with idiopathic chronic pancreatitis (Masson et al., 2008). Thus, in two other patients with the CFTR [delta]F508/G91G and [delta]F508/2789 + 5G > A genotype we identified the CTRC V235I and G217S missense mutations, respectively. Rosendahl et al. investigated the functional consequences of these two CTRC missense mutations through transient transfections in HEK 293 T cells (Rosendahl et al., 2008). They demonstrated that the G217S causes a loss-of-function of the CTRC protein, whereas the V235I results in normal or slightly reduced function, respectively. Moreover, data observed in another report suggest a role for the V235I mutation in triggering the pancreatic phenotype in a patient with CF (Tomaiuolo et al., 2015). Rosendahl identified the G217S mutation also in a healthy control and similarly, in our study we found it in a patient with CF without RP/CP thus, we cannot conclude on the pathogenic role of such mutation.
In addition, we found two missense mutations in CASR: the V149I and the N189D; both the mutations were considered pathogenic by bioinformatic tools and by the very low frequency in the general population.
Finally, we found two missense mutations in KRT8. The G62C was identified in two PS patients: the first case had the R553X/2789 + 5G > A CFTR genotype and the second had the [delta]F508/2789 + 5G > A CFTR genotype in addition to the c.592-24C > T mutation in PRSS1. Initially, the KRT8 G62C mutation was considered pathogenic by Cavestro and coworkers (Cavestro et al., 2003). Later, Witt et al. observed that the frequency of the mutation did not differ between patients with acute or chronic pancreatitis, pancreatic adenocarcinoma and control individuals (Witt et al., 2006). Also in our study, this mutation was found in a healthy subject. Thus, we cannot conclude on the pathogenic role of such mutation. Finally, the KRT8 A358V novel mutation was identified in a patient with the [delta]F508/D110H CFTR genotype and we speculate on its pathogenetic role on the basis of the bioinformatic prediction and of its absence in alleles from the general population. Interestingly, in a recent study it was demonstrated an interaction between KRT8 and the CFTR protein that could influence the function of CFTR (Treiber et al., 2006).
All the genes discussed so far are involved in the premature intra-pancreatic activation of trypsin. This pathway plays a pivotal role in triggering the activation cascade of all pancreatic digestive zymogens caused by the breaking of the interactions of these proteins in pancreas leading to injury of acinar cells and consequently recurrent attacks of pancreatitis (Sofia et al., 2016; Chen & Férec, 2009).
Moving to patients with CF without pancreatitis, we found two cases with mutations IPAT genes: a patient had the PRSS1/PRSS2 hybrid and a the CTRC G217S, a mutation previously identified in a normal subject by Rosendahl et al. (Rosendahl et al., 2008). In healthy subjects, we identified three individuals with mutations in SPINK1, CASR and KRT8 genes. The N34S mutation identified in SPINK1 was described by Threadgold et al. as a variation associated with a familial pattern of idiopathic chronic pancreatitis (Threadgold et al., 2002). Actually, it is considered not disease causing being found in normal control too with an average prevalence of 2.5% and an allele frequency of 1.25% (Premchandar & s, 2017). The missense A558S identified in CASR is a novel mutation considered potentially pathogenic by bioinformatic analysis despite a frequency of about 1% in the general population, while the KRT8 G62C mutation found in another healthy control was identified also in healthy subjects by Witt et al. (Witt et al., 2006).
All mutations found in the second group of 23 genes PSP in the three groups of subjects were classified as possibly damaging by the three bioinformatic tools and all but two had a frequency in the general population < 0.1%. However, even if there is a trend of higher frequency of such mutations in patients with CF and RP/CP as compared to the patients with CF without pancreatitis and to healthy controls, the difference is not significant. To be noted that 6 out of 14 patients affected by CF with RP/CP with mutations in genes encoding proteins potentially involved in premature intra-pancreatic activation of trypsin also have mutations in genes of the pancreatic secretion pathway, in particular those belonging to the Ca2+ signalling, pancreatic secretion and autophagy pathways, further reinforcing the concept that trans-heterozygous mutations in different genes may have a synergic effect in the pathogenesis of RP/CP.
Conclusions
Our data strongly suggest that the trans-heterozygosity for mutations in CFTR and in genes encoding proteins involved in IPAT and PSP may enhance the risk for RP/CP in patients with CF, as we previously demonstrated in subjects with idiopathic RP/CP (Sofia et al., 2016). Further studies are called, to define if patients with trans-heteroygous mutations have a more severe outcome of pancreatitis (the small number of cases limited such evaluation in the present study) and functional studies are necessary to elucidate the pathogenetic mechanism of pancreatitis in patients bearing mutated genes/proteins.
Abbreviations
- CF:
-
cystic fibrosis
- CFTR:
-
cystic fibrosis transmembrane conductance regulator
- CP:
-
chronic pancreatitis
- IPAT:
-
intra-pancreatic activation of trypsin
- PI:
-
pancreatic insufficiency
- PS:
-
pancreatic sufficiency
- PSP:
-
pancreatic secretion pathways
- RP:
-
recurrent pancreatitis
References
Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Berni Canani R, Terrin G, Cardillo G, Tomaiuolo R, Castaldo G. Congenital diarrheal disorders: improved understanding of gene defects is leading to advances in intestinal physiology and clinical management. J Pediatr Gastroenterol Nutr. 2010;50:360–6.
Bishop MD, et al. The cystic fibrosis transmembrane conductance regulator gene and ion channel function in patients with idiopathic pancreatitis. Hum Genet. 2005;118:372–81.
Castellani C, Assael BM. Cystic fibrosis: a clinical view. Cell Mol Life Sci. 2017;74:129–40.
Cavestro GM, et al. Association of keratin 8 gene mutation with chronic pancreatitis. Dig Liver Dis. 2003;35:416–20.
Chen JM, Férec C. Chronic pancreatits: genetics and pathogenesis. Annu Rev Genomics Hum Genet. 2009;10:63–87.
Chen JM, Mercier B, Audrezet MP, Férec C. Mutational analysis of the human pancreatic secretory trypsin inhibitor (PSTI) gene in hereditary and sporadic chronic pancreatitis. J Med Genet. 2000;37:67–9.
Desmet FO, et al. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67.
Durno C, Corey M, Zielenski J, Tullis E, Tsui LC, Durie P. Genotype and phenotype correlations in patients with cystic fibrosis and pancreatitis. Gastroenterology. 2002;123:1857–64.
Farrell PM, et al. Diagnosis of cystic fibrosis: consensus guidelines from the Cystic Fibrosis Foundation. J Pediatr. 2017;181:1–58.
Felderbauer P, et al. A novel mutation of the calcium sensing receptor gene is associated with chronic pancreatitis in a family with heterozygous SPINK1 mutations. BMC Gastroenterol. 2003;3:34–41.
Fortunato F, Kroemer G. Impaired autophagosome-lysosome fusion in the pathogenesis of pancreatitis. Autophagy. 2009;5:850–3.
Gukovskaya AS, Gukovsky I. Autophagy and pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2012;303:G993–1003.
Hicks S, Wheeler DA, Plon SE, Kimmel M. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum Mutat. 2011;32:661–8.
Jin CX, Hayakawa T, Ko SB, Ishiguro H, Kitagawa M. Pancreatic stone protein/ regenerating protein family in pancreatic and gastrointestinal diseases. Intern Med. 2015;50:1507–16.
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–205.
Keiles S, Kammesheidt A. Identification of CFTR, PRSS1, and SPINK1 mutations in 381 patients with pancreatitis. Pancreas. 2006;33:221–7.
Kumar S, Ooi CY, Werlin S, Abu-El-Haija M, Barth B, Bellin MD, et al. Pediatric acute recurrent and chronic pancreatitis: lessons from INSPPIRE. JAMA Pediatr. 2016;170:562–9.
Lew D, Afghani E, Pandol S. Chronic Pancreatitis: Current Status and Challenges for Prevention and Treatment. Dig Dis Sci. 2017;62:1702–12.
Lupia E, et al. Ablation of phosphoinositide 3-kinase-gamma reduces the severity of acute pancreatitis. Am J Pathol. 2004;165:2003–11.
Mahurkar S, et al. Association of cathepsin B gene polymorphisms with tropical calcific pancreatitis. Gut. 2006;55:1270–5.
Masson E, Chen JM, Scotet V, Le Maréchal C, Férec C. Association of rare chymotrypsinogen C (CTRC) gene variations in patients with idiopathic chronic pancreatitis. Hum Genet. 2008;123:83–91.
Mathe E, et al. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res. 2006;34:1317–25.
Morinville VD, Husain SZ, Bai H, Barth B, Alhosh R, Durie PR, et al. Definitions of pediatric pancreatitis and survey of present clinical practices. J Pediatr Gastroenterol Nutr. 2012;55:261–5.
Multigner L, Sarles H, Lombardo D, De Caro A. Pancreatic stone protein. II. Implication in stone formation during the course of chronic calcifying pancreatitis. Gastroenterology. 1985;89:387–91.
Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–74.
Ohmuraya M, Yamamura K. Autophagy and acute pancreatitis: a novel autophagy theory for trypsinogen activation. Autophagy. 2008;4:1060–2.
Ooi CY, Dorfman R, Cipolli M, Gonska T, Castellani C, Keenan K, et al. Type of CFTR mutation determines risk of pancreatitis in patients with cystic fibrosis. Gastroenterology. 2011;140:153–61.
Premchandar A, et al. New insights into interactions between the nucleotide-binding domain of CFTR and keratin 8. Protein Sci. 2017;26:343–54.
Ramnath RD, Sun J, Bhatia M. PKC delta mediates pro-inflammatory responses in a mouse model of caerulein-induced acute pancreatitis. J Mol Med. 2010;88:1055–63.
Rehm HL, et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med. 2013;15:733–47.
Robinson JT, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6.
Rosendahl J, et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet. 2008;40:78–82.
Schneider A, Löhr JM, Singer MV. The M-ANNHEIM classification of chronic pancreatitis: introduction of a unifying classification system based on a review of previous classifications of the disease. J Gastroenterol. 2007;42:101–19.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. Mutation taster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–6.
Sharma A, et al. Protection against acute pancreatitis by activation of protease-activated receptor-2. Am J Physiol Gastrointest Liver Physiol. 2005;288:G388–95.
Shwachman H, Lebenthal E, Khaw KT. Recurrent acute pancreatitis in patients with cystic fibrosis with normal pancreatic enzymes. Pediatrics. 1975;55:86–95.
Singhi AD, et al. The histopathology of PRSS1 hereditary pancreatitis. Am J Surg Pathol. 2014;38:346–53.
Sofia VM, et al. Extensive molecular analysis suggested the strong genetic heterogeneity of idiopathic chronic pancreatitis. Mol Med. 2016;26:300–9.
Stevens T, Conwell DL, Zuccaro G. Pathogenesis of chronic pancreatitis: an evidence-based review of past theories and recent developments. Am J Gastroenterol. 2004;99:2256–70.
Szmola R, Sahin-Toth M. Chymotrypsin C (caldecrin) promotes degradation of human cationic trypsin: identity with Rinderknecht’s enzyme Y. Proc Natl Acad Sci U S A. 2007;104:11227–32.
Thorvaldosdottir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92.
Threadgold J, et al. The N34S mutation of SPINK1 (PSTI) is associated with a familial pattern of idiopathic chronic pancreatitis but does not cause the disease. Gut. 2002;50:675–81.
Tomaiuolo AC, et al. Relationship between CFTR and CTRC variants and the clinical phenotype in late-onset cystic fibrosis disease with chronic pancreatitis. J Mol Diagn. 2015;17:171–8.
Treiber M, et al. Keratin 8 sequence variants in patients with pancreatitis and pancreatic cancer. J Mol Med. 2006;84:1015–22.
Walkowiak J, Glapa A, Nowak JK, Bober L, Rohovyk N, Wenska-Chyży E, et al. Pancreatic Elastase-1 Quick Test for rapid assessment of pancreatic status in cystic fibrosis patients. J Cyst Fibros. 2016;15:664–8.
Walkowiak J, Lisowska A, Blaszczyński M. The changing face of the exocrine pancreas in cystic fibrosis: pancreatic sufficiency, pancreatitis and genotype. Eur J Gastroenterol Hepatol. 2008;20:157–60.
Whitcomb DC, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet. 1996;14:141–5.
Willemer S, Kloppel G, Kern HF, Adler G. Immunocytochemical and morphometric analysis of acinar zymogen granules in human acute pancreatitis. Virchows Arch A Pathol Anat Histopathol. 1989;415:115–23.
Williams JA. Receptor-mediated signal transduction pathways and the regulation of pancreatic acinar cell function. Curr Opin Gastroenterol. 2008;24:573–9.
Witt H, et al. A degradation-sensitive anionic trypsinogen (PRSS2) variant protects against chronic pancreatitis. Nat Genet. 2006;38:668–73.
Acknowledgements
We acknowledge the Ministero della Salute (Rome, Italy) L.548/93 for the regional research funding quote of years 2007–15.
We acknowledge the Società Italiana per lo studio della Fibrosi Cistica (SIFC) that stimulated this multicentric study. We are also grateful to Dr. Gianfranco Savoldi for his precious suggestions in reviewing genetic data.
Availability of data and materials
Data and materials were available if requested.
Author information
Authors and Affiliations
Contributions
The manuscript was written by GC and Adriano Angioni. VMS and CS performed NGS of pancreatic genes, variants analysis and validation, interpretation of the data and revised critically the manuscript. LDS contributed to design of pancreatic gene panel and data analysis. VMS, CS, and ACT contributed to experimental performance and data interpretation. VT, FA, Antonella Angiolillo, CB, NC, CC, ADL, RP, SQ, VR, GT, FZ, VL, and ML recruited patients, collected biological samples, and performed clinical evaluations. Antonella Angiolillo, NC, RP, AN, VL, and ML revised critically the manuscript. GC and Adriano Angioni had a main role in conception and design, analyses, interpretation of the data and revised critically the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethical Committee (Scientific Board of “Bambino Gesù” Children’s Hospital, IRCCS, Rome, Italy) and was conducted in accordance with the Helsinki Declaration.
Consent for publication
The informed consent to participate at this study and for publication of the results was obtained from all patients or from the parents or guardians of minors.
Competing interests
The authors declare they have no competing interests or other interests that might be perceived to influence the results and discussion reported in this paper.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. List of mutations in genes involved in the intrapancreatic activation of trypsin (IPAT) and pancreatic secretion pathway (PSP) and allelic frequency (%) in the three groups of subjects studied (A: CF with RP; B: CF without RP; C: healthy controls) and D: in the general population (ExAC tool). (DOCX 17 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sofia, V.M., Surace, C., Terlizzi, V. et al. Trans-heterozygosity for mutations enhances the risk of recurrent/chronic pancreatitis in patients with Cystic Fibrosis. Mol Med 24, 38 (2018). https://doi.org/10.1186/s10020-018-0041-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10020-018-0041-6