
Abstract
Genetic features of chronic pancreatitis (CP) have been investigated extensively, mainly by testing genes associated to the trypsinogen activation pathway. However, different molecular pathways involving other genes may be implicated in CP pathogenesis. A total of 80 patients with idiopathic chronic pancreatitis (ICP) were investigated using a Next-Generation Sequencing (NGS) approach with a panel of 70 genes related to six different pancreatic pathways: premature activation of trypsinogen, modifier genes of cystic fibrosis phenotype, pancreatic secretion and ion homeostasis, calcium signaling and zymogen granules (ZG) exocytosis, autophagy and autoimmune pancreatitis-related genes. We detected mutations in 34 out of 70 genes examined; of the 80 patients, 64 (80.0%) were positive for mutations in one or more genes and 16 (20.0%) had no mutations. Mutations in CFTR were detected in 32 of the 80 patients (40.0%) and 22 of them exhibited at least one mutation in genes of other pancreatic pathways. Of the remaining 48 patients, 13/80 (16.3%) had mutations in genes involved in premature activation of trypsinogen and 19/80 (23.8%) had mutations only in genes of the other pathways: 38 (59.3%) of the 64 patients positive for mutations showed variants in two or more genes. Our data, although to be extended with functional analysis of novel mutations, suggest a high rate of genetic heterogeneity in CP and that trans-heterozygosity may predispose to the ICP phenotype.
Similar content being viewed by others
Introduction
Chronic pancreatitis (CP) is a persistent inflammatory disease in which exocrine acinar tissue is gradually replaced by fibrotic tissue (1). The annual incidence of CP in adults is estimated to be 5–12 per 100,000 inhabitants each year in Japan, Europe and the United States (2). Data on the incidence in children are not available (3). In adults, CP is frequently secondary to alcohol abuse or to gallstones (2). Such causes are uncommon in pediatric pancreatitis, where a percentage of the cases are due to congenital pancreatic malformations. More rare causes include trauma and drugs (4).
Furthermore, in the last decade, hereditary pancreatitis (HP), pancreatitis with no other etiology that appears in more members of the family (5), has been described and is frequently associated to mutations in genes encoding proteins related to zymogen granules (ZG) activation (6) such as protease serine 1 (PRSS1) and serine protease inhibitor Kazal-type 1 (SPINK1).
However, mutations in such genes were observed also in patients with idiopathic pancreatitis (IP) without family history. Mutations in the PRSS1 gene were found in about 1.9% of IP patients ranging from 0.2% to 10% (7,8), while the prevalence of the most common mutation N34S in SPINK1 gene is about 2% (9) in such cases. Furthermore, 43% and 11% of patients with idiopathic recurrent pancreatitis or CP carried one and two cystic fibrosis transmembrane conductance regulator (CFTR) mutations, respectively, even if symptoms suggestive for cystic fibrosis (CF) are absent in most cases (10–12). Conversely, about 1.2% of CF patients develop CP (13), and the risk increases for patients with residual pancreatic function (about 10% of cases). However, also excluding all such causes, about one-third of recurrent/chronic pancreatitis remains idiopathic, and this number is higher in children, who seldom report the classical risk factors observed in adults (8).
All forms of pancreatitis are primarily due to the altered pathway of activation and/or inhibition of ZG within the pancreatic acinar cells, in particular the premature activation of trypsinogen and a myriad of proteins are involved in regulating such processes (14). Only a few of these proteins have been studied extensively, and pathogenic mutations were identified in encoding genes. In fact, different molecular mechanisms are implicated in CP pathogenesis and, interestingly, the interplay among all pathways contributes to the development of the disease. The best-studied mechanisms are the premature intrapancreatic activation of trypsin, increased concentration of intracellular calcium (Ca2+), impaired autophagy in acinar cells and autoimmune reactions.
Therefore, we planned to extend the molecular analysis to 70 genes encoding proteins involved in several pathways related to ZG activation and secretion in a cohort of ICP.
Materials and Methods
Patients
Informed consent was obtained from all patients or from the parents or guardians of minors. The study was approved by the Ethical Committee (Scientific Board of Bambino Gesù Children’s Hospital) and was conducted in accordance with the Helsinki Declaration. This study included 80 patients (38 males and 42 females) with idiopathic chronic pancreatitis (ICP), age range: 3–61 years and median: 11 years. Inclusion criteria was CP according to Rosemont’s criteria (15,16). All cases presented an early onset (17–19), that is, they were diagnosed within the first three decades of life. Exclusion criteria were any known etiology of CP and particularly: (i) alcohol abuse (excluded by negative anamnesis and normal values of serum γ-glutamyltransferase); (ii) gallstones, pancreatic calcifications or congenital malformations (excluded by ultrasound scanning); (iii) trauma or protracted assumption of drugs (negative anamnesis in all patients); (iv) metabolic diseases (excluded by clinical evaluation); (v) chronic viral infections (excluded by clinical evaluation and serology: one HIV-positive patient was excluded from this study); (vi) familial pancreatitis (no patients included in this study had other members of the family affected); (vii) autoimmune pancreatitis (all the patients included in this study had serum IgG4 within reference intervals) and (viii) CF (one patient homozygous for ΔF508 CFTR mutation was identified and excluded from this study). Surprisingly, the patient had not been diagnosed for CF because he had a very mild clinical expression of CF with CP that usually does not appear in patients with two CFTR severe mutations. Furthermore, five ICP patients were diagnosed with CFTR-related disorder after this analysis, (20) having CP as the lone symptom, a borderline sweat test and at least a mild CFTR mutation detected after our sequencing analysis. In addition to cases of pancreatitis, we studied 50 unrelated controls. Finally, we referred the frequency of mutations to that reported in the 1000 Genomes Project Consortium (21).
MiSeq Panel Genes
We selected 70 genes encoding proteins related to the pancreatic activation of ZG. Such genes were selected among the genes annotated in the “Pancreatic Secretion Pathway” (map04972), available in the KEGG database (22). The 70 genes selected were classified into six groups according to the activity of the encoded protein or their role in the pathogenesis of pancreatitis: (i) genes encoding proteins potentially involved in premature intrapancreatic activation of trypsin (CFTR, PRSS1, PRSS2, SPINK1, CTRC, CTSB, KRT8 and CASR) (23–30); (ii) CF modifier genes (MBL2, SCNN1G, SERPINA1, TGFB1 and TNFRSF1A) (31,32); (iii) genes encoding proteins involved in pancreatic secretion and ion homeostasis (PPY, F2RL1, TMPRSS15, SLC4A2, SLC4A4, SLC26A3, CELA3B, CPB1, REG1A, REG3A, REG1B, and CLPS) (33–37); (iv) genes encoding proteins involved in calcium (Ca2+) signaling and ZG granule exocytosis (CCKAR, GNA12, GNA13, GNAQ, RAB3D, RAB27B, RAP1A, RAP1B, RHOA, RAC1A, PRKCD, ITPR2, ITPR3, DMBT1, VAMP2, VAMP8, GP2, TRPC1, TRPC3, TRPC6, STIM1, ORAI1, ATP2A2, ATP2C2, TPCN2, TRPV1, TRPV5, TRPV6 and PIK3CG) (38–41); (v) genes encoding proteins involved in autophagy (HSP90AA1, HSPA8, ATP6V1A, ATG5, VMP1, LAMP2, SQSTM1, USP9X, MAP1LC3A, MAP1LC3B, MAPLC3B2 and MAP1LC3C) (42–47) and (vi) autoimmune pancreatitis-related genes (CA2, CA4, LTF and ABCF1) (48,49). To search mutations in such genes, we used the targeted resequencing performed by a uniquely customized design: TruSeq Custom Amplicon (Illumina) with the MiSeq sequencing platform (Illumina). The probe design (locus-specific oligos) was carried out by entering target genomic regions into Design Studio software (Illumina). The design was performed over a cumulative target region of 313,445 bp and generated a panel of 1,269 amplicons with coverage of 90% of the cumulative region. Library preparation and sequencing runs have been performed according to the manufacturer’s procedure. Only the PRSS2 gene was analyzed by Sanger sequencing because its genomic sequence was updated in the University of California Santa Cruz (UCSC) genome database after the design of the resequencing panel.
Data and Bioinformatics Analysis
The MiSeq system includes a data analysis software; the MiSeq Reporter software performs secondary analysis on the base calls and quality score (Qscore) generated by Real-Time Analysis software during the sequencing run and provides a list of all detected variants compared with the reference genome (Homo sapiens, hg19, build 37.2). Each single variant reported in the output file has been evaluated for the coverage and the Qscore and visualized via the Integrative Genome Viewer (50). Based on the guidelines of the American College of Medical Genetics and Genomics (51), all regions that have been sequenced with a sequencing depth <30 have been considered not suitable for the analysis. Furthermore, we established a minimum threshold in Qscore of 30 (base call accuracy of 99.9%). All identified variants were analyzed with bioinformatics software that evaluates the impact of the change in amino-acidic structure on protein functionality with several parameters, and we filtered all variants to retain those alterations with high disease-causing potential. We used four tools based on different parameters: PolyPhen-2 (52), Align-GVGD (53,54), SIFT (55) and MutationTaster (56). To facilitate the analysis of the different splicing mutations, we used Human Splicing Finder to predict the effects of mutations on splicing signals or motifs in any human sequence (57). Variants that have been predicted as “damaging” by at least three of the software have been validated by Sanger sequencing using standard protocols.
Results
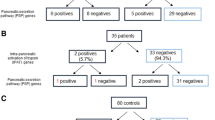
We analyzed 80 patients (160 alleles) with ICP and 50 healthy control participants (100 alleles). All individuals were investigated for 70 genes encoding proteins involved in six different pathways already reported to contribute to the pathogenesis of pancreatitis. The experiments showed a good reproducibility of the data, and the results were evaluated on the basis of their Qscore (Q > 30), the coverage (sequencing depth > 20) and the frequency of variants. Missense mutations were analyzed by four bioinformatics software systems (PolyPhen-2, Align-GVGD, SIFT and MutationTaster) to predict the pathogenicity of amino acid substitutions and their possible consequences on the protein function. Variants in intronic regions were predicted as dangerous by specific bioinformatics tools. Finally, we found 96 potentially pathogenic mutations in 34 genes with a frequency <0.1% in the 1000 Genomes tool, all absent in the 50 control participants. Such mutations include: 80 missense, 6 nonsense, 2 synonymous, 6 splicing variants, 1 small deletion and 1 large deletion (Tables 1–3). All the patients carrying mutations in genes of all pathways of the panel were heterozygous, except for the CFTR gene for which we found seven patients compound heterozygous for two different CFTR mutations and four patients heterozygous for a complex allele (i.e., two mutations in cis on the same allele). As detailed in Table 1 (A), mutations were detected in all the genes encoding proteins potentially involved in premature intrapancreatic activation of trypsin: 37 missense (19 in CFTR, 1 in SPINK1, 4 in PRSS1, 4 in PRSS2, 3 in CTRC, 2 each in CASR, CTSB and KRT8); 5 splicing variants (3 in CFTR and 2 in PRSS1); 1 large deletion, 1 small deletion, 1 synonymous (CFTR) and 2 nonsense (1 in CFTR and 1 in PRSS2).
Table 1 (B) reports the mutations found in CF modifier genes. Only four genes carried mutations: five missense (one in MBL2, two in SERPINA1, one each in TNFRSF1A and SCNN1G) and one nonsense (SERPINA1).
As shown in Table 2, 10 genes encoding proteins involved in calcium signaling pathways and ZG exocytosis showed 16 missense mutations (four in ATP2C2, three in STIM1 and TRPV6, two in DMBT1 and one each in TRPV5 ITPR3, PRKCD and GP2), one nonsense (TRPC3), one synonymous (TRPV1) and one splicing variant (ITPR3).
In the pancreatic secretion and ion homeostasis pathway genes (Table 3 (A)), we recognized 16 missense mutations: one each in CLPS, F2RL1, SLC4A2 and RAP27B; two each in CPB1, SLC4A4 and SLC26A3 and six each in TMPRSS15 and one nonsense mutation in PPY and CPB1.
In the autophagy pathway (Table 3 (B)), we detected three missense mutations in three different genes: HSP90AA1, LAMP2 and MAP1LC3B.
Finally, among the autoimmune pancreatitis-related genes, we identified three missense mutations in two genes: two mutations in CA4 and one mutation in ABCF1 (Table 3 (C)).
Of the 80 patients, 64 (80%) included in the study showed one or more mutations, which are described in detail here. Seven patients were positive for two CFTR mutations and were diagnosed as having CFTR-related disorder. Among these seven patients, two had mutations both in genes encoding proteins potentially involved in premature intrapancreatic activation of trypsin and in genes of the other pancreatic pathways, three had only genetic variants of the latter and two had CFTR mutations alone. Furthermore, 25 patients were heterozygous for a single CFTR mutation even though four cases had a complex allele (i.e., two mutations in cis). Of these 25 patients, eight had only one CFTR mutation, two had mutations also of PRSS2 or SPINK1 and four had mutations both in genes encoding proteins potentially involved in premature intrapancreatic activation of trypsin and in genes of the other pancreatic pathways, while 11 patients showed a CFTR mutation and only variants in genes of the other pancreatic pathways (Table 4).
Of the 48 cases negative to CFTR mutations, 12 had at least one mutation in genes encoding proteins potentially involved in premature intrapancreatic activation of trypsin (eight of them had also at least one mutation in genes of the other pancreatic pathways) (Table 4). Of the remaining 36 cases, 20 presented mutations in at least one gene of the other pancreatic pathways of the panel and, finally, 16 (20%) of the 80 ICP patients had no mutations (Table 5).
Discussion
About 80% of patients with ICP show potentially pathogenic variants in one or more of 34 different genes belonging to different pathways described in the pathogenesis of pancreatitis. The most relevant mechanism of the disease is the premature intrapancreatic activation of trypsin. Other pathways, such as the activation and release of pancreatic ZG and autophagy, contribute to the development of the disease.
The role of genes encoding proteins involved in the premature intrapancreatic activation of trypsin (CFTR, PRSS1, PRSS2, SPINK1, CTRC, CTSB, KRT8 and CASR) in the pathogenesis of pancreatitis has been studied extensively. We found mutations in at least one of these genes in 44 out of 80 patients (55%) as shown in Figure 1. Among such genes, CFTR is the most represented. In fact, 32 (40%) of the 80 patients have CFTR mutations (Table 4), and this agrees with the percentage of 39% reported by Masson et al. (27), who studied a cohort of 253 young patients with ICP. In our series, seven patients were compound heterozygous for two CFTR mutations. None of them met the diagnostic criteria of CF, while all of them were diagnosed as having CFTR-related disorder, defined as having at least one mild CFTR mutation, borderline sweat test or CP as lone symptom. Twenty-five patients were heterozygous for a CFTR mutation. Of note, 10 of the 32 patients present only mutations in CFTR, while the remaining 22 patients have one or two CFTR mutations in addition to variants in genes encoding proteins involved in the premature intrapancreatic activation of trypsin and in other pancreatic pathway. In particular, among the 25 heterozygous cases for CFTR mutations, only two patients have a variant in PRSS1 or SPINK1, while four of them have variants also in genes belonging to the other pancreatic pathways. Based on this data, we suggest that in most patients with CFTR mutations, other genes may act as modulators, increasing the risk for CP. Therefore, unlike current opinions, CFTR mutations alone, particularly in heterozygosity, do not significantly increase the risk for CP. Moreover, only 12 patients have at least one mutation in genes encoding proteins potentially involved in premature intrapancreatic activation of trypsin, but not in CFTR. This data suggest that such genes, initially related to HP, also have a relevant role as risk factors for ICP. In fact, mutations in some of these genes were previously found in patients with ICP (e.g., SPINK1 and PRSS1 mutations were found in 57% and 9% of 75 ICP Chinese patients, respectively) (58), SPINK1, PRSS1 and CTRC mutations alone or combined were identified in about 24% of 253 young French ICP patients, and CTRC mutations that reduce the protective role of the CTRC protein toward trypsin degradation were considered a risk factor for CP in other studies (59,60). Similarly, CASR mutations (even if different from those recorded in two patients from this study) were considered a risk factor for ICP (61): this risk increased further if it was associated with SPINK1 mutations (62), and the same has been confirmed for KRT8 mutations detected in four patients. Mutations were found in CTSB in two patients, all of them with CFTR mutations. Therefore, such genes, previously related to tropical calcific pancreatitis (18), may also act as adjunctive risk factors for ICP. Of note, nine of the 12 patients with mutations in genes encoding proteins potentially involved in premature intrapancreatic activation of trypsin also have variants in genes of other pancreatic pathways related to the release of ZG and autophagy, thus suggesting a role of these latter as adjunctive risk factors for ICP.

Flowchart of the results of molecular analysis in 80 patients affected by recurrent/chronic pancreatitis.
Finally, 36 patients were negative to mutations in genes encoding proteins potentially involved in premature intrapancreatic activation of trypsin. Among these patients, only 20 have variants in at least one of the genes of the other pancreatic pathway panel (Table 5) and 16 were without mutation (Figure 1). Therefore, we suppose that other genes can be involved in contributing to onset of pancreatitis. A total of 23 patients have variants in genes encoding proteins of pancreatic secretion and ion homeostasis (PPY, F2RL1, TMPRSS15, SLC4A2, SLC4A4, SLC26A3, CPB1 and CLPS). The pancreatic polypeptide (PPY) is an important regulator of both exocrine and endocrine secretions of the pancreas and inhibits the juice pancreatic secretion that contains all the digestive enzymes, including colipase (CLPS) and procarboxypeptidase B1 (CPB1). In both acute and chronic pancreatitis, changes in the plasma concentration of PPY and its regulation have been reported, but the clinical value remains to be elucidated. In acute pancreatitis, inappropriate and premature activation of trypsin from trypsinogen within the acinar cell leads to autodigestion of the pancreas. The conversion of the proenzyme is due to enterokinase (encoded by TMPRSS15). Mutations in this gene cause enterokinase deficiency, a malabsorption disorder characterized by diarrhea and failure to thrive (33). In the duodenum, trypsin activates the protease-activated receptor 2 (PAR2, encoded by F2RL1) that modulates acinar cell secretion of digestive enzymes and the duct cell ion channel function. Experimental animal model demonstrated that PAR2 has a protective role against the damaging effects of intrapancreatic trypsinogen activation in acute pancreatitis (34). The exocrine function of the pancreas is carried out also by pancreatic duct cells that secrete fluid and ions (mainly Na+ and HCO3−), to neutralize the gastric acid contents of the duodenum. Several channels and specific transporters, including CFTR, SLC26A3, SLC4A2 and SLC4A4, are crucial to maintaining the ion homeostasis that is essential, as pancreatic fluid in predisposed individuals may be lithogenic, leading to a protein plug and stone formation. Moreover, HCO3− and H+ are produced by a carbonic anhydrase, such as CA4, from CO2 derived from the blood. The CA4 subfamily F member 1 (ABCF1) as well as the ATP-binding cassette have been reported to be antigens in the autoimmune pancreatitis that represents the 2%–4% of cases of CP (48,49) and two patients present a variant in one of these two genes.
We also found variants in genes encoding proteins involved in calcium (Ca2+) signaling and ZG exocytosis (ITPR3, STIM1, TRPC3, ATP2C2, PIK3CG, PRKCD, TRPV1, TRPV5, TRPV6, GP2, RAB27B and DMBT) in a total of 26 patients. In pancreatitis, the excess of intracellular Ca2+ concentration triggers an early activation of trypsin within the ZG that digests the acinar cells and destroys the parenchymal tissue. The hyperstimulation of the cholecystokinin (CCK) receptors with the analogue cerulein modifies vesicular transport and leads to intracellular proteolytic enzyme activation and ultimately cell death (38). The CCK triggers the release of Ca2+ in the cytoplasm from the endoplasmic reticulum and internal stores via IP3 receptor activation, principally ITPR2 and ITPR3. The equilibrium between intracellular and extracellular Ca2+ levels and the Ca2+ stored in the endoplasmic reticulum is crucial for the acinar cell and is regulated by several proteins and channels, including STIM1, TRPC3, TRPC6 and ATP2C2. Intracellular Ca2+ also activates the protein kinase C (PKC) isoform delta (PRKCD) that triggers ZG activation and secretion in acinar cells and mediates bicarbonate (HCO3−) secretion in duct pancreatic cells. Other important proteins for granule secretion are localized in the ZG membrane, including the glycoprotein GP2 and RAB27B. They regulate the intracellular vesicular trafficking and Ca2+-mediated exocytosis. Moreover, the muclin protein (DMBT1, deleted in malignant brain tumors 1) interacts with pancreatic ZG in a pH-dependent manner and may act as a Golgi cargo receptor in the regulated secretory pathway of the pancreatic acinar cell. In muclin-deficient mice, the trafficking fundamental for exocrine pancreatic function was impaired (41).
Moreover, Gukovskaya and Gukovsky used PI3Kγ-deficient mice to investigate the role of PI3K in CCK-induced responses in isolated pancreatic acinar cells, and they observed a decrease in the CCK-induced calcium response in pancreatic acini by inhibiting both intracellular calcium mobilization and calcium influx (42). Other authors showed that the ablation of PI3KCG can also reduce the severity of acute pancreatitis by blocking neutrophil infiltration within the pancreatic tissue at an early stage of the disease, thus suggesting a double role for IP3KCG (40). The severity of pancreatitis is also due to the generation of proinflammatory mediators such as substance P that activates PKC (34) and is released by primary sensory neurons via depolarization mediated by activation of transient receptor potential vanilloid (TRPV1, TRPV5 and TRPV6).
Finally, we found variants in genes encoding proteins involved in autophagy (LAMP2 and MAP1LC3B) in a total of three patients. In acute pancreatitis, the autophagy is impaired due to different mechanisms described by several studies in experimental models (42). In LAMP2-null mice, the lysosomal/autophagic dysfunction causes spontaneous pancreatitis manifested by acinar cell vacuolation, progressive inflammatory infiltration in the pancreas and acinar cell necrosis. The defective decreased autophagosome-lysosome fusion may create a large pool of autophagosomes, which may contribute to causing the disease (42–44). This process requires numerous proteins and their interactions, including microtubule-associated protein 1A/1B-light chain 3 (MAPLC3 or LC3) (44). The accumulation of large vacuoles is also accompanied by increased pancreatic levels of LC3-II in experimental models of pancreatitis. Of note, SPINK1 is strongly elevated in pancreatitis and the elevation correlates with the severity of disease. In addition, mutations in the SPINK1 gene were associated to CP. A recent study also demonstrated the role of SPINK1 as a negative regulator of autophagy in mice-deficient SPINK3 (a mouse homologue gene of human SPINK1) (47).
On the other hand, 38 patients had mutations in two or more genes of different pathways. Previous studies on these genes are not available and the influence of such genes should be further evaluated. The importance of digenic genotypes or trans-heterozygosity (two pathogenic genotypes in two different genes) has been clearly evidenced for ICP patients in two large series (39). Depending on the gene and type of sequence variation, digenic genotypes have been considered able in some instances to explain how the disease developed. Considering the large and innovative panel of genes that we selected to investigate pancreatitis and the poor data reported in literature, our conclusions on this point may be only speculative. However, we can hypothesize that (i) mutations in such genes could reasonably be considered pathogenic as the sequence variations found in ICP patients had not been identified in 100 control alleles from our study, they have a low frequency (if any) in subjects from the general population studied within the 1000 Genomes Project, and they have been predicted in silico to have an impact on protein function; (ii) all these genes encode proteins involved in pancreatic enzyme activation and release through different pathways that may interact and contribute all together to the pathogenesis of the disease and (iii) in 38 patients, mutations in two different genes may combine their effect.
Conclusion
A limit of this study is that it is difficult to define clear pathogenic relationships between ICP and mutations found in genes of the pancreatic pathway panel in the absence of a functional analysis on the effect of each mutation. However, we selected a group of genes that may potentially contribute to enhancing the risk for ICP, calling for future studies that will elucidate such a relationship. In any case, this is the first study in which a large series of genes potentially related to pancreatic secretion pathways were studied in patients with ICP without a family history. Our data suggest that a genetic predisposition may exist in most patients (i.e., 80%), but the genes potentially involved are strongly heterogeneous (up to 34 different genes bear mutations in the 64 patients) and, in most patients, mutations in different genes (up to 7 in 1 case) may interact in contributing to the onset of the ICP phenotype.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Brock C, Nielsen LM, Lelic D, Drewes AM. (2013) Pathophysiology of chronic pancreatitis. World J. Gastroenterol. 19:7231–40.
Yadav D, Lowenfels AB. (2013) The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology. 144:1252–61.
Morinville VD, et al. (2012) Definitions of pediatric pancreatitis and survey of present clinical practices. J. Pediatr. Gastroenterol. Nutr. 55:261–5.
Bai HX, et al. (2011) Novel characterization of drug-associated pancreatitis in children. J. Pediatr. Gastroenterol. Nutr. 53:423–8.
Whitcomb DC. (2001) Hereditary pancreatitis: A model for understanding the genetic basis of acute and chronic pancreatitis. Pancreatology. 1:565–70.
Whitcomb DC. (2010) Genetic aspects of pancreatitis. Annu. Rev. Med. 61:413–24.
Simon P, et al. (2002) Spontaneous and sporadic trypsinogen mutations in idiopathic pancreatitis. JAMA. 288:2122.
Keim V, Teich N. (2003) Idiopathic vs hereditary pancreatitis. JAMA. 289:983.
Whitcomb DC. (2002) How to think about SPINK and pancreatitis. Am. J. Gastroenterol. 97:1085–8.
Bishop MD, et al. (2005) The cystic fibrosis transmembrane conductance regulator gene and ion channel function in patients with idiopathic pancreatitis. Hum. Genet. 118:372–81.
Sharer N, et al. (1998) Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N. Engl. J. Med. 339:645–52.
Cohn JA, et al. (1998) Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N. Engl. J. Med. 339:653–8.
De Boeck K, Weren M, Proesmans M, Kerem E. (2005) Pancreatitis among patients with cystic fibrosis: Correlation with pancreatic status and genotype. Pediatric. 115:e463–9.
Sah RP, Dawra RK, Saluja AK. (2013) New insights into the pathogenesis of pancreatitis. Curr. Opin. Gastroenterol. 29:523–30.
Tandon M, Topazian M. (2001) Endoscopic ultrasound in idiopathic acute pancreatitis. Am. J. Gastroenterol. 96:705–9.
Banks PA, et al. (2013) Classification of acute pancreatitis-2012: Revision of the Atlanta classification and definitions by international consensus. Gut. 62:102–11.
Catalano MF, et al. (2009) EUS-based criteria for the diagnosis of chronic pancreatitis: The Rosemont classification. Gastrointest. Endosc. 69:1251–61.
Keller J, Layer P. (2008) Idiopathic chronic pancreatitis. Best Pract. Res. Clin. Gastroenterol. 22:105–13.
Derikx MH, Drenth JP. (2010) Genetic factors in chronic pancreatitis; implications for diagnosis, management and prognosis. Best Pract. Res. Clin. Gastroenterol. 24:251–70.
Dequeker E, Stuhrmann M, Morris MA, et al. (2009) Best practice guidelines for molecular genetic diagnosis of cystic fibrosis and CFTR-related disorders-updated European recommendations. Eur. J. Hum. Genet. 17:51–65.
1000 Genomes Project Consortium, et al. (2012) An integrated map of genetic variation from 1,092 human genomes. Nature. 491:56–65.
Kanehisa M, Goto S, Sato Y, Furumichi M, and Tanabe M. (2014) Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 42:D199–205.
Nemeth BC, Sahin-Toth M. (2014) Human cationic trypsinogen (PRSS1) variants and chronic pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 306:G466–73.
Masson E, et al. (2008) Trypsinogen copy number mutations in patients with idiopathic chronic pancreatitis. Clin. Gastroenterol. Hepatol. 6:82–8.
Witt H, et al. (2000) Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat. Genet. 25:213–6.
Whitcomb DC. (2013) Genetic risk factors for pancreatic disorders. Gastroenterology. 144: 1292–302.
Masson E, Chen JM, Audrezet MP, Cooper DN, Férec C. (2013) A conservative assessment of the major genetic causes of idiopathic chronic pancreatitis: Data from a comprehensive analysis of PRSS1, SPINK1, CTRC and CFTR genes in 253 young French patients. PLoS One. 8:73522.
Cavestro GM, et al. (2003) Association of keratin 8 gene mutation with chronic pancreatitis. Dig. Liver Dis. 35:416–20.
Treiber M, et al. (2006) Keratin 8 sequence variants in patients with pancreatitis and pancreatic cancer. J. Mol. Med. (Berl). 84:1015–22.
Mahurkar S, et al. (2006) Association of cathepsin B gene polymorphisms with tropical calcific pancreatitis. Gut. 55:1270–75.
Tomaiuolo R, et al. (2009) An MBL2 haplotype and ABCB4 variants modulate the risk of liver disease in cystic fibrosis patients: A multicentre study. Dig. Liver Dis. 41:817–22.
Bartlett JR, et al. (2009) Genetic modifiers of liver disease in cystic fibrosis. JAMA. 302:1076–83.
Berni Canani R, et al. (2010) Congenital diarrheal disorders: Improved understanding of gene defects is leading to advances in intestinal physiology and clinical management. J. Pediatr. Gastroenterol. Nutr. 50:360–6.
Sharma A, et al. (2005) Protection against acute pancreatitis by activation of protease-activated receptor-2. Am. J. Physiol. Gastrointest. Liver Physiol. 288:G388–95.
Stevens T, Conwell DL, Zuccaro G. (2004) Pathogenesis of chronic pancreatitis: An evidence-based review of past theories and recent developments. Am. J. Gastroenterol. 99:2256–70.
Multigner L, Sarles H, Lombardo D, De Caro A. (1985) Pancreatic stone protein. II. Implication in stone formation during the course of chronic calcifying pancreatitis. Gastroenterology. 89:387–91.
Jin CX, Hayakawa T, Ko SB, Ishiguro H, Kitagawa M. (2011) Pancreatic stone protein/regenerating protein family in pancreatic and gastrointestinal diseases. Intern. Med. 50:1507–16.
Williams JA. (2008) Receptor-mediated signal transduction pathways and the regulation of pancreatic acinar cell function. Curr. Opin. Gastroenterol. 24:573–9.
Ramnath RD, Sun J, Bhatia M. (2010) PKC delta mediates pro-inflammatory responses in a mouse model of caerulein-induced acute pancreatitis. J. Mol. Med. (Berl). 88:1055–63.
Lupia E, et al. (2004) Ablation of phosphoinositide 3-kinase-gamma reduces the severity of acute pancreatitis. Am. J. Pathol. 165:2003–11.
De Lisle RC, Xu W, Roe BA, Ziemer D. (2008) Effects of Muclin (Dmbt1) deficiency on the gastrointestinal system. Am. J. Physiol. Gastrointest Liver Physiol. 294:G717–27.
Gukovskaya AS, Gukovsky I. (2012) Autophagy and pancreatitis. Am. J. Physiol. Gastrointest Liver Physiol. 303:G993–1003.
Willemer S, Kloppel G, Kern HF, Adler G. (1989) Immunocytochemical and morphometric analysis of acinar zymogen granules in human acute pancreatitis. Virchows Arch. A Pathol Anat Histopathol. 415:115–23.
Fortunato F, Kroemer G. (2009) Impaired autophagosome-lysosome fusion in the pathogenesis of pancreatitis. Autophagy. 5:850–3.
Grasso D, et al. (2011) Zymophagy, a novel selective autophagy pathway mediated by VMP1-USP9x-p62, prevents pancreatic cell death. J. Biol. Chem. 286:8308–24.
Kim JN, et al. (2011) Heat shock proteins and autophagy in rats with cerulein-induced acute pancreatitis. Gut Liver. 5:513–20.
Ohmuraya M, Yamamura K. (2008) Autophagy and acute pancreatitis: A novel autophagy theory for trypsinogen activation. Autophagy. 4:1060–2.
Lohr JM, et al. (2010) Autoantibodies against the exocrine pancreas in autoimmune pancreatitis: Gene and protein expression profiling and immunoassays identify pancreatic enzymes as a major target of the inflammatory process. Am. J. Gastroenterol. 105:2060–71.
Braganza JM, Lee SH, McCloy RF, McMahon MJ. (2011) Chronic pancreatitis. Lancet. 377:1184–97.
Thorvaldsdottir H, Robinson JT, Mesirov JP. (2013) Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 14:178–92.
Rehm HL, et al. (2013) ACMG clinical laboratory standards for next-generation sequencing. Genet. Med. 15:733–47.
Adzhubei IA, et al. (2010) A method and server for predicting damaging missense mutations. Nat. Methods. 7:248–9.
Mathe E, et al. (2006) Computational approaches for predicting the biological effect of p53 missense mutations: A comparison of three sequence analysis based methods. Nucleic Acids Res. 34:1317–25.
Hicks S, Wheeler DA, Plon SE, Kimmel M. (2011) Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum. Mutat. 32:661–8.
Ng PC, Henikoff S. (2001) Predicting deleterious amino acid substitutions. Genome Res. 11:863–74.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. (2010) MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 7:575–6.
Desmet FO, et al. (2009) Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37:67.
Wang W, et al. (2013) Comprehensive screening for PRSS1, SPINK1, CFTR, CTRC and CLDN2 gene mutations in Chinese paediatric patients with idiopathic chronic pancreatitis: A cohort study. BMJ Open. 3:003150.
Rosendahl J, et al. (2013) CFTR, SPINK1, CTRC and PRSS1 variants in chronic pancreatitis: Is the role of mutated CFTR overestimated? Gut. 62:582–92.
Tomaiuolo AC, Sofia VM, Surace C, et al. (2015) Relationship between CFTR and CTRC variants and the clinical phenotype in late-onset cystic fibrosis disease with chronic pancreatitis. J. Mol. Diagn. 17:171–8.
Felderbauer P, et al. (2003) A novel mutation of the calcium sensing receptor gene is associated with chronic pancreatitis in a family with heterozygous SPINK1 mutations. BMC Gastroenterol. 3:34.
Murugaian EE, Premkumar RM, Radhakrishnan L, Vallath B (2008) Novel mutations in the calcium sensing receptor gene in tropical chronic pancreatitis in India. Scand. J. Gastroenterol. 43:117–21.
Acknowledgments
This study was supported financially by Bambino Gesù Children’s Hospital.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (http://creativecommons.org/licenses/by-nc-nd/4.0/)
About this article

Cite this article
Sofia, V.M., Da Sacco, L., Surace, C. et al. Extensive Molecular Analysis Suggested the Strong Genetic Heterogeneity of Idiopathic Chronic Pancreatitis. Mol Med 22, 300–309 (2016). https://doi.org/10.2119/molmed.2016.00010
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2016.00010