
Abstract
Background
Stenotrophomonas maltophilia is a ubiquitous Gram-negative bacterium previously named as Xanthomonas maltophilia. This organism is an important nosocomial pathogen associated with infections in immunocompromised patients. Clinical isolates of S. maltophilia are mostly resistant to multiple antibiotics and treatment of its infections is becoming problematic. Several virulent bacteriophages, but not temperate phage, of S. maltophilia have been characterized.
Results
In this study, a temperate myophage of S. maltophilia (Smp131) was isolated and characterized. Sequence analysis showed that its genome is 33,525-bp long with 47 open reading frames (ORFs). Its similarity to P2-like phages and prophages in S. maltophilia and several Xanthomonas pathovars includes genomic organization, arrangement of several operons, and possession of a slippery sequence T7G for translational frameshifting in tail assembly genes. Smp131 encodes a tyrosine family integrase that shares low degrees of similarity with those of other phages and a lysin belonging to family 19 chitinase that is observed in plants and some bacteria, although not in phages. tRNA are the preferred sites for host integration of Smp131 and the related phages: tRNA-Thr for Smp131 and prophage of S. maltophilia K279a; tRNA-Lys for prophages of X. campestris pv. campestris ATCC33913, X. oryzae pv. oryzae strains MAFF311018, and KACC10331; and tRNA-Asn for prophage of X. oryzae pv. oryzae PXO99A and remnant of X. axonopodis pv. citri 306. Regions flanking the prophages are varied highly in nucleotide sequence and rich in transposase genes, suggesting that frequent insertion/excision had occurred.
Conclusions
Prevalence of closely related prophages in Stenotrophomonas and Xanthomonads may have contributed to the diversity of these closely related species owing to possible horizontal gene transfer mediated by the phages.
Similar content being viewed by others


Background
Stenotrophomonas maltophilia, previously named as Pseudomonas maltophilia and then Xanthomonas maltophilia[1], is an aerobic, Gram-negative, rod-shaped bacterium common in different environments. S. maltophilia can cause various types of nosocomial infections, resulting in high morbidity and mortality in severely immunocompromised and debilitated patients [2, 3]. This organism is increasingly prevalent in hospitals worldwide; in Taiwan, it is ranked one of the highest occurring nosocomial infections [4]. In addition, isolates obtained from hospitalized patients show significant genetic diversity, suggesting that they can be derived from various sources [5]. Recently, treatment of S. maltophilia infections has become more difficult because of the high prevalence of multiple resistance to antibiotics of this organism [6].
Phage therapy has attracted significant attention for its effectiveness in treating bacterial infections [7]. Some S. maltophilia phages have been reported including i) two lytic phages (phiSMA5 and Smp14) from our laboratory that resemble members of Myoviridae in morphology with a genome of approximately 250 and 160 kb, respectively [4, 8], ii) a T7-like phage lytic to pan-resistant S. maltophilia and a phage that has large burst size and unique plaque polymorphism, with their genomes being sequenced [9, 10], iii) a phage remnant in S. maltophilia strain P28 that is capable of producing a novel phage tail-like bacteriocin, designated as maltocin P28 [11], iv) detection of a phage genome carrying a zonula occludens like toxin gene [12], and v) three filamentous phages [13, 14]. In addition, we have described a novel lysozyme encoded by a Xanthomonas oryzae phage, phiXo411, that is active against both Xanthomonas and Stenotrophomonas[15]. Although the lytic phages, the lysozyme and the maltocin P28 are potentially useful in treating S. maltophilia infection, feasible testing has yet been reported.
In spite of the above mentioned efforts in phage study, no temperate phage of S. maltophilia has been reported. In this study, we isolated a temperate phage of S. maltophilia and designated as Smp131. Since acquisition of external DNA by horizontal gene transfer and gene loss are major driving-forces of bacterial genome evolution and integration and excision of temperate bacteriophages contribute actively to such evolution [16], we deemed it worthy to study this phage. The phage genome was sequenced and sequence analysis revealed that Smp131 is similar to phage P2 and shares high degrees of identity with prophages of Stenotrophomonas and xanthomonads.
Results and discussion
Phage Smp131 is a temperate myophage infecting S. maltophilia
In this study, temperate phages were detected by spotting culture supernatants from 86 clinical isolates of S. maltophilia onto lawns formed separately by all other isolates. The culture supernatant from S. maltophilia strain T13 was observed to cause clearing zones on 3 of the samples (ATCC 13637, BCRC 11901, and T16). Following 3 rounds of single plaque isolation, Smp131 was obtained and used for further study. Less turbid plaques were formed on lawns of strain T16; therefore, this strain was used as the host for phage propagation and indicator host in titering the phage.
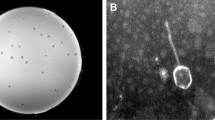
Cultures of S. maltophilia T13 released from 1 × 104 to 1 × 106 PFU/ml of Smp131 and treatment by adding mitomycin C (1 μg/ml) into the cultures produced titers of approximately 7 × 108 PFU/ml. Electron microscopy showed that Smp131 has an icosahedral head approximately 60 nm in diameter and a contractile tail 100–120 nm in length and 20–30 nm in width (Figure 1), resembling members of Myoviridae phages.

Transmission electron micrograph of Smp131. Samples were stained with 2% uranyl acetate. Scale bar represents 50 nm.
In SDS-polyacrylamide gel (10%) electrophoresis, phage particles purified by CsCl ultracentrifugation displayed more than 15 distinct protein bands, with molecular masses ranging from 16 to 120 kDa, upon staining the gel with Coomassie brilliant blue. Four bands, with molecular masses of 44, 39.5, 38, and 21 kDa, were more abundant than the others. The 38-kDa protein was the most abundant and is likely the major capsid protein.
Host range testing showed that only the three S. maltophilia strains, ATCC 13637, BCRC 11901, and T16, were sensitive to Smp131 as indicated by the formation of single plaques. Several reasons are possible for the phage resistance, including immunity, impaired adsorption and block at later stages during phage infection, and further study is needed to test these possibilities. With such a narrow host range, Smp131 apparently has limited use in control of S. maltophilia infection. Spot tests and plaque assays were also tested on bacteria other than S. maltophilia strains, including Escherichia coli (n = 14, with n being the number of isolates), Serratia marcescens (n =33), Enterobacter cloacae (n = 12), Klebsiella pneumoniae (n = 10), Proteus mirabilis (n = 11), Pseudomonas aeruginosa (n = 7), Xanthomonas campestris pv. campestris (n = 7), X. axonopodis pv. citri (n = 1), X. axonopodis pv. dieffenbachiae (n = 1), X. axonopodis pv. glycines (n = 1), X. axonopodis pv. phaseoli (n = 1), X. axonapodis pv. vesicatoria (n = 46), and X. oryzae pv. oryzae (n = 2). None of these bacteria were sensitive to Smp131, indicating that this phage has a narrow host range. This is different from phage P2 that can infect several enteric bacterial species [17].
The circular Smp131 genome has a cohesive region conserved in P2-like phages
Restriction endonucleases AvaI, EcoRI, EcoRV, HincII, KpnI, NcoI, NotI, PstI, PvuII, and SphI were tested and found to be capable of cutting the Smp131 genomic DNA into distinct fragments. Sequencing of the Smp131 genome showed 33,525 bp, and 47 ORFs were identified (Additional file 1: Table S1). Nucleotide sequence comparison revealed that Smp131 had a region similar to the 55-bp cos region conserved in P2 and the related phages required for phage packaging [18]; GC-rich 19-nt 5′-extruding cohesive ends (5′-GGCGTGGCGGGGAGACGAG-3′) similar to those of P2-related phages (5′-GGCGAGGCGGGGAAAGCAC-3′) were observed in the cos region of Smp131 (Figure 2) [19]. By analogy to the P2 case, the extruding regions were set as the ends of the Smp131 genome.

Smp131 cos region deduced by analogy to those of P2-related phage. The Smp131 sequence is aligned with the known cos regions of Enterobacteria phages P2 (GenBank:NC_001895) and P4 (GenBank:NC_001609), with arrowheads indicating cos cleavage sites [12]. Also aligned are corresponding regions from Enterobacteria phages 186 (GenBank:U32222) and PSP3 (GenBank:NC_005340), and Pseudomonas phage phiCTX (GenBank:NC_003278). CLUSTAL X1.83 was used for alignment. Letters with black and grey backgrounds are nucleotides identical in all and four or more sequences, respectively.
The circularity of the Smp131 genome was demonstrated as follows. As shown in Additional file 2: Figure S1A, when displayed in a circular form, the left- and right-hand 19-nt extruding ends of the Smp131 genome would be paired. The genome had 6 EcoRI and 12 EcoRV sites, which were numbered from E1 to E6 and V1 to V12, respectively. Based on this predicted map, we isolated and sequenced a 2.5 kb EcoRI fragment (Additional file 2: Figure S1A). Results showed that this fragment was 2501-bp long, identical in nucleotide sequence to the E6-E1 region in the genome, and indeed contained the 19-bp cos site. To confirm circularity of the genome, fragment V12-E6 was used as the probe for Southern hybridization to probe a 4.7-kb EcoRV fragment (V12-V1). As anticipated, a 4.7-kb fragment was detected in the hybridization (Additional file 2: Figure S1B). These results indicate that Smp131 has a circular genome.
Smp131 is similar to prophages in Stenotrophomonas and Xanthomonas
Sequence analysis shows that Smp131 shares similarity with several prophages in genome organization and encoded proteins. They included 1) a 27-kb prophage remnant in X. axonopodis pv. citri strain 306; 2) a prophage each in X. campestris pv. campestris strain ATCC33913 (37 kb) and X. oryzae pv. oryzae strains KACC10331 (40 kb), MAFF311018 (37 kb) and PXO99A (42 kb); and 3) a 35-kb prophage in S. maltophilia K279a (Figure 3, Additional file 3: Table S2). Additionally, most Smp131-encoded proteins were similar to those encoded by several P2-like temperate phages (see below).

Genome organization of phage Smp131, phage P2, and P2-like prophages in Stenotrophomonas and Xanthomonas . Colored arrows indicate the directions and categories (denoted below) of the genes. The numbers or letters near the arrows indicate the names or locus_tags of the genes; red numbers indicate the homologues not found in Smp131. Bent arrows indicate the six promoters identified in phage P2 and those predicted for Smp131 by analogy. Abbreviations: P2, Enterobacteria phage P2; Xac306, prophage remnant in X. axonopodis pv. citri 306; Xcc33913, prophage in X. campestris pv. campestris ATCC33913; K279a, prophage in S. maltophilia K279a; KACC10331, PXO99A and MAFF311018, prophages in X. oryzae pv. oryzae strains KACC10331, PXO99A, MAFF311018, respectively.
Similarity between Smp131 and prophages in Xanthomonas and Stenotrophomonas can be summarized as follows (Additional file 3: Table S2). First, genomes of these prophages (defined as the regions flanked by attL and attR, see below) were slightly larger than that of Smp131 (Figure 3), suggesting that some insertions in these prophages (Figure 3, numbered in red) and deletions (in/del) from Smp131 had occurred during evolution. Most of these in/dels encode hypothetical proteins. It is apparent that those absent from Smp131 are nonessential genes. Second, some Smp131 genes (orf01, 02, 03, 05, 22, 29, 36, 38, 41, 44, 45, and 46) were absent from one or more of the other prophages (remnant in X. axonopodis pv. citri strain 306 lacked orf 01, 02, 03, 23–40, and orf 44–46). Third, there were transposase genes associated with the Xanthomonas prophages and remnant (Figure 3): 1) two in the upstream region and three in the downstream flanking region of the remnant, 2) four in the downstream flanking region of X. oryzae pv. oryzae KACC10331 prophage, 3) one in the upstream flanking region and three in the upstream of X. oryzae pv. oryzae PXO99A prophage, and 4) five in the downstream flanking region of X. oryzae pv. oryzae MAFF311018 prophage. Fourth, identity in amino acid sequence between corresponding proteins of Smp131 and these prophages ranged between 30% and 94%, with the majority falling above 50% (Additional file 3: Table S2). However, because none of their encoded proteins had been characterized, sequence comparison with proteins of these prophages did not lead to the identification of Smp131 gene functions.
Among the prophage harboring strains of Stenotrophomonas and Xanthomonas, X. campestris pv. campestris ATCC33913 was the only strain available to us. Spot test showed that the culture supernatant from X. campestris pv. campestris ATCC33913 did not form lysis zones on lawns of X. campestris pv. campestris strains Xc11 and Xc17, indicating that this strain may not release phage particles.
The majority of Smp131-encoded proteins are similar to those of P2-like phages
No homologues were identified for proteins encoded by orf1, orf2, and orf3 in the database, whereas orf4 and orf5 encoded a site-specific DNA methyltransferase and a hypothetical protein, respectively. Cluster orf06 to orf11 encoding capsid and packaging proteins was organized in the same order as P2 genes QPONML; orf12 was similar to P2 gene X, annotated as tail protein (Additional file 1: Table S1, Figure 3).
Proteins encoded by orf13 and orf14 possessed three transmembrane domains similar to Class I holins [20]. The product of orf13 had a highly charged C terminus, which is characteristic of members of Class I, whereas ORF14 contained a slightly lower charged C terminus. Orf15 was assigned as the endolysin gene. Rather than sharing similarity with phage lysozymes, the orf15 product had a motif (aa 114–127) highly conserved in members of the GH19 chitinases family, [FHY]-G-R-G-[AP]-X-Q-[IL]-[ST]-[FHYW]-[HN]-[FY]-NY, that forms the substrate binding region [21] (Figure 4). Moreover, Glu50/Glu59 of ORF15 were similar to Glu68/Glu77 of Streptomyces coelicolor chitinase G experimentally identified as the active sites [22]. Family GH19 chitinases have long been identified in plants [23] and recently in bacteria [22, 24–27], although not in phages; this Smp131 enzyme appears to be the first reported for phages.

Alignment of predicted Smp131 lysin with family 19 chitinases that have determined catalytic domains. Identical residues are highlighted, with the conserved glutamate residues involved in catalysis indicated by downward arrowheads. The conserved sequence motif, [FHY]-G-R-G-[AP]-X-Q-[IL]-[ST]-[FHYW]-[HN]-[FY]-NY, that forms the substrate binding region is boxed. Abbreviations: Smp131, lysin encoded by orf15 of Smp131; K279a, lysin encoded by prophage in S. maltophilia K279a (GenBank:YP_001970233); Xcc, lysin encoded by prophage in X. campestris pv. campestris ATCC33913 (GenBank:NP_638326); ChiC, chitinase C encoded by Streptomyces griseus (GenBank:YP_001824912); ChiG, chitinase G encoded by S. coelicolor (GenBank:BAA75648).
Proteins encoded by orf17 and orf18 were homologous to R and S of P2, the tail completion proteins essential for stable head joining [28]. Proteins encoded by orf19, orf20, orf23, and orf24 were homologous to that of the P2 J, I, V, and W (clustered with H and G as VWJIHG), respectively, whereas the position of orf21 and orf22 is similar to that of P2 H and G. Among the P2 genes, VWJI code for baseplate assembly proteins; H for putative tail fiber protein; and G for probable tail fiber assembly protein [29, 30]. The difference in gene order suggests that rearrangement of these genes had occurred during evolution.
Orf25 to orf31, except orf29 that encoded a possible membrane protein, encoded tail proteins, whereas orf32 encoded a late gene control protein. These genes corresponded to the P2 operon F I F II EE’TUD (Figure 3, Additional file 1: Table S1; [31]). In P2, E’ overlaps the start of gene T, lacks a potential ribosome binding site, and extends 37 nt back into E in the -1 reading frame. A run of 6 T residues (T6G slippery sequence) was located 20 nt upstream of the possible GUG start of E’ and an extension of gene E following a -1 translational frameshift has been designated as E + E’[31]. The arrangement of E and E’ genes within the tail gene cluster and their coupling through a translational frameshift is conserved among P2-related phages as well as in several other phages such as lambda although they share no similarity in amino acid sequence [31–33]. Near the 3′-end of orf27, there is a T7G similar to the conserved T6G slippery sequence [31], nt 288–295 relative to the orf27 start codon. Thus, by analogy, a -1 translational frameshift may occur here during translation, thereby producing a protein product of orf27.1 (Additional file 4: Figure S2A). Instead of the T7G, a predicted T7C slippery sequence was observed in the corresponding tail genes of prophages of S. maltophilia K279a, X. campestris pv. campestris 33913, X. oryzae pv. oryzae strains KACC10331, MAFF311018, and PXO99A (Additional file 4: Figure S2B). These findings indicate that this type of arrangement may be conserved in all P2-like phages.
The protein predicted for orf33 was a phage-related protein similar to gp17 of phage BcepMu; orf34 encoded a protein similar to that of P2 regulatory protein Ogr (see below); the products predicted for orf35-46 were all hypothetical proteins, except that orf39 and orf43 encoded a DNA primase-like protein and a tyrosine family integrase, respectively. Tyrosine family integrases are responsible for DNA cleavage, strand exchange, and religation steps with a covalently bound phosphotyrosine intermediate [34]. As shown in Additional file 5: Figure S3, similarity search based on domain architecture [35] and sequence alignments showed that the predicted protein of orf43 possessed 4 residues of the pentad conserved residues (R241, K264, H348 and H366) and the possible catalytic site Tyr375 (Additional file 5: Figure S3). However, no significant similarity in amino acid sequence was observed between the N-terminal region of Smp131 integrase and those of other integrases.
Varied degrees of identity were shared by Smp131 proteins with the analogous proteins from phages encompassing a wild host range (Figure 3, Additional file 6: Table S3). These homologues include 23 encoded by Pseudomonas phage phiCTX (27% to 73% identity), 22 by Burkholderia phage KL3 (34% to 62% identity), and 20 by Enterobacteria phage P2 (26% to 60% identity). The majority of these homologues cluster within the regions coding for capsid and packaging proteins (ORF06 to ORF11) as well as tail related proteins (ORF17 to ORF32, except ORF21 and ORF22). The cluster encoding lysis related proteins (ORF13 to ORF16) and the phage tail fiber protein (ORF21) shared lower degrees of identity, while ORF22 (hypothetical protein) shared no appreciable homology. The very recently reported P2-like phage remnant in S. maltophilia strain P28 possesses 23 orf s [11], nine of the deduced proteins share 31% to 53% identities with the Smp131 encoded proteins (Additional file 6: Table S3).
Smp131 late genes may be regulated in a manner similar to that in P2
P2 has four late promoters, PP, PO, PV, and PF, possessing the consensus sequence TGT-N12-ACA and controlling PQ, ONMLKRS, VWJIHG, and F I F II EE’TUD operons, respectively [36, 37]. Transcription of these operons depends on the Ogr protein, a zinc-finger containing transcriptional activator with a conserved cysteine motif, CX2CX22CX4C, where a zinc atom coordinates with four cysteine residues [38, 39]. In Smp131, four putative late promoters were observed with sequences similar to TGT-N12-ACA, which were designated as PP’, PO’, PJ’, and PV’ located at nt 4398–4381, 4381–4398, 10,964-10,981, and 14,928-14,946 in the genome, respectively (Figure 3). Operons presumably controlled by PP’ and PO’ were analogous to those by P2 PP and PO, respectively, but those by PJ’ and PV’ had some exchanged members due to gene rearrangement, that is, VWJIHG and F I F II EE’TUD in P2 versus orf19-orf22 (homologous to JIHG) and orf23-orf32 in Smp131 (Figure 3). Additionally, the protein encoded by Smp131 orf34, which had a relative position similar to that of the P2 Ogr gene, had a conserved CX2CX22CX4C motif, although overall similarity shared by the two proteins was low. Thus, similarity in genome organization, promoter sequence, and a regulatory protein suggests that Smp131 late genes are regulated in a manner similar to that in P2.
tRNA genes are the preferred sites for integration of P2-like prophages of Xanthomonas and Stenotrophomonas
It is known that in E. coli i) P2 can integrate at over 10 different loci, with locI (attB site containing the core sequence, 5′-AAAAAATAAGCCCGTGTAAGGGAGATT-3′, which is identical to the attP sequence) being preferred over any other sites in E. coli C, ii) this site is occupied by a remnant of a P2 prophage in E. coli K-12 and P2 therefore will integrate into secondary sites, and iii) the P2 integrase accepts at least up to 37% mismatches within the core sequence [40]. Searching for a region similar to the P2 attP site in Smp131 genome revealed no such region. We then turned to identify putative attR and attL at the ends of prophage sequences from Stenotrophomonas and Xanthomonas and observed a 46-nucleotide perfect direct repeat at the extremities of the integrated prophage of S. maltophilia K279a, apparently representing attL and attR of the prophage (GenBank:NC_010943, Figure 3 and Additional file 7: Table S4). This 46-nucleotide sequence corresponded to the 3′-end of an intact tRNA-Thr gene. Nucleotide sequence comparison showed that a region identical to the att regions of the S. maltophilia K279a prophage was present in bp 30,738-30,783 (orf43/orf44 intergenic region) of the Smp131 genome (Additional file 7: Table S4). This region, situated downstream of the integrase gene and similar in location to those in P2-like phages (phiCTX, GenBank:NC_003278; 186, GenBank:U32222), was thus predicted to be the attP site for Smp131 (Figure 3).
Based on the position of attP, we predicted that upon integration via attP, orf44 and orf43 would become flanked by attL and attR, respectively. In addition, an NaeI and a HincII restriction sites were located 644 bp and 667 bp relative to the orf43 and orf44 start codons, respectively, in the Smp131 genome (Additional file 8: Figure S4). Sequencing revealed that the amplicons were 1,092 bp and 704 bp containing attL and attR, respectively, which had a sequence identical to that of the Smp131 attP. To verify the att-flanking sequences, primers L3/L4 and R2/R3 were used to amplify the junctions of attL and attR regions, respectively (Additional file 8: Figure S4). Sequencing of these 2 replicons confirmed that our inverse PCR reactions had faithfully amplified the targeted regions. The result revealed that a segment of a possible defective integrase gene (480 bp) downstream of the attL was similar to that of Burkholderia thailandensis E264 (GenBank:YP_441483), whereas a 177-bp long host chromosomal region upstream of the attR was highly similar to the sequence adjacent to the tRNA-Thr of S. maltophilia strains (K279a and R551-3). These results suggest that upstream regions of tRNA-Thr are conserved in different strains of S. maltophilia, whereas the downstream regions are not. It was also noticed that upon integration, an intact tRNA-Thr that included the attR was regained, similar to the target site duplication observed by Rocco et al. [41].
In addition to S. maltophilia strain K279a (GenBank:NC_010943), the genome sequence has been determined for strain R551-3 (GenBank:NC_011071) [42, 43]; they each had only one copy of tRNA-Thr located near one o’clock relative to the origin of chromosome replication (ori), as identified by containing DnaA boxes and genes involved in the initiation of bacterial chromosome replication [44]. Therefore, it is highly probable that this tRNA-Thr is the preferred site for Smp131 integration. Sequence analysis of junctions of integrated Xanthomonas prophage suggests that 1) prophages of X. campestris pv. campestris strain ATCC33913, and X. oryzae pv. oryzae strains MAFF311018 and KACC10331 integrated into a 45-bp region corresponding to 3′-end of a tRNA-Lys gene (GenBank:XCC3013, GenBank:XOO_r26, GenBank:XOO4676), 2) prophage of X. oryzae pv. oryzae PXO99A integrated into a 46-bp region corresponding to the 3′ end of a tRNA-Asn (GenBank:PXO_rna33), and 3) prophage remnant of X. axonopodis pv. citri 306 used the same sequence (GenBank:XAC2627) as that of X. oryzae pv. oryzae PXO99A prophage for integration, except that only attL was retained (Figure 3, and Additional file 7: Table S4). All identified attB sites for Xanthomonas are also located near one o’clock on the bacterial chromosomes.
Host integration of P2-like phages involves binding of integrase to the two arm-binding sites flanking the imperfect repeat, each having two direct repeats [45]. Careful examination of the Smp131 sequence revealed a pair of perfect direct repeats (5′-AATTTTACCGG-3′, bp 30635–30645 and bp 30647–30657) and an inverted repeat (5′-AAAAAGGCCAGCGCACCGCGCTGGCCTTTTT-3′, bp 30665–30695) in the upstream of attP (after the integrase gene, orf43), but no such sequences were found between attP and orf44. By analogy, it is possible that these repeats are involved in recognition by Smp131 integrase for host integration. However, lack of conserved repeats in the downstream suggests that the Smp131 integrase may be less demanding for sequence conservation in the downstream region for the function.
Conclusions
This study is the first to isolate a temperate phage of S. maltophilia, Smp131. It is identified as a P2-like phage based on similarities to P2 in amino acid sequences of the encoded proteins, genomic organization, arrangement of several operons, and possession of a slippery sequence T7G for translational frameshifting in tail assembly genes. Smp131 is able to infect only S. maltophilia, different from phage P2 that can infect several enteric bacterial species. Several P2-like prophages in S. maltophilia and xanthomonads are also identified by bioinformatic analyses. In contrast to P2 that can integrate into several loci of the host chromosome, with certain loci being favoured and none of them being t-RNA gene, single t-RNA genes are found to be the locus for integration of these Stenotrophomonas and xanthomonads prophages. In addition, the regions flanking the prophages are rich in transposase-like genes, suggesting frequent exchange of genes during evolution. Existence of closely related prophages in Stenotrophomonas and xanthomonads is consistent with the close relatedness of these bacteria and the previous classification including Stenotrophomonas in genus Xanthomonas. Prevalence of the phages may have contributed to diversity of these closely related species owing to possible horizontal gene transfer mediated by the phages. With a narrow host range, the value to use Smp131 for controlling S. maltophilia infection is apparently limited.
Methods
Bacterial strains and growth conditions
Bacterial strains used in this study have been described previously [4]. S. maltophilia strains ATCC13637, BCRC 11901 and BCRC 15678 were used as reference strains [4]. Strain T16 was the host for propagation of phage Smp131 and as the indicator host in plaque assay. Luria-Bertani (LB) broth and LB agar plate were the general-purpose media [46] used to cultivate S. maltophilia (30°C), Escherichia coli (37°C), Serratia marcescens (37°C), Enterobacter cloacae (37°C), Klebsiella pneumoniae (37°C), Proteus mirabilis (37°C), Pseudomonas aeruginosa (37°C), and Xanthomonas strains (28°C).
Spot test, isolation of bacteriophage and plaque assay
To detect the presence of phage in the culture supernatants and the phage sensitivity of a bacterium, spot tests were performed as described previously [4], except that LB broth and LB agar plates were used. The top agar containing the clearing zones was picked and soaked for 30 min in 100 μl of LB broth. Following appropriate dilution, the suspensions were plated for single plaque formation. Two more rounds of single-plaque isolation were performed to obtain the pure phage culture. To determine the phage titers, double-layered bioassays were performed on LB agar plates in which the top and bottom layers contained 0.75% and 1.5% agar, respectively. One-tenth of a milliliter each of a phage suspension after serial dilutions and cells of S. maltophilia strain from an overnight culture were mixed with 3 ml of molten soft agar and poured onto the bottom solidified agar (12 ml). Numbers of plaques were counted after the plates were incubated overnight. The same method was used to confirm phage susceptibility with the cells of different bacteria as the indicator hosts.
Purification of phage particles
High-titer lysates of Smp131 (400 ml, approximately 1.0 × 1010 PFU/ml) were centrifuged (10,000 × g, 20 min at 4°C). The supernatants were passed through a membrane filter (0.45 μm pore size) and then centrifuged (15,000 × g at 4°C) for 2 hr. The phage pellets were suspended in 1.0 ml of the SM buffer (50 mM Tris–HCl, pH 7.5, containing 100 mM NaCl, 10 mM MgSO4, and 0.01% gelatin) and loaded on the block gradient of CsCl (1.2, 1.35, 1.45, 1.50, and 1.70 g/ml), followed by ultracentrifugation (28,000 rpm for 2 h at 4°C) with rotor TH641 (Sorvall OTD Combi) [15]. The phage particles concentrated into a zone were recovered and dialyzed against the SM buffer.
DNA techniques
Phage particles purified following ultracentrifugation were treated with sodium dodecyl sulfate (SDS, 1%) and 20 U of proteinase K (Sigma P-2308) at 58°C for 1 h. An equal volume of phenol/chloroform (1:1) was then added to remove the proteinaceous materials. Phenol/chloroform extraction was repeated twice and the DNA was precipitated as described previously [47]. Restriction enzyme digestion of the phage DNA was performed in accordance with supplier instructions. DNA fragments were separated in 0.7% agarose gels in a TAE buffer (40 mM Tris acetate, pH, 8.0, containing 2 mM EDTA). Isolation of DNA fragments from agarose gel was performed using commercial kits (Qiagen). Standard protocols were followed for blotting DNA fragments onto the membrane (NEN catalog number NEF988), preparation of probes by labeling with [α-32P] dCTP (Du Pont. NEN), and Southern hybridization.
DNA sequencing and bioinformatics
Processes for purification and shearing of phage DNA, cloning the DNA fragments into pBluescript II SK, and determination of the nucleotide sequence were performed as detailed in previous research [48]. Gaps were closed by primer walking with PCR-amplification on Smp131 genomic DNA as the template using primers designed according to available sequences. Programs used for DNA sequence analysis and similarity search based on domain architecture were selected according to previous research [49]. Possible ORFs were searched in 6 reading frames on both strands of the Smp131 genomic DNA, which used ATG or GTG as the start codon, consisted of longer than 50 amino acid residues, and had a putative ribosomal binding site in the upstream region.
The 33,525-bp DNA sequence determined in this study for phage Smp131 has been deposited in GenBank under accession number JQ809663.
Cloning of the attL and attR regions flanking the Smp131 prophage
To clone the junction regions containing attL and attR, an inverse PCR-based strategy was employed. The chromosome prepared from S. maltophilia T13, the Smp131 lysogenic strain, was cleaved with NaeI and HincII separately and self-ligated to circularize the DNA molecules. Inverse PCR was performed using the circularized HincII and NaeI fragments as the templates with primer pairs L1/L2 (for amplification of the attL-containing region) and R1/R2 (for amplification of the attR- containing region), respectively. The amplicons obtained were sequenced for comparison.
Separation of virion proteins by SDS-polyacrylamide gel electrophoresis
Following dialysis, phage particles (approximately 1 × 108 PFU) purified by ultracentrifugation were boiled in a loading buffer for 3 min and separated in SDS-PAGE (10% polyacrylamide and 0.1% SDS). Protein bands were visualized by staining the gel with Coomassie brilliant blue (Bio-Rad) [47].
Electron microscopy
Phage Smp131 was examined by electron microscopy of negatively stained preparations as described previously [4] using a JEM-1200 EX II transmission electron microscope (JEOL, Peabody, Mass) operated at 120 kV.
References
Palleroni NJ, Bradbury JF: Stenotrophomonas, a new bacterial genus for Xanthomonas maltophilia (Hugh 1980) Swings et al. 1983. Int J Syst Bacteriol. 1993, 43: 606-609. 10.1099/00207713-43-3-606.
Roilides E, Butler KM, Husson RN, Mueller BU, Lewis LL, Pizzo PA: Pseudomonas infections in children with human immunodeficiency virus infection. Pediatr Infect Dis J. 1992, 11: 547-553. 10.1097/00006454-199207000-00008.
Vartivarian SE, Papadakis KA, Anaissie EJ: Stenotrophomonas (Xanthomonas) maltophilia urinary tract infection. A disease that is usually severe and complicated. Arch Intern Med. 1996, 156: 433-435. 10.1001/archinte.1996.00440040111012.
Chang HC, Chen CR, Lin JW, Shen GH, Chang KM, Tseng YH, Weng SF: Isolation and characterization of novel giant Stenotrophomonas maltophilia phage phiSMA5. Appl Environ Microbiol. 2005, 71: 1387-1393. 10.1128/AEM.71.3.1387-1393.2005.
Caylan R, Kaklikkaya N, Aydin K, Aydin F, Yilmaz G, Ozgumus B, Koksal I: An epidemiological analysis of Stenotrophomonas maltophilia strains in a university hospital. Jpn J Infect Dis. 2004, 57: 37-40.
Milne KE, Gould IM: Combination antimicrobial susceptibility testing of multidrug-resistant Stenotrophomonas maltophilia from cystic fibrosis patients. Antimicrob Agents Chemother. 2012, 56: 4071-4077. 10.1128/AAC.00072-12.
Harper DR, Enright MC: Bacteriophages for the treatment of Pseudomonas aeruginosa infections. J Appl Microbiol. 2011, 111: 1-7. 10.1111/j.1365-2672.2011.05003.x.
Chen CR, Lin CH, Lin JW, Chang CI, Tseng YH, Weng SF: Characterization of a novel T4-type Stenotrophomonas maltophilia virulent phage Smp14. Arch Microbiol. 2007, 188: 191-197. 10.1007/s00203-007-0238-5.
Huang Y, Fan H, Pei G, Fan H, Zhang Z, An X, Mi Z, Shi T, Tong Y: Complete genome sequence of IME15, the first T7-like bacteriophage lytic to pan-antibiotic-resistant Stenotrophomonas maltophilia. J Virol. 2012, 86: 13839-13840. 10.1128/JVI.02661-12.
Fan H, Huang Y, Mi Z, Yin X, Wang L, Fan H, Zhang Z, An X, Chen J, Tong Y: Complete Genome Sequence of IME13, a Stenotrophomonas maltophilia bacteriophage with large burst size and unique plaque polymorphism. J Virol. 2012, 86: 11392-11393. 10.1128/JVI.01908-12.
Liu J, Chen P, Zheng C, Huang YP: Characterization of maltocin P28, a novel phage tail-like bacteriocin from Stenotrophomonas maltophilia. Appl Environ Microbiol. 2013, 79: 5593-5600. 10.1128/AEM.01648-13.
Hagemann M, Hasse D, Berg G: Detection of a phage genome carrying a zonula occludens like toxin gene (zot) in clinical isolates of Stenotrophomonas maltophilia. Arch Microbiol. 2006, 185: 449-458. 10.1007/s00203-006-0115-7.
Liu J, Liu Q, Shen P, Huang YP: Isolation and characterization of a novel filamentous phage from Stenotrophomonas maltophilia. Arch Virol. 2012, 157: 1643-1650. 10.1007/s00705-012-1305-z.
Petrova M, Shcherbatova N, Kurakov A, Mindlin S: Genomic characterization and integrative properties of phiSMA6 and phiSMA7, two novel filamentous bacteriophages of Stenotrophomonas maltophilia. Arch Virol. 2013, [Epub ahead of print]
Lee CN, Lin JW, Chow TY, Tseng YH, Weng SF: A novel lysozyme from Xanthomonas oryzae phage ϕXo411 active against Xanthomonas and Stenotrophomonas. Protein Expr Purif. 2006, 50: 229-237. 10.1016/j.pep.2006.06.013.
Wagner PL, Waldor MK: Bacteriophage control of bacterial virulence. Infect Immun. 2002, 70: 3985-3993. 10.1128/IAI.70.8.3985-3993.2002.
Bertani LE, Six EW: The P2-like phages and their parasite. The bacteriophages, Volume 2. Edited by: Calendar R. 1988, New York, N.Y: Plenum Publishing Corp, 73-143. 4
Ziermann R, Calendar R: Characterization of the cos sites of bacteriophages P2 and P4. Gene. 1990, 96: 9-15. 10.1016/0378-1119(90)90334-N.
Padmanabhan R, Wu R, Calendar R: Complete nucleotide sequence of the cohesive ends of bacteriophage P2 deoxyribonucleic acid. J Biol Chem. 1974, 249: 6197-6207.
Savva CG, Dewey JS, Deaton J, White RL, Struck DK, Holzenburg A, Young R: The holin of bacteriophage lambda forms rings with large diameter. Mol Microbiol. 2008, 69: 784-793. 10.1111/j.1365-2958.2008.06298.x.
Huet J, Rucktooa P, Clantin B, Azarkan M, Looze Y, Villeret V, Wintjens R: X-ray structure of papaya chitinase reveals the substrate binding mode of glycosyl hydrolase family 19 chitinases. Biochemistry. 2008, 47: 8283-8291. 10.1021/bi800655u.
Hoell IA, Dalhus B, Heggset EB, Aspmo SI, Eijsink VG: Crystal structure and enzymatic properties of a bacterial family 19 chitinase reveal differences from plant enzymes. FEBS J. 2006, 273: 4889-4900. 10.1111/j.1742-4658.2006.05487.x.
Collinge DB, Kragh KM, Mikkelsen JD, Nielsen KK, Rasmussen U, Vad K: Plant chitinases. Plant J. 1993, 3: 31-40. 10.1046/j.1365-313X.1993.t01-1-00999.x.
da Silva AC, Ferro JA, Reinach FC, Farah CS, Furlan LR, Quaggio RB, Monteiro-Vitorello CB, Van Sluys MA, Almeida NF, Alves LM, do Amaral AM, Bertolini MC, Camargo LE, Camarotte G, Cannavan F, Cardozo J, Chambergo F, Ciapina LP, Cicarelli RM, Coutinho LL, Cursino-Santos JR, El-Dorry H, Faria JB, Ferreira AJ, Ferreira RC, Ferro MI, Formighieri EF, Franco MC, Greggio CC, Gruber A, et al.: Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature. 2002, 417: 459-463. 10.1038/417459a.
Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, McKenney K, Sutton G, FitzHugh W, Fields C, Gocayne JD, Scott J, Shirley R, Liu L, Glodek A, Kelley JM, Weidman JF, Phillipps CA, Spriggs T, Hedblom E, Cotton MD, Utterback TR, Hanna MC, Nguyen DT, Saudek DM, Brandon RC, et al.: Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995, 269: 496-512. 10.1126/science.7542800.
Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, Gill SR, Nelson KE, Read TD, Tettelin H, Richardson D, Ermolaeva MD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, McDonald L, Utterback T, Fleishmann RD, Nierman WC, White O, Salzberg SL, Smith HO, Colwell RR, Mekalanos JJ, et al.: DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature. 2000, 406: 477-483. 10.1038/35020000.
Kawase T, Saito A, Sato T, Kanai R, Fujii T, Nikaidou N, Miyashita K, Watanabe T: Distribution and phylogenetic analysis of family 19 chitinases in Actinobacteria. Appl Environ Microbiol. 2004, 70: 1135-1144. 10.1128/AEM.70.2.1135-1144.2004.
Linderoth NA, Julien B, Flick KE, Calendar R, Christie GE: Molecular cloning and characterization of bacteriophage P2 genes R and S involved in tail completion. Virology. 1994, 200: 347-359. 10.1006/viro.1994.1199.
Haggard-Ljungquist E, Halling C, Calendar R: DNA sequences of the tail fiber genes of bacteriophage P2: evidence for horizontal transfer of tail fiber genes among unrelated bacteriophages. J Bacteriol. 1992, 174: 1462-1477.
Lengyel JA, Goldstein RN, Marsh M, Calendar R: Structure of the bacteriophage P2 tail. Virology. 1974, 62: 161-174. 10.1016/0042-6822(74)90312-2.
Christie GE, Temple LM, Bartlett BA, Goodwin TS: Programmed translational frameshift in the bacteriophage P2 FETUD tail gene operon. J Bacteriol. 2002, 184: 6522-6531. 10.1128/JB.184.23.6522-6531.2002.
Levin ME, Hendrix RW, Casjens SR: A programmed translational frameshift is required for the synthesis of a bacteriophage lambda tail assembly protein. J Mol Biol. 1993, 234: 124-139. 10.1006/jmbi.1993.1568.
Xu J, Hendrix RW, Duda RL: Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol Cell. 2004, 16: 11-21. 10.1016/j.molcel.2004.09.006.
Grainge I, Jayaram M: The integrase family of recombinase: organization and function of the active site. Mol Microbiol. 1999, 33: 449-456. 10.1046/j.1365-2958.1999.01493.x.
Geer LY, Domrachev M, Lipman DJ, Bryant SH: CDART: protein homology by domain architecture. Genome Res. 2002, 12: 1619-1623. 10.1101/gr.278202.
Christie GE, Calendar R: Bacteriophage P2 late promoters. II. Comparison of the four late promoter sequences. J Mol Biol. 1985, 181: 373-382. 10.1016/0022-2836(85)90226-8.
Julien B, Calendar R: Purification and characterization of the bacteriophage P4 delta protein. J Bacteriol. 1995, 177: 3743-3751.
Lee TC, Christie GE: Purification and properties of the bacteriophage P2 ogr gene product. A prokaryotic zinc-binding transcriptional activator. J Biol Chem. 1990, 265: 7472-7477.
Pountney DL, Tiwari RP, Egan JB: Metal- and DNA-binding properties and mutational analysis of the transcription activating factor, B, of coliphage 186: a prokaryotic C4 zinc-finger protein. Protein Sci. 1997, 6: 892-902.
Barreiro V, Haggard-Ljungquist E: Attachment sites for bacteriophage P2 on the Escherichia coli chromosome: DNA sequences, localization on the physical map, and detection of a P2-like remnant in E. coli K-12 derivatives. J Bacteriol. 1992, 174: 4086-4093.
Rocco F, De Gregorio E, Colonna B, Di Nocera PP: Stenotrophomonas maltophilia genomes: a start-up comparison. Int J Med Microbiol. 2009, 299: 535-546. 10.1016/j.ijmm.2009.05.004.
Crossman LC, Gould VC, Dow JM, Vernikos GS, Okazaki A, Sebaihia M, Saunders D, Arrowsmith C, Carver T, Peters N, Adlem E, Kerhornou A, Lord A, Murphy L, Seeger K, Squares R, Rutter S, Quail MA, Rajandream MA, Harris D, Churcher C, Bentley SD, Parkhill J, Thomson NR, Avison MB: The complete genome, comparative and functional analysis of Stenotrophomonas maltophilia reveals an organism heavily shielded by drug resistance determinants. Genome biol. 2008, 9: R74-10.1186/gb-2008-9-4-r74.
Taghavi S, Garafola C, Monchy S, Newman L, Hoffman A, Weyens N, Barac T, Vangronsveld J, van der Lelie D: Genome survey and characterization of endophytic bacteria exhibiting a beneficial effect on growth and development of poplar trees. Appl Environ Microbiol. 2009, 75: 748-757. 10.1128/AEM.02239-08.
Yen MR, Lin NT, Hung CH, Choy KT, Weng SF, Tseng YH: oriC region and replication termination site, dif, of the Xanthomonas campestris pv. campestris 17 chromosome. Appl Environ Microbiol. 2002, 68: 2924-2933. 10.1128/AEM.68.6.2924-2933.2002.
Yu A, Haggård-Ljungquist E: Characterization of the binding sites of two proteins involved in the bacteriophage P2 site-specific recombination system. J Bacteriol. 1993, 175: 1239-1249.
Miller JH: Experiments in molecular genetics. 1972, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory
Sambrook J, Russell DW: Molecular cloning: a laboratory manual. 2001, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory, 3
Lee CN, Hu RM, Chow TY, Lin JW, Chen HY, Tseng YH, Weng SF: Comparison of genomes of three Xanthomonas oryzae bacteriophages. BMC genomics. 2007, 8: 442-10.1186/1471-2164-8-442.
Lee CN, Lin JW, Weng SF, Tseng YH: Genomic characterization of the intron-containing T7-like phage phiL7 of Xanthomonas campestris. Appl Environ Microbiol. 2009, 75: 7828-7837. 10.1128/AEM.01214-09.
Acknowledgements
This work was supported by grant No. NSC101-2313-B-005-033 and NSC99-2321-B-005-010-MY3 from the National Science Council of the Republic of China.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SFW designed the experiments. CNL and HCC carried out the wet lab. TTT and CNL performed bioinformatic analyses. JWL and TTT edited the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12866_2013_2203_MOESM2_ESM.ppt
Additional file 2: Figure S1: Strategy employed to test whether Smp131 has a circular form of genome. Lines: 1, restriction map deduced from the Smp131 sequence determined in this study; 2, fragments E1-3 (2.5 kb) and E5B1 (0.7 kb) used as probes for Southern hybridization; 3 and 4, 4.7-kb AvaI fragment (A1) and 4.7-kb EcoRV fragment (B5), respectively, that would hybridize to probes EI-3 and E5BI should the genome be circular. (B) Southern hybridization of AvaI and EcoRV digests from Smp131 genome using E1-3 and E5B1 separately as probes. (PPT 234 KB)
12866_2013_2203_MOESM3_ESM.xls
Additional file 3: Table S2: Comparison of proteins deduced from prophages or remnant in Xanthomonas and Stenotrophomonas. (XLS 43 KB)
12866_2013_2203_MOESM4_ESM.ppt
Additional file 4: Figure S2: Predicted T7G translational frameshift sites in Smp131 and closely related prophages from Xanthomoas and Stenotrophomonas. (A) T7G (enclosed by a rectangle) and the surrounding regions including genes p27, p27.1 and p28 of Smp131. Stop codons are denoted by three dots after the amino acids. Predicted start codon ATG of p27.1 is underlined, whereas ribosomal binding site AGAGG for gene p28 is in gray background. (B) DNA sequence alignment of the regions surrounding T7G translational frameshift sites (enclosed in rectangles) from Smp131 and the related prophages from X. campestris pv. campestris 33913, X. oryzae pv. oryzae strains KACC10331, MAFF311018 and PXO99A. An asterisk indicates identical nucleotides in all phages. (PPT 1 MB)
12866_2013_2203_MOESM5_ESM.ppt
Additional file 5: Figure S3: Comparison of tyrosine integrase of Smp131 and its homologues. Identical residues found in more than 3 residues are highlighted. Active sites determined for XerD are indicated by downward arrowhead and the RKHRH pentad conserved residues are indicated above. The α-helix (empty rectangle) and β-sheet (empty arrow) structural motifs under the alignments are based on the crystal structure of E. coli XerD. Abbreviations: Smp131, integrase deduced from Smp131 orf43; P2, integrase of Enterobacteria phage P2 (GenBank:P36932); 186, integrase of Enterobacteria phage 186 (GenBank:P06723); XerD, site-specific recombinase of E. coli (GenBank:1A0P_A). (PPT 2 MB)
12866_2013_2203_MOESM6_ESM.xls
Additional file 6: Table S3: Identities of amino acid sequence shared between the proteins deduced from Smp131 and those from bacteriophages. (XLS 44 KB)
12866_2013_2203_MOESM7_ESM.xls
Additional file 7: Table S4: Positions and sequences of att sites and tRNA of Smp131 and prophages in Xanthomonas and Stenotrophomonas.(XLS 26 KB)
12866_2013_2203_MOESM8_ESM.ppt
Additional file 8: Figure S4: Strategy for cloning the host-prophage junctions from Smp131-lysogenized S. maltophilia T13. (A) Sketch depicting the circular Smp131 genome and genes near the predicted attP site. Arrows represent the genes and predicted attP site. (B) Sketch showing the host S. maltophilia T13 chromosome and its attB site. (C) Map showing relative positions of genes after Smp131 integration into host S. maltophilia T13. Primers used in PCR were: L1; 5′-TGAAAGGTGCCATGACCACACG-3′; L2, 5′-GCGTTGCCAAGGTCAGATCGG-3′; L3; 5′-CGCATCGCACTCTAGGAAGTGAAG-3′; L4, 5′-AACTGCCAGAACCTCTGCAGTG-3′; R1, 5′-CTCTTGTCCTCGCTGTCGGT-3′; R2, 5′-TGATAGCCCTATTTTCAAGGGC-3′; R3, 5′-AGGCCCAGCAGCGCA-3′; R4, 5′-TGCCTGCCGCCAGCT-3′. S. maltophilia T13 chromosome containing prophage Smp131 was digested with HincII and NaeI. The fragments were self-ligated and the circularized DNA was then used as the templates for inverse PCR. Amplicons obtained were sequenced for comparison. (PPT 183 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lee, CN., Tseng, TT., Chang, HC. et al. Genomic sequence of temperate phage Smp131 of Stenotrophomonas maltophilia that has similar prophages in xanthomonads. BMC Microbiol 14, 17 (2014). https://doi.org/10.1186/1471-2180-14-17
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-14-17