
Abstract
Background
Mapping expression quantitative trait loci (eQTL) of targeted genes represents a powerful and widely adopted approach to identify putative regulatory variants. Linking regulation differences to specific genes might assist in the identification of networks and interactions. The objective of this study is to identify eQTL underlying expression of four gene families encoding isoflavone synthetic enzymes involved in the phenylpropanoid pathway, which are phenylalanine ammonia-lyase (PAL; EC 4.3.1.5), chalcone synthase (CHS; EC 2.3.1.74), 2-hydroxyisoflavanone synthase (IFS; EC1.14.13.136) and flavanone 3-hydroxylase (F3H; EC 1.14.11.9). A population of 130 recombinant inbred lines (F5:11), derived from a cross between soybean cultivar ‘Zhongdou 27’ (high isoflavone) and ‘Jiunong 20’ (low isoflavone), and a total of 194 simple sequence repeat (SSR) markers were used in this study. Overlapped loci of eQTLs and phenotypic QTLs (pQTLs) were analyzed to identify the potential candidate genes underlying the accumulation of isoflavone in soybean seed.
Results
Thirty three eQTLs (thirteen cis-eQTLs and twenty trans-eQTLs) underlying the transcript abundance of the four gene families were identified on fifteen chromosomes. The eQTLs between Satt278-Sat_134, Sat_134-Sct_010 and Satt149-Sat_234 underlie the expression of both IFS and CHS genes. Five eQTL intervals were overlapped with pQTLs. A total of eleven candidate genes within the overlapped eQTL and pQTL were identified.
Conclusions
These results will be useful for the development of marker-assisted selection to breed soybean cultivars with high or low isoflavone contents and for map-based cloning of new isoflavone related genes.
Similar content being viewed by others


Background
Soy food has been taken as a functional food because it contains many health beneficial molecules such as isoflavones [1]. Studies on human nutrition have shown that soybean isoflavones play an important role in preventing a number of chronic diseases [2, 3]. Equally, isoflavones are critical factors in defending soybean crops against pests [4, 5], in promoting nodulation by rhizobia [6], and in changing or adjusting the microorganisms around plant roots [7]. The major bioactive components of soybean isoflavones in human nutrition are daidzein (DZ), genistein (GT) and glycitein (GC). Isoflavone contents in soybean seed are inherited as complex quantitative traits [8–11]. Since soy seed isoflavones are regulated by multiple genetic factors, their concentrations in seed are highly variable [1, 12–14]. Over fifty QTLs underlying individual and/or total soybean isoflavone content have been reported [8, 15–23]. However, only 12 of these QTLs were in genomic regions encoding isoflavone synthesis enzymes.
A group of enzymes in the phenylpropanoid pathway lead to the biosynthesis of DZ, GT and GC [11]. Phenylalanine ammonia lyase (PAL; EC 4.3.1.5), chalcone synthase (CHS; EC 2.3.1.74) and flavanone 3-hydroxylase (F3H; EC 1.14.11.9) [24] are the first three enzymes that convert the amino acid phenylalanine into p-Coumaroyl-CoA in this pathway [11]. In the isoflavonoid biosynthetic pathway [25], the co-catalytic action of CHS [26, 27] with chalcone reductase (CHR; EC 2.3.1.170) [28] produces isoliquiritigenin and naringenin chalcone, which are isomers of the central isoflavanone intermediates naringenin and liquiritigenin, respectively. Isoliquiritigenin and naringenin chalcone are respectively converted into liquiritigenin and naringenin by chalcone isomerase (CHI; EC 5.5.1.6) [29]. These two products are the precursors of DZ and GT, which are formed after the catalysis of the precursors by the key enzyme 2-hydroxyisoflavanone synthase (IFS; EC 1.14.13.136) [30, 31]. The enzyme F3H, that competes with IFS in utilizing naringenin, catalyzes the conversion of flavanones to dihydroflavonols, which are intermediates in the biosynthesis of flavonols, anthocyanidins, catechins and proanthocyanidins [32, 33]. For the synthesis of GC, isoliquiritigenin is likely a precursor to form GC after several biochemical steps, which are not entirely known yet [34]. However, seed isoflavone concentrations in soybean can be regulated by metabolic engineering of the complex phenylpropanoid biosynthetic pathways [35].
Regulating transcript abundance is an effective approach to improve phenotypes [36]. The integrated analysis of genotype and transcript abundance data for association with complex traits can be used to identify novel genetic pathways involved in complex traits. ‘Expression QTL’ (eQTL), first defined by Jansen and Nap [37], could identify the genetic determinants of transcript abundances and is widely used for investigating gene regulation pathways. This approach treats transcript abundance of individual genes as quantitative traits in a segregating population. The eQTL map information enables genetic regulatory networks to be modeled that can provide a better understanding of the underlying phenotypic variation. It has been successfully applied in humans [38–40], plants [41–44], yeasts [45, 46], worms [47], flies [48], mice [49, 50], pigs [51] and rats [52] populations. These studies showed that transcript abundance was highly heritable and could be linked to either a local locus (cis-eQTL) or a distant locus (trans-eQTL). Cis-eQTL is mapped to the same genomic location like an expressed gene (within 5 Mb), and trans-eQTL is mapped to a different genomic location from an expressed gene (>5 Mb or on different chromosomes) [40, 53]. In general, cis-eQTL tends to produce stronger statistical associations than does by trans-eQTL [54]. This phenomenon is regarded as evidence of greater biological plausibility for the existence of true functional cis-eQTL [55]. Trans-eQTL could occur individually at a single genomic locus or could occur collectively as part of eQTL trans-bands [55]. This genomics approach has been employed to identify eQTL related genes in soybean [36, 56–58]. To date, no information concerning eQTLs underlying soybean isoflavone synthetic enzyme genes is available.
It has been proved that many enzymes in the phenylpropanoid pathway underlie QTLs that determine the accumulation of isoflavone contents in soybean seeds [11]. Meanwhile, the modification of enzyme encoded genes that are involved in phenylpropanoid pathway could promote the biosynthesis of isoflavone [31, 35]. In this study, PAL, CHS, IFS and F3H in the phenylpropanoid pathway were selected as the target genes (TGs) to analyze isoflavone-relative eQTL. Potential candidate genes underlying the accumulation of isoflavone contents in soybean seed were also evaluated. In addition, overlapped loci both for eQTL and phenotypic QTL (pQTL) were identified.
Results
Total and individual isoflavone contents, target gene transcript abundance and correlation analysis
Transcript abundances of target genes (TGs) between parents from R3 to R8 developmental stages were compared. Total and individual isoflavone contents and transcript abundances of TGs at R6 stage of soybean development were measured in the F5:11 population. The results showed that significant differences among the transcript abundances of TGs between the two parents existed at the R6 stage. The phenotypic variation of individual and total isoflavones showed a continuous distribution (Table 1).
GT showed a high positive correlation coefficient with DZ (r = 0.762, P < 0.01; Table 2). The transcript abundance of PAL was positively correlated with both GT and TI, but exhibited no significant correlation with DZ and GC. The transcript abundance of CHS was positive correlated with DZ, GT and TI, but negatively associated with GC amount. The transcript abundance of IFS displayed a positive correlation with DZ, but showed no correlation with other isoflavone components. The transcript abundance of F3H showed significantly negative correlation with individual and total isoflavone contents.
Identification of genomic region for target genes
Through BLAST searches (http://www.phytozome.net/soybean), the PAL has six homologous regions (E ≤ 0), which are located on Gm10 (LG O, PAL1/ PAL2), Gm13 (LG F, PAL1), Gm03 (LG N, PAL1), Gm19 (LG L, PAL1), Gm20 (LG I) and Gm02 (LG D1b, PAL1). Homologous regions encoding CHS (E-value ≤ 1.0E-05) are located on Gm11 (LG B1, CHS8), Gm01 (LG D1a, CHS6/CHS7), Gm08 (LG A2, CHS1/CHS2/CHS3/CHS4/CHS5/CHS9), Gm05 (LG A1, CHS2), Gm02 (LG D1b), Gm09 (LG K, CHS6), Gm19 (LG L) and Gm13 (LG F). Genes that encode F3H are located on Gm02 (LG D1b, F3H1/F3H2), Gm16 (LG J), Gm01 (LG D1a), Gm11 (LG B1), Gm18 (LG G) and Gm19 (LG L). Genes encoding IFS are located on Gm07 (LG M IFS1), Gm13 (LG F, IFS2), Gm10 (LG O), Gm03 (LG N), Gm12 (LG H), Gm19 (LG L), Gm17 (LG D2) and Gm11 (LG B1). Genes encoding IFS have the function of P450 cytochromes [27] and might have additional functional homologs.
eQTL analysis for four TGs
The linkage map that included 194 SSR markers (accepted by Molecular Biology Reports) and covered 2,312 cM with mean distance of about 12 cM between markers was used to identify eQTLs associated with the expression of the four TGs. Thirty-three eQTLs that appeared to underlie transcript abundance of the four TGs are detected and located on fifteen LGs (Table 3, Figure 1). Regarding to the locational relationships between the eQTL and the genes, thirteen of the eQTLs were cis-acting (within 5 Mb upstream or downstream of the genes) and twenty of the eQTLs were trans-acting (more than 5 Mb away or on different chromosomes) [40, 53].

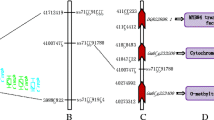
Summary of eQTL and QTL locations detected in the soybean genome. eQTL/ QTL represented by bars were shown on the left of the linkage groups, close to their corresponding markers. The lengths of the bars were proportional to the confidence intervals of the corresponding eQTL/QTL in which the inner line indicates the position of maximum LOD score.
Among the identified eQTLs (Table 3), qPALB2_1 and qPALD2_1 were associated with PAL transcript abundance, and could explain 8.11% and 6.67% of the phenotypic variation, respectively. Eight eQTLs, underlying CHS transcript abundance, were located on six LGs, and could explain 2.07-15.65% of the phenotypic variation. qCHSDla_1 (Satt436-Sat_345, Gm01) was detected with a higher LOD score (8.64) in the regions where cis-elements and CHS family genes were located.
Two eQTLs (qCHSDlb_1, qCHSDlb_2), located in the interval of Satt459 and Satt546, could explain 2.13% and 3.90% of phenotypic variance and overlap with qGCD1b_1. qCHSF_1 (Satt149-Sat_234), associated with CHS and IFS transcript abundance, were overlapped with the marker interval of qGTF_2, and could explain 3.57% of phenotypic variance. qCHSL_1 (Satt278-Sat_134) and qCHSL_2 (Sat_134-Sct_010) were associated with the same SSR marker (Sat_134), and contributed 16.12% and 17.97% of the variation of IFS transcript abundance.
Twelve eQTLs were associated with IFS expression. Of them, qIFSD2_1 (Satt186-Satt226) explained 16.67% of the phenotypic variation. qIFSF_1 (Satt423-Satt569, R2 = 15.84%) shared the same SSR marker Satt569 with other three QTLs (qDZF_2, qGTF_1, qTIF_2). qIFSN shared the same SSR marker (Satt530) with qGCN_1 (Table 3, Figure 1).
Eleven eQTLs were associated with F3H expression (Table 3, Figure 1). Of them, four eQTLs were located on Gm02 (LG Dlb), and explained 5.54-14.32% of the phenotypic variation. qF3HDlb_2 (Sat_135-Sat_096) had higher LOD score and explained 14.32% of the phenotypic variation. qF3HE_1 (R2 = 4.85%) had the same interval (Sat_112- Sat_380) with qGCE_1, qGTE_1 and qTIE_1, meanwhile, qF3HF_1 and qDZF_1 shared the same marker interval (Sat_262- Sat_103) (Figure 1).
Identification of candidate genes underlying the overlapped loci of pQTL and eQTL
Thirty four pQTLs for both individual and total seed isoflavone contents of soybean were compared with eQTLs to identify the overlapped loci. Five eQTL intervals were overlapped with pQTLs, and a total of eleven candidate genes within the overlapped eQTL and pQTL were identified (Table 4). Two genes, C4H (Glyma02g40290.1) and PAL1 (Glyma02g47940.1), were identified on Gm02 (LG D1b) between Satt546-Satt459. CHI (Glyma17g34430.1) and DFR (dihydroflavonol reductase; EC 1.1.1.219) were identified on Gm17 (LG D2) between Satt186-Satt226. Genes encoding 4-coumarate-CoA ligase (EC 6.2.1.12; Glyma13g01080.1/2), FLS (Glyma13g02740.1) and CHS (Glyma13g09640.1) were identified on Gm13 (LG F) between Satt423-Satt569. Additionally, CHS (Glyma13g24200.1) and IFS (Glyma13g09640.1) was found within another eQTL/pQTL interval (Satt149-Sat_234).
Discussion
Soybean isoflavones have been broadly used in food, medicine, cosmetics and animal husbandry [59]. Increasing and decreasing seed isoflavone content will be an important target of soybean breeding. MAS based on genotype selection rather than solely on phenotype selection provides additional power for the selections during soybean breeding [60]. Cultivar ‘Zhongdou 27’ proved to have high-isoflavone content (3,791 μg/g isoflavone in seed) as reported previously [16]. Meng et al. [19] identified two QTL underlying resistance to soybean aphid through leaf isoflavone-mediated antibiosis in soybean cultivar ‘Zhongdou 27’. A number of pQTLs associated with seed isoflavone were identified in multiple environments from cultivar ‘Zhongdou 27’ using 194 SSR markers (accepted by Molecular Biology Reports). Therefore, ‘Zhongdou 27’ should be given more attention as an elite germplasm to improve soybean seed isoflavone concentration, disease and pest resistances.
In our previous studies, some identified QTLs associated with individual/total isoflavone contents showed higher contribution to phenotypic variation. Some specific copies of genes (PAL, CHS, IFS, F3H) in the phenylpropanoid pathway were near or falling into these quantitative trait loci by browsing the reference genome sequence of Williams 82 (http://www.phytozome.net/soybean).
To investigate the regulation mechanism of isoflavone synthetic enzyme genes, the transcript abundances of PAL, CHS, IFS and F3H in the mapping population were examined, and the genomic regions affecting the expression of the TGs were identified using the eQTL methodology [61]. A global microarray eQTL analysis of a limited number of samples can be used for exploring functional and regulatory gene networks and for scanning cis-eQTL, whereas the subsequent analysis of a subset of likely cis-regulated genes by real-time RT-PCR in a larger number of samples may identify QTL region by targeting these positional candidate genes [62]. In this study, real-time PCR reactions were used to analyze the transcript abundance variations of the four TGs in the F5:11 RI lines.
When combined with classical QTL phenotypes, correlation analysis can directly provide an overview of potential genes underlying isoflavone traits [63, 64]. Through the comparison of the transcript abundances of the four TGs (PAL, CHS, IFS and F3H), the parents (‘Zhongdou 27’ and ‘Jiunong 20’) showed different patterns at the R6 stage. This observation was consistent with the previous report by Sarah et al. [65]. Significant correlations between the transcript abundances of TGs and isoflavone contents were found in developing seeds at the R6 stage, indicating that these genes could affect total and individual isoflavone accumulations (Table 2).
Previously, two major QTLs that affect isoflavone content across multiple environments were mapped on Gm05 (LG A1) and Gm08 (LG A2) by Gutierrez et al. [17] and Yang et al. [20], respectively. In the present work, one eQTL qIFSA2_1 (Sat_129-Sat_181) was mapped close to qGCA2_1 on Gm08 (LG A2) (Figure 1, Table 5). This result suggested that qIFSA2_1 might be a cis-enzyme related locus. Some of these identified eQTLs associated with seed isoflavone content did not coincide with the TGs, suggesting that the differences in TGs transcript abundances might be caused by several trans-acting factors [66].
In this study, since the 194 markers were not uniformly distributed, large gaps appeared with low marker density on chromosomes Gm02, 04, 13, 16 and 18, implying that more markers should be developed among these gaps and the authenticity of pQTL or eQTL should be further clarified. Among these gaps, special attention should be paid to eQTL qF3HDlb_2 on chromosome Gm02 and qIFSC1_1 on chromosome Gm04 because of their higher LOD score and contribution to phenotypic variation (Table 3). Overlapped loci of qF3HF_1 and qDZF_1, and genes that fall into this region should also be further clarified with more markers. Consequently, fine mapping on these intervals with more SSR or SNP markers and to determine the authenticity of these loci as well as the underlying genes were extremely essential in the future work.
The analysis of eQTL overlapped with pQTL suggested that the candidate genes or elements among the marker intervals could affect phenotypic traits [49, 67, 68]. Therefore, overlapped loci of eQTLs and pQTLs were analyzed to find the potential candidate genes affecting the accumulation of isoflavone contents in soybean seed. Five eQTL intervals were overlapped with pQTLs according to the comparison of genomic regions between pQTLs and eQTLs (Table 5). These results indicated that some candidate genes or elements in these intervals could regulate the biosynthesis of isoflavone components, and affect their accumulation. Additionally, some eQTLs overlapped with other eQTLs or shared the same markers with pQTLs, suggesting that some candidate genes or elements were located near these loci.
Several genes involved in isoflavone accumulation in soybean seed had been identified [22, 27, 31]. 11 candidate genes falling into the overlapped intervals of pQTL and eQTL were found (Table 4). Bolon et al. [58] identified eQTL for genes with seed-specific expression and discovered striking eQTL hotspots at distinct genomic intervals on chromosome Gm13. A chalcone isomerase (CHI3) and IFS2 gene were located in the same region identified by qGEN13 on Gm13 [11]. Another QTL for GC that encoded PAL and 4CL paralog was also reported on Gm13 [10, 11]. In the present work, seven candidate genes on Gm13 (LG F) were identified, implying that there could be a hotspot of gene cluster that regulated seed isoflavone content on Gm13. Among them, CHS (Glyma13g09640.1) and FLS (Glyma13g02740.1) were identified on three overlapped loci, implying that they could interact or trans-regulate other genes in the phenylpropanoid pathway. Furthermore, PAL1 (Glyma13g20800.1) and IFS (Glyma13g24200.1) paralogs were identified within two overlapped loci. In the marker interval (Satt149-Sat_234) associated with qCHSF_1, qIFSF_2 and qGTF_2, both Glyma13g24200.1 and Glyma13g09640.1 were found to encode CHS and IFS, indicating that they could be the potential candidate genes. It was supposed that Glyma13g09640.1 could interact or trans-regulate the expression of IFS. However, the function of these potential candidate genes should be tested in future works.
Although open questions about the biology and applications of eQTL mapping still exist [69], there are considerable advances in the eQTL studies. Detailed analysis of eQTL combined with cluster analysis of transcript abundance and eventually gene expression patterns could assist map-based cloning of genes underlying these traits. Markers based on underlying genes are also desirable for MAS in soybean breeding programs. The mechanism underlying seed isoflavone synthesis and its accumulation may contribute to the development of marker-assisted selection for soybean cultivars with high or low isoflavone contents.
Conclusions
A total of thirty three eQTLs (thirteen cis-eQTLs and twenty trans-eQTLs) were identified on fifteen chromosomes. Five eQTL intervals were overlapped with pQTLs and a total of eleven candidate genes within the overlapped eQTL and pQTL were identified. These results might be beneficial for the development of marker-assisted selection to breed soybean cultivars with high isoflavone contents.
Methods
Plant materials and growing conditions
The mapping population of 130 F5:11 recombinant inbred (RI) lines were derived through single-seed-descent from the cross between ‘Zhongdou 27’ (developed by the Chinese Academy of Agricultural Sciences, Beijing, China) and ‘Jiunong 20’ (developed by Jilin Academy of Agricultural Sciences, Jilin, China). ‘Zhongdou 27’ contains high individual and total isoflavone (TI) contents in seed (daidzein, DZ, 1,865 μg/g; genistein, GT, 1,614 μg/g; glycitein, GC, 311 μg/g and total isoflavone, TI, 3,791 μg/g), whereas ‘Jiunong 20’ has low individual and TI contents (DZ, 844 μg/g; GT, 1,046 μg/g; GC, 193 μg/g and TI, 2,061 μg/g).
To detect eQTL, the parents and the 130 F5:11 RI lines were planted at Harbin, Heilongjiang Province, China, in 2011. Randomized complete block designs were used for all experiments with rows 3 m long, 0.65 m apart, and a space of 6 cm between plants. Mature and immature seeds in the reproductive stages (from soybean growth stage R3 to R8) [70] were harvested from a bulked sample collected from three plants in each plot. These samples were quantified for individual and total seed isoflavone contents and transcript abundances.
Isoflavone extraction and quantification
Approximately 150 g of soybean seed samples were ground to a fine power using a commercial coffee grinder. Isoflavones were extracted from flour and separated using HPLC as described previously [16]. Measurements were done as micrograms of isoflavone per gram of seeds plus and minus the standard deviations (μg/g ± SD).
Synthesis of cDNA, Real-Time PCR and data collection
To investigate the expressions of four TGs, total RNA was isolated from soybean seed samples from R3 to R8 stages using plant RNA purification reagent Kit (D9108A, TaKaRa, Japan). RNAs were transcribed to cDNA using the first strand DNA synthesis reagent Kit (D6110A, TaKaRa, Otsu, Shiga, Japan). Four TGs (PAL, GenBank accession: GQ220305; CHS, GenBank accession: EU526827; IFS, GenBank accession: FJ770473 and F3H, GenBank accession: AY595420) in the phenylpropanoid pathway, were selected to analyze the transcript abundance variations in the F5:11 RI line population. These four TGs were analyzed by real-time PCR (Kit DRR081A, TaKaRa, Japan). Gene-specific primers for expression analysis of the four TGs were listed in Table 6. Primer specificity was confirmed based on each primer pair sequence against soybean genome sequences by BLASTing (http://www.phytozome.net/soybean) using the BLASTN algorithm. Moreover, through the BLASTN of the sequences of the TGs, PAL2 (located on Gm10 (LG O)) of the PAL gene family, CHS8 (located on Gm11 (LG B1)) of the CHS gene family, IFS1 (located on Gm07 (LG M)) of the IFS gene family, and F3H1 and F3H2 (located on Gm02 (LG D1b)) of the F3H gene family were amplified [11].
PCR amplification was performed as follows: 95°C for 60 s, followed by 40 cycles of 95°C for 11 s, 60°C for 12 s and 72°C for 18 s. The soybean actin4 (GenBank accession: AF049106) gene was used as a reference to quantify the expression levels of the target genes [71]. Three replicates for each reaction were performed. The relative transcript abundance of TGs in different samples was calculated using 2-ΔΔCt method [72], defined as: ΔCt = Ct (target) – Ct (actin). Pearson correlations between total/individual isoflavone contents and the expression of the four TGs in F5:11 RILs were evaluated using SAS 8.2 (Cary, NC, USA) [73].
Identification of genomic region of target genes
The whole genome sequence Glyma1 assembly for Williams 82 [74] provided a powerful tool for interrogating QTL data. Previously reported genes for isoflavone biosynthesis [75] were used in BLAST searches against the whole genome sequence to identify homologous regions in the genome with assigned or putative functions. All twenty soybean chromosomes have regions sharing a high percentage of homology with genes of known function in the phenylpropanoid pathway [11]. The coding regions of TGs were compared with genome of Williams 82 through BLAST (E-value ≤ 1.0E-05, http://www.phytozome.net/soybean) to identify homologous regions.
eQTL analysis
In previous work, fifteen QTL underlying seed isoflavone contents of soybean were identified based on RI line populations derived from a cross between ‘Zhongdou 27’ (high isoflavone) and ‘Jiunong 20’ (low isoflavone) through a genetic linkage map including 99 SSR markers [16]. Another 95 SSR markers were added to the map of Zeng et al. [16] to identify novel phenotypic QTLs (pQTLs) associated with seed isoflavone contents of soybean (accepted by Molecular Biology Reports). In this study, 194 polymorphic markers were assembled onto the 20 linkage groups (LGs) by Mapmaker 3.0b with the Kosambi mapping function [76]. WinQTLCart2.1 [77] was used to detect eQTL between marker intervals by 1,000 permutations at significance (P ≤ 0.05). The genetic linkage map was constructed using Mapchart 2.1 [78]. The nomenclature of the eQTLs/pQTLs included four parts following the recommendations of the Soybean Germplasm Coordination Committee. For example, qCHSF_1, q, CHS, F and 1 represent eQTL, trait (CHS), linkage group name and eQTL order in the linkage group, respectively.
Identification of candidate genes underlying overlapped loci of pQTL and eQTL
Coincident genetic locations of eQTL and pQTL may be available to identify important regulatory genes underlying traits, and lead to the identification of molecular mechanisms [49, 67, 68]. Previous studies have combined eQTL and pQTL mapping to gain insight into regulatory pathways involved in determining phenotypic traits [49, 68, 79–81]. eQTL located in the same marker intervals of pQTL might contribute to significant phenotypic variations [49, 67, 68]. In this study, thirty four phenotypic QTL (pQTL) identified with the 194 SSR markers were compared with eQTL to identify overlapped loci. Genetic map positions were estimated by identifying the nearest flanking SSR markers using the genome browser (http://www.soybase.org). The candidate genes underlying overlapped loci of pQTL and eQTL were identified by browsing after using BLAST search of flanking markers against the whole genome sequence of Williams 82 (available at: http://www.phytozome.net/soybean).
References
Tsukamoto C, Shimada S, Igita K, Kudou S, Kokubun M, Okubo K, Kitamura K: Factors affecting isoflavone content in soybean seeds: Changes in isoflavones, saponins, and composition of fatty acids at different temperatures during seed development. J Agric Food Chem. 1995, 43: 1184-1192.
Munro IC, Harwood M, Hlywka JJ, Stephen AM, Doull J, Flamm WG, Adlercreutz H: Soy isoflavones: a safety review. Nutr Rev. 2003, 61: 1-33.
Cederroth CR, Nef S: Soy, phytoestrogens and metabolism: a review. Mol Cell Endocrinol. 2009, 304: 30-42.
Benhamou N, Nicole M: Cell biology of plant immunization against microbial infection: the potential of induced resistance in controlling plant diseases. Plant Physiol Biochem. 1999, 37: 703-719.
Subramanian S, Graham MY, Yu O, Graham TL: RNA interference of soybean isoflavone synthase genes leads to silencing in tissues distal to the transformation site and to enhanced susceptibility to Phytophthora sojae. Plant Physiol. 2005, 137: 1345-1353.
Subramanian S, Stacey G, Yu O: Distinct, crucial roles of flavonoids during legume nodulation. Trends Plant Sci. 2007, 12: 282-285.
Lozovaya VV, Lygin AV, Zernova OVLISX, Hartman GL, Widholm M: Isoflavonoid accumulation in soybean hairy roots upon treatment with Fusarium solani. Plant Physiol Biochem. 2004, 42: 671-679.
Meksem K, Njiti VN, Banz WJ, Iqbal MJ, Kassem MM, Hyten DL, Yuang J, Winters TA, Lightfoot DA: Genomic regions that underlie soybean seed isoflavone content. J Biomed Biotechnol. 2001, 1 (1): 38-44.
Kassem MA, Meksem K, Iqbal MJ, Njiti VN, Banz WJ, Winters TA, Wood A, Lightfoot DA: Definition of soybean genomic regions that control seed phytoestrogen amounts. J Biomed Biotechnol. 2004, 1: 52-60.
Primomo VS, Poysa V, Ablett GR, Jackson CJ, Gijzen M, Rajcan I: Mapping QTL for individual and total isoflavone content in soybean seeds. Crop Sci. 2005, 45: 2454-2462.
Gutierrez GJJ, Wu XL, Gillman JD, Lee JD, Zhong R, Yu O, Shannon G, Ellersieck M, Nguyen HT, Sleper DA: Intricate environment-modulated genetic networks control isoflavone accumulation in soybean seeds. BMC Plant Biol. 2010, 10: 105-120.
Hoeck JA, Fehr WR, Murphy PA, Welke GA: Influence of genotype and environment on isoflavone contents of soybean. Crop Sci. 2000, 40: 48-51.
Dhaubhadel S, McGarvey BD, Williams R, Gijzen M: Isoflavonoid biosynthesis and accumulation in developing soybean seeds. Plant Mol Biol. 2003, 53: 733-743.
Murphy SE, Lee EA, Woodrow L, Seguin P, Kumar J, Rajcan I, Ablett GR: Genotype × Environment interaction and stability for isoflavone content in soybean. Crop Sci. 2009, 49: 1313-1321.
Kassem MA, Shultz J, Meksem K, Cho Y, Wood AJ, Iqbal MJ, Lightfoot DA: An updated ‘Essex’ by ‘Forrest’ linkage map and first composite interval map of QTL underlying six soybean traits. Theor Appl Genet. 2006, 113: 1015-1026.
Zeng G, Li D, Han Y, Teng W, Wang J, Qiu L, Li W: Identification of QTL underlying isoflavone contents in soybean seeds among multiple environments. Theor Appl Genet. 2009, 118: 1455-1463.
Gutierrez GJJ, Vuong TD, Zhong R, Yu O, Lee JD, Shannon G, Ellersieck M, Nguyen HT, Sleper DA: Major locus and other novel additive and epistatic loci involved in modulation of isoflavone concentration in soybean seeds. Theor Appl Genet. 2011, 123: 1375-1385.
Liang HZ, Yu YL, Wang SF, Lian Y, Wang TF, Wei YL, Gong PT, Liu XY, Fang XJ, Zhang MC: QTL mapping of isoflavone, oil and protein contents in soybean (Glycine max L. Merr.). Agric Sci China. 2010, 9: 1108-1116.
Meng FL, Han YP, Teng WL, Li YG, Li WB: QTL underlying the resistance to soybean aphid (Aphis glycines Matsumura) through isoflavone-mediated antibiosis in soybean cultivar ‘Zhongdou 27’. Theor Appl Genet. 2011, 123: 1459-1465.
Yang K, Moon JK, Jeong N, Chun HK, Kang ST, Back K, Jeong SC: Novel major quantitative trait loci regulating the content of isoflavone in soybean seeds. Genes Genom. 2011, 33: 685-692.
Zhang JY, Ge YN, Sun JM, Han FX, Yu FK, Yan SR, Yang H: Identification of QTLs for major isoflavone components among multiple environments in soybean seeds. Sci Agric Sin. 2012, 45: 3909-3920.
Akond M, Richard B, Ragin B, Herrera H, Kaodi U, Akbay C, Kantartzi SK, Njiti V, Barakat A, Meksem K, Lightfoot DA, Kassem MA: Additional quantitative trait loci and candidate genes for seed isoflavone content in soybean. J Agric Sci. 2013, 5: 20-33.
Akond M, Liu SM, Kantartzi SK, Meksem K, Bellaloui N, Lightfoot DA, Yuan JZ, Wang DC, Kassem MA: Quantitative trait loci for seed isoflavone contents in ‘MD96-5722’ by ‘Spencer’ recombinant inbred lines of soybean. J Agric Food Chem. 2014, 62: 1464-1468.
Wellmann E: UV dose-dependent induction of enzymes related to flavonoid biosynthesis in cell suspension cultures of parsley. FEBS Lett. 1975, 51: 105-107.
Elio GWM,CH, Arjen J, Arnaud G: Modification of flavonoid biosynthesis in crop plants. Phytochemistry. 2004, 65: 2631-2648.
Austin MB, Noel JP: The chalcone synthase superfamily of type III polyketide synthases. Nat Prod Rep. 2003, 20: 79-110.
Du H, Huang YB, Tang YX: Genetic and metabolic engineering of isoflavonoid biosynthesis. Appl Microbiol Biotechnol. 2010, 86: 1293-1312.
Forkmann G, Martens S: Metabolic engineering and applications of flavonoids. Curr Opin Biotechnol. 2001, 12: 155-160.
Joung JY, Kasthuri M, Park JY, Kang WJ, Kim HS, Yoon BS, Joung H, Jeon JH: An overexpression of chalcone reductase of Pueraria montana var. lobata alters biosynthesis of anthocyanin and 50-deoxyflavonoids in transgenic tobacco. Biochem Biophys Res Commun. 2003, 3003: 326-331.
Jung W, Yu O, Lau SC, O’Keefe DP, Odell J, Fader G, McGonigle B: Identification and expression of isoflavone synthase, the key enzyme for biosynthesis of isoflavones in legumes. Nat Biotechnol. 2000, 18: 208-212.
Hao C, Oliver Y, Deyue Y: Polymorphisms of IFS1 and IFS2 gene are associated with isoflavone concentrations in soybean seeds. Plant Sci. 2008, 175: 505-512.
Britsch L, Dedio J, Saedler H, Forkmann G: Molecular characterization of flavanone 3 beta-hydroxylases. Consensus sequence, comparison with related enzymes and the role of conserved histidine residues. Eur J Biochem. 1993, 217: 745-754.
Cheng H, Wang J, Chu S, Yan H-L, Yu D: Diversifying selection on flavanone 3-hydroxylase and isoflavone synthase genes in cultivated soybean and its wild progenitors. PLoS One. 2013, 8: e54154-
Yu O, McGonigle B: Metabolic engineering of isofavone biosynthesis. Adv Agron. 2005, 86: 147-190.
Yu O, Shi J, Hession AO, Maxwell CA, McGonigle B, Odell JT: Metabolic engineering to increase isoflavone biosynthesis in soybean seed. Phytochemistry. 2003, 63: 753-763.
Yin Z, Meng F, Song H, Wang X, Xu X, Yu D: Expression quantitative trait loci analysis of two genes encoding RUBISCO activase in soybean. Plant Physiol. 2010, 152: 1625-1637.
Jansen R, Nap J: Genetical genomics: the added value from segregation. Trends Genet. 2001, 17: 388-391.
Cheung VG, Conlin LK, Weber TM, Arcaro M, Jen KY, Morley M, Spielman RS: Natural variation in human gene expression assessed in lymphoblastoid cells. Nat Genet. 2003, 33: 422-425.
Goring HH, Curran JE, Johnson MP, Dyer TD, Charlesworth J, Cole SA, Jowett JBM, Abraham LJ, Rainwater DL, Comuzzie AG, Mahaney MC, Almasy L, MacCluer JW, Kissebah AH, Collier GR, Moses EK, Blangero J: Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nat Genet. 2007, 39: 1208-1216.
Sasayama D, Hori H, Nakamura S, Miyata R, Teraishi T, Hattori K, Ota M, Yamamoto N, Higuchi T, Amano N, Kunugi H: Identification of single nucleotide polymorphisms regulating peripheral blood mRNA expression with Genome-Wide Significance: an eQTL study in the Japanese population. PLoS One. 2013, 8: e54967-
DeCook R, Lall S, Nettleton D, Howell SH: Genetic regulation of gene expression during shoot development in Arabidopsis. Genetics. 2006, 172: 1155-1164.
Jordan MC, Somers DJ, Banks TW: Identifying regions of the wheat genome controlling seed development by mapping expression quantitative trait loci. Plant Biotechnol J. 2007, 5: 442-453.
Potokina E, Druka A, Luo Z, Wise R, Waugh R, Kearsey M: Gene expression quantitative trait locus analysis of 16,000 barley genes reveals a complex pattern of genome-wide transcriptional regulation. Plant J. 2008, 53: 90-101.
Chen X, Hackett CA, Niks RE, Hedley PE, Booth C, Druka A, Marcel TC, Vels A, Bayer M, Milne I, Morris J, Ramsay L, Marshall D, Cardle L, Waugh R: An eQTL analysis of partial resistance to Puccinia hordeii in barley. PLoS One. 2010, 5 (1): e8598-
Brem RB, Yvert G, Clinton R, Kruglyak L: Genetic dissection of transcriptional regulation in budding yeast. Science. 2002, 296: 752-755.
Brem RB, Kruglyak L: The landscape of genetic complexity across 5,700 gene expression traits in yeast. Proc Natl Acad Sci U S A. 2005, 102: 1572-1577.
Li Y, Alvarez OA, Gutteling EW, Tijsterman M, Fu J, Riksen JAG, Hazendonk E, Prins P, Plasterk RHA, Jansen RC, Breitling R, Kammenga JE: Mapping determinants of gene expression plasticity by genetical genomics in C. elegans. PLoS Genet. 2006, 2: 2155-2161.
Hughes KA, Ayroles JF, Reedy MM, Drnevich JM, Rowe KC, Ruedi EA, Caceres CE, Paige KN: Segregating variation in the transcriptome: cis regulation and additivity of effects. Genetics. 2006, 173: 1347-1355.
Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, Colinayo V, Ruff TG, Milligan SB, Lamb JR, Cavet G, Linsley PS, Mao M, Stoughton RB, Friend SH: Genetics of gene expression surveyed in maize, mouse and man. Nature. 2003, 22: 297-302.
Ghazalpour A, Doss S, Zhang B, Wang S, Plaisier S, Castellanos R, Brozell A, Schadt EE, Drake TA, Lusis AJ, Horvath S: Integrating genetic and net-work analysis to characterize genes related to mouse weight. PLoS Genet. 2006, 2: e130-
Çinar MU, Fan H, Neuhoff C, Groβe-Brinkhaus C: eQTL Analysis and association of MYF6 mRNA expression with meat quality traits in pigs. Kafkas Universitesi Veteriner Fakultesi Dergisi. 2012, 18: 235-242.
Hubner N, Wallace CA, Zimdahl H, Petretto E, Schulz H, Maciver F, Mueller M, Hummel O, Monti J, Zidek V, Musilova A, Kren V, Causton H, Game L, Born G, Schmidt S, Muller A, Cook SA, Kurtz TW, Whittaker J, Pravenec M, Aitman TJ: Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nat Genet. 2005, 37: 243-253.
Kliebenstein D: Quantitative Genomics: analyzing intraspecific variation using global gene expression polymorphisms or eQTL. Annu Rev Plant Biol. 2008, 60: 93-114.
Doss S, Schadt EE, Drake TA, Lusis AJ: Cis-acting expression quantitative trait loci in mice. Genome Res. 2005, 15: 681-691.
Gatti DM, Harrill AH, Wright FA, Threadgill DW, Rusyn I: Replication and narrowing of gene expression quantitative trait loci using inbred mice. Mamm Genom. 2009, 20: 437-446.
Yin Z, Meng F, Song H, Wang X, Chao M, Zhang G, Xu X, Deng D, Yu D: GmFtsH9 expression correlates with in vivo Photosystem II function: Chlorophyll a fluorescence transient analysis and eQTL mapping in soybean. Planta. 2011, 234: 815-827.
Song H, Yin Z, Chao M, Ning L, Zhang D, Yu D: Functional properties and expression quantitative trait loci for phosphate transporter GmPT1 in soybean. Plant Cell Environ. 2014, 37: 462-472.
Bolon YT, Hyten DL, Orf JH, Vance CP, Muehlbauer GJ: eQTL networks reveal complex genetic architecture in the immature soybean seed. Plant Genom. 2014, 7: 1-14.
Brouns F: Soya isoflavones: a new and promising ingredient for the health food sector. Food Res Int. 2002, 35: 187-193.
Bernardo R: Molecular markers and selection for complex traits in plants: learning from the last 20 years. Crop Sci. 2008, 48: 1649-1664.
Sladek R, Hudson TJ: Elucidating cis-and trans-regulatory variation using genetical genomics. Trends Genet. 2006, 22: 245-250.
Ponsuksili S, Murani E, Phatsara C, Schwerin M, Schellander K, Wimmers K: Expression quantitative trait loci analysis of genes in porcine muscle by quantitative real-time RT-PCR compared to microarray data. Heredity. 2010, 105: 309-317.
Druka A, Potokina E, Luo ZW, Bonar N, Druka I, Zhang L, Marshall DF, Steffenson BJ, Close TJ, Wise RP, Kleinhofs A, Williams RW, Kearsey MJ, Waugh R: Exploiting regulatory variation to identify genes and loci underlying quantitative traits in barley. Theor Appl Genet. 2008, 117: 261-272.
Holloway B, Li B: Expression QTLs: applications for crop improvement. Mol Breed. 2010, 26: 381-391.
Sarah IJ, Delkin OG, Lila OV: Flux of transcript patterns during soybean seed development. BMC Genom. 2010, 11: 136-150.
Liu P, Wang CM, Li L, Sun F, Yue GH: Mapping QTL for oil traits and eQTL for oleosin genes in jatropha. BMC Plant Biol. 2011, 11: 132-140.
Wang J, Yu H, Xie W, Xing Y, Yu S, Xu C, Zhang Q: A global analysis of QTLs for expression variations in rice shoots at the early seedling stage. Plant J. 2010, 63: 1063-1074.
Chen X, Guo W, Liu B, Zhang Y, Song X, Cheng Y, Zhang L, Zhang T: Molecular mechanisms of fiber differential development between G. barbadense and G. hirsutum revealed by genetical genomics. PLoS One. 2012, 7: e30056-
Gilad Y, Rifkin SA, Pritchard JK: Revealing the architecture of gene regulation: the promise of eQTL studies. Trends Genet. 2008, 24: 408-415.
Fehr WR, Caviness CE: Stages of Soybean Development. Special Report 80, Cooperative Extension Service, Agriculture and Home Economic Experiment Station. 1977, Ames, Iowa: Iowa State University, 1-11.
Iqbal MJ, Yaegashi S, Njiti VN, Ahsan R, Cryder KL, Lightfoot DA: Gene locus pyramids alter transcript abundance in soybean roots inoculated with Fusarium solani f.sp. glycines. Mol Genet Genomic. 2002, 268: 407-417.
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001, 25: 402-408.
Zhao G, Wang J, Han Y, Teng W, Sun G, Li W: Identification of QTL underlying the resistance of soybean to pod borer, Leguminivora glycinivorella (Mats. Obraztsov), and correlations with plant, pod and seed traits. Euphytica. 2008, 164: 275-282.
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L, Gill N: Genome sequence of the palaeopolyploid soybean. Nature. 2010, 463: 178-183.
Zhang J, Yu O: Metabolic engineering of isoflavone biosynthesis in seeds. In Modification of seed composition to promote health and nutrition. Agronomy Monograph Series. Edited by: Krishnan H. 2009, 151-177.
Lander ES, Green P, Abrahamson J, Barlow A, Daly M, Lincoln S, Newburg L: Mapmaker: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics. 1987, 1: 174-181.
Zeng Z: Theoretical basis of separation of multiple linked gene effects on mapping quantitative trait loci. Proc Natl Acad Sci U S A. 1993, 90: 10972-10976.
Voorrips R: MapChart: software for the graphical presentation of linkage maps and QTL. J Hered. 2002, 93: 77-
Mehrabian M, Allayee H, Stockton J, Lum PY, Drake TA, Castellani LW, Suh M, Armour C, Edwards S, Lamb J, Lusis AJ, Schadt EE: Integrating genotypic and expression data in a segregating mouse population to identify 5-lipoxygenase as a susceptibility gene for obesity and bone traits. Nat Genet. 2005, 37: 1224-1233.
Sonderby IE, Hansen BG, Bjarnholt N, Ticconi C, Halkier BA, Kliebenstein DJ: A systems biology approach identifies a R2R3 MYB gene subfamily with distinct and overlapping functions in regulation of aliphatic glucosinolates. PLoS One. 2005, 2: e1322-
Moscou MJ, Lauter N, Steffenson B, Wise RP: Quantitative and qualitative stem rust resistance factors in barley are associated with transcriptional suppression of defense regulons. PLoS Genet. 2011, 7: e1002208-
Acknowledgements
This study was conducted in Soybean Research & Development Center (CARS) and Collaborative Innovation Center of Grain Production Capacity Improvement in Heilongjiang Province, financially supported by National Core Soybean Genetic Engineering Project (2014ZX08004-003), National 863 Project (2013AA102602), National 973 Project (2012CB126311), National 863 Project (2012AA101106-1-9), and Provincial/National Education Ministry for the team of soybean molecular design.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WL conceived the project and its components, contributed to the original concept of the project. YW and YH performed the SSR marker screening, eQTL analyses and paper writing. WT, XZ, YL, LW and DL collected samples and performed the phenotype analyses. All authors read and approved the final manuscript.
Yan Wang, Yingpeng Han contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Wang, Y., Han, Y., Teng, W. et al. Expression quantitative trait loci infer the regulation of isoflavone accumulation in soybean (Glycine max L. Merr.) seed. BMC Genomics 15, 680 (2014). https://doi.org/10.1186/1471-2164-15-680
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-680