
Abstract
Odorant-binding proteins (OBPs) in insects bind to volatile chemical cues that are important in regulating insect behavior. It is hypothesized that OBPs bind with specificity to certain volatiles and may help in transport and delivery to odorant receptors (ORs), and may help in buffering the olfactory response and aid the insect in various behaviors. Honeybees are eusocial insects that perceive olfactory cues and strongly rely on them to perform complex olfactory behaviors. Here, we have identified and annotated odorant-binding proteins and few chemosensory proteins from the genome of the dwarf honey bee, Apis florea, using an exhaustive homology-based bioinformatic pipeline and analyzed the evolutionary relationships between the OBP subfamilies. Our study confirms that the Minus-C subfamily in honey bees has diverged from the Classic subfamily of odorant-binding proteins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Insects are a diverse class of Arthropods with a highly sensitive olfactory system. Olfactory information helps in mate selection, mating and oviposition, foraging for food, and social behavior (Hildebrand and Shepherd 1997). Odorant-binding proteins (OBPs) are abundantly present in the sensillar lymph of insects with the presence of at least 50 OBP genes reported in some species like Drosophila (Hekmat-Scafe et al. 2002), and in the nasal mucus of many vertebrate animal species (Bianchet et al. 1996; Loebel et al. 2000; Mastrogiacomo et al. 2014; Scaloni et al. 2001; Tegoni et al. 1996; White et al. 2009; Zhu et al. 2017; Manikkaraja and Bhavika et al. 2020). Despite their abundance and diversity, the role of OBPs in olfactory coding is yet to be completely explored (Larter et al. 2016).
Insect OBPs are small, soluble globular proteins, 10–30 kDa, that are further characterized by alpha-richness, and the presence of six highly conserved cysteine residues (C1–C6) with conserved disulfide spacing (Vogt et al. 1981; Pelosi and Maida, 1990) that stabilize its tertiary structure. Alpha helix-rich OBPs found in insects do not show structural homology with vertebrate OBPs, characterized with a classical lipocalin fold (Flower 1996). It has been hypothesized that OBPs bind to ligands and solubilize them to aid transport and delivery towards odorant receptors.
Genome-wide surveys to identify odorant-binding proteins in insect orders have been previously performed for various insect species in existing literature. Previous studies have predicted the presence of odorant-binding proteins in various species including Apis mellifera (order: Hymenoptera) (Forêt and Maleszka 2006), Drosophila melanogaster (order: Diptera) (Hekmat-Scafe et al. 2002; Graham and Davies 2002), Anopheles gambiae (order: Diptera) (Manoharan et al. 2013), and Periplaneta americana (order: Blattodea) (He et al. 2017) using homology-based bioinformatic approaches as a typical start point. Previous work in our laboratory (Karpe et al. 2016) has identified odorant receptors (ORs) in Apis florea using an exhaustive genomic pipeline. In order to complement the search of ORs (Karpe et al. 2016) towards a better understanding of odor coding (Missbach et al. 2015), this study investigated odorant-binding proteins (OBPs) in Apis florea.
Apis florea or the red dwarf honey bee exhibits the complex behavior of eusociality, where there is a reproductive division of labor within a colony that comprises a female queen, male drones, and female worker bees. While worker bees perform important tasks such as foraging, guarding the colony hive, maintenance, and other diverse tasks for the colony, the queen and drone perform reproductive roles (Page and Robinson 1991).
Members of the species exhibit haplodiploidy (Halling et al. 2001) system of genetic inheritance, where the males in this species are haploid, possessing half the number of chromosomes as diploid females. Apis florea is geographically distributed with a preference for warm climate (Otis 1991) in regions such as mainland Asia, the southern border of the Himalayas, the plateau of Iran, Oman and Vietnam, southeast China, and peninsular Malaysia (Hepburn et al. 2005; Oldroyd and Nanork 2009; Moritz et al. 2010) and display open nesting typically on low-lying tree branches in shaded regions (Wongsiri et al. 1997; Hepburn et al. 2005). Apis florea are important pollinators of tropical and ornamental plants as well as agricultural crops. They primarily feed on pollen and nectar from flowering plants. Like other honeybees, the body of Apis florea is studded with various types of sensilla among which olfactory sensilla (sensilla basiconica and sensilla chaetica) are prominent structures (Gupta 1986, 1992). The antenna of the insect is typically the main site for olfactory receptors (Wigglesworth 1965). The antennae of Apis florea harbor hair-like sensillae trichodea types I, II, III, IV, sensilla basiconica, sensilla placodea, and sensilla ampullaceal (Gupta 1992; Kumar et al. 2014; Suwannapong et al. 2011).
Insect OBPs, although highly divergent, are classified on the basis of conserved cysteine signature into Classic (six cysteines), Minus-C (loss of two conserved cysteines), Plus-C (additional cysteine residues and one proline) (Zhou et al. 2004), and atypical (~ 10 cysteines and long C-terminus) (Hekmat-Scafe et al. 2002; Xu et al. 2003) and Dimer OBPs (two cysteine signatures). Rapid identification of repertoires of putative OBPs across various insect genomes has been suggestive of the idea that the ecological niche of an insect species may correlate with an abundance of OBPs and social behavior (Zhou et al. 2020). While reference Dipteran fruitfly Drosophila melanogaster and Japanese encephalitis vector Culex quinquefasciatus have been found to have 51 and 110 putative OBPs respectively (Hekmat-Scafe et al. 2002; Manoharan et al. 2013), previous studies in Hymenopteran OBPs have also found species-specific differences in OBPs including 21 OBPs in eusocial Apis mellifera (Foret et al. 2006), 7 OBPs in fig wasp Ceratosolen solmsi (Wang et al. 2014) that lives in closed spaces, and 90 in diamondback moth Plutella xylostella (Vieira et al. 2012) that lives in open spaces. Using Apis mellifera as a closely related reference genome and a revised annotation of Apis mellifera OBPs, we thus investigated the identification, annotation, and subfamily-based classification of putative OBPs from the genome of Apis florea and examined their evolutionary relationships using in silico approaches (Karpe et al. 2017; Mam and Sowdhamini, 2020).
2 Materials and methods
2.1 Preparing query dataset from Apis mellifera OBPs
The genome of Apis florea, Aflo_1.1 (Fouks et al. 2021), was obtained from the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov).
In order to have a standard query set from Apis mellifera, overlapping and unique hits from three sources were retained in one final dataset. Unique hits were those that were not reciprocal best hits across datasets, and further depending on sequence identity were labelled as isoforms or distinct. Based on homology with the final AmelOBP dataset, AfloOBPs were identified, scored, and annotated (explained in detail in later sections).
AmelOBPs were pooled from the NCBI non-redundant protein database (29 putative AmelOBPs) and a previous study (Foret et al. 2007; 21 AmelOBPs) to obtain a filtered set of query protein sequences. The filtered dataset contained sequences that were common to both or unique to either dataset (Supplementary Data). Reciprocal homology was performed using the filtered query set obtained and AmelOBPs from a recent study (Vieira and Rozas 2011; 21 AmelOBPs). An e-value cutoff of e− 10 was used. The resultant matches as well as unmatched OBPs (putative OBPs with no reciprocal hit; 10 protein sequences) resulted in a final dataset of annotated AmelOBPs (Supplementary Table 1).
2.2 Preparing query dataset from Insecta OBPs
Protein sequences annotated as OBPs were obtained for organisms within Insecta from literature (Supplementary Tables 2 and 3). A non-redundant dataset was prepared by using CD-hit (Li and Godzik, 2006) with a filtering threshold of 95% sequence identity.
2.3 Query protein to subject genome alignments
Genomic alignments were obtained using Exonerate (Slater and Birney 2005) with intron sizes of 500, 2000, 5000, and 10,000 respectively with BLOSUM62 (Henikoff and Henikoff 1992) as the substitution matrix. In order to identify phylogenetically distant orthologs, PAM250 (Dayhoff et al. 1978) was also used as a substitution matrix.
The genomic alignments were processed as per the methodology in a previous in-house study from a lab (Karpe et al. 2016, 2017, 2021). The pipeline involves thoroughly scanning and scoring alignments to the genome based on length, degree of similarity, and the best match of the scaffold location in the subject genome to the query sequence. The unique set of genomic alignments was then processed further to translate amino acids from corresponding in-frame codons. The resultant set of gene models and protein sequences were also manually corrected for missing start and stop codons and missing N-terminal and C-terminal amino acids, and annotated as “complete,” “partial,” or “pseudogene.”
For the purpose of further evolutionary analysis, only Apis florea OBPs obtained from Apis mellifera as the query (Sect. 2.1) have been discussed (Supplementary Table 4). The results of Apis florea OBPs obtained by querying OBPs in other insect orders (Sect. 2.2) have been provided as supplementary information (Supplementary Table 5).
2.4 Homology-based validation and nomenclature
The predicted Apis florea OBPs (AfloOBP) were subjected to reciprocal homology with our manually curated AmelOBP dataset, as explained above. The final dataset of predicted AfloOBPs comprised resultant matches as well as unique sequences with no corresponding reciprocal hits found in the AmelOBP dataset. The AfloOBP predicted protein sequence dataset was thus annotated with respect to AmelOBP homolog, if present as well as its status as “complete” or “partial.”
2.5 Secondary structure prediction
The secondary structure of the protein sequences was predicted using neural network-based PSIPRED v3.2 (Buchan et al. 2013).
2.6 Detection of signal peptide and subcellular localization
N-terminal signal peptide was detected using SignalP 4.1 (Nielsen et al. 1997; Petersen et al. 2011) and SignalP 6.0 (Teufel et al. 2022). This algorithm uses neural networks and Hidden Markov Models to determine signal peptides in each protein sequence. The predicted signal peptide for a given sequence was cleaved off and the “mature” sequence was used for multiple sequence alignment and phylogeny. Prediction of subcellular localization was performed through DeepLoc (Armenteros et al. 2017), an algorithm based on deep neural networks.
2.7 Preparing dataset of insect OBPs for rooted and unrooted phylogeny
In order to prepare an outgroup for the rooted phylogeny, annotated chemosensory proteins of Apis mellifera (AmelCSPs) were obtained from a previous study (Forêt et al. 2007), namely, AmelCSP1, AmelCSP2, AmelCSP3, AmelCSP4, AmelCSP5, and AmelCSP6.
In order to construct the phylogeny, protein sequences of OBPs from 11 insect orders from representative insect species were obtained from previous literature and UniProt (The UniProt Consortium 2019) database. The insect orders, corresponding species, and the number of species-specific OBPs have been tabulated as in Supplementary Table 1.
2.8 Structure-based sequence alignment and phylogenetic analysis
A structure-based seed template was obtained from the PASS2.5 database (Gandhimathi et al. 2012) with the SCOP ID of the fold as 47,565. PASS2 is a database of alignments of proteins organized as structural superfamilies. Such structure-guided alignments (seed templates) are useful for guiding sequence alignments of protein families that are diverse in sequence but are conserved at the level of their structures, e.g., insect OBPs. After the removal of signal peptides, the dataset of “mature” AfloOBP sequences was aligned against the seed template using the G-INS-i algorithm, BLOSUM 30 substitution matrix, and 1000 iterations in MAFFT (Katoh et al. 2002, 2013). The output of the multiple sequence alignment was checked for the best-fit model of evolution using ProtTest v.3.4.2 (Darriba et al. 2011; Guindon and Gascuel 2003) as determined by the AIC and BIC scores. Phylogeny was constructed with RAxML (Stamatakis 2006, 2014) using the maximum likelihood method with 1000 bootstraps and LG matrix (Le and Gascuel 2008) of amino substitution, the proportion of invariant sites, and the gamma rate heterogeneity (“LG + I + G”) model of evolution.
Tips labelled as seed are structure-based templates of insect OBPs derived from the PASS 2.5 database (Gandhimathi et al. 2012). They were used to verify the clustering observed in the sequence-based phylogeny, as insect OBPs are known to have low sequence identity among themselves. A seed tip label contains the following information in this order: (a) abbreviated form of the insect species, (b) letter “d,” (c) PDB ID, and (d) PDB chain ID.
The phylogenetic tree was visualized and annotated using iTOL (Letunic and Bork 2006, 2016).
2.9 Validation of gene models using RNA-seq exon data from NCBI
Gene models that were obtained through our in-house in silico homology-based pipeline and those obtained from literature (Fouks et al. 2021) were validated and/or corrected using publicly available RNA-seq exon data from the NCBI Apis florea Annotation Release 102.
3 Results and discussion
We filtered and re-annotated OBPs from closely related reference genome Apis mellifera using a homology-based approach (see Sect. 2). The final dataset (Supplementary Table 1) comprised 25 AmelOBP protein sequences.
A genome-wide survey of Apis florea using these AmelOBPs revealed 22 novel OBP protein sequences with 15 complete and 7 partial sequences either towards the N-terminus, C-terminus, or both with an average exon number of 5 (Supplementary Tables 4, 6). A parallel study has recently provided annotation of 22 OBPs in Apis florea (Fouks et al. 2021).
We also checked whether the addition of OBPs from other insects while performing a genome-wide survey in A. florea would increase the number of identified AfloOBP genes. For this purpose, our final query dataset of AmelOBPs was used as input against several Hymenopteran genomes (Sect. 2; Supplementary Table 5) to obtain gene models and predict OBPs in each such genome using the methodology detailed in Sect. 2.3. Here, through an extensive filtering strategy, we identified 30 putative OBPs orthologous to other social bees (Apis dorsata, Apis cerana, Melipona quadrifasciata), facultatively social bee Eufrisea mexicana, solitary bee (Megachile rotundata), social wasp (Polistes dominula), and bumblebees (Bombus terrestris and Bombus impatiens). Of these 30 putative sequences from the family Apidae (Supplementary Table 5), twenty-two genes were very similar in gene structure to those identified using only AmelOBPs. The remaining 8 OBPs were a result of remote homology searches (with PAM250 matrix or allowing very long intronic regions), and hence 5 of these also had significant overlap with previously mentioned good-quality 22 gene models (Supplementary Table 5)In the end, expanding the search space by using many insect OBPs and remote homology searches yielded only 3 novel OBPs on the NW_003789197.1 scaffold. These three hits were not the best hits in bidirectional blast with their respective query sequences. As such remote homologs may sometimes belong to another related protein family, we decided to not include them in our further analysis.
3.1 Chemosensory proteins in Apis florea
Furthermore, a similar approach yielded 8 putative chemosensory proteins (CSPs) in the Apis florea genome, out of which 7 putative CSPs showed significant e-value for the presence of OS-D domain (Supplementary Table 5). The 7 CSPs identified have orthology with Atta colombica (leafcutter ant), Bombus ignitus (bumblebee), Apis cerana cerana (Asian honey bee), Camponotus floridanus (Florida carpenter ant), Trachymyrmex zeteki (fungus-farming ant), and Trichogramma pretiosum (endoparasitoid wasp) (Supplementary Table 5). The CSP gene family is highly conserved and shows an expansion in the flour beetle Tribolium castaneum (20 genes; Forêt et al. 2007) and a reduction in bees, wasps (10 genes; Werren et al.), and Dipterans (4 in fruit flies and 8 in mosquitoes; Vieira and Rozas 2011). Although 2 CSP genes have been identified in Apis cerana (Diao et al. 2018), studies have confirmed 6 CSPs in honeybee species (Wanner et al. 2004; Forêt and Maleszka 2006; Forêt et al. 2007; Fouks et al. 2021) such as Apis florea, Apis mellifera, and Apis dorsata. Our finding is thus in agreement with the knowledge of insect CSPs in current literature. Although CSPs have been typically detected in pheromone glands (e.g., mandibular glands) and reproductive organs (Zhu et al. 2019), members of CSPs, like OBPs, have also been associated with non-sensory functions. CSPs have been involved in embryonic development (Maleszka et al. 2007), insecticide resistance (Liu et al. 2014), and limb regeneration (Nomura et al. 1992).
3.2 Comparative study on AfloOBP predicted protein sequences
Finally, out of 22 OBP genes, 15 complete AfloOBP genes (i.e., annotated as having a start and stop codon), and 16 translated protein sequences were predicted to have signal peptide sequences. The average length of signal peptides predicted in our AfloOBP dataset was 19 amino acids. Cleavage position ranged from 16 to 24th amino acids in the sequence. Secondary structure analysis revealed an alpha-rich state of OBPs with high confidence. Typically, 6–7 alpha helices per complete AfloOBP sequence were predicted.
We used a reciprocal BLASTp approach to compare protein sequences of AfloOBPs obtained through our pipeline with that of the recent study (also reporting 22 OBPs) (Fouks et al. 2021). Our analysis reports 20 OBPs as reciprocal best-hit matches (Supplementary Table 8). However, triangular associations are likely with more sequences in their dataset. For example, AfloOBP17-like shows complete query coverage and identity with OBP17 (Fouks et al. 2021) but OBP17 shows identity only to AfloOBP17. Similarly, isoform AfloOBP19-like did not have reciprocal matches with OBPs in Fouks et al. 2021 and has been annotated as a long non-coding RNA (Supplementary Table 9). OBP20PSE (61 amino acids) showed full query coverage with AfloOBP19 in our dataset. The exon coordinates in the gene model for OBP20PSE were also found in our prediction from protein to genome alignments using Exonerate (Sect. 2.3; Supplementary Table 6).
Interestingly, OBP22PSE (216 amino acids) was annotated in a recent and parallel study (Fouks et al. 2021) and corresponds to a gene under negative selection (Fouks et al. 2021). It did not find reciprocal matches in our dataset. Additionally, we observed that OBP22PSE was positive for PBP/GOBP domain against the Pfam database with a considerably higher e-value (3.4e − 06) than other OBPs. With a length of 216 amino acids, we expected it to be a double-domain or atypical OBP but, interestingly, observed only 1 domain with a relatively weak e-value. As our study created a revised annotation of Apis mellifera OBPs combining three studies (Sect. 2.1) before using it for identifying OBPs in Apis florea, therefore, we retain our annotation independently.
3.3 Comparative study on gene models of AfloOBPs and revised annotation using RNA-seq data
We had performed the GWS work independently and deposited the manuscript in BioRxiv in 2020 prior to the 2021 release of the publication by Fouks et al. group (2021). We present a detailed comparative analysis of the gene models below using a combination of RNA-seq data from NCBI and our computational genome-wide survey.
We have used publicly available RNA-seq data from NCBI Apis florea Annotation Release 102. We report that OBPs in Apis florea have 5 exons with the exception of OBP1, OBP8_2, OBP10, and OBP11 having 6 exons. OBPs predicted computationally by our study and Fouks et al. (2021) have been named identically through our RBH analysis except the following: [our annotation (annotation by Fouks et al.)] OBP3_l2 (OBP03), OBP8_2 (OBP06), OBP8_2like (OBP8), OBP15-like (OBP15), OBP21-like (OBP21), OBP21 (OBP18).
We have annotated and identified splice variants for AfloOBP7, AfloOBP8_2, AfloOBP10, AfloOBP12, AfloOBP15-like, and OBP22PSE gene models. Furthermore, we have also added signal peptide-coding exons that were missing in few gene models obtained through predictive analysis-OBP06 (Fouks), OBP1 (our study), OBP8_2 (our study), OBP15-like (our study), and OBP16 (our study).
Fouks et al. (2021) annotated gene models for OBP02, OBP03, OBP04, OBP09, OBP11, OBP13, OBP16, 17, OBP18, OBP19, and OBP21 lacked stop codons that have been now added to the revised models (Supplementary Table 9).
As discussed earlier, the study by Fouks et al. (2021) also annotated two genes AfloOBP20PSE and AfloOBP22PSE. The annotation for AfloOBP20PSE by Fouks et al. (2021) comprised two exons only. We have revised the model to a five-exon model using RNA-seq exon data and corrected the exon–intron boundaries. AfloOBP22PSE is a novel gene model predicted by Fouks et al. (2021). However, we report the presence of two splice variants, and present corrected gene boundaries for the same.
Recent research on long non-coding RNA in Hymenoptera reveals their involvement in various neuronal processes in ants (Shields et al. 2018) and adult honeybee workers (Sawata et al. 2004; Kiya et al. 2012) as well as olfactory behaviors (Liu et al. 2019) in honeybees. Their potential involvement in regulating behavioral plasticity, ovary activity, and division of labor in honeybees through targeting mRNAs is yet to be fully understood (Chen et al. 2017, Chen and Shi 2020). We are the first to report the annotation of long non-coding RNA (lncRNA) sequence in Apis florea through both our in silico homology-based GWS pipeline and publicly available RNA-seq exon data. The sequence contains a peroxisomal targeting signal and has a weak PBP/GOBP domain. The novel sequence AfloOBP19-like at scaffold NW_003791127.1 indicates a weak PBP/GOBP domain but is a long non-coding RNA. It is a two-exon gene (289 bases) with a peroxisomal targeting signal.
In general, using RNA-seq exon data, we have corrected gene boundaries in most models in both Fouks et al. (2021) and our in silico prediction to maintain in-frame translation of protein sequences. There have been frame shifts due to gene boundaries annotated causing issues with the translated amino acid sequence. These have been corrected throughout. For example, we have improved gene boundaries for the last exon in Fouks et al.’s (2021) annotated OBP19. On the other hand, the C-terminus has been extended in the case of OBP3. We have tabulated the differences among the models in Supplementary Table 9.
AfloOBP1 is encoded by six exons with a well-defined N-terminal signal peptide and C-terminus. This is an improvement on the gene boundaries in the homology-based annotations by ours and Fouks et al. 2021 (Supplementary Table 9). Our homology-based annotation lacked a signal peptide and the C-terminal exon. The gene model for OBP1 by Fouks et al. (2021) has slightly differing gene boundaries at the third and fifth exons respectively that translate to frame shifts and do not align with their predicted protein sequence. The RNA-Seq-based annotation for OBP1 overcomes these limitations (Supplementary Table 9). AfloOBP2 is encoded by five exons and is characterized as complete in our in silico analysis. In AfloOBP3, the amino terminus has been extended further and gene boundaries have been corrected to correct frame shifts. OBP7 X1 splice variant has been annotated by both homology-based studies—ours and Fouks et al.’s (2021) pipelines. We have corrected the gene boundaries using publicly available RNA-seq exon data and have annotated the AfloOBP7 X2 isoform as well. For AfloOBP8_2, splice variants X1 and X2 have been annotated. For both OBP10 and OBP12, X1 isoform was originally annotated through our GWS, whereas X2 has been annotated by Fouks et al.’s (2021) study. In the case of OBP12 gene model, exon 1 (886,573–886,608) predicted is absent from the RNA-seq exon data. We did not find transcriptomic evidence for OBP17-like that was a predicted partial gene in our in silico pipeline. Despite partial overlap with OBP17, the exon–intron boundary (1,817,764–1,817,790) of OBP17-like did not correlate well with transcriptomic data.
3.4 Evolution of OBP subfamilies in honeybees
Sequences AfloOBP1-AfloOBP13 were found to display the conserved Cysteine signature of Classic and Minus-C subfamilies, as their orthologs in Apis mellifera, and comparable to that of AgamOBP (Figure 1). Multiple sequence alignment revealed conserved Cysteine profiles specific to Classic and Minus-C subfamilies in the Apis florea genome (Figure 2). Sequences AfloOBP14-AfloOBP21 were found to show the conserved Minus-C cysteine signature where Cysteine residues in the conserved second and fifth positions are missing. Our analysis shows that the conserved cysteine signature for both subfamilies in Apis florea is similar to the representative signature observed in a previous study (Xu et al. 2009). The conserved cysteine signature for the Classic subfamily for the Hymenopteran insect order was determined as C1-X 23:35-C2-X3-C3-X 27:45-C4-X 7:14-C5-X8-C6 (Xu et al. 2009). Our study has identified 13 Classic and 9 Minus-C OBPs in Apis florea. We observe the Classic cysteine signature to be conserved similarly as C1-X 27:37-C2-X3:4-C3-X 33:43-C4-X 9:13-C5-X8-9-C6.

Cysteine signatures across insect orders are comparable. Cysteine residue positioning and inter-disulfide spacing are conserved in Apis florea (Hymenoptera) genome across Classic and Minus-C subfamilies. The cysteine signature of OBPs from Anopheles gambiae is given for reference

Multiple sequence alignment of OBPs from Apis florea (AfloOBPs). The alignment also contains OBPs from its phylogenetic neighbor Apis mellifera (AmelOBP). Chemosensory proteins from Apis mellifera (AmelCSPs) constitute the outgroup
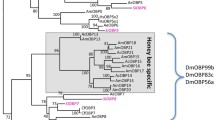
We tested various models of evolution on our insect OBP data (Darriba et al. 2011; Guindon and Gascuel 2003). Out of these, the most optimal model of evolution was parameters “LG + I + G.” We generated our phylogenetic tree using these parameters with 1000 iterations. Phylogenetic inference revealed the clustering of Minus-C OBPs as a subclade of the Classic OBP subfamily comprising members of both Apis mellifera and Apis florea OBPs (Figure 3A). Moreover, a conserved cysteine signature specific to the chemosensory protein (CSP) family was observed in the outgroup chemosensory proteins (AmelCSP) (Figure 2). AmelCSPs used as outgroup clustered distinctly (Figure 3A) from the odorant-binding proteins input to the phylogeny with 100% bootstrap value. Minus-C OBPs were found to cluster together with a 60% bootstrap value closest to AfloOBP9, annotated as a Classic OBP. However, the topology of the Minus-C clade with Classic AfloOBP13 outgroup has good bootstrap support in general. OBPs of the Minus-C subfamily, AfloOBP 14–20, emerge closest to AfloOBP13, a Classic OBP with an observed six cysteine signature. Interestingly, all the other Classic OBPs cluster distinctly in a clade corresponding to the insect Classic subfamily; however, AfloOBP13 clusters closely with the Minus-C group in a distinct subclade with high confidence (Figure 3B). Interestingly, antennal OBP (MsexABP1) (Vogt et al. 2015) from Lepidopteran insect Manduca sexta clustered close to the Minus-C clade along with other bee species (Hymenopteran) with high bootstrap support of 97%. We found that most Classic OBPs in Apis florea are phylogenetically distant from Minus-C (bee OBPs) than clades representing atypical OBPs in Dipterans and Plus-C insect OBPs. It is possible that Minus-C OBPs in honey bees may have evolved from a single ancestral Classic OBP (similar to AfloOBP13, AmelOBP13) of its species by deletion/mutation of second and fifth cysteines. However, we also acknowledge that we have used representative organisms covering 11 insect orders with a focus on Hymenoptera. It is also plausible that Minus-C OBPs in insects may have evolved independently under positive selection pressure. The evolution and insect order-specific occurrence of Minus-C, Plus-C, and atypical subfamilies of insect OBPs may have functional roles and would be interesting to investigate.

Phylogenetic tree of Apis florea OBPs. Rooted phylogeny (A) of OBPs from sister species, Apis florea (Aflo; in bright green) and Apis mellifera (Amel; in purple). Members of the alignment template are labelled with the prefix “seed” and colored based on order, whereas the outgroup consisting of Apis mellifera chemosensory proteins (AmelCSP) is labelled in black. The Minus-C subfamily has been indicated in a rectangular box. The bootstrap values of the branches are indicated on the nodes in percentage values. Unrooted phylogeny (B) of OBPs from representative members of 11 insect orders represents phylogenetic clades. Inner branches and leaf labels denoting insect species are colored based on order whereas the outer strips denote the OBP subfamily. Subfamily annotation label for a sequence has been provided only if the criteria of high confidence in annotation have been met (i.e., phylogeny and cysteine pattern conservation). Subfamily labels have been left blank in case of limited or no information about subfamily classification for the OBP sequence. Classic subfamily is colored in brown, Minus-C in dark blue, Plus-C in dark green, two-domains in bright pink, and atypical in red. The outer circle denotes members’ clades. The inner branch colors and label colors are colored as per order. Hymenoptera is denoted in violet. The bootstrap values of the branches are indicated on the nodes in percentage values. An interrogative, user-friendly version of the phylogeny will be provided at http://caps.ncbs.res.in/download/Apid
Taken together, our observations from a comprehensive bioinformatic analysis strongly suggest that Minus-C OBPs are likely to have evolved from a Classic OBP subfamily member in honeybees. Similar co-clustering of Classic and Minus-C OBPs across other insects suggests a recurrent need for such variation. It is possible that the evolution of a subfamily could be an adaptation to the local niche of the insect species for functional specificity (Zhou et al. 2020).
4 Conclusion
In a step towards understanding the role of OBPs in insects, a bioinformatics-based approach was used. We have curated OBPs from Apis mellifera from three sources and used them to query the genome of Apis florea. To study the evolutionary relationships, we used OBPs from 11 insect orders from diverse habitats (Supplementary Table 3). Our phylogeny was constructed using a novel structural template-based approach (Gandhimathi et al. 2012) that addresses the challenges faced in sequence alignments due to low sequence identities.
A total of 22 OBPs including isoforms have been identified and annotated from the genome of eusocial Asian red dwarf honeybee, Apis florea, using a modified in-house pipeline. Our results include AfloOBPs that have been previously identified by the automated pipeline of NCBI with a query coverage and identity of 100% each with their respective subjective sequences (AfloOBP9 and AfloOBP11) (Supplementary Table 7). Our annotated data includes complete OBPs that were identified as having incomplete exons in N-termini and C-termini or/and labelled as uncharacterized by the automated pipeline of NCBI. We also observe that a number of OBP genes in Apis florea (22) and the western honeybee, Apis mellifera (25), are similar despite the differences in respective ecological niches.
We have analyzed the characteristic conserved features of these OBPs using computational methods and phylogeny resulting in the discovery of new gene models as well as improvement on existing gene models from NCBI. Presence of conserved cysteine pattern, disulfide spacing, domain analysis, size, and predicted secondary structure further strengthen their identity as putative insect OBPs.
The Classic OBP subfamily clade appears to have expanded to Minus-C OBPs in our study on the dwarf honeybee, Apis florea, and also in a few other insect orders (Vieira et al. 2007; Sanchez-Gracia and Rozas 2008).
It is suggested that the expansion of OBPs in the last common ancestor of honeybees explains their unique chemosensory behavior. Although a study on bumble bee OBP (Sadd et al. 2015) identified only the Classic subfamily (16 OBPs in Bombus terrestris), and its eight orthologs in Apis mellifera, previously, the C-minus subfamily in honeybees was identified by Foret and Maleszka (2006). Using phylogeny, our study also shows the evolution of Classic OBP, AfloOBP13, and its Minus-C relatives, AfloOBP14-AfloOBP21. The exon length of B. terrestris is typically 4 or 5 exons, and is similar to Apis mellifera and Apis florea. However, Classic OBP AfloOBP10 (ortholog to AmelOBP10) has 6 exons. The strong phylogenetic similarity between Apis mellifera and Apis florea OBPs strongly suggests that a similar birth and death model of evolution occurs in the OBP gene repertoire in Apis florea as well.
Among other corbiculate bees, the whole genomes of honey bees (Apis mellifera) have a comparable number of OBPs (21) (Robertson and Wanner, 2006; Croset et al. 2010; Foret and Maleszka 2006), stingless bees (Melipona quadrifasciata) (14), orchid bees (Eufrisea mexicana) (16) (Brand and Ramirez 2017), and Euglossa dilemma (15) (Brand and Ramirez 2017). However, C-minus OBPs are missing from the genomes of bees other than honey bees (Brand and Ramirez 2017).
Interestingly, AmelOBP3 was found to be under positive selection in Apis melifera (Fouks et al. 2021). Inhibition in OBP7 in response to glyphosate stress was also accompanied by a decrease in metabolites contribution to the chemosensory pathways such as L-malic acid, histamine, and gamma-aminobutyric acid. There is an increase in differentially expressed OBPs in response to glyphosate and commercial formulation-based stress in both A. cerana cerana and A. melifera ligustica (Zhao et al. 2020). Significant changes in the expression of OBP4, OBP16, OBP18, and OBP21 are observed in A. mellifera as a stress response to sublethal doses of imidacloprid, a nicotine-mimic insecticide. It is worthwhile to note that AmelOBP16, AmelOBP18, and AmelOBP21 belong to the Minus-C subfamily and could explain the positive selection of the C-minus subfamily. This strengthens the possibility of testing the response of their respective orthologs (Supplementary Table 4) to various abiotic stress factors in Apis florea.
We used a query set for Apis mellifera OBPs derived from three studies, and used this revised query set to annotate OBPs in Apis florea using our in silico homology-based approach as described in the present study. Furthermore, we used available RNA-seq exon data to refine our in silico predictions of exon–intron boundaries for each gene model. We have also considered AfloOBP gene models in a recent study (Fouks et al. 2021). Overall, we corrected exon boundaries, annotated novel isoforms, and missing exons. We also report a long non-coding RNA in Apis florea. We have provided an improved annotation for Apis florea OBPs that incorporates all the above features into our work (Supplementary Table 9).
Data availability
All the main data generated or analyzed during this study are included in this published article [and its supplementary information files]. Related datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
Not applicable.
Change history
22 March 2023
A Correction to this paper has been published: https://doi.org/10.1007/s13592-023-01004-0
References
Armenteros JJA, Sønderby CK, Sønderby SK, Nielsen H, Winther O (2017) DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics 33(21):3387–3395. https://doi.org/10.1093/bioinformatics/btx431
Bianchet MA, Bains G, Pelosi P, Pevsner J, Snyder SH, Monaco HL, Amzel LM (1996) The three-dimensional structure of bovine odorant binding protein and its mechanism of odor recognition. Nat Struct Biol 3(11):934–939. https://doi.org/10.1038/nsb1196-934
Brand P, Ramírez SR (2017) The evolutionary dynamics of the odorant receptor gene family in corbiculate bees. Genome Biol Evol 9(8):2023–2036. https://doi.org/10.1093/gbe/evx149
Buchan DWA, Minneci F, Nugent TCO, Bryson K, Jones DT (2013) Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res 41(W1):W349–W357. https://doi.org/10.1093/nar/gkt381
Chen X, Shi W (2020) Genome-wide analysis of coding and non-coding RNAs in ovary of honey bee workers and queens. Apidologie 51:777–792. https://doi.org/10.1007/s13592-020-00760-7
Chen X, Ma C, Chen C, Lu Q, Shi W, Liu Z, Wang, H, Guo H (2017) Integration of lncRNA-miRNA-mRNA reveals novel insights into oviposition regulation in honey bees. Peer J 5:e3881. https://doi.org/10.7717/PEERJ.3881.
Croset V, Rytz R, Cummins SF, Budd A, Brawand D, Kaessmann H, Gibson TJ, Benton R, Stern DL (2010) Ancient protostome origin of chemosensory ionotropic glutamate receptors and the evolution of insect taste and olfaction. PLoS Genet 6(8):e1001064. https://doi.org/10.1371/journal.pgen.1001064
Dayhoff MO, Schwartz RM, Orcutt, BC (1978) In atlas of protein sequence and structure, (Dayhoff, M.O. ed.), vol. 5 suppl. 3 pp 345–352, NBRF, Washington. https://doi.org/10.2307/2412074
Darriba D, Taboada GL, Doallo R, Posada D (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165. https://doi.org/10.1093/bioinformatics/btr088
Diao Q, Sun L, Zheng H, Zeng Z, Wang S, Xu S, Zheng H, Chen Y, Shi Y, Wang Y, Meng F, Sang Q, Cao L, Liu F, Zhu Y, Li W, Li Z, Dai C, Yang M et al (2018) Genomic and transcriptomic analysis of the Asian honeybee Apis cerana provides novel insights into honeybee biology. Sci Rep 8(1):822. https://doi.org/10.1038/s41598-017-17338-6
Flower DR (1996) The lipocalin protein family: structure and function. Biochem 318(Pt 1):1–14. https://doi.org/10.1042/bj3180001
Forêt S, Maleszka R (2006) Function and evolution of a gene family encoding odorant binding-like proteins in a social insect, the honeybee (Apis mellifera) Genome Res 16(11):1404–1413. https://doi.org/10.1101/gr.5075706
Forêt S, Wanner KW, Maleszka R (2007) Chemosensory proteins in the honey bee: Insights from the annotated genome comparative analyses and expressional profiling. Insect Biochemistry and Molecular Biology 37(1):19–28. https://doi.org/10.1016/j.ibmb.2006.09.009
Fouks et al (2021) The genomic basis of evolutionary differentiation among honeybees. Genome Res 31(7):1203–1215. https://doi.org/10.1101/gr.272310.120
Gandhimathi A, Nair AG, Sowdhamini R (2012) PASS2 version 4: an update to the database of structure-based sequence alignments of structural domain superfamilies. Nucleic Acids Res: D531–4. https://doi.org/10.1093/nar/gkr1096.
Graham LA, Davies PL (2002) The odorant-binding proteins of Drosophila melanogaster: annotation and characterization of a divergent gene family. Gene 292(1–2):43–55. https://doi.org/10.1016/s0378-1119(02)00672-8
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704. https://doi.org/10.1080/10635150390235520
Gupta M (1986) A quantitative study and ultrastructure of flagellar sensillae of Apis florea F. (Hymenoptera: Apidae). Proc Indian Acad Sci (Anim. Sci.) 95: 5: 595–603
Gupta M (1992) Scanning electron microscopic studies of antennal sensilla of adult worker Apis florea F (Hymenoptera: Apidae). Apidologie, Springer Verlag 23(1):47–56
Halling L, Oldroyd B, Wattanachaiyingcharoen W et al (2001) Worker policing in the bee Apis florea. Behav Ecol Sociobiol 49:509–513. https://doi.org/10.1007/s002650100325
He P, Li ZQ, Zhang YF, Chen L, Wang J, Xu L, Zhang YN, He M (2017) Identification of odorant-binding and chemosensory protein genes and the ligand affinity of two of the encoded proteins suggest a complex olfactory perception system in Periplaneta americana. Insect Mol Biol 26:687–701. https://doi.org/10.1111/imb.12328
Hekmat-Scafe DS, Scafe CR, McKinney AJ, Tanouye MA (2002) Genome-wide analysis of the odorant-binding protein gene family in Drosophila melanogaster. Genome Res 12(9):1357–1369. https://doi.org/10.1101/gr.239402
Henikoff S, Henikoff JG (1992) Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci 89(22):10915–10919. https://doi.org/10.1073/pnas.89.22.10915
Hepburn RH, Radloff S, Otis G, Fuchs S, Verma L, Tan K et al (2005) Apis florea: morphometrics, classification and biogeography. Apidologie 36(3):359–376. https://doi.org/10.1051/apido:2005023
Hildebrand JG, Shepherd GM (1997) Mechanisms of olfactory discrimination: converging evidence for common principles across phyla. Annu Rev Neurosci 20:595–631. https://doi.org/10.1146/annurev.neuro.20.1.595
Karpe SD, Jain R, Brockmann A, Sowdhamini R (2016) Identification of complete repertoire of Apis florea odorant receptors reveals complex orthologous relationships with Apis mellifera. Genome Biol and Evol 8:2879–2895. https://doi.org/10.1093/gbe/evw202
Karpe SD, Dhingra S, Brockmann A, Sowdhamini R (2017) Computational genome-wide survey of odorant receptors from two solitary bees Dufourea novaeangliae (Hymenoptera: Halictidae) and Habropoda laboriosa (Hymenoptera: Apidae). Sci Rep 7:10823. https://doi.org/10.1038/s41598-017-11098-z
Karpe SD, Tiwari V, Ramanathan S, Papanicolaou A (2021) InsectOR—webserver for sensitive identification of insect olfactory receptor genes from non-model genomes. PLoS One 16(1):e0245324. https://doi.org/10.1371/journal.pone.0245324
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. https://doi.org/10.1093/nar/gkf436
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Kiya T, Ugajin A, Kunieda T, Kubo T (2012) Identification of kakusei, a nuclear non-coding RNA, as an immediate early gene from the honeybee, and its application for neuroethological study. Int J Mol Sci. https://doi.org/10.3390/ijms131215496
Kumar N, Nayyar K, Sharma R, Anudeep (2014) Ultramorphology of antennal sensilla of open-nesting honeybees Apis florea F. and Apis dorsata F. (Hymenoptera: Apidae) J Appl & Nat Sci 6(1):315–319. https://doi.org/10.31018/jans.v6i1.421
Larter NK, Sun JS, Carlson JR (2016) Organization and function of Drosophila odorant binding proteins. Elife 5:e20242. https://doi.org/10.7554/eLife.20242
Le SQ, Gascuel O (2008) An improved general amino acid replacement matrix. Mol Biol Evol 25(7):1307–1320. https://doi.org/10.1093/molbev/msn067
Letunic I, Bork P (2006) Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23(1):127–128. https://doi.org/10.1093/bioinformatics/btl529
Letunic I, Bork P (2016) Interactive Tree Of Life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44(W1):W242–W245. https://doi.org/10.1093/nar/gkw290
Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22(13):1658–1659. https://doi.org/10.1093/bioinformatics/btl158
Liu GX, Xuan N, Chu D, Xie HY, Fan ZX, Bi YP, Picimbon J-F, Qin YC, Zhong ST, Li YF, Gao ZL, Pan WL, Wang GY, Rajashekar B (2014) Biotype expression and insecticide response of Bemisia tabaci chemosensory protein-1. Arch Insect Biochem Physiol 85(3):137–151. https://doi.org/10.1002/arch.21148
Liu F, Shi T, Qi L, Su X, Wang D, Dong J, Huang ZY (2019) lncRNA profile of Apis mellifera and its possible role in behavioural transition from nurses to foragers. BMC Genomics 20(1):393. https://doi.org/10.1186/s12864-019-5664-7
Loebel D, Scaloni A, Paolini S, Fini C, Ferrara L, Breer H, Pelosi P (2000) Cloning, post-translational modifications, heterologous expression and ligand-binding of boar salivary lipocalin. Biochemical J 350 Pt 2(2):369–379. https://doi.org/10.1042/bj3500369
Maleszka J, Forêt S, Saint R, Maleszka R (2007) RNAi-induced phenotypes suggest a novel role for a chemosensory protein CSP5 in the development of embryonic integument in the honeybee (Apis mellifera). Dev Genes Evol 217(3):189–196. https://doi.org/10.1007/s00427-006-0127-y
Mam B, Sowdhamini R (2020) SoCCer: a pipeline to identify classes of soluble proteins in chemical communication in insect genomes. PROTOCOL (Version 1) available at Protocol Exchange. https://doi.org/10.21203/rs.3.pex-1095/v1
Manikkaraja M, Bhavika M, Singh R, Nagarathnam B, George G, Gulyani A, Archunan G, Sowdhamini R (2020) Molecular and functional characterization of buffalo nasal epithelial odorant binding proteins and their structural insights by in silico and biochemical approaches. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1854117
Manoharan M, Ng Fuk Chong M, Vaïtinadapoulé A, Frumence E, Sowdhamini R, Offmann B (2013) Comparative genomics of odorant binding proteins in Anopheles gambiae, Aedes aegypti, and Culex quinquefasciatus. Genome Biol Evol 5(1):163–180. https://doi.org/10.1093/gbe/evs131
Mastrogiacomo R, D’Ambrosio C, Niccolini A, Serra A, Gazzano A, Scaloni A, Pelosi P (2014) An odorant-binding protein is abundantly expressed in the nose and in the seminal fluid of the rabbit. PloS One 9(11):e111932. https://doi.org/10.1371/journal.pone.0111932
Missbach C, Vogel H, Hansson BS, Groβe-Wilde E (2015) Identification of odorant binding proteins and chemosensory proteins in antennal transcriptomes of the jumping bristletail Lepismachilis y-signata and the firebrat Thermobia domestica: evidence for an Independent OBP–OR Origin. Chem Senses 40(9):615–626. https://doi.org/10.1093/chemse/bjv050
Moritz RF et al (2010) Invasion of the dwarf honeybee Apis Florea into the Near East. Biol Invasions 12:1093–1099. https://doi.org/10.1007/s10530-009-9527-z
Nielsen H, Engelbrecht J, Brunak S, von Heijne G (1997) Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng 10:1–6. https://doi.org/10.1093/protein/10.1.1
Nomura A, Kawasaki K, Kubo T, Natori S (1992) Purification and localization of p10, a novel protein that increases in nymphal regenerating legs of Periplaneta americana (American cockroach). Int J Dev Biol 36(3):391–398
Oldroyd B, Nanork P (2009) Conservation of Asian honeybees. Apidologie 40(3):296–312. https://doi.org/10.1051/apido/2009021
Otis GW (1991) Revised distribution of three recently recognised species of honeybees in Asia. Honeybee Sci 15:167–170
Page RE, Robinson G (1991) The genetics of division of labour in honeybee colonies. Adv Insect Physiol 23:117–169. https://doi.org/10.1016/S0065-2806(08)60093-4
Pelosi P, Maida R (1990) Odorant-binding proteins in vertebrates and insects: similarities and possible common function. Chem Senses 15(2):205–215. https://doi.org/10.1093/chemse/15.2.205
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. https://doi.org/10.1038/nmeth.1701
Robertson HM, Wanner KW (2006) The chemoreceptor superfamily in the honey bee, Apis mellifera: expansion of the odorant, but not gustatory, receptor family. Genome Res 16(11):1395–1403. https://doi.org/10.1101/gr.5057506
Sadd BM, Barribeau SM, Bloch G, de Graaf DC, Dearden P, Elsik CG, Gadau J, Grimmelikhuijzen CJP, Hasselmann M, Lozier JD, Robertson HM, Smagghe G, Stolle E, Van Vaerenbergh M, Waterhouse RM, Bornberg-Bauer E, Klasberg S, Bennett AK, Câmara F et al (2015) The genomes of two key bumblebee species with primitive eusocial organization. Genome Biol 16(1):76. https://doi.org/10.1186/s13059-015-0623-3
Sanchez-Gracia A, Rozas J (2008) Divergent evolution and molecular adaptation in the Drosophila odorant-binding protein family: inferences from sequence variation at the OS-E and OS-F genes. BMC Evol Biol 8:323. https://doi.org/10.1186/1471-2148-8-323
Sawata M, Takeuchi H, Kubo T (2004) Identification and analysis of the minimal promoter activity of a novel noncoding nuclear RNA gene, AncR-1, from the honeybee (Apis mellifera L.). RNA 10:1047–1058. https://doi.org/10.1261/rna.5231504
Scaloni A, Paolini S, Brandazza A, Fantacci M, Bottiglieri C, Marchese S, Navarrini A, Fini C, Ferrara L, Pelosi P (2001) Purification, cloning and characterisation of odorant- and pheromone binding proteins from pig nasal epithelium. Cell Mol Life Sci 58(5–6):823–834. https://doi.org/10.1007/pl00000903
Shields EJ, Sheng L, Weiner AK, Garcia BA, Bonasio R (2018) High-quality genome assemblies reveal long non-coding RNAs expressed in ant brains. Cell Rep 23:3078–3090. https://doi.org/10.1016/j.celrep.2018.05.014
Slater GSC, Birney E (2005) Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6:31. https://doi.org/10.1186/1471-2105-6-31
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. https://doi.org/10.1093/bioinformatics/btl446
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Suwannapong G, Seanbualuang P, Benbow ME (2011) Using sensillum potential analysis to quantify pheromone sensing of the antennal sensilla of Apis florea Fabricius (1787), foragers and guards. Journal of Asia-Pacific Entomology 14(1):7–10. https://doi.org/10.1016/j.aspen.2010.11.001
Tegoni M, Ramoni R, Bignetti E, Spinelli S, Cambillau C (1996) Domain swapping creates a third putative combining site in bovine odorant binding protein dimer. Nature Struct Biol 3(10):863–867. https://doi.org/10.1038/nsb1096-863
Teufel F, Almagro Armenteros JJ, Johansen AR et al (2022) SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat Biotechnol 40:1023–1025. https://doi.org/10.1038/s41587-021-01156-3
The UniProt Consortium (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47:D506-515
Vieira FG, Sanchez-Gracia A, Rozas J (2007) Comparative genomic analysis of the Odorant-binding protein family in 12 Drosophila genomes: purifying selection and birth-and-death evolution. Genome Biol 8:R235. https://doi.org/10.1186/gb-2007-8-11-r235
Vieira FG, Rozas J (2011) Comparative genomics of the odorant-binding and chemosensory protein gene families across the Arthropoda: origin and evolutionary history of the chemosensory system. Genome Biol Evol 3:476–490. https://doi.org/10.1093/gbe/evr033
Vieira FG, Forêt S, He X, Rozas J, Field LM, Zhou JJ (2012). Unique features of odorant-binding proteins of the parasitoid wasp Nasonia vitripennis revealed by genome annotation and comparative analyses. PLoS One.; 7(8):e43034. doi: https://doi.org/10.1371/journal.pone.0043034.
Vogt RG, Riddiford LM (1981) Pheromone binding and inactivation by moth antennae. Nature 293(5828):161–163. https://doi.org/10.1038/293161a0
Vogt GR, Große-Wilde E, Zhou JJ (2015) The Lepidoptera Odorant Binding Protein gene family: gene gain and loss within the GOBP/PBP complex of moths and butterflies. Insect Biochem Mol Biol 62:142–153. https://doi.org/10.1016/j.ibmb.2015.03.003
Wang N, Wang NX, Niu LM, Bian SN, Xiao JH, Huang DW (2014) Odorant-binding protein (OBP) genes affect host specificity in a fig-pollinator mutualistic system. Insect Mol Biol 23(5):621–631. https://doi.org/10.1111/imb.12110
Wanner KW, Willis LG, Theilmann DA, Isman MB, Feng Q, Plettner E (2004) Analysis of the insect OS-D-like gene family. J Chem Ecol 30(5):889–88l. https://doi.org/10.1023/B:JOEC.0000028457.51147.d4
White SA, Briand L, Scott DJ, Borysik AJ (2009) Structure of rat odorant-binding protein OBP1 at 1.6 A resolution. Acta Crystallogr D Biol Crystallogry 65(Pt 5):403–410. https://doi.org/10.1107/S090744490900420X
Wigglesworth VB (1965) The principles of insect physiology. Methuen Co Ltd, London, 6th edn
Wongsiri S et al (1997) Comparative biology of Apis andreniformis and Apis florea in Thailand. Bee World 78(1):23–35. https://doi.org/10.1080/0005772X.1997.11099328
Xu PX, Zwiebel LJ, Smith DP (2003) Identification of a distinct family of genes encoding atypical odorant-binding proteins in the malaria vector mosquito Anopheles gambiae. Insect Mol Biol 12(6):549–560. https://doi.org/10.1046/j.1365-2583.2003.00440.x
Xu YL, He P, Zhang L, Fang SQ, Dong SL, Zhang YJ, Li F (2009) Large-scale identification of odorant-binding proteins and chemosensory proteins from expressed sequence tags in insects. BMC Genomics 10:632. https://doi.org/10.1186/1471-2164-10-632
Zhao H, Li G, Guo D, Wang Y, Liu Q, Gao Z, Wang H, Liu Z, Guo X, Xu B (2020) Transcriptomic and metabolomic landscape of the molecular effects of glyphosate commercial formulation on Apis mellifera ligustica and Apis cerana cerana. Sci Total Environ 744:140819
Zhou J-J, Huang W, Zhang G-A, Pickett JA, Field LM (2004) “Plus-C” odorant-binding protein genes in two Drosophila species and the malaria mosquito Anopheles gambiae. Gene 327(1):117–129. https://doi.org/10.1016/j.gene.2003.11.007
Zhou YN, Xie S, Chen JN et al (2020) Expression and functional characterization of odorant-binding protein genes in the endoparasitic wasp Cotesia vestalis. Insect Sci 28(5):1354–1368. https://doi.org/10.1111/1744-7917.12861
Zhu J, Arena S, Spinelli S, Liu D, Zhang G, Wei R, Cambillau C, Scaloni A, Wang G, Pelosi P (2017) Reverse chemical ecology: olfactory proteins from the giant panda and their interactions with putative pheromones and bamboo volatiles. Proc Natl Acad Sci USA 114(46):E9802–E9810. https://doi.org/10.1073/pnas.1711437114
Zhu J, Iovinella I, Dani FR, Pelosi P, Wang G (2019) Chemosensory proteins: a versatile binding family. In: Picimbon JF (ed) Olfactory concepts of insect control - alternative to insecticides. Springer, Cham. https://doi.org/10.1007/978-3-030-05165-5_6
Acknowledgements
BM acknowledges Prof. (Dr.) Axel Brockmann and Prof. (Dr.) Shannon Olsson for valuable comments during the study. All authors acknowledge NCBS for infrastructural support.
Funding
BM received support from NCBS-TIFR and the Tata Education and Development Trust. SDK received support from Shyama Prasad Mukherjee Fellowship from the Council of Scientific and Industrial Research (CSIR) and Bridging Postdoctoral Fellowship from NCBS-TIFR. RS received support and funding from JC Bose fellowship (JBR/2021/000006) and from the DBT-Bioinformatics Centre (BT/PR40187/BTIS/137/9/2021).
Author information
Authors and Affiliations
Contributions
RS and SDK conceived this research and designed experiments; BM co-designed experiments for protein sequence annotation, performed experiments and analysis, and wrote the first draft of the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Manuscript editor: Klaus Hartfelder.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mam, B., Karpe, S.D. & Sowdhamini, R. Minus-C subfamily has diverged from Classic odorant-binding proteins in honeybees. Apidologie 54, 12 (2023). https://doi.org/10.1007/s13592-022-00988-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13592-022-00988-5