
Abstract
Background
The shift from solitary to social behavior is one of the major evolutionary transitions. Primitively eusocial bumblebees are uniquely placed to illuminate the evolution of highly eusocial insect societies. Bumblebees are also invaluable natural and agricultural pollinators, and there is widespread concern over recent population declines in some species. High-quality genomic data will inform key aspects of bumblebee biology, including susceptibility to implicated population viability threats.
Results
We report the high quality draft genome sequences of Bombus terrestris and Bombus impatiens, two ecologically dominant bumblebees and widely utilized study species. Comparing these new genomes to those of the highly eusocial honeybee Apis mellifera and other Hymenoptera, we identify deeply conserved similarities, as well as novelties key to the biology of these organisms. Some honeybee genome features thought to underpin advanced eusociality are also present in bumblebees, indicating an earlier evolution in the bee lineage. Xenobiotic detoxification and immune genes are similarly depauperate in bumblebees and honeybees, and multiple categories of genes linked to social organization, including development and behavior, show high conservation. Key differences identified include a bias in bumblebee chemoreception towards gustation from olfaction, and striking differences in microRNAs, potentially responsible for gene regulation underlying social and other traits.
Conclusions
These two bumblebee genomes provide a foundation for post-genomic research on these key pollinators and insect societies. Overall, gene repertoires suggest that the route to advanced eusociality in bees was mediated by many small changes in many genes and processes, and not by notable expansion or depauperation.
Similar content being viewed by others

Background
Social living, and in particular eusociality (the social system in which many individuals forego reproduction), represents one of the major transitions in evolution [1], where a balance between cooperation and conflict must be met [2,3]. Eusociality has arisen multiple times [2,4]. Although the selective bases of the evolution of eusociality are relatively well understood [2,5,6], the evolutionary origins and dynamics of the molecular mechanisms underpinning eusociality remain obscure [7,8], making the understanding of the genomics of eusocial species a priority. Genome sequencing of social and eusocial species is expanding [9-17], but existing data do not span the spectrum of sociality or the phylogenetic diversity of social taxa.
In the Hymenoptera, the honeybees (tribe Apini), stingless bees (Meliponini), and certain ant species display advanced eusocial traits, including a permanent reproductive division of labor between queen and worker castes, worker females that show high degrees of task specialization, and, sometimes, caste polymorphism, and large perennial colonies with complex communication and organization [18,19]. In addition to these advanced eusocial species, the Hymenoptera include species with a spectrum of social traits [2]. The four tribes of corbiculate bees, Apini, Meliponini, Bombini, and Euglossini, are thought to have shared a primitively eusocial ancestor. Subsequently, the Meliponini and Apini evolved advanced eusociality independently, while the predominantly solitary behavior of the Euglossini was secondarily derived [18]. Although rare overall, advanced eusociality has arisen twice in this group, once following the split of honeybee and bumblebee lineages (approximately 77 to 95 million years ago (mya)), and once following the split of stingless bee and bumblebee lineages (approximately 66 to 82 mya) [18].
The ‘primitively eusocial’ bumblebees (Bombus spp., Bombini) share some traits with advanced eusocial bees, yet lack particular aspects that would qualify them as advanced eusocial organisms (Table 1). In comparison to honeybees, they have queen-worker caste differentiation based mainly on body size and physiology, annual colonies of hundreds rather than many thousands of individuals, and worker offspring that have lost the ability to mate, but can reproduce readily by laying haploid (male) eggs [20]. Bumblebees typically exhibit an annual colony cycle (Figure 1), although perennial colonies have been recorded in some bumblebee species such as the neotropical B. atratus [21], and social parasitic cuckoo bumblebees do not found their own colonies. There is a clear value to investigating bumblebees as they hold a key, intermediate position on the eusocial spectrum.

An illustrative colony cycle of bumblebee species living in temperate regions (a). This is representative of the colony cycles of Bombus terrestris (b) and B. impatiens (c). Queen bumblebees emerge from hibernation, establish a nest as a single foundress and provision it with pollen and nectar. Egg batches are laid that develop into female worker offspring. Once these offspring have developed and emerged as adults they take over foraging duties from the queen, and tend to developing brood. After sustained colony growth, males and new queens are produced. These sexuals leave the colony and mate. The new queens hibernate while males and the remainder of the colony perish.
Bumblebees are natively found around the globe, except for sub-Saharan Africa and Oceania, but reach their highest diversity in temperate, alpine, and arctic zones [20]. Two species of bumblebee, B. terrestris and B. impatiens, have in particular been the focus of research activity in a wide variety of fields. These include, among others, social evolution and organization [22-24], caste-structured development [25], learning [26], color vision [27], host-parasite interactions [28,29], plant-pollinator interactions [30], and community ecology [31,32]. The lineages leading to B. terrestris and B. impatiens separated approximately 18 mya [33,34]. B. terrestris is native to the Palaearctic and is common in many parts of Europe, North Africa, and parts of Western Asia, while the Nearctic B. impatiens is frequent in bumblebee communities of eastern North America. The species are placed in distinct sub-genera (B. (Bombus) terrestris and B. (Pyrobombus) impatiens) within the genus Bombus [33] and have some important biological differences (Table 1).
As a major component of the wild pollinator assemblage, bumblebees provide vital ecosystem services [35,36] and are also important for the productivity of agroecosystems [37]. Due to their effective pollination abilities, which are distinct from those of honeybees, bumblebees have also been employed in commercial pollination services [38]. B. terrestris and B. impatiens are both reared commercially and distributed internationally. The key role of bumblebees as pollinators is highlighted by losses in managed honeybee populations placing an increasing pollination burden on alternative pollinators [39]. However, many bumblebee species have also experienced marked population decreases recently [40,41]. Nonetheless, to date, B. terrestris and B. impatiens have proven to be relatively resilient to these declines. Relative abundances of B. terrestris in bumblebee communities in Europe have increased [42], while their absolute abundance has remained relatively stable [43]. B. impatiens has likewise increased in relative abundance in bumblebee communities in North America, since its populations have remained stable against a trend of declines in other bumblebee species [40,44]. In addition to remaining stable in its native range, B. terrestris has also proven to be an effective invader, further expanding its range as the result of human introductions [41,45,46]. Threats to both bumblebees and honeybees come from a variety of sources [47], and recently it has come to light that there is significant overlap of important pathogens between the two [48].
Genomic data form a rich platform on which comparative biology can be based. Comparative analyses of the genomes of honeybees and bumblebees will be crucial for understanding the relationships between these species, the dynamics of the evolution of eusociality, their resistances to pathogens, and their susceptibilities to other threats to pollinator health. Genomes of pollinators, such as bumblebees, will not only help understanding of the pollinator organisms themselves, but will also aid understanding of interactions between pollinators and plants [49]. For both B. terrestris and B. impatiens some genetic, genomic, and transcriptomic resources already exist [8,50-54].
Here we present high-quality draft genome sequences of two bumblebees, B. terrestris (Bter_1.0, accession AELG00000000.1) and B. impatiens (BIMP_2.0, accession AEQM00000000.2), and analyses that highlight both shared and divergent features compared to the honeybee (A. mellifera), other Hymenoptera, and further selected insects.
-
We present high sequence coverage assembled genomes of B. terrestris (249 Mb) and B. impatiens (248 Mb).
-
The two bumblebee genomes exhibit extensive synteny, with limited rearrangements over the estimated 18 My of divergence between the two lineages.
-
We find relatively few repetitive elements and a low diversity of transposable elements, although there is some evidence of recent activity.
-
Orthology and protein domain analysis uncovered bee- and bumblebee-specific genes and domains, with hints of evolutionary processes differentially acting upon aspects relating to chemosensation and muscle function in the bumblebee lineage.
-
B. terrestris and B. impatiens are extremely similar in terms of gene content related to developmental pathways in molting, metamorphosis, and exoskeleton dynamics. This gene repertoire shows striking similarities among social and non-social Hymenoptera.
-
A similar set of genes underlying haplo-diploid sex determination is present relative to honeybees, despite an alternative primary signal for sex determination being employed.
-
Genes involved in behavior, neurophysiology, and endocrinology are broadly conserved between A. mellifera and bumblebees, yet limited differences do exist, and in particular among Juvenile Hormone Binding Proteins this may be connected to functional differences between these species.
-
Xenobiotic detoxifying enzymes were found to be depauperate, as in A. mellifera, which has consequences for the ability of these species to deal with novel environmental xenobiotics, such as insecticides.
-
Genes involved in chemoreception show expected complex patterns of gene birth and death. However, surprisingly, the gene repertoire of B. terrestris suggests that, relative to honeybees, bumblebees emphasize gustation over olfaction.
-
Venom constituents, in general, are highly similar between honeybees and bumblebees.
-
While components of all major immune pathways are present, as in A. mellifera, the complement of immune genes in the bumblebees is much reduced relative to Dipteran models, suggesting this is not a honeybee-specific characteristic, nor is it linked to advanced eusociality. Rather, it is likely that a reduced immune repertoire is basal to the bee lineage.
-
RNAi core genes, RNA editing, and DNA methylation genes and genome wide patterns are highly conserved between A. mellifera and the two bumblebees.
-
MicroRNAs (miRNAs) show a distinct pattern between the bumblebees and honeybees. Unique miRNAs were identified in both groups as well as potentially functionally relevant changes in conserved miRNAs. These are excellent candidates that may tune key biological differences between advanced eusocial honeybees and primitively eusocial bumblebees.
Results and discussion
Genome sequence and organization
We sequenced and de novo assembled the genomes of B. terrestris and B. impatiens from DNA derived from haploid males. B. terrestris sequence reads were assembled into a draft genome containing 236 Mb of sequence and spanning 249 Mb including estimated gaps (Table 2). Further genome information and statistics for Bter_1.0 can be found in Additional file 1. The B. impatiens genome was sequenced and de novo assembled to produce an assembly containing 243 Mb of sequence spanning 248 Mb including estimated gaps (Table 2). These genomes can also be accessed through BeeBase ([55], hymenopteragenome.org), which provides Genome Browser capabilities and BLAST searches against scaffolds and all gene predictions of both bumblebee genomes. Both genomes had high completeness as judged by presence of conserved gene sets (Additional file 1) and representation of independent transcriptome data.
Synteny between the bumblebee genomes
Large-scale synteny is observed between the 18 chromosomes of B. terrestris and their corresponding B. impatiens scaffolds and contigs (details of coordinates in Additional file 2). Ninety-four B. terrestris scaffolds from the 18 chromosomes with the addition of 11 unplaced scaffolds (average length, 2.34 Mb; median, 1.34 Mb; range, 0.7 kb to 13.65 Mb; total length, 220.2 Mb) are covered by 101 synteny blocks formed from B. impatiens scaffolds and contigs (average synteny block length, 2.25 Mb; median, 1.16 Mb; range, 1.7 kb to 12.9 Mb) spanning 226.9 Mb. Thus, 89% of the investigated B. terrestris assembly is covered by synteny blocks representing 91% of the B. impatiens assembly. While synteny is high, it is likely an underestimate, being constrained by the fragmented genome assemblies of the two species. The existence of 14 large-sized synteny blocks (>5 Mb) corroborates this. Moreover, only eight and 10 cases were detected of intra- and inter- chromosomal rearrangements, respectively. The finding of a high degree of synteny between both bumblebee genomes is striking, as it is known that several social bees, including B. terrestris, have high genomic recombination rates [53,56]. This would lead to the expectation of higher frequencies of genomic rearrangements. However, these results concur with comparative linkage map based analyses suggesting a high conservation of genetic architecture within the Apidae [53].
Repetitive elements in the bumblebee genomes
The B. terrestris and B. impatiens genomes were found to have 1,043 and 1,688 de novo predicted repetitive elements, respectively, of which 812 and 1,304 were validated by annotation of at least one complete copy. In total, 14.8% (36.2 Mb) of the B. terrestris assembly and 17.9% (44.6 Mb) of the B. impatiens assembly was found to be repetitive, with the diversity and abundance of transposable (interspersed) elements appearing similar across the two species (Additional file 1). Class I retroid elements and derivatives make up a large proportion of the genomes (8.5% in B. terrestris, 12.2% in B. impatiens). Gypsy is the most common long terminal repeat (LTR) retrotransposon covering 2.4 Mb in B. terrestris and 4.8 Mb in B. impatiens. Non-LTR retroid long interspersed elements (LINEs) have a similar cumulative length, with the majority being Jockey-like (2.6 Mb in both bumblebee species). Short interspersed elements (SINEs) are scarce. A major fraction of retroid elements (13.8 Mb in B. terrestris and 18.8 Mb in B. impatiens) were classified as large retrotransposon derivatives (LARD) or terminal repeat retrotransposons in miniature (TRIM). Class II DNA transposons were less frequent, with the majority being terminal inverted repeat (TIR) transposons, of which only Mariner and PiggyBac elements were common. Numerous repeat elements could not be assigned to a class (3.9 Mb in B. terrestris and 6.7 Mb in B. impatiens), and require further investigation.
The majority of the repeat elements appear shared between the two bumblebees. A large fraction of the Gypsy and Mariner elements were very similar to previously known transposable elements (two Gypsy, two Mariner) in B. terrestris (RepBase v17.01, [57]). The few R2 clade elements show a more distant similarity to the single previously described R2 element in A. mellifera. Other classified retroid elements show similarities to elements in other insect species, including the wasp Nasonia vitripennis, the ants, mosquitoes, and Drosophila. Interestingly, some of the bumblebee PiggyBac elements showed high similarities to other such elements from the beetle Tribolium castaneum or the moth Bombyx mori. Despite the lineage divergence time of 18 My, we found high degrees of sequence similarity between subsets of Gypsy, Mariner, and PiggyBac elements in B. terrestris and B. impatiens, suggesting a recent invasion by horizontal transfer into both species. A number of transposable elements are present in potentially active copies, with a high copy number indicating recent activity.
The two bumblebee genomes have an overall low number of transposable elements, together with a low diversity relative to other sequenced arthropods that typically have much higher percentages of repetitive DNA with higher diversity. For example, repetitive DNA in the genome comprises of 48.8% in the centipede Strigama maritima [58], 30% in the beetle T. castaneum [59], 38% in the aphid Acyrthosiphon pisum [60], over 30% in the wasp N. vitripenis [61], and typically greater than 20% in Drosophila [62] and ant species genomes [9,11,13,15,63]. The two bumblebee genomes contain only very small fractions or are almost devoid of Copia, BelPao, Retrovirus, and many of the LINE retroid elements, which are more common in other genomes [13,60,61]. Similarly, many superfamilies of DNA transposons are absent or only found in small quantities. For example, hAT and P elements, frequent in other insects [13,60,64], are scarce in these bumblebees. Some ant species have similar percentages of repetitive DNA in their genomes as the bumblebee genomes presented here [9,12], but only extremely specialized insects, the figwasp Ceratosolen solmsi (6.4%) [65] and an Antarctic midge Belgica antarctica (0.49%) [66], exhibit greatly reduced percentages in comparison. Within the Arthropoda, but outside of the Insecta, Daphnia pulex has a genomic repeat content of 9.4% [67]. A low percentage in A. mellifera (9.5%) also [16], however, suggests that the overall low number of transposable elements and low diversity in bumblebees is more deeply rooted in the Apinae.
Predicted protein coding genes in the bumblebee genomes
Protein-coding genes were predicted from the Bter_1.0 and BIMP_2.0 assemblies using a diversity of de novo pipelines (NCBI RefSeq and Gnomon pipelines, AUGUSTUS, SGP2, GeneID, Fgenesh++ and N-SCAN; see Additional file 1). A merged gene set for each species was produced using GLEAN [68]. Targeted manual annotation was used to validate 657 gene models from B. terrestris and 346 gene models from B. impatiens. Approximately one-quarter of the automated models (24%) were edited, usually only to correct start and stop codon usage and intron-exon boundaries using transcriptome and comparative evidence (see Additional file 2 for details of these genes, along with gene information of species-specific gene names reported in some subsequent sections (for example, Bter_ or Bimp_)). Analyses focusing on specific gene families and pathways identified additional issues with some automated predictions (see below, for example, chemoreceptors).
Analysis of orthology relationships of bumblebee genes
Ortholog analysis was carried out with OrthoDB6 [69,70] based on the Refseq gene sets of B. impatiens and B. terrestris. As expected, the vast majority of genes have orthologous relationships across the Hymenoptera (Figure 2). Ortholog sets that are only found in particular lineages are likely to play important roles in lineage-specific biological traits, and thus deserve further attention. The addition of the bumblebee genomes allowed for the identification of 38 orthologs specific to bees (B. terrestris, B. impatiens, A. florea, A. mellifera) (Additional file 2). Of greater interest for bumblebee biology are the 118 orthologs that, at this time, are found solely in the bumblebee lineage (Additional file 2). These bumblebee-specific ortholog groups were in general poorly annotated with InterPro domains [71], but 13 of the 24 orthologs in which domains could be found contained an olfactory receptor domain (IPR004117). Further, one bumblebee lineage specific gene ortholog (OrthoDB group: EOG6VDNJ0) has likely duplicated in B. terrestris. Genes containing this domain are seven-transmembrane proteins and are candidate odorant receptors in other species. These preliminary insights suggest alterations in chemosensation in the bumblebees, further explored below. However, considerable work will be required to discover how these uncovered bumblebee-specific genes relate to bumblebee biology.

Bumblebee orthology with selected Hymenopterans and representative species from other insect orders. The maximum likelihood phylogenetic tree built from the concatenated alignments of 2,294 single-copy orthologs recovers the expected phylogeny rooted with the human body louse, Pediculus humanus. The tree highlights the pairs of closely-related bumblebees (Bombus terrestris and Bombus impatiens), honeybees (Apis mellifera and Apis florea), and fungus-growing ants (Atta cephalotes and Acromyrmex echinatior). It also shows slow average rates of molecular evolution in the Hymenopterans, similar to the flour beetle, Tribolium castaneum, but much slower than the silk moth, Bombyx mori, the malaria mosquito, Anopheles gambiae, and the fruit fly, Drosophila melanogaster. The bars represent the total gene counts in each species partitioned according to their homology to genes in the other species and other arthropods: from universally present single-copy orthologs (dark blue, left) to lineage-specific orthologs, and genes with no detectable orthology (gray, right). A small fraction made up of about 100 to 150 genes in each of the bee and ant species exhibit orthology only to genes from the most closely-related species (red, green, purple). The inset boxplots show the distributions of percent amino acid identities between pairs of Bombus, Apis, and Attini (ants) universal single-copy orthologs, where the identity is much higher between the bumblebee orthologs than between the honeybees or the ants.
Patterns of protein domain evolution in B. terrestris
The evolutionary dynamics of protein domains are often distinct from dynamics on the gene level, and sometimes signals of adaptation only become apparent at the protein domain level. Protein domains of B. terrestris were compared to several reference species (A. mellifera, Atta cephalotes, Culex cinquefasciatus, Drosophila melanogaster, Daphnia pulex, Harpegnathos saltator, Pediculus humanus, T. castaneum) (Additional file 1). Domains found uniquely in one species compared to closely related species can indicate horizontal gene transfer. One B. terrestris domain not found in the other arthropods was an RNA-dependent RNA-polymerase (Pfam domain PF00978). This domain is found on predicted gene au6.g7950 between 97 and 133 kb of scaffold CM001185.1. The predicted gene has 17 exons, three of which are annotated with Pfam domains, with PF00978 located in the eighth exon. This domain is normally part of RNA viruses, and its presence in the B. terrestris genome may be a nuclear insertion of genetic material from a viral infection, but no longer encode a functional RNA-dependent RNA-polymerase.
Expansions and contractions of domains relate to lineage-specific changes in domain copy numbers. These can be linked to gene duplication or loss, but can also be independent of this process. When comparing B. terrestris domains to all reference species, only two domains were significantly different in their occurrences (P <0.05, Fisher’s exact test). Both of these domains (PF07530 (P <0.001) and PF07727 (P = 0.001)) appear related to transposable elements [72,73]. In comparison to A. mellifera alone, 16 expansions or contractions were found in B. terrestris, with most being related to transposable elements (Additional file 2) or the zinc finger receptor family. However, three of these domain changes are of further interest. A major royal jelly protein (PF03022 (P = 0.007)), involved in honeybee larval nutrition and caste differentiation [74] is contracted in B. terrestris, with six copies in comparison to the 22 in A. mellifera. PF07993 (P = 0.01), involved in age-related decreases of transcript levels in D. melanogaster [75], is expanded to six copies in B. terrestris, but is not present in A. mellifera. Three seven-transmembrane receptors involved in chemoreception (PF02949 (P <0.001), PF00001 (P = 0.016), PF08395 (P = 0.016)) are contracted in B. terrestris based on analyses of these domains, and are found at 12%, 53%, and 0%, respectively, of their abundances in A. mellifera. A comparison of the bees (B. terrestris and A. mellifera) to the ants (A. cephalotes and H. saltator) revealed seven significant domain expansions or contractions, with four being related to transposable elements. The others were, in bees relative to ants, a contracted trypsin domain (PF00089), an expanded trypsin inhibitor domain (PF01826), and an expanded zinc finger domain (PF13912).
Domain repeat numbers within a protein often change rapidly, with multiplication of a domain within a protein potentially indicating a strong signal of selection. Repeat numbers of domains within proteins were compared between B. terrestris and A. mellifera. Nine domains were found to differ significantly in their repeat numbers. Several of these domains have functions related to muscle, and hint at potential selection pressure on the muscle apparatus of bumblebees. These adaptations could be related to distinct muscle features of bumblebees, for example, in warming up the flight muscles [20].
Development related genes in the bumblebee genomes
Examination of both bumblebee genomes for developmental genes indicates that the developmental gene complement of A. mellifera, with its loss of specific developmental genes, is shared with bumblebees, strengthening the finding that these are indeed missing from the A. mellifera genome and indicating that early development may have evolved differently in the corbiculate bees as a whole.
The complement of genes involved in segmentation and dorsoventral patterning are identical to those of A. mellifera. Both bumblebees have all components found in the A. mellifera for Notch, Wingless, hedgehog, and TGF-beta signaling. They also have similar runt [76], E(spl) [77], and Hox complexes [78] to A. mellifera. An interesting finding from the A. mellifera genome sequence was that a set of genes that act in early patterning of Drosophila embryos were absent [78]. This set of genes (torso, trunk, gurken, bicoid, swallow, and oskar) is also completely lacking in the bumblebee genomes. While some of these genes arose in the dipteran lineage (for example, bicoid and swallow) [78-80], others are found in the genomes of hemimetabolous insects such as the pea aphid (torso) [81], or in other Hymenoptera (for example, torso, oskar) [82]. This indicates within-Hymenoptera lineage-specific loss of at least the torso and oskar genes.
Hemocyanin derived genes include hexamerins (hex), involved in metamorphic molting [83], and prophenoloxidases (PPO), associated with melanisation and exoskeleton pigmentation [84]. As in A. mellifera, four hex genes, with the genomic clustering of three of these genes also being conserved, and a single copy of PPO, with transcriptomic evidence for alternative splicing, were found. Other genes with a single copy and likely one-to-one orthologous relationships include many involved in post-embryonic development, including ftz-f1, Broad-complex, calponin (Chd64), eclosion homone (EH), ecdysis triggering hormone (ETH), bursicon α and β, cuticular peroxidase, dopa-decarboxylase (DDC), chitinase, and tyrosine hydroxylase (TH). Predicted gene models and/or transcriptional evidence indicate alternative isoforms for most of these genes in bumblebees. Tweedle, apidermins, and other cuticular proteins (such as CPLCP and CPF) have the same numbers as A. mellifera [85]. Multicopper oxidases (MCOs), including laccases, have roles in development and a wide variety of other biological processes [86]. Seven MCO genes are found in both bumblebee genomes, while there are only five in A. mellifera and 11 in Nasonia spp. These genes show clear orthology, but also species-specific expansion (for example, Nasonia spp.) and loss (for example, A. mellifera). CPR cuticular proteins are encoded by 37 to 58 genes across sequenced Hymenopterans, which is considerably fewer than are found in other insects (86 to 241). Other genes involved in development, including members of the basic Helix-Loop-Helix/Per-Arnt-Sim (bHLH-PAS) transcriptional factors, show deep conservation within the insects.
Genes encoding proteins that are important for development and differentiation of the central nervous system and the brain, cell polarity, axon guidance, Malphigian tubule morphogenesis, eye development, and pathways like the Notch signaling pathway are conserved in the bumblebees. The genes achaete, scute, lethal of scute, and asense are part of the achaete-scute complex of D. melanogaster [87] and encode transcriptional activators of the bHLH class. They are important proneural genes that instruct clusters of cells to become competent to form neuroblasts [88] and play a critical role in the formation of the central nervous system of the embryo and the peripheral nervous system (sensory bristles) of adults [87]. Of the four genes of the achaete-scute complex, only one copy is present in B. terrestris while two copies are found in A. mellifera [89]. This finding suggests that a single gene of the achaete-scute gene family is sufficient in B. terrestris to form the neuroblasts and the CNS. Another gene involved in the development of the CNS, escargot [90] is not present in B. terrestris. In D. melanogaster, escargot acts with redundant function with other members of the snail protein family (snail and worniu) to control embryonic central nervous system development [90]. The homeobox containing genes ladybird late and ladybird early encode transcription regulators, which play an important role in neurogenesis, myogenesis, and cardiogenesis [91], and are also missing in B. terrestris.
Haplo-diploid sex determination in the bumblebee genomes
Hymenoptera species do not possess sex-specific chromosomes, but instead employ a haplodiploid mode of sex determination, which typically leads to males arising from unfertilized eggs (haploid) and females from fertilized eggs (diploid). Single-locus sex determination has been extensively studied in A. mellifera, where the initial signal has been identified to be the multiallelic gene complementary sex determiner (csd) [92]. Bumblebees share orthologs for numerous genes known to be involved in Drosophila and Apis sex determination such as doublesex (dsx), transformer 2 (tra 2), fruitless (fru), and transformer (tra)/feminizer (fem). The single sex determination locus described for A. mellifera harbors the tandemly arrayed paralogous genes csd and fem [93], whereas in B. impatiens and B. terrestris the paralog of fem, feminizer 1 (fem1), is located on different chromosomes. In contrast to the allelic variability of Apis csd, fem1 lacks allelic variability based on sequences from natural B. terrestris populations, suggesting that fem1 is unlikely to be an allelic factor acting as a primary signal in the sex determination pathway (Hasselmann et al., unpublished). In other Hymenopteran genomes than Apis and Bombus, paralogous copies of fem (tra) have been found in ants and the Halictid bee Lasioglossum albipes, suggesting this gene duplication as a potentially ancestral event in the early evolutionary history of Hymenopteran species [15,94,95]. However, analysis provides evidence that csd and fem1 are not orthologs, and originated independently by gene duplication from the fem gene in A. mellifera, the bumblebees, and ants [96,97]. Bumblebee dsx and fem are sex-specifically spliced, consistent with the evolutionary conservation of the pathway at this level. Alternative splice variants are also found for fem1, which is not the case for csd in A. mellifera (Hasselmann et al., unpublished). For three genes (sisterless A, outstreched, suppressor of variegation 3-7) no potential orthologs were identified in the bumblebee or any other Hymenopteran genome. All genes involved in dosage compensation in D. melanogaster have orthologs in the bumblebee genomes, despite the lack of sex chromosomes. These genes might have additional, for example chromatin-related, functions in the bumblebee. It is also tempting to speculate about an association to haplodiploidy given the complexity by which those genes orchestrate with the transcription regulating machinery [98]. Consequently, for example, these orthologs could fine-tune the transcription of maternally and paternally provided genetic material in fertilized eggs, compensating allele-specific differences.
Behavior, neurophysiology, and endocrinology related genes in the bumblebee genomes
Advanced eusociality requires extensive behavioral coordination, and castes typically differ in their behavioral phenotypes, spatially or temporally. In the primitively eusocial bumblebees, while behavioral differentiation is present, it is not as distinct as in the advanced eusocial honeybees, making the landscape of genes involved in behavior and the neuronal and physiological processes underlying behavioral phenotypes an interesting avenue of investigation.
Innate circadian clocks govern the daily timing of many organismal processes, from gene expression to behavior. The set of clock genes in bumblebees is highly similar to those of A. mellifera [99]. Only a timeout (Tim2), but no timeless (Tim1), and a mammalian-like cryptochrome (Cry-m), but not a Drosophila-like cryptochrome (Cry-d) were found in the bumblebee genomes. Thus, the core circadian feedback loop is mammalian-like rather than Drosophila-like, a finding also recently described in ants [100].
The Takeout/juvenile hormone binding proteins (To/JHBP), present only in insects, share a defining domain thought to bind small lipophilic molecules such as juvenile hormone (JH) [101], yet the exact ligands are mostly unknown. In Drosophila, To is linked to circadian rhythms, with To mutants showing abnormal locomotor activity rhythms and rapid death on starvation [102,103]. In A. mellifera, eight genes of the To/JHBP family have been identified [104]. The genomes of the two bumblebees each contain 11 putative To/JHBP family genes (Additional file 1). Interestingly, two of these genes have no true orthologs in A. mellifera (GB13060_1, and GB17010), but orthologs are present in Nasonia, suggesting the loss of these two genes since the separation of the honeybee and bumblebee lineages (Additional file 1). It would be interesting to test if these differences in To/JHBP complements are related to different physiologies of honeybees and bumblebees, such as the apparent differences in JH signaling. In bumblebees JH regulates fertility and female reproductive physiology [105], whereas in adult honeybees JH influences worker division of labor but not fertility and reproduction [106]. All protein models of the identified genes contain significant JH-binding domains, and signal peptides were identified by at least one of the two methods used in all except Bter_GB17010 and Bimp_GB17010 (Additional file 1). All putative To/JHBPs are co-localized on B. terrestris LG B09, apart from Bter_GB19811, which is located on LG B08. The location of the JHBPs in the genome of B. terrestris is comparable to that in A. mellifera, suggesting high synteny for this group of genes, which is consistent with the premise that the To/JHBP family was created by ancient duplication events.
The genes of the cys-loop ligand-gated ion channel (cysLGIC) superfamily mediate synaptic transmission in insects. The genomes of B. terrestris and B. impatiens both contain the same complement of 21 cysLGIC genes, with 11 of these genes encoding putative nicotinic acetylcholine receptor subunits (nAChRs), while the remainder of the bumblebee cysLGIC superfamily include genes for ion channels gated by gamma-aminobutyric acid (GABA receptors), glutamate (GluCls), and histamine. This complement of 21 cysLGIC genes is the same as found in A. mellifera [107]. Studies in honeybees have shown that nAChRs, GABA receptors, and GluCls play key roles in behavior, such as olfactory learning and memory [108,109]. Insect cysLGICs are also of importance as they are targets of widely used insecticides, examples of which are fipronil (which acts on GABA receptors and GluCls) and neonicotinoids (which act on nAChRs) [110]. The bumblebee nAChRs may mediate sublethal effects of neonicotinoid pesticides on foraging behavior and colony traits [111-113]. CysLGIC sequence information from diverse species, including key pollinating insects, is a valuable starting point for understanding the interaction of insecticides with their targets, and may prove instructive in the future design and development of improved insecticides with enhanced specificity and reduced effects on non-target beneficial species.
Biogenic amines, neuropeptides, protein hormones, and their G-protein-coupled receptors (GPCRs) play a central role in the physiology of insects and control many important processes, including behavior, development, feeding, and reproduction [114]. Insects have 16 to 22 biogenic amine GPCRs for identified insect biogenic amines (acetylcholine, adenosine, dopamine, octopamine, tyramine, and serotonin) [115,116]. Both bumblebees have a similar set of 20 biogenic amine GPCRs (Additional file 1). Compared to other sequenced insects, one octopamine receptor was found to be duplicated in the two bumblebees, as was also found in A. mellifera. The two bumblebees have a similar set of 34 neuropeptide preprohormone genes coding for approximately 65 different neuropeptides (Additional file 1). However, functionality of corazonin, thought to be involved in copulation behavior [117], carbohydrate and lipid mobilization [118], and stress [119], is likely different in B. impatiens, as the preprohormone cannot be cleaved to a functional corazonin (Additional file 1). Twelve neuropeptide genes found in other arthropods are absent in both bumblebee species, with their GPCRs also absent where they are known. Interestingly, the two bumblebee species have a similar, but still unique, neuropeptide suite compared to A. mellifera, with sulfakinin found only in A. mellifera and trissin found only in bumblebees (Additional file 1). This unique suite of neuropeptides is likely to underlie bumblebee-specific physiology and behavior.
TRP (Transient Receptor Potential) channels are activated by diverse stimuli and function as the primary integrators of sensory information such as vision, thermosensation, olfaction, hearing, and mechanosensation. The TRP superfamily is divided into seven subfamilies (TRPA, TRPC, TRPM, TRPML, TRPN, TRPP, and TRPV) [120]. The bumblebee genomes contain the same set of TRP channel genes (5 TRPA, 3 TRPC, 1 TRPM, 1 TRPML, 1 TRPN, and 2 TRPV subfamily members) as A. mellifera and N. vitripennis. Both bumblebee species lack TRPA1, but maintain TRPA5 (a TRPA subfamily member lost in Diptera) and HsTRPA. Three other conserved TRPA channels, Painless, Pyrexia, and Wtrw, are present. Thus, the pattern of TRPA subfamily members is conserved between these two bumblebees, A. mellifera, and N. vitripennis.
Xenobiotic detoxifying enzymes and related genes in the bumblebee genomes
Overall bumblebees, similar to honeybees, have a reduced set of detoxification enzymes. Being mutualistic pollinators, bumblebees are not faced with a plethora of toxic plant secondary metabolites that require detoxification, as herbivores are. This, and the potential of low incidence of xenobiotics due to their social lifestyle, could account for why these xenobiotic detoxifying enzymes are so impoverished in these species. However, xenobiotics are now encountered in the form of systemic insecticides, and investigations of how a general lack of detoxification related genes, along with species-specific alterations, influences susceptibility is vital to understand one of the major purported threats to pollinator health.
Glutathione-S-transferases (GSTs), carboxyl/cholinesterases (CCEs), and cytochrome P450 monooxygenases (p450s) are involved in the detoxification of xenobiotics, along with playing roles in key physiological pathways. These gene families were found to be extremely depauperate in the A. mellifera genome [121].
GST and CCE numbers are comparable in the two bumblebees to those numbers found in A. mellifera (Table 3). Thus, the reduction in numbers in A. mellifera [121] is not unique, but rather taxonomically more widespread. Despite a similarity in overall numbers, there are key changes in CCEs between the bumblebees and honeybees on the level of clades and their classes. In bumblebees, relative to A. mellifera, dietary/detoxification associated CCE genes are reduced in number, while hormone and semiochemical processing associated CCE genes are increased.
The bumblebee genomes contain 44 putatively functional cytochrome P450 monooxygenase genes (P450s) and seven pseudogenes, very similar to the complement of 46 P450s encoded in the genome of A. mellifera, but considerably smaller than the number of P450s in the genomes of most other holometabolous insects (Table 3) [12,59,121,122]. The bumblebee genomes include all other expected orthologous P450s in the CYP2 and mitochondrial clans, which are involved in ecdysteroid hormone synthesis and breakdown [123]. The insect steroid hormone, 20-hydroxyecdysone (20E), controls and coordinates insect development through the ecdysteroid-signaling cascade. Enzymes responsible for 20E synthesis are a group of cytochrome P450s (Additional file 1). To date, four P450 enzymes, namely CYP306A1 (Phantom, Phm), CYP302A1 (Disembodied, Dib), CYP315A1 (Shadow, Sad) and CYP314A1 (Shade, Shd), involved in ecdysteroid biosynthesis have been identified and characterized. Additionally, a group of paralogous CYPs (CYP307A1 (Spook, Spo), CYP307A2 (Spookier, Spok), the paralog gene of Spo, and CYP307B1 (Spookiest, Spot)) are identified. They are all involved in the initial conversion process from 7-dehydrochoresterol into ketodiol, but their biochemical functions are not well understood [124]. They are called the Halloween genes. All Halloween genes present in A. mellifera are found in B. terrestris and B. impatiens (Additional file 1). Similar to A. mellifera, no ortholog for Spo, which is present in multiple other hemimetabolous and holometabolous insects, was found. Phylogenetic analysis demonstrated the identity of the Spot paralog and also confirmed the identity of the other Halloween genes (Additional file 1).
Honeybees and bumblebees are uniquely depauperate in the CYP4 P450s, as each bee genome encodes just four well-conserved orthologs in this clan, while other insect genomes contain a great diversity of genes in this group. Lack of CYP4 P450 diversity in the bees is somewhat surprising because this group has been associated with pheromone synthesis and breakdown [125]. Two of the four CYP4 P450s shared by bees, the pair of CYP4G orthologs, are known to be involved in the synthesis of cuticular hydrocarbons in other insects [126] and may be involved in the production of secreted wax in bumblebees and honeybees.
The CYP3 group members are the only P450s in the sequenced bees that do not display clear 1:1 orthology with other insects. This clan shows evidence of recent gene duplication and divergence in species specific ‘blooms’ [123]. Members of the CYP3 clan detoxify pesticides and natural xenobiotics in honeybees and other insects [127,128]. While the A. mellifera and two bumblebee genomes appear to encode similar numbers of CYP3 P450s, this gene count masks gene birth and death events occurring in each genome. Apis mellifera has three CYP9Q P450s, which metabolize synthetic insecticides [127]. While B. impatiens also has three CYP9Qs, B. terrestris has a single putatively functional CYP9Q P450.
Chemoreceptors in the bumblebee genome
Chemosensation plays a major role in social interactions in insect societies, and is critical to the ecological interactions of bees. The odorant receptor (OR) family of seven-transmembrane proteins in insects mediates most of insect olfaction [129], with additional contributions from a subset of the distantly related gustatory receptor (GR) family, for example, the carbon dioxide receptors in flies [130], and a subset of the unrelated ionotropic receptors (IRs) [131].
Odorant receptors (ORs)
The A. mellifera genome revealed an expansion of the OR family relative to previously sequenced fly genomes [132], with a total of 177 genes (updated in [11,12]). This expansion has been even greater in other sequenced Hymenoptera, including Nasonia wasps with around 300 OR genes [133] and several ant species with around 400 OR genes [11,12,15,134]. Analysis of Bombus ORs indicates that they have a slightly less diverse OR family than A. mellifera, with 164 genes (Additional file 1). There are just five pseudogenes (3%), which is even lower than the 5% in A. mellifera. The result is 159 apparently intact OR proteins, although there is a small subfamily (BtOr128-139, related to AmOr97-105) with an additional short coding exon for the start codon, which usually could not be confidently identified, so their functionality remains uncertain. As expected, there is a single conserved ortholog of the DmOr83b protein, now called Orco [135], sharing 92% amino acid identity with AmOrco (Or2) and 63% with DmOrco (Or83b). There are no other orthologous relationships of bee ORs to the Drosophila ORs [132]. Comparing B. terrestris and A. mellifera ORs there is a combination of single orthologs for many genes, duplications of genes in one or both species, several large species-specific gene lineage expansions, and at least 22 gene losses, reflecting the birth-and-death gene family evolution typical of these receptors (Additional file 1). The largest known tandem duplication of insect chemoreceptors is AmOr1-61 with equivalents in Bombus of BmOr1-46, and this large subfamily contains the only Hymenopteran OR for which a ligand is known, AmOr11, which perceives the major bee queen pheromone component 9-ODA [136].
Another large subfamily is the 9-exon gene subfamily, totaling 49 genes (BtOr116-164 and AmOr97-113, 122–139, 140, 159, 172–177), which all share the same gene structure, except that AmOr97-113 and BtOr128-139 have an additional very short 5’ exon containing the start codon. The major expansion of this subfamily in ants was suggested to indicate that it comprises the cuticular hydrocarbon receptors involved in nestmate and kin recognition [11,12,134]. This subfamily has the largest species-specific expansions in both B. terrestris and A. mellifera (Additional file 1). Based on branch lengths, these also appear to be among the most rapidly evolving ORs. Meanwhile, the oldest lineages in this subfamily, AmOr 159, 176, and 177 and BtOr156, 157, and 159, each appear to have been lost from the other species (indeed a non-functional fragment of an AmOr177 ortholog remains in the bumblebee genome), suggesting that their functions are being outlived. This 9-exon subfamily also contains the first clear case of trans-splicing observed in the insect chemoreceptor family, something that is becoming better known in other insect genes [137]. The lineage of BtOr161-163 and AmOr140 has the first coding exon in the appropriate location upstream of the remaining exons, but in reverse orientation. While this unusual arrangement was recognized for AmOr140, it was discounted and that gene was previously treated as having an unrecognized N-terminus (AmOr140NTE) [132]. Discovery of the same arrangement for this exon in the related bumblebee genes makes it clear that these are trans-spliced genes.
Gustatory receptors (GRs)
The GR family of seven-transmembrane proteins in insects mediates most of insect gustation [129], as well as some aspects of olfaction. In contrast to the OR family, the GR repertoire was considerably reduced in A. mellifera compared with flies, at just 12 genes [11,12,132], which is far fewer than the number found in the other available Hymenoptera [11,12,133,134]. Compared to A. mellifera, somewhat surprisingly, B. terrestris has 25 GR genes (Additional file 1). Of these, 23 are apparently intact proteins. The phylogenetic tree (Additional file 1) reveals the relationships of B. terrestris and A. mellifera GRs in relation to those of D. melanogaster. As is the case for A. mellifera [132] and other Hymenoptera examined to date [138], there are no B. terrestris orthologs for the carbon dioxide receptors (DmGr21a and 63a). B. terrestris has conserved orthologs for the two candidate sugar receptors in A. mellifera, BtGr1/2 [139]. The BtGr3/AmGr3 lineage is the ortholog of the DmGr43a receptor that has recently been shown to be a fructose receptor that also functions as a brain nutrient receptor [140]. The AmGr4/5 lineage appears to be an Apis-specific duplication, because there is only one gene in B. terrestris, and it is now a pseudogene (BtGr4PSE). Remaining B. terrestris and A. mellifera GRs have no convincing relationships with D. melanogaster GRs to allow for functional inference, but show expected patterns of birth-and-death typical of the chemoreceptor and other environmentally-relevant gene families. While Gr6 and Gr7 are simple orthologs, the others provide interesting comparisons. BtGr5 is an intact relative of the large set of highly degraded pseudogenes in the A. mellifera genome, represented here by the AmGrX-Z constructs. This lineage apparently both expanded and completely pseudogenized within the Apis and related bee lineages. Gr10 and Gr11 are pseudogenes in B. terrestris and Apis, respectively, so their respective functions have been lost. The AmGr8/9 pair of duplicated genes have experienced a repeated set of duplications as a pair in B. terrestris, yielding five genes each (BtGr8/9, and 14–21), although BtGr17 is a pseudogene. The newly recognized AmGr12 is also repeatedly duplicated in B. terrestris, again yielding five genes, all still intact (BtGr12 and 22-25). Finally, a truncated version of a highly divergent GR was recognized in each genome, called Gr13. Gr13 is missing the usually conserved C-terminus, but is otherwise a seemingly intact gene.
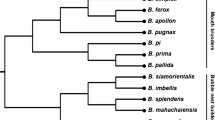
The most impressive feature of the GR gene family in B. terrestris is the expansion of three A. mellifera GRs into 15 genes in B. terrestris (Figure 3). These expansions are all very recent, being unique to Bombus, and have short branches to each new gene. Thus, while the total GR family size in B. terrestris is considerably larger than that of A. mellifera, the difference is not an ancient one involving the loss of Apis genes, but rather a lineage-specific and recent expansion in bumblebees. Unfortunately, there is little information on what ligands these novel Bombus-specific GRs might detect, but it is likely that they are bitter taste receptors [141], perhaps related to the more diverse nest-building habits of bumblebees.

Section of the phylogenetic tree of the Bombus terrestris, Apis mellifera, and Drosophila melanogaster gustatory receptors (GRs) showing the impressive B. terrestris-specific expansion. This is a corrected distance tree. B. terrestris and A. mellifera proteins are highlighted in orange and blue, respectively, as are the branches leading to them to emphasize gene lineages. Bootstrap support level in percentage of 10,000 replications of uncorrected distance analysis is shown above major branches. The full phylogenetic tree of GRs can be found in Additional file 1.
Ionotropic receptors (IRs)
The IR family also contributes to insect olfaction and gustation. Apis mellifera, Nasonia, and various ants do not differ much in their repertoire sizes [11,12,131], and the IR family in B. terrestris is comparable, with 22 genes (Additional file 1). The IR family contains several conserved orthologous genes shared across insects. The co-receptor IR8a and 25a genes are unusually highly conserved and cluster confidently with the ionotropic glutamate receptors from which they clearly evolved [131]. They were therefore used as an outgroup to root a phylogenetic tree of IRs (Additional file 1). Somewhat surprisingly, IR25a has a duplicate gene copy in B. terrestris (BtIR25a.2) that is rapidly evolving, and encodes only the second half of the protein. While this could be a pseudogenic copy, it has the hallmarks of a functional gene, and is the only known instance of a duplication of IR25a to date. The other orthologous lineages are rather more rapidly evolving, including IR93a, 76b, and 68a. A. mellifera and B. terrestris have several highly divergent IRs, only one of which (IR218) was noted in Croset et al. [131]. The new genes are IR328-339, which mostly have simple orthologs in both species, except that AmIR338 is a pseudogene in B. terrestris, while B. terrestris has a paralog of IR332, numbered IR333, which has been lost from A. mellifera.
Odorant binding proteins (OBPs)
Finally, OBPs are involved in the initial transport of odorants from the air to the sensory neuron dendrites in olfactory sensilla. The classic OBPs were the only sub-family identified in A. mellifera, and this is also the case for B. terrestris. While A. mellifera have 21 OBPs [142], there are 16 members in B. terrestris (Additional file 1), with eight of the 16 being simple orthologs with eight of A. mellifera OBPs. There is some species-specific gene duplication and loss (Additional file 1). Most impressive, however is the evolution of AmOBP13 and its relatives AmOBP14-21. As noted in [142], the latter are a large tandemly duplicated set derived from AmOBP13. In B. terrestris there are just three genes duplicated from an ortholog to AmOBP13. However, the functions of these proteins in honeybee and bumblebee biology are largely unknown.
Thus, the chemosensory repertoire of bumblebees appears to emphasize gustation over olfaction relative to honeybees. In addition, within each chemoreceptor family there has been an expected pattern of gene birth and death, even when gene numbers are comparable between A. mellifera and B. terrestris. However, the ligand specificity of just one A. mellifera OR is known [136], so extensive work will be required to identify precisely how their chemosensory abilities have changed to suit their different social and ecological situations.
Defense and venom constituents in the bumblebees
Defense and venom constituents in the bumblebees were characterized by incorporating information from a venom proteome of B. terrestris workers and the bumblebee genomes. The proteomic analysis revealed 519 unique peptides and provided evidence for 57 venom proteins in B. terrestris. Of these, 52 were previously not described for B. terrestris venom. Manual annotation of genes supported by the venom peptides (Additional file 2) showed that most venom genes are fully (72%) or partially (23%) covered by transcriptomic evidence. Venom proteome evidence was not found for several well-characterized honeybee venom compounds, although based on searches for syntenic regions and homology, five of these missing components were recovered from the B. terrestris genome (Additional file 2). However, apamin and tertiapin, two neurotoxic honeybee venom compounds, were not found in the bumblebee genomes, including when searching initial sequence reads. Highly similar protein sequences to those identified in B. terrestris were identified in B. impatiens (Additional file 2). Additional details can be found in [143].
Immune components and responses in the bumblebees
Given the perceived high risk of disease in densely packed social groups, it was surprising when A. mellifera was found to have only one-third as many immune-related genes as solitary Dipteran model insects [144]. Dietetic differences, artifacts of honeybee breeding, and advanced eusociality allowing for complex group-based defenses or hygienic behavior, were all possible reasons advanced for the presence of this depauperate immune complement. Comparatively examining immune genes in bumblebees addresses some of these hypotheses. Furthermore, given the potential role of parasites in concerning declines of some bumblebee species [40,48], understanding the architecture of the bumblebee immune system has a clear importance.
Both bumblebee genomes contain components of all major immune pathways described in insects and exhibit a similar immune repertoire to A. mellifera (Additional file 2). The total number of immune genes in bumblebees is similar to A. mellifera, and therefore also considerably lower than in Dipteran model species (Figure 4). While numeric representation of immune components is similar, the bumblebee immune repertoire is not however completely undifferentiated from that of the honeybee. Both Bombus species have only a single copy of the antimicrobial peptide (AMP) defensin, which is present in two copies in A. mellifera, and have an expanded set of serine protease inhibitors. In B. terrestris, there are five, highly similar (average 75% sequence similarity), putative serpin 3/4-like genes, while only a single ortholog is identified in A. mellifera. A homolog of the apoptosis-involved caspase decay, which has not been described in either A. mellifera or the parasitoid wasp N. vitripennis, and a Hymenoptera-specific clade of caspases that are most similar to Ice in Drosophila are also present. A recently duplicated species-specific peptidoglycan receptor protein (PGRP) is present in B. impatiens. Further in-depth analyses are reported in a companion paper on immune genes [145]. Quantitative expression analyses in B. terrestris confirm expression changes of many immune-related genes following immune-stimulation. Interacting with parasites, including those that are co-evolving, make immune genes an interesting focus of molecular evolution studies. In the bumblebees, patterns of evolutionary selection differ across immune system components, with certain genes showing lineage-specific patterns of selection. Broadly however, the comparative analysis of immune genes present in the two bumblebee genomes show a reduced immune complement is not the result of honeybee-specific traits or those relating to complex social defenses in advanced eusocial organisms, such as hygienic behavior, but is instead basal in the bees and independent of the level of sociality [145].

Immune gene counts in bumblebees relative to selected insects. Number of genes belonging to 29 categories of immune genes are presented in the cells. Heat colours in a cell reflect the number of genes in that category relative to those other species (light blue: fewer - dark red: more). The tree represents a clustering analysis using Euclidean distances based on the number of genes within these groups. AMP = Antimicrobial peptide, APHAG = Autophagy, CASP = Caspase, CASPA = Caspase A, CAT = Catalase, CLIP = CLIP serine protease, CTL = C-type lectin, FREP = Fibrinogen-like, GALE = Galectin, GNBP = Gram-negative binding protein/Beta-glucan recognition protein, IAP = IAP repeat, IGG = Immunoglobulin, IMDPATH = Imd pathway, JAKSTAT = JAK/STAT pathway, LYS = Lysozyme, ML = MD-2-related lipid recognition, NIMROD = nimrod, PGRP = Peptidoglycan recognition protein, PPO = Prophenoloxidase, PRDX = Peroxidase, REL = Relish, SCR = Scavenger receptor, SOD = Superoxide dismutase, SPZ = Spatzle, SRPN = Serine protease inhibitor, SRRP = Small RNA regulatory pathway, TEP = Thioester-containing protein, TOLL = Toll genes, TOLLPATH = Toll pathway.
Bumblebee queen hemolymph proteome
To further improve our understanding of the immune potential of B. terrestris queens and to cross-check the draft genome with further proteomic data, a proteomic analysis of the hemolymph, an important transporter of metabolic and immune components, and site of immune activity, was conducted. In total 821 peptides were identified (Additional file 2) representing 98 proteins, 46 of which had associated immune function. Gene ontology mapping also indicated that the hemolymph proteome comprised proteins associated with primary and secondary metabolism, protein transport, olfaction, chemosensory processes, and venom. The presence of venom and chemosensory proteins in the hemolymph may represent novel functions and processes for these proteins.
A relatively large proportion of the hemolymph proteome (17 of 98 proteins) comprises proteins of unknown function. These genes were provisionally annotated as hemolymph associated proteins (HAP 1-14) with three proteins grouped into a novel protein family (HAP family A1-3). Homology searches indicated that many of the HAPs show restricted taxonomic distribution including genes specific to Bombus (n = 2), Apidae (n = 2), Apoidea (n = 1), Hymenoptera (n = 7), and Hexapoda (n = 5) (Additional file 1). A comparison of proteomic data for A. mellifera queen hemolymph [146] identified ubiquitous proteins involved in defense-related processes, such as immunity and antioxidant activity. However, a significant proportion (over 45%) of the proteomes vary in composition, with the majority of this variation being attributed to the B. terrestris HAPs indicating a potential role in the behavioral, physiological, and social differences observed between these two species. The mass spectrometry data generated here was utilized in a proteogenomic capacity to identify missed protein coding genes (12 genes in total) and correct inaccurate gene models.
Regulation of gene expression in the bumblebee genomes
Gene regulation is likely to be especially important in eusocial organisms, where it will shape behavioral and developmental differences between castes.
RNAi
RNAi leads to sequence-specific gene silencing, which plays a role in immunity against viruses and mobile genetic elements, gene regulation, and cellular development [147]. The functionality of the RNAi machinery has been demonstrated in bumblebees [148], but the genes involved had, as yet, not been identified. In both bumblebee species homologs for genes encoding the core RNAi machinery proteins were found (that is, dicer, drosha, argonaute, aubergine, pasha, R2D2, loquacious) (Additional file 1). In addition, genes involved in the uptake and spread of the silencing signal were found to be consistent with the findings from A. mellifera, with Snipper and sid-1 homologs being present, but a sid-2 homolog being absent. Phylogenetic analysis of the bumblebee SID homologs shows they cluster with SID proteins from other Hymenoptera (Additional file 1).
MicroRNAs
MicroRNAs (miRNAs) are a class of small non-coding RNAs that regulate gene expression at the post-transcriptional level, and several studies have shown that miRNAs are implicated in the regulation of social behavior in social insects in general [9,14]. In honeybees, miRNAs have been associated with development [149], queen-worker caste differentiation [150], and task specialization and polyethism in worker bees [151,152]. Most recently, miRNAs have been shown to play a role in response to some of the physiological changes associated with vitellogenin in worker bees [153].
Here, a diverse complement of methods was used to identify miRNAs in the bumblebee genomes, including sequencing of miRNAs in B. terrestris, a homology search of A. mellifera miRNAs from miRBase [154], and miRNA prediction using miRCat [155] and miR-abela [156]. These methods identified 130 and 115 miRNAs in B. terrestris and B. impatiens, respectively (Additional file 2), including a number of previously uncharacterized miRNAs in B. terrestris. Of these bumblebee miRNAs, 17 miRNAs had not previously been identified in A. mellifera. Comparing new miRNAs back to the A. mellifera genome revealed that two miRNAs are conserved across the two bumblebee species and A. mellifera, but 14 miRNAs were unique to one of the two bumblebee species, with five being unique to B. terrestris and one potentially representing a unique duplicated miRNA in B. impatiens (Figure 5).

Venn diagram of the distribution of unique and shared miRNAs across the two bumblebee species investigated and Apis mellifera. A total of 116 miRNAs were found in the genomes of all three species. Strikingly, 103 miRNAs of the 219 in A. mellifera were not found in the genome of either bumblebee species.
Those miRNAs with homologs in A. mellifera and the two bumblebees are highly conserved, being identical or near identical. This fits with expectations from models of miRNA evolution [157]. However, mir-263a and mir-3736 showed substitutions in bases 2 to 8 of the mature miRNA. This is the ‘seed region’, that is, where miRNAs bind to the target transcript, so substitutions here are most likely to change the function of the mature miRNA. Historically, the most expressed sequence has been classified as the mature miRNA and this is the sequence that is assumed to have a regulator function, while the other sequence (historically termed the *sequence) is lost or degraded. It is becoming increasingly clear that sometimes these sequence arms switch between species, so the *sequence in one species might not be the same in another [158]. In addition, it is clear that, for some miRNAs, both arms of the duplex have biological activity, and the most highly expressed will vary in different tissues. Ten miRNAs were found to have switched arms, with the most abundantly expressed arm in B. terrestris being the opposite of that in A. mellifera. This implies that while the sequences of the miRNAs themselves were conserved, functional changes could have happened due to such switches.
In spite of the conservation of several miRNAs between A. mellifera and the bumblebees, there is a large disparity with 103 of the 217 A. mellifera miRNAs apparently being exclusively found in the A. mellifera genome (Figure 5). A. mellifera has 217 miRNAs identified in miRBase, but for at least 97 of these there is very little or no evidence that they are present in the genomes of the two bumblebee species, and neither is there evidence (according to miRBase) that they are present in other species with sequenced miRNAs. Furthermore, six A. mellifera miRNAs are duplicated in the A. mellifera genome, but have only one copy in the bumblebee genomes, meaning there are currently 103 miRNA genes that appear exclusively in the A. mellifera genome. These differences in miRNA numbers between honeybees and bumblebees are surprising given that new RNAs are thought to evolve and be maintained at a low rate [157]. For example, one study of miRNA evolution in Drosophila found that species diverged by up to 60 My were far more similar in their miRNA expression profiles [159]. However, insects appear to have a particularly high rate of miRNA generation and disappearance [159] and an especially high rate in A. mellifera (but not Bombus) could explain the disparity in numbers with bumblebees. An alternative is that there are a high number of false positives in the Apis miRNA set, an issue that was guarded against in the identification of unique bumblebee miRNAs by taking into account several parameters in the prediction process [155]. However, this would not account for the miRNA duplications in A. mellifera.
The results of the miRNA analysis show that despite conservation of a core set of miRNAs between bumblebees and honeybees, there are important differences that likely affect miRNA functionality. Given the role that miRNAs have been shown to have in traits relating to social behavior, these differences are striking and may underlie key biological differences between advanced eusocial honeybees and primitively eusocial bumblebees.
DNA methylation
DNA methylation plays an important role across taxa in epigenetic modification that alters expression patterns, and in this way it can impact on development, behavior, learning, memory formation, and phenotypic plasticity [160]. In honeybees, DNA methylation has an important role underlying eusocial characteristics, influencing developmental divergence of queens and workers, and changes in worker behavior [161-163]. Thus, DNA methylation is an important process to investigate and understand in bumblebees. While not as distinct as in honeybees, caste differences in bumblebees are also marked, and in B. terrestris methylation is associated with plastic reproductive division of labor [164]. DNA methylation in the two bumblebee genomes has many defining features that are similar to those of A. mellifera (Additional file 1). This indicates that DNA methylation may play an equally important role in directing caste differentiation and behavioral divergence in bumblebees as it does in their advanced eusocial relatives. Both bumblebee genomes have a complement of DNA methyltransferases (DNMTs) similar to A. mellifera, including two copies of the maintenance methyltransferase (DNMT1) and one de novo DNA methyltransferase (DNMT3) (Additional file 1). Enzymes functioning in DNA methylation targeting (PIWI) and removal (TET) are also present. A computational prediction of the methylation landscape of the bumblebee genomes based on CpG depletion demonstrated that it is very similar to that of A. mellifera, and half of all genes can be methylated, with DNA methylation primarily targeted to exons (Additional file 1). Based on gene ontology terms, genes with metabolism and ubiquitous housekeeping functions were significantly enriched for genes with predicted methylation (Additional file 1). The similarity in methylation between A. mellifera and the bumblebees was further confirmed by DNA methylation quantification, with 0.5 to 0.6% of all CpGs being methylated (Additional file 1).
Selenoproteins and selenoprotein-related genes in the bumblebee genomes
Selenoproteins are a class of proteins that include selenocysteine (Sec), an unusual amino acid inserted through the recoding of a UGA codon (normally a translation stop). The number of selenoproteins encoded in genomes varies among eukaryotic lineages [165]. While other animals show extensive conservation of selenoproteins, some insect lineages have been reported to be devoid [166]. Interestingly, this is attributed to multiple independent events of loss in the different lineages. The bumblebee genomes, like all other Hymenoptera investigated thus far, lack selenoproteins. They have also lost part of the machinery necessary to build and insert selenocysteine. The bumblebee genomes possess no tRNAsec, eEFsec, and pstk, while they retain SecS, secp43, and SBP2. These losses fit with the mosaic pattern of selenoprotein loss through multiple independent events in insects due to relaxed selective constraints, with Lepidopteran, Hymeopteran, Dipteran, and Coleopteran lineages retaining varying complements of selenoprotein machinery [166]. Given their conservation across multiple Hymenopteran species, the retained proteins have probably acquired a function unrelated to selenocysteine. There is conservation in the number of Sec machinery genes found, and in their exonic structure, between the bumblebees and A. mellifera. The extinction of selenoproteins in the Hymenoptera opens an interesting field of research for the study of evolution of selenoprotein genes and selenium metabolism in insects.
Using the bumblebee genomes: an example of genome-wide patterns of diversity based on SNPs in B. impatiens
High quality genomes provide important resources for post-genomic research, including population-level assays of single nucleotide polymorphism (SNP) variation that can be challenging when no reference is available. The newly sequenced Bombus genomes will provide a major resource for studies of genetic diversity, demographic history, natural selection, and genome-wide associations with disease, for example, that may promote understanding of factors involved in pollinator declines. To illustrate the value of the Bombus genome for next-generation SNP analysis, RAD-tag sequencing [167] was carried out on 22 B. impatiens worker samples collected throughout the geographic range of this species (Additional file 1), and reads were mapped to the B. impatiens genome. After stringent filtering, 9,607 SNPs were identified across the 22 diploid individuals over 1.113 Mb of sequence. Genome-wide diversity was estimated at θ pi = 0.0014 per site, consistent with previous de novo analyses of RAD-tags [168]. The SNP positioning information from the B. impatiens genome enables population genetic assessment of linkage disequilibrium (LD) that was not possible for de novo RADseq analyses. Overall LD was low and declined rapidly with distance between SNPs: average r 2 within 10 kb was 0.15 and decayed to 0.05 over larger distances. Such weak LD is consistent with A. mellifera subspecies that experience recombination rates of 19 cM/Mb [169] and a similarly high rate is also present in bumblebees [56]. Weak LD is expected for the large population sizes typical for B. impatiens [40]. The two bumble bee genomes will thus provide an excellent reference that enables resequencing studies in B. impatiens and B. terrestris, and furthermore, because of the substantial synteny revealed here, will also become powerful resources for positioning and annotating data in other closely related Bombus species.
Conclusions
In addition to providing an excellent basis for future post-genomic studies, the two bumblebee genomes presented here illuminate key aspects of the biology of these important pollinator insects, and, based on comparisons with honeybees, offer an insight into potential foundations of advanced eusociality. Given the plethora of differences between the two bumblebees investigated here and A. mellifera (Table 1), it is surprising that in general the gene repertoires of the species are highly similar. Some of these similarities are informative in themselves, and demonstrate that certain genome characteristics found in A. mellifera are not unique. For example, depauperate complements of xenobiotic detoxification and immune genes in comparison with many other insects are not honeybee-specific. With regard to protein-coding genes, striking differences relate to chemosensation, with bumblebees emphasizing gustation relative to olfaction, which is likely tied to specific ecology of these species.
While mechanisms of gene regulation are known to have an important role in insect societies, the surprising divergence in miRNA complement and potential functionality represents a major difference between the primitively eusocial bumblebees and advanced eusocial honeybees. It may well be that these elements are what tune complex behavior and organization in the advanced eusocial bees. However, selection on gene regulation, potentially driven or limited by environmental constraints (for example, temperate and tropical, annual and perennial) could underlie other key biological differences aside from the level of eusociality.
In general, the gene repertoires reported here for two key bumblebee species suggest that the evolution of advanced eusociality in bees did not occur through large leaps involving notable gene expansions and/or depauperation between lineages. Rather, the route to advanced social living was mediated by many small changes in many genes and processes.
Methods
Genome sequencing and assembly
Bombus terrestris
DNA from a single haploid drone was used for XLR fragment data. Pools of haploid drones were used for the 8 kb and 20 kb libraries. Drones were provided by the ETH group of Paul Schmid-Hempel, Switzerland. These drones were the offspring of field-caught queens collected in the spring of 2008 in Northern Switzerland. DNA extraction was carried out using Genomic tips and the blood and cell culture DNA kit (Qiagen). A whole genome shotgun sequence was generated using the Roche 454 platform, with fragment (NCBI SRA: SRX016989) and mate-pair (NCBI SRA: SRX016990, SRX016992, SRX016991) DNA libraries. Library preparation and sequencing protocols were carried out as previous described [14]. The B. terrestris assembly was produced by assembling the approximately 14.3 million Roche 454 reads representing approximately 21× coverage of the genome. Sequences were combined with the Newbler-assembler (2.3-PreRelease-10/19/2009) and then reads from each Newbler generated scaffold were grouped, along with any missing mate-pairs, and reassembled using Phrap in an attempt to close the gaps within each Newbler scaffold.
B. impatiens
DNA extracted using a standard chloroform/phenol procedure was used from a single drone taken from a colony obtained from Koppert USA. Sequence was generated on an Illumina GAIIx sequencer at the University of Illinois. Libraries of 500 bp, 3 kb, and 8 kb were prepared using standard Illumina protocols and sequenced, producing a total of 497 million reads and representing approximately 108× raw coverage of the genome (NCBI SRA: SRX679085, SRX679084, SRX679082). Read lengths were in the range of 100 to 124 bp. Duplicates were removed and all reads were error-corrected with the Quake program [170]. After error correction and duplicate removal, 276 million reads remained which were assembled using both CABOG and SOAPdenovo. The final assembly used SOAPdenovo release 1.9 with a k-mer size of 47. The final assembly contained 5,559 scaffolds ≧200 bp, of which 1,505 were >1,000 bp. The 5,559 scaffolds contained 16,215 contigs, while the 1,505 scaffolds contained 12,033 contigs.
Synteny
Both the Bter_1.0 and the BIMP_2.0 assemblies were scanned for microsatellite loci and compared based on sequence similarity to identify loci shared (that is, ‘homologous’) between both species. The procedure is described in detail in [171]. The relative positions and order of homologous loci were compared. Using the Bter_1.0 assembly as a reference, homologous scaffolds were identified, ordered, and oriented. If two scaffolds had consecutive homologous loci at their ends, they were considered linked. Single loci, missing in the consecutive order or those homologous to a distinct scaffold than the surrounding loci in the scaffold were ignored, whether at the ends or within scaffolds. As an exception to this, loci homologous to unplaced scaffolds were included, but only where several consecutive loci justified the position of the unplaced scaffold within gaps or at scaffold ends. This way, based on marker order and distances, previously unmapped small scaffolds and contigs could be putatively placed and were considered in the analysis of the synteny lengths if they contributed to an extension or linkage between scaffolds.
Repetitive elements
Repetitive elements were detected and annotated with the REPET software package ([172], version 2.0). First, repeated sequences were detected by similarity (all-by-all blast using BLASTER) and LTR retrotransposons were detected by structural search (LTRharvest). The similarity matches were clustered with GROUPER, RECON, and PILER, the structural matches with single-linkage NCBI BLASTCLUST. From each cluster a consensus sequence is generated by multiple alignment with Map. The consensus sequences were analyzed for terminal repeats (TRsearch), tandem repeats (TRF), open reading frames (dbORF.py, REPET), and poly-A tails (polyAtail, REPET). Furthermore, the consensuses were screened for matches to nucleotide and amino acid sequences from known transposable elements (RepBase 17.01, [57]) using BLASTER (TBLASTX, BLASTX) as well as searched for HMM profiles (Pfam database 26.0, [173]) using hmmer3. Based on the detected structural features and homologies, the consensuses are classified by PASTEC according to [174]. Redundancies are removed (BLASTER, MATCHER) as well as elements classified as SSRs (>0.75 SSR coverage) or unclassified elements built from less than 10 fragments. This set of de novo detected repetitive elements was used to mine the genome in the second pipeline with BLASTER (NCBI BLAST, sensitivity 4, followed by MATCHER), RepeatMasker (NCBI BLAST/CrossMatch, sensitivity q, cutoff at 200) and CENSOR (NCBI BLAST). False positive matches were removed by an empirical statistical filter. Satellites were detected with TRF, MREPS, and RepeatMasker and were then merged. Furthermore the genomic sequences were screened for matching nucleotide and amino acid sequences from known transposable elements (RepBase 17.01) via BLASTER (TBLASTX, BLASTX) followed by MATCHER. Finally a removal of TE doubletons, removal of SSR annotations included into TE annotations and ‘long join procedure’ to connect distant fragments was performed. Sequences from the de novo repetitive element library found to have at least one perfect match in the genome were then used to rerun the whole analysis. To ensure compatibility and to avoid bias, a manual curation or clustering of the de novo detected elements was not performed before mining the genome. However, post hoc all elements were manually analyzed that were previously classified into class I retrotransposon or class II DNA transposon elements or unclassified elements with detected coding element features (similarity to known transposable elements) due to potential chimeric insertion. At this stage derivative elements (LARD, TRIM, MITE) were excluded from detailed further inspection unless carrying such a feature. Elements classified as ‘potential Hostgene’ or unclassified elements (noCat) were also excluded. Manual inspection was carried out with ORF Finder (NCBI), CDD search (NCBI, [175]), with a search in the most up to date online RepBase database (accessed December 2012 to February 2013) via CENSOR [176] and phylogenetic analysis for LINE RT domains with RTclass1 [177] in order to achieve a detailed classification for each element, determine its potential relation to a family of known elements, to evaluate the completeness, and to detect potential active elements. Elements were defined as complete if they possessed the relevant coding parts with the element-typical domains and the structural features (LTR, TIR). The potential activity was defined according to the region an intact ORF, if present, covered. If an intact ORF seemed to cover a complete region including the typical domains (for example, GAG, POL, Tase) then the element is considered to potentially active. If a Tase domain is covered by a truncated ORF or the Tase itself appears to be truncated but is covered by an intact ORF, or if the RT domain is covered by an active ORF but not the remaining element-typical domains, then the element is considered to be potentially active. During the manual classification to at least superfamily level, novel transposable element types not covered by the system of [174] were also considered: Kolobok, Sola, Chapaev, Ginger, Academ, Novosib, and ISL2EU class II DNA transposons [178,179]. Simple sequence repeats and other low complexity regions were extracted from the REPET pipeline database and processed to calculate the total coverage of these types of repetitive DNA, while omitting those overlapping with transposable element annotation.
Gene predictions
NCBI Refseq and Gnomon
Bter_1.0 and BIMP_2.0 assemblies were annotated with NCBI’s eukaryotic genome annotation pipeline (v.3, see [180]). Evidence used for B. terrestris included Refseq protein annotation of A. mellifera, 214 k TSA assemblies of Illumina RNAseq reads from B. terrestris queen heads (NCBI SRA: SRX090531), queen ovaries (NCBI SRA: SRX090532) and male heads (NCBI SRA: SRX090533), Roche 454 B. terrestris RNAseq reads (NCBI SRA: SRX040734 and ERP000936), and additional cross-species protein alignments. Evidence used for B. impatiens included the above, plus 406 k of Roche 454 RNAseq reads (NCBI SRA: SRX040732).
AUGUSTUS
AUGUSTUS can be used as an ab initio gene prediction tool, but can also integrate extrinsic evidence from various sources [181]. Training gene structures for B. terrestris were generated using transcriptome data and an AUGUSTUS parameter set for A. mellifera [16]. RNAseq data mentioned above were mapped to the genome using BLAT [182] and alignments were integrated into gene predictions using AUGUSTUS. RNAseq data were mapped to predicted genes and fully covered transcripts selected as training genes to optimize a species-specific parameter set, with the flanking region being set to 10,000 nucleotides and UTR parameters adopted from A. mellifera. Final gene predictions were made using the B. terrestris parameter set, the above-mentioned RNAseq evidence, available peptides [143] and repeat information [183]. Greater weight was given to informing sequences from the target species. Genes in B. impatiens were predicted using the B. terrestris parameter set. Extrinsic evidence was generated as described for B. terrestris, without the peptide data, and with B. impatiens repeat information [184].
Fgenesh++
Predictions were made using FGENESH 3.1.1 [185]. RNAseq data for B. impatiens and B. terrestris described above were incorporated, along with the GenBank NR database to predict genes similar to known proteins.
GeneID
GeneID [186] is an ab initio gene prediction program used to find potential protein-coding genes in anonymous genomic sequences. An initial training set, as used in AUGUSTUS, was used to develop a B. terrestris specific parameter file based on a method employed to obtain a D. melanogaster parameter file [187].
SGP2
SGP2 [188] combines ab initio gene prediction (GeneID) combined with TBLASTX searches between genomes. Genomes of Nasonia giraulti, N. longicornis, and N. vitripennis were used as reference to develop the B. terrestris parameter file. The B. terrestris-specific parameter file was produced based on the methodology described to obtain a human sgp2 parameter file [189].
N-SCAN
The N-SCAN package [190] was used to leverage conservation between the target genome, B. impatiens or B. terrestris, and genomes of two informant bee species, A. mellifera (Amel_4.5) and the other Bombus species (Bter_1.0 or BIMP_2.0, respectively). The target Bombus species was masked for simple sequence repeats using RepeatMasker [191]. LASTZ [192] was run using default parameters with the target Bombus genome and each informant genome. For B. terrestris, iParameterEstimation was used to generate both a Bter_1.0-Amel_4.5 specific parameter set as well as a Bter_1.0-BIMP_2.0 specific parameter set using the training set described for AUGUSTUS, including UTR features. N-SCAN was run using each of the B. terrestris specific parameter sets with the respective LASTZ informant genome alignments to produce two N-SCAN gene prediction sets, one based on Amel_4.5 and the other based on BIMP_2.0 as the informant genomes. The Amel_4.5 as the informant set was chosen as the best prediction set based on Eval analysis [193] against the RefSeq and Gnomon annotations for B. terrestris. Insufficient B. impatiens transcriptome data were available to generate BIMP_2.0-Amel_4.5 and BIMP_2.0-Bter_1.0 specific parameter sets, so the parameter files generated for Amel_4.5-Bter_1.0 (where Amel_4.5 was the target species and Bter_1.0 the informant species), Bter_1.0- BIMP_2.0, and Bter_1.0-Amel_4.5 were used to evaluate Bter_1.0 and Amel_4.5 as informant genomes for BIMP_2.0. N-SCAN was run using the Amel_4.5-Bter_1.0 parameter set with the LASTZ alignments between BIMP_2.0 and Amel_4.5, the Bter_1.0- BIMP_2.0 parameter set with the LASTZ alignments between BIMP_2.0 and Bter_1.0, and the Bter_1.0-Amel_4.5 parameter set with the LASTZ alignments between BIMP_2.0 and Amel_4.5. The set using the Amel_4.5-Bter_1.0 parameter set and Amel_4.5 as the informant genome was chosen as the best prediction set based on Eval analysis against the RefSeq and Gnomon annotations for B. impatiens.
GLEAN
Gene sets described above were combined with GLEAN, also using assembled transcript sequences described above and protein homologs. Transcript sequences were aligned to the Bter_1.0 and BIMP_2.0 genome assemblies using MAKER2 v2.15, which uses WU-BLAST [194] and Exonerate est2genome [195], with minimum 80% alignment coverage and 95% identity. Protein homolog alignments included SwissProt Metazoa homologs [196], D. melanogaster (r5.31) [197], A. mellifera (OGSv3.2) [16], N. vitripennis (OGSv1.2) [61], and the ants: Acromyrmex echinatior (OGSv3.8) [63], A. cephalotes (OGSv1.1) [13], Camponotus floridanus (OGSv3.3), H. saltator (OGSv3.3) [9], Linepithema humile (OGSv1.1) [11], Pogonomyrmex barbatus (OGSv1.1) [12], and Solenopsis invicta (OGSv2.2.3) [15]. Proteins in the SwissProt dataset annotated as transposable elements were removed prior to alignment. Protein sequences were aligned to the Bter_1.0 and BIMP_2.0 genome assemblies using Exonerate protein2genome with a minimum 60% identity and 60% alignment coverage.
Manual annotation
The annotation consortium used tools available at BeeBase (hymenopteragenome.org) and elsewhere to manually check certain gene models. Gene models and transcriptomic evidence were viewed and edited in Apollo [198].
Orthology analysis
Orthology assignments were retrieved from OrthoDB [69]. OrthoDB6 includes a total of 45 arthropods with the following gene sets for the selected species: Pediculus humanus PhumU1.2 and A. gambiae AgamP3.6 from VectorBase; N. vitripennis Nvit_OGSv2.0, A. mellifera Amel_OGSv3.2, A. cephalotes Acep_OGSv1.2, and A. echinatior Aech_OGSv3.8 from Hymenoptera Genome Database; B. impatiens Bimp_RefSeq, B. terrestris Bter_RefSeq, and A. florea Aflor_Augustus from NCBI; T. castaneum Tcas_3.0 from BeetleBase; B. mori Bmor_GLEAN from SilkDB; and D. melanogaster Dmel_r5.45 from FlyBase. The maximum likelihood phylogeny was built using RAxML [199] from the concatenated multiple sequence alignments of 2,294 single-copy orthologs aligned with MUSCLE [200] and trimmed with TrimAl [201]. The superalignment contained 666,462 amino acids with 215,542 distinct alignment patterns.
Protein domain analysis
Gene sets of all species (Additional file 1) were annotated with Pfam-Scan (based on HMMR3 [202]) against the Pfam A database (version of 4 October 2012) [173]. If there were different splicing variants, only the longest transcript was used.
Unique domains: A domain was considered as unique if it appears only in B. terrestris and in no other reference species. Expansion and contraction of domains and arrangements: All domains were counted just once for each gene within which they appear. Arrangements are considered as the combination of domains present in one protein, where the number and order of domains are not taken into account. Repeats: For the detection of repeats, genes were clustered according to their domain arrangement, again without considering the number and order of domains (just for the clustering). For each cluster in A. mellifera and B. terrestris the original repeat number was extracted. Each arrangement was analyzed for the minimal and maximal repeat count for each domain. If two domains cover one PFAM model consecutively, they are counted as one domain. Only arrangements appearing in both of the analyzed species were considered, and an arrangement needed to occur at least twice in at least one species.
Development
Orthology of developmental genes was assigned using reciprocal BLASTP or TBLASTN searches using the T. castaneum, A. mellifera, and D. melanogaster protein sequences. Where necessary HMMer [202] was used to identify potential orthologs of fast-evolving genes in the bumblebee genomes.
Sex determination
Orthologs for sex determination, germline development, and dosage compensation were identified in Refseq proteins and assembly scaffolds of the two bumblebee species by using BLASTP and TBLASTN using sequences from A. mellifera, N. vitripennis, and D. melanogaster. Manual annotation was performed by comparing Refseq bumblebee genes against available insect genomes in Apollo [198].
Behavior, neurophysiology, and endocrinology
Circadian clock genes: Putative circadian clock genes were identified via TBLASTN searches of A. mellifera clock gene coding sequences against the bumblebee genome assembly. These putative homologs were then manually annotated in detail by comparison with homologs from other Hymenopteran species as well as expression datasets. Multiple sequence alignments were carried out with ClustalW [203]. Take-out/Juvenile hormone binding proteins: Refseq proteins and assembled genome scaffolds of the two bumblebee species were searched with BLAST for homologs of the D. melanogaster To gene or to JHBP genes of various insects. The SMART server [204] was used to demarcate JHBP domains and signal peptides of sequences, and SignalP server [205] was used to confirm putative signal peptides. Only domains with an E-value <0.1 were considered significant. Multiple sequence alignments were carried out with ClustalW. Cys-loop ligand-gated ion channel gene superfamilies: Putative Bombus cys-loop ligand-gated ion channel subunits were identified by TBLASTN using protein sequences of every member of the A. mellifera cys-loop ligand-gated ion channel superfamily [107] and then were manually annotated. Bioamines/neuropeptides: To identify neuropeptides, protein hormones, and their receptors, and biogenic amine receptors, TBLASTN searches were performed, using known insect, or arthropod sequences for these proteins. TRP channel genes: TRP channel genes in the bumblebee genomes were identified in the same way as previously for other insects [206].
Xenobiotic detoxification enzymes and related genes
Cytochrome P450s, GSTs, and CCEs were manually annotated by comparing the genome sequence of each bumblebee with all annotated P450, GST, and CCE protein sequences from A. mellifera, N. vitripennis, and D. melanogaster using TBLASTN in a method similar to that used to annotate these genes in the N. vitripennis genome [122]. GSTs and CCEs were additionally compared to A. florea, and P450s to P. barbatus, T. castaneum, and D. melanogaster.
Chemoreceptors
Odorant receptors (ORs): The OR family in B. terrestris was manually annotated using methods employed before for other insect genomes [132,133]. The BtOrs were numbered independently of their AmOr relatives, because while some are orthologs, much duplication and some gene losses make using the AmOr numbers for the BtOr genes impossible. The numbering does start with the conserved ortholog of AmOr1 as BtOr1, but diverges from there, in part because AmOr2 is now called Orco. Genome assembly problems associated with this gene family are noted in Additional file 1. Pseudogenes were translated as best possible to provide an encoded protein that could be aligned with the intact proteins for phylogenetic analysis, and attention was paid to the number of pseudogenizing mutations in each pseudogene. A 200 amino acid minimum was enforced for including pseudogenes in the analysis (roughly half the length of a typical insect OR). For phylogenetic analysis, the poorly aligned and variable length N-terminal and C-terminal regions were excluded (specifically 10 amino acids before the conserved GhWP motif in the N-terminus and 10 after the conserved SYFT motif in the C-terminus), as was a major internal region of length differences, specifically a long length difference region between the longer DmOr83b orthologs, now known as Orco proteins [135] and most of the other ORs. Other regions of potentially uncertain alignment between these highly divergent proteins were retained, because while potentially misleading for relationships of the subfamilies (which are anyway poorly supported), they provide important information for relationships within subfamilies. Phylogenetic analysis of this set of 342 proteins was carried out in the same fashion as for previous OR analyses [132,133]. Gustatory receptors (GRs): GRs were identified using the basic protocol referenced above for ORs. Numbering of the BtGrs is complicated. The names Gr1-4 and 6/7 and 10/11 were employed for the 1:1 orthologs of these genes in A. mellifera. However, while A. mellifera has paralogs Gr4/5, B. terrestris only has a single gene, so the BtGr5 name was employed for the intact ortholog of a large set of highly degraded pseudogenes in the A. mellifera genome, represented by constructs AmGrX, Y, and Z. Bombus terrestris has three very recent sets of duplicated genes, related to AmGr8, 9, and 12. These were named for their orthologs and then with additional numbers. Assembly problems related to genes in this family are noted in Additional file 1. For phylogenetic analysis, the poorly aligned and variable length N-terminal and C-terminal regions were excluded (specifically from 10 amino acids before the conserved GhWP motif in the N-terminus and five amino acids after the conserved TYhhhhhQF motif in the C-terminus), as was a major internal region of length differences involving DmGr66a. Including 68 GRs from D. melanogaster, phylogenetic analysis of a set of 107 total proteins was carried out in the same fashion as for previous GR analyses [132,133]. Ionotropic receptors (IRs): IRs were identified using the basic protocol above. Additionally, iterative searches were also conducted with each new B. terrestris protein as query until no new genes were identified in each major subfamily or lineage. Naming and numbering of the B. terrestris IRs is not simple. Following the example [131], the conserved orthologs of several IRs in other insects are given those names, specifically 8a, 25a, 93a, 76b, and 68a. Assembly problems related to genes in this family are noted in Additional file 1. The A. mellifera, B. terrestris, and D. melanogaster IRs were aligned in CLUSTALX v2.0 [207] using default settings. For phylogenetic analysis, the poorly aligned and variable length N-terminal and C-terminal regions were excluded, along with several internal regions of highly length-variable sequence. Other regions of potentially uncertain alignment were retained, because while potentially misleading for relationships of the subfamilies, they provide important information for relationships within subfamilies. Phylogenetic analysis of this set of proteins was carried out in the same fashion as for previous IR analyses [132,133]. Odorant binding proteins (OBPs): OBPs were identified as above for chemoreceptors. Because their phylogenetic relationships with the A. mellifera proteins are somewhat complicated, they were not named for their A. mellifera orthologs, but rather according to their locations in the genome, although the relatively conserved OBP1 genes are orthologous. Assembly problems related to genes in this family are noted in Additional file 1. Phylogenetic analysis employed corrected distance methods (see ORs methods) and only the mature proteins with signal sequences removed, as well as the different length C-termini.
Defense and venom constituents
Venom proteome data [143] were used to search against the au5 (AUGUSTUS) and NCBI Refseq gene predictions for Bter_1.0, and genome six-frame translation databases using Mascot (v2.3, Matrix Science). Setting the significance threshold at P <0.01 leads to a peptide false discovery rate (FDR) of 5.34% for the au5 and 2.88% for the NCBI Refseq searches. Mass spectra data generated from all combinatorial peptide ligand library (CPLL) flow-through fractions, and the CPLL elution fractions of the Tris-glycine- and Tris-tricine-SDS-PAGE gel were separately searched against the genome six-frame translation database resulting in FDRs of 0.86%, 0.68% and 3.17%, respectively. Significant and top ranking peptides from the Mascot output with an ion score ≥30 were retained in the final peptide lists. All peptides found in the separate genome six-frame translation database searches were merged in one list and double peptides were removed. Identified B. terrestris venom proteins were used in BLAST searches against the B. impatiens Refseq database. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium [208] via the PRIDE partner repository with the dataset identifier PXD001623 and 10.6019/PXD001623.
Immune components and responses
Using OrthoDB6 [69] we identified orthologs from the two bumblebees of previously characterized immune genes from other arthropods. To complement the orthology searches, we searched for homologs of known immune proteins in the two bumblebees using BLASTP against RefSeq proteins. To confirm the absence of any proteins that appeared to be missing, we searched the genome assemblies and short reads archive with TBLASTN. Further details, including evolutionary analyses using Apis spp. and Megachile rotundata are described in the companion paper [145]. Queen hemolymph proteome: Post-diapausing queen hemolymph proteome data obtained from a Thermo Scientific LTQ ORBITRAP XL mass spectrometer were searched against protein datasets derived from NCBI reference sequences (downloaded August 2013), an AUGUSTUS (au6) analysis of the genome and a transcriptome assembly [50] using MaxQuant (version 1.2.2.5; [209]). FDRs were set to 0.01 for both peptides and proteins, and proteins were considered identified when more than one unique peptide was observed. The queen hemolymph proteomic data have been deposited to the ProteomeXchange Consortium [208] via the PRIDE partner repository with the dataset identifier PXD001644 and 10.6019/PXD001644. Uploaded search result files were generated by searching mass spectrometry data against NCBI reference sequences using Proteome Discoverer (v1.4.0.288) and converted to pride.xml format using PRIDE Converter 2 [210]. Identified proteins were functionally annotated using Blast2GO v2.5 [211,212] and assigned gene ontology terms relating to biological processes, molecular function, and cellular component. Functional domain analysis was performed using InterProScan [213]. BLASTP searches were conducted in Blast2Go to determine the phylogenetic distribution of the hemolymph-associated proteins of unknown function. A reciprocal BLAST search (BLASTP, E-value cutoff of 1e-10, sequence similarity >25%) against protein data for A. mellifera hemolymph [146] was performed to identify homologous/orthologous proteins between the two hemolymph sets. All protein coding gene models were inspected and corrected in Apollo and missing genes were added to the manually curated gene set.
Regulation of gene expression
RNAi: Genes putatively involved in RNAi from other insects (D. melanogaster, A. mellifera, B. mori, T. castaneum) were used to search both bumblebee genomes using TBLASTN. microRNAs (miRNAs): miRNAs were isolated by high-throughput sequencing of cDNA libraries from total RNA extracted from female larvae from four B. terrestris colonies. Libraries were prepared using the Illumina Trusec 2.0 kit, with modifications to reduce the risk of inherent sequencing biases [214]. Eight libraries (two from each colony) were prepared and sequenced by BaseClear B.V (Leiden, the Netherlands). Libraries were combined and mapped to the B. terrestris genome. MiRNA precursor sequences, identified by their characteristic hairpin-loop secondary structures, are processed into approximately 22 bp mature miRNAs that have biological function. To identify the mature miRNAs, first precursor sequences were identified by their secondary structures. For this purpose, miRNA prediction software miRCat [155] was used, employing sequence data and genomic context of the mapped sequences. Predicted miRNAs in B. terrestris were compared to the known miRNAs of A. mellifera published in miRBase [154]. Blast v.2.2.15 was used to search miRNAs not previously described from A. mellifera against the A. mellifera and B. impatiens genomes. To identify more miRNAs, including in B. impatiens, and the miRNAs not expressed in B. terrestris larvae, precursor sequences of all published miRNAs for A. mellifera from miRBase were used in a BLAST search of the B. terrestris and B. impatiens genomes. The miRNA prediction tool miR-abela [156] was used to identify hairpin-loop structures in the 500 bp regions around each of the identified homolog sequences. Finally, all Bombus homologs of the miRNAs that had been published in A. mellifera, but were not predicted by either miRCat or miR-abela, were assessed based on (a) showing a high mature sequence similarity to A. mellifera (>85%), (b) showing a clear hairpin secondary structure in their putative precursor sequences, and (c), in the case of B. terrestris, had been sequenced more than 100 times in the high-throughput sequencing libraries. These thresholds were selected to reduce the numbers of putative miRNAs that were false positives when identified from the BLAST searches. Sequence data used in these miRNA analyses are deposited in the NCBI Gene Expression Omnibus [GSE64512]. DNA methylation: A. mellifera or human proteins with known DNA methylation functions were used in a BLAST search of the bumblebee genomes. Methylated genes were predicted based on CpG depletion (CpG[O/E] <1) in the B. terrestris (n = 3,393) and B. impatiens (n = 3,671) genomes. The CpG[O/E] value (=#CpGs observed/#CpGs expected) was used to predict the presence of DNA methylation in a genomic region [215]. Global DNA methylation patterns in B. terrestris and B. impatiens were measured using the MethylFlash DNA quantification kit (Fluorometric) from Epigentek. We used thoraxes of newly emerged B. terrestris and B. impatiens workers, with similar A. mellifera samples as controls.
Selenoproteins
The program Selenoprofiles [216] was used to search for all known selenoprotein families and Sec synthesis machinery genes. The program SECISearch3 [217] was run with permissive criteria to scan for SECIS elements (selenocysteine insertion sequences) downstream of potential selenoprotein candidates. All results were manually inspected and compared to other available insect genomes.
SNP production and mapping in B. impatiens
To reduce genome complexity and enable sequencing from the same fraction of the genome across multiple individuals, we used restriction-site associated DNA marker (RAD) sequencing [167]. Genomic DNA was isolated from 22 B. impatiens workers from sites throughout the species’ geographic range. Samples were submitted to Floragenex (Oregon) for library preparation, sequencing, and preliminary bioinformatics [218-220]. Briefly, samples were digested with SgrAI, uniquely barcoded, and pooled. Fragments were sequenced from cut sites using single-end chemistry on an Illumina HiSeq 2000. Samples were demultiplexed and trimmed to a length of 90 bp. RAD-tag reads were mapped using the B. impatiens BIMP2.0 assembly. Sequences were aligned using BOWTIE 0.11.3 [221], taking into account sequence quality, allowing up to three mismatches, and ignoring reads that mapped to more than one location in the genome. Single nucleotide polymorphisms (SNPs) were called using SAMTOOLS 0.1.12a [222] with custom Floragenex scripts. Only variants with a minimum phred score of 15, sequence coverage of 10, and a missing data rate of <15% were considered. The resulting 10,966 SNP candidates were filtered to remove variants with more than two alleles and those invariant in the 22 samples but different from the BIMP2.0 reference and those with >500 reads per individual. Remaining SNPs were tested for Hardy-Weinberg deviations using vcftools 0.1.9 [223] and loci with significance <0.01 were removed. This resulted in a final dataset containing 9,607 SNPs in the 22 diploid individuals (average coverage of 151× per individual per site). Linkage disequilibrium (LD) between pairs of SNPs (minor allele frequency >0.05) within the same scaffold was estimated using r 2 on allele counts (geno-r2 in vcftools). BAM alignments were then processed using ANGSD 0.577 to estimate θ pi = 3Nμ from per-site nucleotide diversity across all sequenced sites (1.113 × 106) using a genotype-likelihood based approach that does not rely on SNP calling [224,225]. This method incorporates genotype uncertainty inherent to sequence depth and quality variation from next-generation sequencing, and analyzes all sites as opposed to estimating diversity from SNPs alone. The site frequency spectrum was estimated using SAMTOOLS genotype likelihood estimation, requiring a minimum of 20 individuals sequenced per site, a minimum base quality score of 20, and map quality score of 10. The folded site frequency spectrum was EM optimized for 22 individuals and θ pi estimated across sites and individuals. RAD sequence data in the form of BAM alignments to AEQM02.fasta have been uploaded to Genbank Sequence Read Archive [NCBI SRA: SRP051027], and SNP data in vcf format are available from the DRYAD digital repository: http://dx.doi.org/10.5061/dryad.52hj2.
Data availability
The genome assemblies and raw sequence data generated in this study are available at NCBI under the BioProject IDs PRJNA45869 for B. terrestris and PRJNA61101 for B. impatiens. Illumina RNAseq reads generated during this study include, from B. terrestris, queen heads (NCBI SRA: SRX090531), queen ovaries (NCBI SRA: SRX090532) and male heads (NCBI SRA: SRX090533). Illumina RAD sequence of 22 B. impatiens samples is available in the Genbank Sequence Read Archive (NCBI SRA: SRP051027). Illumina RNAseq data from B. terrestris used for miRNA mining are deposited in the NCBI Gene Expression Omnibus (GSE64512). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium [208] via the PRIDE partner repository with the dataset identifier PXD001623 and 10.6019/PXD001623 for the B. terrestris venom proteome and dataset identifier PXD001644 and 10.6019/PXD001644 for the B. terrestris queen hemolymph proteome. Principally, comparisons were made with the honeybee A. mellifera (NCBI: GCA_000002195.1), but in addition, depending on the purpose of the analysis the following arthropod genomes were also used for comparative analysis. Bees: A. florea (NCBI: GCA_000184785.1) and M. rotundata (NCBI: GCA_000220905.1). Wasps: N. vitripennis (NCBI: GCA_000002325.2), N. giraulti (NCBI: GCA_000004775.1), N. longicornis (NCBI: GCA_000004795.1). Ants: A. echinatior (NCBI: GCA_000204515.1), A. cephalotes (NCBI: GCA_000143395.2), C. floridanus (NCBI: GCA_000147175.1), H. saltator (NCBI: GCA_000147195.1), L. humile (NCBI: GCA_000217595.1), P. barbatus (NCBI: GCA_000187915.1), S. invicta (NCBI: GCA_000188075.1). Flies: D. melanogaster (NCBI: GCA_000001215.2), A. gambiae (NCBI: GCA_000005575.1), C. cinquefasciatus (NCBI: GCA_000209185.1). Moth: B. mori (NCBI: GCA_000151625.1). Aphid: A. pisum (NCBI: GCA_000142985.2). Beetle: T. castaneum (NCBI: GCA_000002335.2). Louse: P. humanus (NCBI: GCA_000006295.1). Waterflea: D. pulex (NCBI: GCA_000187875.1).
References
Maynard Smith J, Szathmary E. The major evolutionary transitions. Nature. 1995;374:227–32.
Bourke AF. The validity and value of inclusive fitness theory. Proc R Soc B. 2011;278:3313–20.
Crozier R, Pamilo P. Evolution of social insect colonies: Sex allocation and kin selection. Oxford: Oxford University Press; 1996.
Hughes WO, Oldroyd BP, Beekman M, Ratnieks FL. Ancestral monogamy shows kin selection is key to the evolution of eusociality. Science. 2008;320:1213–6.
Hamilton WD. The genetical evolution of social behaviour I. J Theor Biol. 1964;7:1–16.
West SA, Griffin AS, Gardner A. Evolutionary explanations for cooperation. Curr Biol. 2007;17:R661–72.
Robinson GE, Grozinger CM, Whitfield CW. Sociogenomics: social life in molecular terms. Nat Rev Genet. 2005;6:257–70.
Woodard SH, Fischman BJ, Venkat A, Hudson ME, Varala K, Cameron SA, et al. Genes involved in convergent evolution of eusociality in bees. Proc Natl Acad Sci U S A. 2011;108:7472–7.
Bonasio R, Zhang G, Ye C, Mutti NS, Fang X, Qin N, et al. Genomic comparison of the ants Camponotus floridanus and Harpegnathos saltator. Science. 2010;329:1068–71.
Simola DF, Wissler L, Donahue G, Waterhouse RM, Helmkampf M, Roux J, et al. Social insect genomes exhibit dramatic evolution in gene composition and regulation while preserving regulatory features linked to sociality. Genome Res. 2013;23:1235–47.
Smith CD, Zimin A, Holt C, Abouheif E, Benton R, Cash E, et al. Draft genome of the globally widespread and invasive Argentine ant (Linepithema humile). Proc Natl Acad Sci U S A. 2011;108:5673–8.
Smith CR, Smith CD, Robertson HM, Helmkampf M, Zimin A, Yandell M, et al. Draft genome of the red harvester ant Pogonomyrmex barbatus. Proc Natl Acad Sci U S A. 2011;108:5667–72.
Suen G, Teiling C, Li L, Holt C, Abouheif E, Bornberg-Bauer E, et al. The genome sequence of the leaf-cutter ant Atta cephalotes reveals insights into its obligate symbiotic lifestyle. PLoS Genet. 2011;7:e1002007.
The Honeybee Genome Sequencing Consortium. Insights into social insects from the genome of the honeybee Apis mellifera. Nature. 2006;443:931–49.
Wurm Y, Wang J, Riba-Grognuz O, Corona M, Nygaard S, Hunt BG, et al. The genome of the fire ant Solenopsis invicta. Proc Natl Acad Sci U S A. 2011;108:5679–84.
Elsik C, Worley K, Bennett A, Beye M, Camara F, Childers C, et al. Finding the missing honey bee genes: lessons learned from a genome upgrade. BMC Genomics. 2014;15:86.
Kocher S, Li C, Yang W, Tan H, Yi S, Yang X, et al. The draft genome of a socially polymorphic halictid bee, Lasioglossum albipes. Genome Biol. 2013;14:R142.
Cardinal S, Danforth BN. The antiquity and evolutionary history of social behavior in bees. PLoS One. 2011;6:e21086.
Grüter C, Menezes C, Imperatriz-Fonseca VL, Ratnieks FLW. A morphologically specialized soldier caste improves colony defense in a neotropical eusocial bee. Proc Natl Acad Sci U S A. 2012;109:1182–6.
Goulson D. Bumblebees: their behaviour and ecology. Oxford: Oxford University Press; 2003.
Cameron S, Jost M. Mediators of dominance and reproductive success among queens in the cyclically polygynous Neotropical bumble bee Bombus atratus Franklin. Insectes Soc. 1998;45:135–49.
Bloch G. Regulation of queen-worker conflict in bumble-bee (Bombus terrestris) colonies. Proc R Soc B. 1999;266:2465–9.
Jandt JM, Dornhaus A. Spatial organization and division of labour in the bumblebee Bombus impatiens. Anim Behav. 2009;77:641–51.
Zanette LRS, Miller SDL, Faria CMA, Almond EJ, Huggins TJ, Jordan WC, et al. Reproductive conflict in bumblebees and the evolution of worker policing. Evolution. 2012;66:3765–77.
Yerushalmi S, Bodenhaimer S, Bloch G. Developmentally determined attenuation in circadian rhythms links chronobiology to social organization in bees. J Exp Biol. 2006;209:1044–51.
Ings T, Raine N, Chittka L. A population comparison of the strength and persistence of innate colour preference and learning speed in the bumblebee Bombus terrestris. Behav Ecol Sociobiol. 2009;63:1207–18.
Skorupski P, Chittka L. Photoreceptor processing speed and input resistance changes during light adaptation correlate with spectral class in the bumblebee. Bombus impatiens. PLoS ONE. 2011;6:e25989.
Otterstatter MC, Thomson JD. Contact networks and transmission of an intestinal pathogen in bumble bee (Bombus impatiens) colonies. Oecologia. 2007;154:411–21.
Schmid-Hempel P. On the evolutionary ecology of host-parasite interactions: addressing the question with regard to bumblebees and their parasites. Naturwissenschaften. 2001;88:147–58.
Gegear RJ, Burns JG. The birds, the bees, and the virtual flowers: can pollinator behavior drive ecological speciation in flowering plants? Am Nat. 2007;170:551–66.
Hatfield RG, LeBuhn G. Patch and landscape factors shape community assemblage of bumble bees, Bombus spp. (Hymenoptera: Apidae), in montane meadows. Biol Conserv. 2007;139:150–8.
Williams PH, Araújo MB, Rasmont P. Can vulnerability among British bumblebee (Bombus) species be explained by niche position and breadth? Biol Conserv. 2007;138:493–505.
Cameron SA, Hines HM, Williams PH. A comprehensive phylogeny of the bumble bees (Bombus). Biol J Linn Soc. 2007;91:161–88.
Hines HM. Historical biogeography, divergence times, and diversification patterns of bumble bees (Hymenoptera: Apidae: Bombus). Syst Biol. 2008;57:58–75.
Fontaine C, Dajoz I, Meriguet J, Loreau M. Functional diversity of plant-pollinator interaction webs enhances the persistence of plant communities. PLoS Biol. 2006;4:e1.
Garibaldi LA, Steffan-Dewenter I, Winfree R, Aizen MA, Bommarco R, Cunningham SA, et al. Wild pollinators enhance fruit set of crops regardless of honey bee abundance. Science. 2013;339:1608–11.
Delaplane KS, Mayer DF. Crop pollination by bees. Cabi: Wallingford; 2000.
Velthuis HHW, van Doorn A. A century of advances in bumblebee domestication and the economic and environmental aspects of its commercialization for pollination. Apidologie. 2006;37:421–51.
Winfree R, Williams NM, Dushoff J, Kremen C. Native bees provide insurance against ongoing honey bee losses. Ecol Lett. 2007;10:1105–13.
Cameron SA, Lozier JD, Strange JP, Koch JB, Cordes N, Solter LF, et al. Patterns of widespread decline in North American bumble bees. Proc Natl Acad Sci U S A. 2011;108:662–7.
Williams PH, Osborne JL. Bumblebee vulnerability and conservation world-wide. Apidologie. 2009;40:367–87.
Bommarco R, Lundin O, Smith HG, Rundlof M. Drastic historic shifts in bumble-bee community composition in Sweden. Proc R Soc B. 2012;279:309–15.
Dupont YL, Damgaard C, Simonsen V. Quantitative historical change in bumblebee (Bombus spp.) assemblages of red clover fields. PLoS One. 2011;6:e25172.
Lozier JD, Strange JP, Stewart IJ, Cameron SA. Patterns of range-wide genetic variation in six North American bumble bee (Apidae: Bombus) species. Mol Ecol. 2011;20:4870–88.
Schmid-Hempel R, Eckhardt M, Goulson D, Heinzmann D, Lange C, Plischuk S, et al. The invasion of southern South America by imported bumblebees and associated parasites. J Anim Ecol. 2014;83:823–37.
Schmid-Hempel P, Schmid-Hempel R, Brunner PC, Seeman OD, Allen GR. Invasion success of the bumblebee, Bombus terrestris, despite a drastic genetic bottleneck. Heredity. 2007;99:414–22.
Vanbergen AJ. Initiative tIP: Threats to an ecosystem service: pressures on pollinators. Front Ecol Environ. 2013;11:251–9.
Furst MA, McMahon DP, Osborne JL, Paxton RJ, Brown MJ. Disease associations between honeybees and bumblebees as a threat to wild pollinators. Nature. 2014;506:364–6.
Clare E, Schiestl F, Leitch A, Chittka L. The promise of genomics in the study of plant-pollinator interactions. Genome Biol. 2013;14:207.
Colgan TJ, Carolan JC, Bridgett SJ, Sumner S, Blaxter ML, Brown MJ. Polyphenism in social insects: insights from a transcriptome-wide analysis of gene expression in the life stages of the key pollinator, Bombus terrestris. BMC Genomics. 2011;12:623.
Sadd BM, Kube M, Klages S, Reinhardt R, Schmid-Hempel P. Analysis of a normalised expressed sequence tag (EST) library from a key pollinator, the bumblebee Bombus terrestris. BMC Genomics. 2010;11:110.
Stolle E, Rohde M, Vautrin D, Solignac M, Schmid-Hempel P, Schmid-Hempel R, et al. Novel microsatellite DNA loci for Bombus terrestris (Linnaeus, 1758). Mol Ecol Resour. 2009;9:1345–52.
Stolle E, Wilfert L, Schmid-Hempel R, Schmid-Hempel P, Kube M, Reinhardt R, et al. A second generation genetic map of the bumblebee Bombus terrestris (Linnaeus, 1758) reveals slow genome and chromosome evolution in the Apidae. BMC Genomics. 2011;12:48.
Wilfert L, Gadau J, Schmid-Hempel P. A core linkage map of the bumblebee Bombus terrestris. Genome. 2006;49:1215–26.
Munoz-Torres MC, Reese JT, Childers CP, Bennett AK, Sundaram JP, Childs KL, et al. Hymenoptera Genome Database: integrated community resources for insect species of the order Hymenoptera. Nucleic Acids Res. 2011;39:D658–62.
Wilfert L, Gadau J, Schmid-Hempel P. Variation in genomic recombination rates among animal taxa and the case of social insects. Heredity. 2007;98:189–97.
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–7.
Chipman AD, Ferrier DEK, Brena C, Qu J, Hughes DST, Schröder R, et al. The first Myriapod genome sequence reveals conservative arthropod gene content and genome organisation in the centipede Strigamia maritima. PLoS Biol. 2014;12:e1002005.
Richards S, Gibbs RA, Weinstock GM, Brown SJ, Denell R, Beeman RW, et al. The genome of the model beetle and pest Tribolium castaneum. Nature. 2008;452:949–55.
The International Aphid Genomics Consortium. Genome sequence of the Pea Aphid Acyrthosiphon pisum. PLoS Biol. 2010;8:e1000313.
Werren JH, Richards S, Desjardins CA, Niehuis O, Gadau J, Colbourne JK, et al. Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science. 2010;327:343–8.
Smith CD, Edgar RC, Yandell MD, Smith DR, Celniker SE, Myers EW, et al. Improved repeat identification and masking in Dipterans. Gene. 2007;389:1–9.
Nygaard S, Zhang G, Schiott M, Li C, Wurm Y, Hu H, et al. The genome of the leaf-cutting ant Acromyrmex echinatior suggests key adaptations to advanced social life and fungus farming. Genome Res. 2011;21:1339–48.
Kaminker J, Bergman C, Kronmiller B, Carlson J, Svirskas R, Patel S, et al. The transposable elements of the Drosophila melanogaster euchromatin: a genomics perspective. Genome Biol. 2002;3:research0084.
Xiao J-H, Yue Z, Jia L-Y, Yang X-H, Niu L-H, Wang Z, et al. Obligate mutualism within a host drives the extreme specialization of a fig wasp genome. Genome Biol. 2013;14:R141.
Kelley JL, Peyton JT, Fiston-Lavier A-S, Teets NM, Yee M-C, Johnston JS, et al. Compact genome of the Antarctic midge is likely an adaptation to an extreme environment. Nat Commun. 2014;5:4611.
Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, Oakley TH, et al. The ecoresponsive genome of Daphnia pulex. Science. 2011;331:555–61.
Elsik C, Mackey A, Reese J, Milshina N, Roos D, Weinstock G. Creating a honey bee consensus gene set. Genome Biol. 2007;8:1–8.
Waterhouse RM, Tegenfeldt F, Li J, Zdobnov EM, Kriventseva EV. OrthoDB: a hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res. 2013;41:D358–65.
OrthoDB: database of orthologous groups. [http://cegg.unige.ch/orthodb6]
Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 2011;40:D306–12.
Doerks T, Copley RR, Schultz J, Ponting CP, Bork P. Systematic identification of novel protein domain families associated with nuclear functions. Genome Res. 2002;12:47–56.
Xiong Y, Eickbush TH. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 1990;9:3353–62.
Buttstedt A, Moritz RFA, Erler S. Origin and function of the major royal jelly proteins of the honeybee (Apis mellifera) as members of the yellow gene family. Biol Rev. 2014;89:255–69.
Zou S, Meadows S, Sharp L, Jan LY, Jan YN. Genome-wide study of aging and oxidative stress response in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2000;97:13726–31.
Duncan E, Wilson M, Smith J, Dearden P. Evolutionary origin and genomic organisation of runt-domain containing genes in arthropods. BMC Genomics. 2008;9:558.
Duncan EJ, Dearden PK. Evolution of a genomic regulatory domain: the role of gene co-option and gene duplication in the Enhancer of split complex. Genome Res. 2010;20:917–28.
Dearden PK, Wilson MJ, Sablan L, Osborne PW, Havler M, McNaughton E, et al. Patterns of conservation and change in honey bee developmental genes. Genome Res. 2006;16:1376–84.
Brown S, Fellers J, Shippy T, Denell R, Stauber M, Schmidt-Ott U. A strategy for mapping bicoid on the phylogenetic tree. Curr Biol. 2001;11:R43–4.
Stauber M, Prell A, Schmidt-Ott U. A single Hox3 gene with composite bicoid and zerknullt expression characteristics in non-Cyclorrhaphan flies. Proc Natl Acad Sci U S A. 2002;99:274–9.
Duncan EJ, Benton MA, Dearden PK. Canonical terminal patterning is an evolutionary novelty. Dev Biol. 2013;377:245–61.
Lynch JA, Ozuak O, Khila A, Abouheif E, Desplan C, Roth S. The phylogenetic origin of oskar coincided with the origin of maternally provisioned germ plasm and pole cells at the base of the Holometabola. PLoS Genet. 2011;7:e1002029.
Martins JR, Nunes FM, Cristino AS, Simoes ZL, Bitondi MM. The four hexamerin genes in the honey bee: structure, molecular evolution and function deduced from expression patterns in queens, workers and drones. BMC Mol Biol. 2010;11:23.
Lourenco AP, Zufelato MS, Bitondi MM, Simoes ZL. Molecular characterization of a cDNA encoding prophenoloxidase and its expression in Apis mellifera. Insect Biochem Mol Biol. 2005;35:541–52.
Willis JH. Structural cuticular proteins from arthropods: Annotation, nomenclature, and sequence characteristics in the genomics era. Insect Biochem Mol Biol. 2010;40:189–204.
Dittmer NT, Kanost MR. Insect multicopper oxidases: diversity, properties, and physiological roles. Insect Biochem Mol Biol. 2010;40:179–88.
Garcia-Bellido A, de Celis JF. The complex tale of the achaete-scute complex: a paradigmatic case in the analysis of gene organization and function during development. Genetics. 2009;182:631–9.
Campos-Ortega JA. Mechanisms of early neurogenesis in Drosophila melanogaster. J Neurobiol. 1993;24:1305–27.
Schlatter R, Maier D. The Enhancer of split and Achaete-Scute complexes of Drosophilids derived from simple ur-complexes preserved in mosquito and honeybee. BMC Evol Biol. 2005;5:67.
Ashraf SI, Hu X, Roote J, Ip YT. The mesoderm determinant snail collaborates with related zinc-finger proteins to control Drosophila neurogenesis. EMBO J. 1999;18:6426–38.
De Graeve F, Jagla T, Daponte JP, Rickert C, Dastugue B, Urban J, et al. The ladybird homeobox genes are essential for the specification of a subpopulation of neural cells. Dev Biol. 2004;270:122–34.
Beye M, Hasselmann M, Fondrk MK, Page RE, Omholt SW. The gene csd is the primary signal for sexual development in the honeybee and encodes an SR-type protein. Cell. 2003;114:419–29.
Hasselmann M, Gempe T, Schiott M, Nunes-Silva CG, Otte M, Beye M. Evidence for the evolutionary nascence of a novel sex determination pathway in honeybees. Nature. 2008;454:519–22.
Schmieder S, Colinet D, Poirie M. Tracing back the nascence of a new sex-determination pathway to the ancestor of bees and ants. Nat Commun. 2012;3:895.
Privman E, Wurm Y, Keller L. Duplication and concerted evolution in a master sex determiner under balancing selection. Proc R Soc B. 2013;280:20122968.
Koch V, Nissen I, Schmitt BD, Beye M. Independent evolutionary origin of fem paralogous genes and complementary sex determination in Hymenopteran insects. PLoS One. 2014;9:e91883.
Geuverink E, Beukeboom LW. Phylogenetic distribution and evolutionary dynamics of the sex determination genes doublesex and transformer in insects. Sex Dev. 2014;8:38–49.
Conrad T, Akhtar A. Dosage compensation in Drosophila melanogaster: epigenetic fine-tuning of chromosome-wide transcription. Nat Rev Genet. 2012;13:123–34.
Rubin EB, Shemesh Y, Cohen M, Elgavish S, Robertson HM, Bloch G. Molecular and phylogenetic analyses reveal mammalian-like clockwork in the honey bee (Apis mellifera) and shed new light on the molecular evolution of the circadian clock. Genome Res. 2006;16:1352–65.
Ingram KK, Kutowoi A, Wurm Y, Shoemaker D, Meier R, Bloch G. The molecular clockwork of the fire ant Solenopsis invicta. PLoS One. 2012;7:e45715.
Debski J, Wyslouch-Cieszynska A, Dadlez M, Grzelak K, Kludkiewicz B, Kolodziejczyk R, et al. Positions of disulfide bonds and N-glycosylation site in juvenile hormone binding protein. Arch Biochem Biophys. 2004;421:260–6.
Dauwalder B, Tsujimoto S, Moss J, Mattox W. The Drosophila takeout gene is regulated by the somatic sex-determination pathway and affects male courtship behavior. Genes Dev. 2002;16:2879–92.
Lazareva AA, Roman G, Mattox W, Hardin PE, Dauwalder B. A role for the adult fat body in Drosophila male courtship behavior. PLoS Genet. 2007;3:e16.
Hagai T, Cohen M, Bloch G. Genes encoding putative Takeout/juvenile hormone binding proteins in the honeybee (Apis mellifera) and modulation by age and juvenile hormone of the takeout-like gene GB19811. Insect Biochem Mol Biol. 2007;37:689–701.
Shpigler H, Amsalem E, Huang ZY, Cohen M, Siegel AJ, Hefetz A, et al. Gonadotropic and physiological functions of juvenile hormone in bumblebee (Bombus terrestris) workers. PLoS One. 2014;9:e100650.
Bloch G, Shpigler H, Wheeler DE, Robinson GE. Endocrine influences on the organization of insect societies. Vol. II. Non-mammalian hormone-behavior systems, non-mammalian invertebrates. In: Pfaff DW, Arnold AP, Etgen AM, Fahrbach SE, Rubin R, editors. Hormones, brain and behavior. 2nd ed. San Diego: Elsevier; 2009. p. 1027–68.
Jones AK, Sattelle DB. The cys-loop ligand-gated ion channel superfamily of the honeybee, Apis mellifera. Invert Neurosci. 2006;6:123–32.
El Hassani AK, Dupuis JP, Gauthier M, Armengaud C. Glutamatergic and GABAergic effects of fipronil on olfactory learning and memory in the honeybee. Invert Neurosci. 2009;9:91–100.
Louis T, Musso PY, de Oliveira SB, Garreau L, Giurfa M, Raymond V, et al. Amelalpha8 subunit knockdown in the mushroom body vertical lobes impairs olfactory retrieval in the honeybee, Apis mellifera. Eur J Neurosci. 2012;36:3438–50.
Raymond-Delpech V, Matsuda K, Sattelle BM, Rauh JJ, Sattelle DB. Ion channels: molecular targets of neuroactive insecticides. Invert Neurosci. 2005;5:119–33.
Fauser-Misslin A, Sadd BM, Neumann P, Sandrock C. Influence of combined pesticide and parasite exposure on bumblebee colony traits in the laboratory. J Appl Ecol. 2014;51:450–9.
Gill RJ, Ramos-Rodriguez O, Raine NE. Combined pesticide exposure severely affects individual- and colony-level traits in bees. Nature. 2012;491:105–8.
Mommaerts V, Reynders S, Boulet J, Besard L, Sterk G, Smagghe G. Risk assessment for side-effects of neonicotinoids against bumblebees with and without impairing foraging behavior. Ecotoxicology. 2010;19:207–15.
Nassel DR, Winther AM. Drosophila neuropeptides in regulation of physiology and behavior. Prog Neurobiol. 2010;92:42–104.
Hauser F, Cazzamali G, Williamson M, Blenau W, Grimmelikhuijzen CJ. A review of neurohormone GPCRs present in the fruitfly Drosophila melanogaster and the honey bee Apis mellifera. Prog Neurobiol. 2006;80:1–19.
Hauser F, Cazzamali G, Williamson M, Park Y, Li B, Tanaka Y, et al. A genome-wide inventory of neurohormone GPCRs in the red flour beetle Tribolium castaneum. Front Neuroendocrinol. 2008;29:142–65.
Tayler TD, Pacheco DA, Hergarden AC, Murthy M, Anderson DJ. A neuropeptide circuit that coordinates sperm transfer and copulation duration in Drosophila. Proc Natl Acad Sci U S A. 2012;109:20697–702.
Kapan N, Lushchak O, Luo J, Nässel D. Identified peptidergic neurons in the Drosophila brain regulate insulin-producing cells, stress responses and metabolism by coexpressed short neuropeptide F and corazonin. Cell Mol Life Sci. 2012;69:4051–66.
Boerjan B, Verleyen P, Huybrechts J, Schoofs L, De Loof A. In search for a common denominator for the diverse functions of arthropod corazonin: a role in the physiology of stress? Gen Comp Endocrinol. 2010;166:222–33.
Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem. 2007;76:387–417.
Claudianos C, Ranson H, Johnson RM, Biswas S, Schuler MA, Berenbaum MR, et al. A deficit of detoxification enzymes: pesticide sensitivity and environmental response in the honeybee. Insect Mol Biol. 2006;15:615–36.
Oakeshott JG, Johnson RM, Berenbaum MR, Ranson H, Cristino AS, Claudianos C. Metabolic enzymes associated with xenobiotic and chemosensory responses in Nasonia vitripennis. Insect Mol Biol. 2010;19:147–63.
Feyereisen R. Insect CYP, genes and P450 enzymes. In: Gilbert LI, editor. Insect molecular biology and biochemistry. London: Academic; 2012.
Ono H, Rewitz KF, Shinoda T, Itoyama K, Petryk A, Rybczynski R, et al. Spook and Spookier code for stage-specific components of the ecdysone biosynthetic pathway in Diptera. Dev Biol. 2006;298:555–70.
Maibeche-Coisne M, Nikonov AA, Ishida Y, Jacquin-Joly E, Leal WS. Pheromone anosmia in a scarab beetle induced by in vivo inhibition of a pheromone-degrading enzyme. Proc Natl Acad Sci U S A. 2004;101:11459–64.
Qiu Y, Tittiger C, Wicker-Thomas C, Le Goff G, Young S, Wajnberg E, et al. An insect-specific P450 oxidative decarbonylase for cuticular hydrocarbon biosynthesis. Proc Natl Acad Sci U S A. 2012;109:14858–63.
Mao W, Schuler MA, Berenbaum MR. CYP9Q-mediated detoxification of acaricides in the honey bee (Apis mellifera). Proc Natl Acad Sci U S A. 2011;108:12657–62.
Mao W, Schuler MA, Berenbaum MR. Honey constituents up-regulate detoxification and immunity genes in the western honey bee Apis mellifera. Proc Natl Acad Sci U S A. 2013;110:8842–6.
Touhara K, Vosshall LB. Sensing odorants and pheromones with chemosensory receptors. Annu Rev Physiol. 2009;71:307–32.
Kwon JY, Dahanukar A, Weiss LA, Carlson JR. The molecular basis of CO2 reception in Drosophila. Proc Natl Acad Sci U S A. 2007;104:3574–8.
Croset V, Rytz R, Cummins SF, Budd A, Brawand D, Kaessmann H, et al. Ancient protostome origin of chemosensory ionotropic glutamate receptors and the evolution of insect taste and olfaction. PLoS Genet. 2010;6:e1001064.
Robertson HM, Wanner KW. The chemoreceptor superfamily in the honey bee, Apis mellifera: expansion of the odorant, but not gustatory, receptor family. Genome Res. 2006;16:1395–403.
Robertson HM, Gadau J, Wanner KW. The insect chemoreceptor superfamily of the parasitoid jewel wasp Nasonia vitripennis. Insect Mol Biol. 2010;19:121–36.
Zhou X, Slone JD, Rokas A, Berger SL, Liebig J, Ray A, et al. Phylogenetic and transcriptomic analysis of chemosensory receptors in a pair of divergent ant species reveals sex-specific signatures of odor coding. PLoS Genet. 2012;8:e1002930.
Vosshall LB, Hansson BS. A unified nomenclature system for the insect olfactory coreceptor. Chem Senses. 2011;36:497–8.
Wanner KW, Nichols AS, Walden KK, Brockmann A, Luetje CW, Robertson HM. A honey bee odorant receptor for the queen substance 9-oxo-2-decenoic acid. Proc Natl Acad Sci U S A. 2007;104:14383–8.
Robertson HM, Navik JA, Walden KK, Honegger HW. The bursicon gene in mosquitoes: an unusual example of mRNA trans-splicing. Genetics. 2007;176:1351–3.
Robertson HM, Kent LB. Evolution of the gene lineage encoding the carbon dioxide receptor in insects. J Insect Sci. 2009;9:19.
Kent LB, Robertson HM. Evolution of the sugar receptors in insects. BMC Evol Biol. 2009;9:41.
Miyamoto T, Slone J, Song X, Amrein H. A fructose receptor functions as a nutrient sensor in the Drosophila brain. Cell. 2012;151:1113–25.
Weiss LA, Dahanukar A, Kwon JY, Banerjee D, Carlson JR. The molecular and cellular basis of bitter taste in Drosophila. Neuron. 2011;69:258–72.
Foret S, Maleszka R. Function and evolution of a gene family encoding odorant binding-like proteins in a social insect, the honey bee (Apis mellifera). Genome Res. 2006;16:1404–13.
Van Vaerenbergh M, Debyser G, Smagghe G, Devreese B, de Graaf D. Unraveling the venom proteome of the bumblebee (Bombus terrestris) by integrating a combinatorial peptide ligand library approach with FTICR-MS. Toxicon. (in press).
Evans JD, Aronstein K, Chen YP, Hetru C, Imler JL, Jiang H, et al. Immune pathways and defence mechanisms in honey bees Apis mellifera. Insect Mol Biol. 2006;15:645–56.
Barribeau SM, Sadd BM, du Plessis L, Brown MJF, Buechel S, Carolan JC, et al. A depauperate immune repertoire precedes evolution of sociality in bees. Genome Biol. 2015;16:83.
Chan QWT, Howes CG, Foster LJ. Quantitative comparison of caste differences in honeybee hemolymph. Mol Cell Proteomics. 2006;5:2252–62.
Jinek M, Doudna JA. A three-dimensional view of the molecular machinery of RNA interference. Nature. 2009;457:405–12.
Kim BY, Lee KS, Yoon HJ, Kim I, Li J, Sohn HD, et al. Expression profile of the iron-binding proteins transferrin and ferritin heavy chain subunit in the bumblebee Bombus ignitus. Comp Biochem Physiol B Biochem Mol Biol. 2009;153:165–70.
Weaver DB, Anzola JM, Evans JD, Reid JG, Reese JT, Childs KL, et al. Computational and transcriptional evidence for microRNAs in the honey bee genome. Genome Biol. 2007;8:R97.
Behura SK, Whitfield CW. Correlated expression patterns of microRNA genes with age-dependent behavioural changes in honeybee. Insect Mol Biol. 2010;19:431–9.
Greenberg JK, Xia J, Zhou X, Thatcher SR, Gu X, Ament SA, et al. Behavioral plasticity in honey bees is associated with differences in brain microRNA transcriptome. Genes Brain Behav. 2012;11:660–70.
Liu F, Peng W, Li Z, Li W, Li L, Pan J, et al. Next-generation small RNA sequencing for microRNAs profiling in Apis mellifera: comparison between nurses and foragers. Insect Mol Biol. 2012;21:297–303.
Nunes FM, Ihle KE, Mutti NS, Simoes ZL, Amdam GV. The gene vitellogenin affects microRNA regulation in honey bee (Apis mellifera) fat body and brain. J Exp Biol. 2013;216:3724–32.
Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–7.
Moxon S, Schwach F, Dalmay T, Maclean D, Studholme DJ, Moulton V. A toolkit for analysing large-scale plant small RNA datasets. Bioinformatics. 2008;24:2252–3.
Sewer A, Paul N, Landgraf P, Aravin A, Pfeffer S, Brownstein MJ, et al. Identification of clustered microRNAs using an ab initio prediction method. BMC Bioinformatics. 2005;6:267.
Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet. 2007;8:93–103.
Griffiths-Jones S, Hui JH, Marco A, Ronshaugen M. MicroRNA evolution by arm switching. EMBO Rep. 2011;12:172–7.
Lu J, Shen Y, Wu Q, Kumar S, He B, Shi S, et al. The birth and death of microRNA genes in Drosophila. Nat Genet. 2008;40:351–5.
Lyko F, Maleszka R. Insects as innovative models for functional studies of DNA methylation. Trends Genet. 2011;27:127–31.
Herb BR, Wolschin F, Hansen KD, Aryee MJ, Langmead B, Irizarry R, et al. Reversible switching between epigenetic states in honeybee behavioral subcastes. Nat Neurosci. 2012;15:1371–3.
Kucharski R, Maleszka J, Foret S, Maleszka R. Nutritional control of reproductive status in honeybees via DNA methylation. Science. 2008;319:1827–30.
Lockett GA, Kucharski R, Maleszka R. DNA methylation changes elicited by social stimuli in the brains of worker honey bees. Genes Brain Behav. 2012;11:235–42.
Amarasinghe HE, Clayton CI, Mallon EB. Methylation and worker reproduction in the bumble-bee (Bombus terrestris). Proc R Soc B. 2014;281:20132502.
Lobanov AV, Hatfield DL, Gladyshev VN. Eukaryotic selenoproteins and selenoproteomes. Biochim Biophys Acta. 2009;1790:1424–8.
Chapple CE, Guigó R. Relaxation of selective constraints causes independent selenoprotein extinction in insect genomes. PLoS One. 2008;3:e2968.
Etter PD, Bassham S, Hohenlohe PA, Johnson EA, Cresko WA. SNP discovery and genotyping for evolutionary genetics using RAD sequencing. Methods Mol Biol. 2011;772:157–78.
Lozier JD. Revisiting comparisons of genetic diversity in stable and declining species: assessing genome-wide polymorphism in North American bumble bees using RAD sequencing. Mol Ecol. 2014;23:788–801.
Whitfield CW, Behura SK, Berlocher SH, Clark AG, Johnston JS, Sheppard WS, et al. Thrice out of Africa: ancient and recent expansions of the honey bee. Apis mellifera Science. 2006;314:642–5.
Kelley DR, Schatz MC, Salzberg SL. Quake: quality-aware detection and correction of sequencing errors. Genome Biol. 2010;11:R116.
Stolle E, Kidner JH, Moritz RF. Patterns of evolutionary conservation of microsatellites (SSRs) suggest a faster rate of genome evolution in Hymenoptera than in Diptera. Genome Biol Evol. 2013;5:151–62.
Flutre T, Duprat E, Feuillet C, Quesneville H. Considering transposable element diversification in de novo annotation approaches. PLoS One. 2011;6:e16526.
Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–301.
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8:973–82.
Marchler-Bauer A, Anderson JB, Derbyshire MK, DeWeese-Scott C, Gonzales NR, Gwadz M, et al. CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res. 2007;35:D237–40.
Kohany O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics. 2006;7:474.
Kapitonov VV, Tempel S, Jurka J. Simple and fast classification of non-LTR retrotransposons based on phylogeny of their RT domain protein sequences. Gene. 2009;448:207–13.
Kapitonov VV, Jurka J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat Rev Genet. 2008;9:411–2.
Yuan YW, Wessler SR. The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies. Proc Natl Acad Sci U S A. 2011;108:7884–9.
NCBI Eukaryotic Genome Annotation Pipeline. [http://www.ncbi.nlm.nih.gov/genome/annotation_euk/process/]
Stanke M, Schoffmann O, Morgenstern B, Waack S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics. 2006;7:62.
Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12:656–64.
Bombus terrestris primary genome assembly. [ftp://ftp.ncbi.nih.gov/genbank/genomes/Eukaryotes/invertebrates/Bombus_terrestris/Bter_1.0/Primary_Assembly/]
Bombus impatiens primary genome assembly. [ftp://ftp.ncbi.nih.gov/genbank/genomes/Eukaryotes/invertebrates/Bombus_impatiens/BIMP_2.0/Primary_Assembly/]
Solovyev V, Kosarev P, Seledsov I, Vorobyev D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 2006;7:S10.11–12.
geneid. [http://genome.crg.es/software/geneid/]
Parra G, Blanco E, Guigo R. GeneID in Drosophila. Genome Res. 2000;10:511–5.
Parra G, Agarwal P, Abril JF, Wiehe T, Fickett JW, Guigo R. Comparative gene prediction in human and mouse. Genome Res. 2003;13:108–17.
van Baren MJ, Koebbe BC, Brent MR. Using N-SCAN or TWINSCAN to predict gene structures in genomic DNA sequences. Curr Protoc Bioinformatics. 2007;Chapter 4:Unit 4.8.
RepeatMasker. [http://www.repeatmasker.org]
Keibler E, Brent MR. Eval: a software package for analysis of genome annotations. BMC Bioinformatics. 2003;4:50.
WU-BLAST. [http://blast.wustl.edu]
Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31.
Consortium UP. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013;41:D43–7.
Marygold SJ, Leyland PC, Seal RL, Goodman JL, Thurmond J, Strelets VB, et al. FlyBase: improvements to the bibliography. Nucleic Acids Res. 2013;41:D751–7.
Lewis SE, Searle SM, Harris N, Gibson M, Lyer V, Richter J, et al. Apollo: a sequence annotation editor. Genome Biol. 2002;3:Research0082.
Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Capella-Gutierrez S, Silla-Martinez JM. Gabaldon T: trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3.
Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–37.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80.
Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci U S A. 1998;95:5857–64.
SignalP. [http://www.cbs.dtu.dk/services/SignalP/]
Matsuura H, Sokabe T, Kohno K, Tominaga M, Kadowaki T. Evolutionary conservation and changes in insect TRP channels. BMC Evol Biol. 2009;9:228.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8.
Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotech. 2014;32:223–6.
MaxQuant documentation. [http://maxquant.org/]
Côté RG, Griss J, Dianes JA, Wang R, Wright JC, van den Toorn HWP, et al. The PRoteomics IDEntification (PRIDE) Converter 2 framework: an improved suite of tools to facilitate data submission to the pride database and the ProteomeXchange Consortium. Mol Cell Proteomics. 2012;11:1682–9.
Conesa A, Gotz S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics. 2008;2008:619832.
Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–35.
Zdobnov EM, Apweiler R. InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–8.
Sorefan K, Pais H, Hall AE, Kozomara A, Griffiths-Jones S, Moulton V, et al. Reducing ligation bias of small RNAs in libraries for next generation sequencing. Silence. 2012;3:4.
Flores KB, Amdam GV. Deciphering a methylome: what can we read into patterns of DNA methylation? J Exp Biol. 2011;214:3155–63.
Mariotti M, Guigo R. Selenoprofiles: profile-based scanning of eukaryotic genome sequences for selenoprotein genes. Bioinformatics. 2010;26:2656–63.
Mariotti M, Lobanov AV, Guigo R, Gladyshev VN. SECISearch3 and Seblastian: new tools for prediction of SECIS elements and selenoproteins. Nucleic Acids Res. 2013;41:e149.
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3:e3376.
Emerson KJ, Merz CR, Catchen JM, Hohenlohe PA, Cresko WA, Bradshaw WE, et al. Resolving postglacial phylogeography using high-throughput sequencing. Proc Natl Acad Sci U S A. 2010;107:16196–200.
Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, Cresko WA. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet. 2010;6:e1000862.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8.
Nielsen R, Korneliussen T, Albrechtsen A, Li Y, Wang J. SNP calling, genotype calling, and sample allele frequency estimation from new-generation sequencing data. PLoS One. 2012;7:e37558.
Korneliussen TS, Moltke I, Albrechtsen A, Nielsen R. Calculation of Tajima’s D and other neutrality test statistics from low depth next-generation sequencing data. BMC Bioinformatics. 2013;14:289.
Acknowledgements
The sequencing, annotation, and assembly of the B. impatiens genome was supported by National Institutes of Health (NIH) Pioneer Award DP1 OD006416 (GER). The B. terrestris genome sequencing, assembly and project coordination was supported by NIH grant U54 HG003273 (RAG). Contributions from members of the CGE lab were supported by Agriculture and Food Research Initiative Competitive grant no. 2010-65106-21301 from the USDA National Institute of Food and Agriculture. GVA was supported by the Research Council of Norway (NFR) and the PEW Charitable Trust. MB was supported by a student fellowship from the University of East Anglia, UK. GB was supported by the Israel Science Foundation (ISF). DHC was supported by a studentship funded by the Biotechnology and Biological Sciences Research Council, UK, and the University of East Anglia. JDL was supported by the University of Alabama College of Arts and Sciences. ZPLS was supported by a São Paulo Research Foundation grant (FAPESP number 11/03171-5). RMW was supported by Marie Curie International Outgoing Fellowship PIOF-GA-2011-303312 and by Swiss National Science Foundation awards 31003A-125350 and 31003A-143936 to EMZ. BMS was supported by a New Faculty Initiative Grant (NFIG) from Illinois State University College of Arts and Sciences. MHasselmann received financial support from the DFG. The work of FC, RG, and MM was supported by the Instituto Nacional de Bioinformática (INB) from ISCIII in Spain. We thank Justin Reese and Christopher Childers for setting up genome annotation tools at Hymenoptera Genome Database.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BMS, PS-H, and KCW led the consortium. RAG, DMM, SR, GER, SES, and KCW led the sequencing projects. SMB, GB, DCdG, PD, CGE, JG, CJPG, MHasselmann, JDL, HMR, GS, ES, MVV, and RMW were coordinators of designated analysis domains. EBB led protein domain analysis, and EBB and SK performed analyses. DP, JQ, SLS, and KCW performed genome assembly. AKB, FC, CGE, RG, KHoff, MMariotti, MM-T, TMurphy, and DS performed gene predictions. ES carried out synteny analysis. RMW and EMZ performed orthology analysis. PKA, KPB, MC, LF, AGH, MHolder, MEH, LJ, JJ, VJ, CK, SLL, RM, TMathew, IFN, RN, GO, CP, PL-L, NS, JS, DNS, RT, AV, KKOW, and WY-Q carried out library preparation and sequencing. GVA, MBeckers, AFGB, DHC, TD, KF, GAL, and FW performed analyses of gene regulation analysis. SH, JHK, BFK, RFAM, and ES carried out repetitive element analysis. DCdG and MVV performed defence gene analysis. CJPG, FH, AKJ and TK carried out neurobiology gene analyses. MBeye, MMGB, RC, OC, AGC, PD, ED, TF, FCPF, TG, KHartfelder, FCH, FI, VK, AK, ML, DSMA, AN, IN, FMFN, AO, MO, DGP, NR, CGS, BDS, CS, ZLPS and MPMS performed development gene analyses. KLC, TJC, HMR and CEJ analyzed chemoreceptor genes. SMB, MJKB, SDB, KC, JCC, OC, TJC, LdP, SE, JE, SH, HMGL, EBM, MMarxer, IM, KN, JN, BMS, PS-H, RS-H, LS, RMW and NY performed immune gene analyses. MBiewer and MHasselmann carried out analyses of sex determination genes. GB, SD, and ECW performed behaviour gene analysis. DFC, LSJ, RMJ, and JGO performed detoxification gene analysis. MLB and TJC carried out transcriptome data analysis. JDL performed SNP analysis. JCC and TJC carried out queen haemolymph proteomic analysis. OR performed recombination gene analysis. MVV provided venom proteome data. GD and BD produced venom proteome data. In addition to all those involved in specific analyses above, CA, JB, ADC, BF, JL, MPO, and SS were involved in gene annotation. BMS wrote the manuscript, with contributions from coordinators and leaders above, and MLB, AFGB, JCC, LC, DHC, and AKJ. All authors read and approved the final manuscript.
Additional files
Additional file 1:
Summary information relating to the B. terrestris genome assembly, details of gene model predictions for B. terrestris and B. impatiens , information on protein domains, bumblebee Juvenile hormone binding protein information, biogenic amine receptor information, bumblebee neuropeptide sequences, a comparison of corazonin between the two bumblebees, neuropeptide gene suites across various Arthropods, Halloween gene (p450s) list and phylogenetic tree, annotated lists, and phylogenetic trees of B. terrestris Odorant Receptors, Gustatory Receptors, Ionotrophic Receptors, and Odorant Binding Proteins, a taxonomic distribution of functionally unknown hemolymph associated proteins, and an overview of core RNAi genes with a phylogenetic tree of SID proteins across insects.
Additional file 2:
Spreadsheet tabs with genomic coordinates of synteny between B. terrestris and B. impatiens (A), a list of manually annotated genes and species-specific names (B), bee-specific (C) and Bombus- specific (D) genes based on ortholog analysis, protein domains relating to transposable elements (E), venom peptide genes in B. terrestris with proteomic support (F), venom proteins without proteomic evidence, and details of their presence/absence in the genome sequence (G), B. impatiens venom protein predictions (H), an ortholog-based list of bumblebee genes in immune related families (I), proteins identified in B. terrestris queen hemolymph by mass spectrometry (J), and miRNAs sequenced in B. terrestris (K), predicted but not sequenced in B. terrestris (L), and predicted in B. impatiens (M).
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Sadd, B.M., Barribeau, S.M., Bloch, G. et al. The genomes of two key bumblebee species with primitive eusocial organization. Genome Biol 16, 76 (2015). https://doi.org/10.1186/s13059-015-0623-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13059-015-0623-3