
Abstract
In amyotrophic lateral sclerosis (ALS), motor neuron degeneration occurs simultaneously with systemic metabolic impairment and neuroinflammation. Playing an important role in the regulation of both phenomena, interleukin (IL)-6, a major cytokine of the inflammatory response has been proposed as a target for management of ALS. Although a pilot clinical trial provided promising results in humans, another recent preclinical study showed that knocking out the IL-6 gene in mice carrying ALS did not improve clinical outcome. In this study, we aimed to determine the relevance of the IL-6 pathway blockade in a mouse model of ALS by using a pharmacological antagonist of IL-6, a murine surrogate of tocilizumab, namely MR16-1. We analyzed the immunological and metabolic effects of IL-6 blockade by cytokine measurement, blood cell immunophenotyping, targeted metabolomics, and transcriptomics. A deleterious clinical effect of MR16-1 was revealed, with a speeding up of weight loss (p = 0.0041) and decreasing body weight (p < 0.05). A significant increase in regulatory T-cell count (p = 0.0268) and a decrease in C-X-C ligand-1 concentrations in plasma (p = 0.0479) were observed. Metabolomic and transcriptomic analyses revealed that MR16-1 mainly affected branched-chain amino acid, lipid, arginine, and proline metabolism. IL-6 blockade negatively affected body weight, despite a moderated anti-inflammatory effect. Metabolic effects of IL-6 were mild compared with metabolic disturbances observed in ALS, but a modification of lipid metabolism by therapy was identified. These results indicate that IL-6 blockade did not improve clinical outcome of a mutant superoxide dismutase 1 mouse model of ALS.
Similar content being viewed by others


Introduction
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive disorder that selectively destroys motor neurons and leads to death 2 to 5years after diagnosis [1]. Metabolic disturbances (i.e., brain, blood, and muscle via the “dying back” phenomenon) play an important role in different tissues in this disease [2]. The clinical heterogeneity and unpredictable prognosis may be due to the interactions between several complex pathophysiological mechanisms. As a result, none of the studies conducted to evaluate ALS pathogenesis has enabled the determination of a successful treatment, except for riluzole, which was approved several years ago [3, 4]. Although markers of denervation precede neuroinflammation in the mutant superoxide dismutase 1 (mSOD1) mouse model [5], the immune system appears to play an active and highly complex role in ALS [6]. Several studies have shown that the immune system can exacerbate the disease [7–9], as well as protect the central nervous system. Regulatory T cells (Tregs) play a protective role in patients with ALS [10, 11] and mSOD1 mice [11, 12]. The progressive loss of Tregs during ALS may be linked to metabolic factors [10, 13, 14]. The signaling cascades activated by inflammatory cytokines can modulate insulin signaling, particularly in adipose tissue, liver, skeletal muscle, and the central nervous system [15, 16].
Interleukin (IL)-6 signaling plays a key role in both Treg differentiation [17, 18] and muscular metabolism [19, 20]. IL-6 may act as an autocrine, paracrine, or endocrine molecule in the insulin-sensitizing effects following exercise [20]. This may be due to the increase of substrate availability, such as glucose and lipids, through enhanced glycogenolysis and lipolysis as reported in isolated skeletal muscle [20]. Tocilizumab (Actemra; Genentech USA, San Francisco, CA, USA), an IgG1 humanized antibody directed to the α chain of the IL-6 receptor (IL-6Rα) [21], may attenuate the proinflammatory effects of mutant SOD1 in vitro [22]. In a pilot clinical study, tocilizumab reduced the inflammation of patients with ALS undergoing a strong inflammatory status [23]. However, mSOD1/IL-6–/– and mSOD1 mice had a similar clinical outcome in a recent study [24].
Therefore, we wished to clarify whether blockade of IL-6Rα may be beneficial in ALS, especially in determining the role of IL-6 anti-inflammatory and metabolic effects. For this purpose, the effects of IL-6 pathway blockade in ALS by a rat antimouse IL-6R antibody (MR16-1) were determined. Metabolomics profile, and immunologic and clinical status of SOD1-G93A transgenic mice (mSOD1) treated with MR16-1 were analyzed and the findings were confirmed by a targeted transcriptomic approach.
Materials and Methods
Animals
Twenty-two SOD1-G93A male mice, on a C57Bl/6 genetic background, and 24 male wild-type C57Bl/6 J (WT) controls were acquired from Charles Rivers Laboratory (Burlington, CA, USA; strains #004435 and #000664, respectively). Mice were genotyped to test for expression of the transgene as previously described [25]. They were housed in groups of 2–6 per cage in a virus-free barrier facility under a 12-h light/dark cycle, with ad libitum access to regular A04 chow and water. All animal experiments followed current European Union regulations (Directive 2010/63/EU), as recommended by the regional ethics committee for animal experimentation (CEEA Val de Loire decision #19).
MR16-1 Treatment
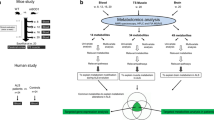
MR16-1, a rat IgG1k monoclonal antibody to the mouse IL-6Rα with neutralizing activity on the effects of IL-6, was graciously provided by Chugai Pharmaceutical (Tokyo, Japan) under a Material Transfer Agreement. A loading dose of 50 mg/kg of body weight was first injected intraperitoneally into 11 transgenic and 7 WT mice 10 weeks after birth, followed by 2 injections of 20 mg/kg per week until 20 weeks after birth. Eleven transgenic and 17 WT control mice received phosphate buffer saline following the same schedule. The administration and sample collection protocol is shown in Fig. 1A and the analysis methodology in Fig. 1B. Metabolomics data from untreated mice have been analyzed for another paper and will not be fully developed herein.

Flowchart for (A) global experimental design and (B) omics data treatment. Mice were divided into 4 groups depending on their genetic status [mutant superoxide dismutase 1 (mSOD1) or not] and the treatment randomly assigned (anti-interleukin-6 receptor monoclonal antibody MR16-1) or phosphate buffer saline (PBS). Blood and tissue samples were collected during the treatment and at sacrifice at when the mice were 20 weeks old. WT = wild type; TS = triceps surae; NMR = nuclear magnetic resonance; HPLC = high-performance liquid chromatography; FIA = flow injection analysis; MS/MS = tandem mass spectroscopy; ALS = amyotrophic lateral sclerosis
Sample Collection
Submandibular collection of blood (150–275 μl or 11 ml/kg body mass) was performed 8, 12, 16, and 20 weeks after birth, without the use of anesthesia. Mice were sacrificed 20 weeks after birth by cervical dislocation. The triceps surae muscles and cerebral cortex were dissected with a scalpel and tweezers and immediately frozen in liquid nitrogen. Tissue samples were then lyophilized in a FreeZone 6-liter benchtop lyophilizer (Labconco, Kansas City, KS, USA). Lyophilisates were crushed and mixed to obtain a powder and stored at –80 °C until batch analysis.
Clinical Evaluation
Disease progression was assessed by monitoring body weight and locomotor performance. From the age of 7 weeks to the time of sacrifice, each animal in all experimental groups was weighed twice a week. The beginning of weight loss was recorded [26].
The rotarod test was used to assess mouse locomotor performance twice a week from 8 weeks of age to the time of sacrifice. Training was performed at 7 weeks of age, on 3 consecutive days. Testing began by placing mice individually on a rod (diameter = 3.5 cm), rotating at a constant speed of 25 rpm (fast rotarod). The time taken before the mice fell off the rod was recorded, and used as a measure of motor function (test limited to 5 min maximum). During testing, 3 trials were performed, and the longest time spent on the rod was recorded. For mice that did not reach a maximum of 5 min on the rod after 3 trials, another attempt was made at a speed of 15 rpm for 2 min (slow rotarod). The age of the mice at the first failure in this last attempt was also recorded and used as another locomotor performance outcome.
Inflammation Assessment
Treg lymphocyte measurements
Blood samples were centrifuged at 3000 × g for 10 min and cell pellets were separated from plasma for flow cytometry analysis. Red blood cells were lysed using hemolysis buffer (NH4Cl 150 mM, KHCO3 10 mM, and ethylenediaminetetraacetic acid 0.1 mM). The leukocytes were stained at the cell surface with the appropriate dilution of the following monoclonal antibodies: R-phycoerythrin-labeled anti-CD3, fluorescein isothiocyanate-labeled anti-CD4, pacific blue-labeled anti-CD8, and PerCP–Cy5.5-labeled anti-CD25 (BD Biosciences, San Jose, CA, USA). Staining of living cells was performed using the LIVE/DEAD Fixable aqua dead cell stain kit from Molecular Probes (Eugene, OR, USA). The cells were then incubated at 4 °C in phosphate-buffered saline containing 5% heat-inactivated fetal calf serum and 0.05 M ethylenediaminetetraacetic acid for 30 min. The allophycocyanin-labeled anti-FoxP3 staining set (eBioscience, San Diego, CA, USA) was used for intracellular staining, according to the manufacturer’s recommendations. Flow cytometry analyses were performed using a MACSQuant Analyzer (Miltenyi Biotec, Bergisch Gladbach, Germany) and VenturiOne software (AppliedCytometry, Sheffield, UK). Fluorescence compensation was performed for each condition using leukocytes from WT mice, and the specificity of intracellular staining was verified by using allophycocyanin-labeled isotype control antibodies (clone eBM2a).
Cytokine measurement
Plasma (20 μl) collected from mice at 20 weeks of age were collected for IL-6, IL-10, IL-17, and mouse keratinocyte-derived chemokine (KC), also referred to as C-X-C ligand-1 (CXCL1), a functional homolog of human IL-8 measurements. They were performed using the Milliplex MAP magnetic bead assay (Millipore, Billerica, MA, USA), with a detection limit of 3.2 pg/ml for each cytokine, using the manufacturer’s protocol. Data were acquired with a Milliplex Analyzer (Luminex, Austin, TX, USA) and analyzed using Milliplex Analyst 5.1 software.
Metabolomics Analysis
Nuclear magnetic resonance spectroscopy
Nuclear magnetic resonance spectroscopy (NMR) was used for metabolomic analysis of plasma and cerebral cortex samples. Twenty microliters of plasma samples were immediately frozen after centrifugation at 3000 × g for 10 min and stored at –80 °C until batch analysis. The plasma samples were thawed on ice. Protein precipitation was performed by adding 150 μl cold methanol to the samples, then vortexing for 2 min and incubating for 15 min at 4 °C. The tubes were then centrifuged and the supernatants collected and evaporated in a Speed Vac system AES spd111v (GMI. Ramsey, MN, USA) at 35 °C for 2 h.
Fifteen milligrams of cerebral cortex lyophilisate was vortexed for 2 min in a 1:1:0.8 (v/v/v) mixture of methanol/dichloromethane/water. The polar phase was collected and evaporated in a Speed-Vac (Biopharma, Winchester, UK) at 35 °C for 2 h.
Plasma and dry tissue residues were dissolved in 8 μl of a 3.2-mmol/l solution of trimethylsilyl propionate (TSP) solution and 192 μl deuterium oxide phosphate buffer pH 7.4 [using monobasic dihydrogen phosphate and dibasic monohydrogen phosphate powder (Sigma-Aldrich, St. Louis, MO, USA)]. We obtained the 1H-NMR spectra with a Bruker Ascend-600 MHz spectrometer (Bruker SADIS, Wissembourg, France). NOESYPR1D sequences were used to acquire NMR data on plasma and cerebral cortex. Spectral data for plasma were obtained on 32 K data points with a spectral width of 7500 Hz, a 90° pulse of 7.67 μs using a repetition delay of 10 s, 256 scans, and 8 dummy scans. Spectral data for tissues were obtained on 32 K data points with a spectral width of 7500 Hz, a 90° pulse of 7.63 μs using a repetition delay of 20 s, 64 scans, and 8 dummy scans. Prior to Fourier transformation, the free induction decays (FIDs) were zero-filled to 64 K data points, which provided sufficient data points for each resonance. Spectra were processed using Topspin version 3.2 software (Bruker Daltonik, Karlsruhe, Germany).
After manual correction of phase distortion and baseline on all spectra, 1H-NMR spectra were automatically reduced to ASCII files using the AMIX software package (Analysis of MIXture, version 3.9.14; Bruker Biospin, Karlsruhe, Germany). The regions containing residual water signal (4.70–5.51 ppm) were removed from each 1H-NMR spectrum to eliminate baseline effects of imperfect water saturation. Spectral intensities were scaled to the internal reference regions corresponding to trimethylsilylpropanoic acid (TSP) (–0.03 to 0.03 ppm) trimethylsilyl propionate region. Spectra annotations were performed using Chenomx 7.1 software (Chenomx Inc., Edmonton, Canada), an in-house database, and Human Metabolome Database (HMDB). All metabolites identified by NMR spectroscopy are listed in Table S1.
Mass spectroscopy analysis
A targeted quantitative approach using a flow injection analysis (FIA), and a high-performance liquid chromatography (HPLC) tandem mass spectrometry assay AbsolutIDQ p180 kit (Biocrates, Innsbruck, Austria) was used for plasma and tissue samples (cortex and muscle). This assay kit allows simultaneous quantification of 188 metabolites (including acylcarnitines, amino acids, hexoses, phospho- and sphingolipids, and biogenic amines). Flow injection analysis enables the semiquantitative measurement of 145 hydrophobic molecules, comprised of acylcarnitines, sphingomyelins, and phospholipids (lyso, diacyl, and acyl-alkyl phosphatidylcholines). Twenty-four indicators based on the nature of fatty acids of these lipids were also analyzed. A set of 39 polar metabolites were measured using HPLC, including 14 metabolites also evaluated by NMR spectroscopy. The results obtained using these 2 platforms were individually analyzed and we retained the integration corresponding to the technique giving less variability.
A detailed list of all analyzed metabolites is provided in Table S1. The AbsoluteIDQ p180 kit contains isotope-labeled internal standards and quality control. A straightforward sample preparation step was used for the assay, as described in the user manual.
Plasma samples were randomly ordered and left to thaw on ice and then vortexed and centrifuged at 13,000 × g. Cerebral cortex lyophilisate (15 mg) and 150 mg of triceps surae (TS) muscle were vortexed for 2 min in methanol, incubated for 15 min at 4 °C, and centrifuged at 13,300 × g. Tissue and blood samples were then identically prepared. Supernatants (10 μl) were loaded onto a filter paper placed on top of the kit plate and dried in a stream of nitrogen. Subsequently, 20 μl of a 5% solution of phenyl-isothiocyanate was added for derivatization. After incubation, the filter spots were dried again using an evaporator. Extraction was achieved by adding 300 μl methanol containing 5 mM ammonium acetate. A centrifugation step allowed extracts to pass through the manufacturer-provided filter paper and to be recovered in a 96-deep well plate in which the extracts were diluted with 600 μl of the kit’s mass spectrometry running solvent. The extracts were delivered to a QTRAP 5500 System (AB Sciex, Framingham, MA, USA) by HPLC using an Ascentis Express C18, 5 Micron HPLC Column (Sigma-Aldrich), followed by the FIA method. Multiple reaction monitoring detection was used for quantification. MetIDQ software (Biocrates) was used to calculate metabolite concentrations according to a standard range.
Transcriptomics Analysis
Approximately 5.0 × 105 white blood cells were prepared following the same procedure as for Treg lymphocyte measurement from blood samples obtained at week 20. Cells were pelleted by centrifugation and then frozen at –80 °C until extraction. Total RNA was extracted using the RNeasy Mini-prep kit (Zymo Research, Irvine, CA, USA). RNA extracts were randomly pooled to obtain 3 pools of 4 mice for each condition, and then concentrated and cleaned up (RNeasy MinElute Cleanup kit; QIAGEN, Hilden, Germany). The spectrophotometric quantification of RNA was performed using NanoDrop system model 2000c (Thermo Scientific, Waltham, MA, USA). Complementary DNA was then prepared from 600 ng total RNA using the RT2 First Strand Kit (QIAGEN) and was added to reverse transcriptase polymerase chain reaction (PCR) reagent SYBR Green Master Mix according to the manufacturer’s protocol (QIAGEN). Ten microliters of the mixture was added to each of the 384 wells of the RT2 Profiler Amino Acid Metabolism I array PAMM-129Z (QIAGEN), which profiles the expression of 84 genes involved in amino acid metabolism (a complete list is given in Table S2). The reverse transcriptase PCR reaction was performed on the Roche LightCycler 480 (Roche, Basel, Switzerland). Data were processed by the threshold cycle (Ct) method. Threshold cycle values were analyzed using the ΔΔCt method with Excel (Microsoft, Redmond, WA, USA). Genes with fold changes (2-ΔΔCt) > 2 or < 0.5 were reported as being up- or downregulated. Reverse transcriptase control, complementary DNA control, and positive PCR control were within the accepted range.
Statistical Analysis
Clinical and immunological data
Body weight data were analyzed as the relative variation in body weight using the weight at the beginning of treatment (week 10) as the reference and calculated as follows:
For each continuous variable, comparisons between subgroups were performed by Student’s t test after verifying the normality of distribution using the Shapiro–Wilk test and the equality of variance by the Fisher–Snedecor F test. The Wilcoxon signed-rank test was used when the Student’s t test was not applicable. The effect of time on continuous variables was analyzed using ANOVA and was also evaluated between subgroups using 2-way ANOVA. The significance threshold was set at 0.05. A correction for multiple tests was applied when necessary.
Metabolomic data
Metabolomic data were analyzed using multivariate analysis by orthogonal partial last square discriminant analysis (OPLS-DA), with Simca P+ version 13.0 (Umetrics, Umeå, Sweden) to 1) evaluate metabolic modifications in mSOD1 mice treated with MR16-1 versus placebo; 2) identify the metabolic modifications induced by MR16-1 and specific to ALS conditions; and 3) determine the metabolites linked to the clinical outcome of treated mSOD1 mice. The third analysis used data obtained from the metabolomics analysis on week 12 on individual treated and untreated mice. The clinical outcomes were the age at first failure in the slow rotarod test and the age at the onset of weight loss.
Multivariate data analysis was preceded by log transformation and unit variance scaling. No data fusion strategy was performed owing to disparate signal intensities, range, and size of the data matrices from the different analytical platforms; we separately analyzed the data matrix from different analytical platforms (NMR, HPLC, FIA) in each condition (time and sample type). Principal component analysis and OPLS-DA were performed for each time point (weeks 8, 12, 16, and 20) and each sample type. The significance of the OPLS-DA model was assessed using the CV-ANOVA test, which uses an F test to determine whether the variance of cross-validated residuals is significantly smaller than the variation of each Yi near the mean value of Y [27]. We used a single automated algorithm to build the best-fitted model for each condition, by stepwise regression, using bidirectional elimination of variables to identify discriminant metabolites for the OPLS-DA models. All OPLS-DA models were cross-validated by withholding one-seventh of the samples in 7 successive simulations such that each sample was omitted once in order to guard against over fitting. Goodness of fit was evaluated for each model by the Q2Ycum, R2Ycum values, and CV ANOVA p value, with a significance threshold set at 0.05.
Univariate analysis was performed on fold-change values, relative to 1, by using the same procedure as for the other types of previously analyzed data. The effect of time was evaluated using ANOVA on fold-change values for each comparison. The significance threshold was adjusted following a Bonferroni correction depending on the number of metabolites measured by each platform (FIA: p < 0.0004; HPLC: p < 0.0012; NMR in plasma: p < 0.002; NMR of cerebral cortex: p < 0.001; indicators from FIA: p < 0.002).
Pathway Analysis
The online tool Metabolomic Pathway Analysis (MetPA) was used to identify the most relevant pathways related to metabolite findings identified using uni- or multivariate analysis [28]. We also used the KEGG metabolism database to research metabolites potentially associated with the pathways identified by MetPA [29].
We built Venn diagrams with inclusion of the most discriminant metabolites from multivariate analysis after variable selection. We collected 4 different plasma samples for each mouse, corresponding to the different time points. Thus, we only considered metabolites that were discriminant in 3/4 time points to be relevant. Among the long list of metabolites specific to only one matrix (blood, muscle, or brain), we represent on the Venn diagram only those that were significant after the univariate test.
Dysregulated genes and discriminating metabolites were mapped using Cytoscape (http://www.cytoscape.org/) [30], according to the KEGG metabolism database. NetworkAnalyzer (http://med.bioinf.mpi-inf.mpg.de/netanalyzer) was applied to analyze the network topological parameters [31], and stress centrality—the number of shortest paths passing through a node—was used as a marker of centrality. In biological networks, the centrality value of a node can be interpreted as the probability of a protein or a metabolite to be functionally capable of organizing others.
Results
Clinical Data
No beneficial effect of MR16-1 on motor performance
We observed a progressive decrease in fast rotarod performance in mSOD1 mice from day 100 to the end of the study (p < 0.0001; not shown). There was no global difference between treated and untreated mSOD1 mice in fast rotarod performance. There was also no difference in the mean age at first failure in the slow rotarod test between treated and untreated mSOD1 mice (133.3 ± 4.8 vs 132.7 ± 8.3 respectively; not shown).
Deleterious effect of MR16-1 on body weight
We observed a global trend of weight loss between treated and untreated mSOD1 transgenic mice over the course of the disease (Fig. 2). Treated mSOD1 transgenic mice lost significantly more weight than untreated mice from week 18.5 to sacrifice at week 20 (p < 0.05) (Fig. 2). The mean age of weight loss was earlier for treated mSOD1 transgenic mice than untreated mice (15.5 ± 0.7 weeks vs 17.0 ± 1.1 weeks; p = 0.0041; Fig. 2). This difference was not observed in treated WT mice, although weight gain was transiently superior in untreated compared with treated WT mice from week 12 to week 16 (p < 0.05; see Fig. S2).

Body weight data of mutant superoxide dismutase 1 (mSOD1) mice. Body weight data were analyzed as the relative change in body weight using the weight at the beginning of the treatment (week 10) as the reference. The beginning of weight loss was used as a clinical outcome and is represented by a red circle for untreated mSOD1 mice and a blue square for MR16-1-treated mSOD1 mice. The change in body weight were compared at each time point. Error bars represent standard error of the mean. *p < 0.05
Inflammatory Effects
Regulatory T-cell counts
Frequency of Tregs among viable cells in peripheral blood significantly decreased with age in untreated mSOD1 mice (p < 0.0001; Fig. 3) and MR16-1 treatment of mSOD1 mice inhibited the decrease in the frequency of Tregs (p = 0.0462; 2-way ANOVA) and we observed a significant increase of Tregs frequency in treated mSOD1 mice relative to untreated mice, at 20 weeks (p = 0.0268; Fig. 3).

Frequency of regulatory T cells (Tregs) among leukocytes. Frequency of Tregs among viable cells in blood samples was measured using flow cytometry. Frequencies were compared for each time point between treated mutant superoxide dismutase 1 (mSOD1) mice (blue dotted line) and untreated mSOD1 mice (red line). Error bars represent standard error of the mean. *p < 0.05
Cytokine measurement
IL-6, IL-10, and IL-17 were under the detection limit of the assay in most of the plasma samples (Table S3). CXCL1 (KC) concentrations were significantly higher in mSOD1 than in WT mice at week 20 (248 ± 200 vs 70 ± 98 pg/ml; p = 0.0066). MR16-1 treatment of mSOD1 mice lowered CXCL1 levels close to those observed in untreated WT mice [248 ± 199 in untreated mSOD1 vs 80 ± 65 pg/ml in treated mSOD1 mice; p = 0.0479 (Table S3)]. MR16-1 treatment had no effect on CXCL1 levels in WT mice (70 ± 98 vs 86 ± 65, untreated and treated, respectively; Table S3).
Metabolic Effect of MR16-1 in mSOD1
The discrimination of all sample types (TS muscle, cerebral cortex, and plasma) from the 4 groups of mice revealed a clearer separation based on the type of mice than the administration of treatment [PCA from muscle samples (Fig. 4), first 2 components accounted for 49% of the total dataset variance]. However, some metabolic changes specifically induced by the treatment have been identified in all sample types (Fig. 5; Tables S4 and S5). Performances of multivariate models were low (Q2 > 0.97 but p > 0.05 in most of the cases; data not shown), thus confirming the moderate effect of MR16-1 on the metabolome. As the aims of these models were not a prediction of mice goups, the identity of metabolites having a discriminant weight, even in a low-performing model were informative. Transcriptomic data revealed that 39 genes were differentially expressed between treated and untreated mSOD1 mice in peripheral white blood cells (Fig. 6A; Table S2). Twenty-eight genes were differentially expressed between treated and untreated WT mice (Table S2). Figure 6B summarizes the main dysregulated metabolites, genes, and pathways between treated and untreated mSOD1 mice.

Principal component analysis plot for triceps surae muscle metabolome measured by flow injection analysis coupled to mass spectroscopy (MS) and obtained from (A) mutant superoxide dismutase 1 (mSOD1) mice (n = 22), treated with MR16-1 (n = 11), in light green, or untreated (n = 11), in dark green; (B) wild-type (WT) mice (n = 24), treated with MR16-1 (n = 7), in blue, or untreated (n = 17), in red. This plot is representative of that obtained using high-performance liquid chromatography coupled to MS. The 2 first components accounted for 49% of the total dataset variance

Venn diagram showing the most discriminant metabolites between treated and untreated mice. An orthogonal partial last square discriminant analysis was performed to identify discriminating metabolites between subgroups for each condition tested and for each analytical platform. Each model was optimized by stepwise regression using automated bidirectional elimination of variables. Metabolites had to be discriminant in 2 of 3 time points tested in plasma to be considered relevant. Metabolites that were discriminant in only 1 matrix are not represented when the univariate test was not significant. *Univariate analysis significant in 1 matrix. (A) Discriminant metabolites between treated and untreated mutant superoxide dismutase 1 (mSOD1) mice. Metabolites in bold were not found discriminant between treated and untreated wild-type (WT) mice. (B) Discriminant metabolites between treated and untreated WT mice. (C) Metabolites from analysis of mSOD1 and WT mice were gathered to identify metabolites specifically involved in discrimination of treated and untreated mSOD1 mice. Triceps Surae (TS), Assymetric Dimethylarginine (ADMA)

Targeted transcriptomic analysis. (A) Heat map of 69 genes related to amino acids metabolism. Up- or down-pointing arrows indicate genes differentially expressed between MR16-1 treated and untreated mutant superoxide dismutase 1 (mSOD1) mice. Scaled expression values are color-coded according to the legend on the left. (B) Diagrammatic representation of dysregulated metabolites (Δ), genes (□), and metabolic pathway (○) in MR16-1-treated mSOD1 mice compared with untreated mice. Arrows connect metabolites to genes, genes to pathway, or metabolites to pathway. Dotted lines connect metabolites involved in a reaction catalysed by an enzyme not schematized. All nodes from this network were sized accordingly to their stress centrality value in the network, calculated using NetworkAnalyzer plugin for Cytoscape
Figure 5A shows the main metabolites involved in discrimination of treated and untreated mSOD1 mice, and Fig. 5B those involved in discrimination of treated and untreated WT mice. Specific metabolites of discrimination in mSOD1 and WT mice are shown in Fig. 5C.
In mSOD1 mice, 18 main discriminating metabolites were identified, among which 15 (83%) were not found discriminant in WT mice. No metabolite discriminated treated and untreated mSOD1 mice in all tissues (Fig. 5A). The most relevant pathways involved in the discrimination of specifically treated and untreated mSOD1 mice were the following: lipid metabolism (glycerol, serine, C4, PC aa C36:3); arginine and proline metabolism (4-hydroxyproline, putrescine); valine, leucine, and isoleucine degradation pathway (isoleucine, valine); taurine and hypotaurine metabolism (taurine, alanine). Importantly, blood serine concentrations decreased in treated versus untreated mSOD1 mice over time (p < 0.0001; Fig. S1A), as well as glycerol (p < 0.0001; Fig. S1B). Some dysregulated genes were related to arginine and proline metabolism (10 genes); valine, leucine, and isoleucine degradation pathway (6 genes), and taurine and hypotaurine metabolism (1 gene: Glud1).
Fifteen metabolites were found to be relevant for the discrimination of treated and untreated WT mice (Fig. 5B), among which 10 (67%) were not found to be discriminant in mSOD1 mice. These metabolites highlighted 2 main pathways: lipid metabolism (acetylcarnitine, PC aa C32:2, PC ae C38:1) and methionine metabolism (methionine, methionine sulfoxide). Transcriptomics analysis confirmed that some genes related to these pathways were dysregulated (Table S2).
Relationship Between the Metabolome and Clinical Outcome of Treated mSOD1 Mice
Univariate analysis showed no significant differences in metabolites concentrations in plasma samples collected at week 12, between mice with a good or poor prognosis. Multivariate analysis to explain the age at first failure of the slow rotarod test and the beginning of weight loss in mSOD1 treated with MR16-1 mice is shown in Table 1. Some metabolites were associated with both weight loss and motor performance in treated mSOD1 mice: 3-methylhistidine, creatinine, D-glucose, hypoxanthine, lysine, tryptophan, and valine.
Discussion
The major findings of this study are 1) the clinical worsening of mSOD1 mice treated with MR16-1, an anti-IL-6R monoclonal antibody; 2) the positive immunological effects of this therapy; and 3) the characterization of metabolic changes due to anti-IL-6 treatment specifically in ALS. Despite the limits of the study inherent to the size of the population, and the use of a preclinical model of ALS not representative of all forms of ALS, this proof of concept of pharmacometabolomics in ALS is very promising.
First Pharmacometabolomics Study in ALS
Pharmacometabolomics studies may be focused on the relation between metabolome findings and efficacy and safety. Major progress in ALS therapeutics will be based on a reassessment of approaches to drug development and pharmacometabolomics may be promising. The metabolic signature at baseline and over the course of the disease may help in understanding the pathophysiology of ALS not only in clinical, but also in preclinical, trials after rigorous standardization [32, 33]. The clinical characteristics to determinate patient subgroups may not be sufficiently sensitive, and adding metabolic profiling may help in patient stratification and in differentiation between responders and nonresponders. This methodology has to be evaluated as a proof of concept in preclinical studies before application in humans, and we provided herein the validation of such approach.
Deleterious Clinical Effect of MR16-1
The onset of weight loss occurred earlier among treated mice, and was more rapid from week 18 to the end of the study. Previous studies reported no effect of MR16-1 therapy on body weight [34–36], and 1 study reported weight gain after MR16-1 treatment in a cancer cachexia mouse model [37]. However, differences in the study design, the metabolic characteristics of ALS mice, and the kinetics of treatment in relation to the clinical evolution of ALS may explain this discrepancy. A similar uncoupled effect of IL-6 blockade on body weight and inflammation was previously reported [38].
Han et al. [24] observed no difference in weight curves of mSOD1/IL-6–/– and mSOD1/IL-6+/+ mice. In the present study, IL-6 blockade was obtained by a different way and modified the weight curve of treated mSOD1 mice. This different effect may be explained by too low concentrations of MR16-1 in the brain. We also may suspect that the timing of drug administration, chosen to mimic clinical situation is not adapted to the neuroinflammation described in ALS [6].
Promising Effect of IL-6 Blockade on Inflammation Status
MR16-1 had a significant effect on inflammation, both on the number of Tregs and cytokine levels. Anti-IL-6R therapy is widely used to treat rheumatoid arthritis because of its strong anti-inflammatory effect [18, 39, 40]. Previous studies reported an effect of MR16-1 or tocilizumab on the Treg count [41, 42], although other studies did not show any effect [43, 44]. In our study, cytokine concentrations were not markedly increased in plasma from mSOD1 mice, except for CXCL1 (KC). Rare studies have reported an increase of inflammatory cytokines in plasma from patients with ALS [22, 45–48]. To our knowledge, CXCL1 (KC) has never been measured in mSOD1 mice. Anti-IL-6R therapy specifically affects neutrophils in humans, with a reduction in neutrophil survival and mediator secretion [49]. Our results suggest that inhibition of the IL-6 pathway in ALS leads to an effective anti-inflammatory effect, associated with an increase in the number of Tregs in the blood, and a decrease of inflammatory cytokines, such as CXCL1 (KC) in the mouse. The dual role of immunity in ALS, articulated in the T helper 2 and T helper 1 immune phase could be considered in relation to the pro- and anti-inflammatory properties of IL-6 to design a new IL-6-based preclinical trial [6, 50].
Moderate Effect on Metabolism
The combined use of mass spectroscopy and NMR platforms in this study broadened the field of analyzed metabolites, and strengthened the results by using overlapping findings generated by 2 analytical platforms. Transcriptomics data also reinforced our findings by confirming the main pathways highlighted by our modeling. Here, we show that IL-6 blockade does not appear to play a major role in the perturbations of the metabolome that occur in ALS, whatever the analyzed matrix (poor performance of the multivariate models).
However, multivariate models identified several metabolites involved in perturbations of the ALS metabolome that may be linked to the IL-6 pathway. Among them, polyamines and especially putrescine was found to be a discriminant metabolite in TS muscle and cerebral cortex samples from mSOD1 transgenic mice, with or without MR16-1treatment. This is consistent with the report showing that IL-6 administration to myeloid cells in culture resulted in the direct downregulation of ornithine decarboxylase [51], the rate-limiting enzyme of polyamine biosynthesis.
Lipid and BCAA metabolism were also affected in MR16-1-treated mSOD1 mice. Lipid metabolism was described as deeply involved in ALS pathophysiology [52, 53]. In particular, several studies reported hyperlipidemia in patients with ALS [53–57], with a potential link with patient survival [53]. Close connections between IL-6 signaling and lipid metabolism exist [58, 59]. At the central level, IL-6 is involved in whole-body energy balance by modulating food intake and leptin levels [60]. In the periphery, IL-6 increases lipolysis in adipose tissue [61] and fatty acid utilization in skeletal muscle [62, 63]. As in the present study mSOD1 mice did not benefit from anti-IL-6 therapy, the involvement of IL-6 signaling in ALS-associated lipid metabolism alterations seems unlikely.
Valine and isoleucine were associated with the discrimination of treated and untreated mSOD1 mice but not of WT mice. Branched chain amino acid (BCAA) metabolism has been largely described in ALS, and we identified this way as potentially interesting for ALS diagnosis and prognosis in previous studies [64]. BCAAs play a crucial role in metabolic signaling [65, 66]. Interestingly, BCAA catabolism can regulate muscular fatty acid β-oxydation via the glyceroneogenesis pathway [67], whose rate-limiting step is the conversion of oxaloacetate to phosphoenolpyruvate by phosphoenolpyruvate carboxykinase. As the expression of the phosphoenolpyruvate carboxykinase gene is strongly inhibited by IL-6 in liver and adipose tissue [68, 69], IL-6 blockade could interfere with plasmatic concentrations of glycerol, serine, and branched chain amino acids, as observed in the present study.
Relationship Between the Metabolome and Clinical Outcome
Concentrations of some metabolites in plasma at disease beginning were found to be associated with biomarkers of mice clinical outcome. Among them, lysine and tryptophan are 2 essential amino acids. Tryptophan is necessary for the synthesis of serotonin whose importance in ALS have been reported [70, 71]. Lysine and tryptophan metabolism may share common mitochondrial enzymes in the central nervous system, whose regulation could be connected to glutamatergic system [72]. Recent study showed that indoleamine-2,3-dioxygenase, a key enzyme in tryptophan catabolism to kynurenine, expression is regulated by the IL-6 signaling pathway [73].
Conclusion
MR16-1 treatment induced a significant anti-inflammatory effect, with an increase of the Treg count in blood and a decrease of the inflammatory cytokine CXCL1. However, this effect was associated with a worsening of disease progression characterized by an acceleration of weight loss in treated mice, and no improvement of motor performance. A pharmacometabolomic approach showed a moderate effect of MR16-1 on metabolic perturbations. Main pathways affected were lipid metabolism, and BCAA, arginine, and proline metabolism. As these pathways have been previously clearly described in ALS, we suspect that IL-6 blockade had a negative effect through a multiparametric effect, including a moderate metabolic effect. We have to pursue the pharmacometabolomics strategy with other Treg-boosting drugs, or with novel treatment kinetics strategies.
References
Chio A, Logroscino G, Hardiman O, et al. Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler 2009;10:310–323.
Dupuis L, Pradat P-F, Ludolph AC, Loeffler J-P. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol 2011;10:75–82.
Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 1994;330:585–591.
Lacomblez L, Bensimon G, Leigh PN, et al. Long-term safety of riluzole in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2002;3:23–29.
Kano O, Beers DR, Henkel JS, Appel SH. Peripheral nerve inflammation in ALS mice: cause or consequence. Neurology 2012;78:833–835.
Murdock BJ, Bender DE, Segal BM, Feldman EL. The dual roles of immunity in ALS: injury overrides protection. Neurobiol Dis 2015;77:1–12.
Alexianu ME, Mohamed AH, Smith RG, Colom LV, Appel SH. Apoptotic cell death of a hybrid motoneuron cell line induced by immunoglobulins from patients with amyotrophic lateral sclerosis. J Neurochem 1994;63:2365–2368.
Butovsky O, Siddiqui S, Gabriely G, et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest 2012;122:3063–3087.
Xiao Q, Zhao W, Beers DR, et al. Mutant SOD1(G93A) microglia are more neurotoxic relative to wild-type microglia. J Neurochem 2007;102:2008–2019.
Beers DR, Henkel JS, Zhao W, et al. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 2011;134:1293–1314.
Henkel JS, Beers DR, Wen S, et al. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med 2013;5:64–79.
Banerjee R, Mosley RL, Reynolds AD, et al. Adaptive immune neuroprotection in G93A-SOD1 amyotrophic lateral sclerosis mice. PLoS One 2008;3:e2740.
Zhao W, Beers DR, Liao B, Henkel JS, Appel SH. Regulatory T lymphocytes from ALS mice suppress microglia and effector T lymphocytes through different cytokine-mediated mechanisms. Neurobiol Dis 2012;48:418–428.
Barbi J, Pardoll D, Pan F. Metabolic control of the Treg/Th17 axis. Immunol Rev 2013;252:52–77.
Arkan MC, Hevener AL, Greten FR, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 2005;11:191–198.
Cai D, Yuan M, Frantz DF, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med 2005;11:183–190.
Korn T, Mitsdoerffer M, Croxford AL, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A 2008;105:18460–18465.
Tanaka T. Can IL-6 blockade rectify imbalance between Tregs and Th17 cells? Immunotherapy 2013;5:695–697.
Van Hall G. Cytokines: muscle protein and amino acid metabolism. Curr Opin Clin Nutr Metab Care 2012;15:85–91.
Mauer J, Denson JL, Brüning JC. Versatile functions for IL-6 in metabolism and cancer. Trends Immunol 2015;36:92–101.
Nishimoto N, Ito K, Takagi N. Safety and efficacy profiles of tocilizumab monotherapy in Japanese patients with rheumatoid arthritis: meta-analysis of six initial trials and five long-term extensions. Mod Rheumatol Jpn Rheum Assoc 2010;20:222–232.
Mizwicki MT, Fiala M, Magpantay L, et al. Tocilizumab attenuates inflammation in ALS patients through inhibition of IL6 receptor signaling. Am J Neurodegener Dis 2012;1:305–315.
Fiala M, Mizwicki MT, Weitzman R, Magpantay L, Nishimoto N. Tocilizumab infusion therapy normalizes inflammation in sporadic ALS patients. Am J Neurodegener Dis 2013;2:129–139.
Han Y, Ripley B, Serada S, Naka T, Fujimoto M. Interleukin-6 deficiency does not affect motor neuron disease caused by superoxide dismutase 1 mutation. PLoS ONE 2016;11:e0153399
Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994;264:1772–1775.
Boillée S, Yamanaka K, Lobsiger CS, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006;312:1389–1392.
Eriksson L, Trygg J, Wold S. CV-ANOVA for significance testing of PLS and OPLS® models. J Chemom 2008;22:594–600.
Xia J, Wishart DS. MetPA: a web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 2010;26:2342–2344.
Kanehisa M, Araki M, Goto S, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res 2008;36:D480–D484.
Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–2504.
Assenov Y, Ramírez F, Schelhorn S-E, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinforma 2008;24:282–284.
Ludolph AC. Motor neuron disease: urgently needed-biomarkers for amyotrophic lateral sclerosis. Nat Rev Neurol 2011;7:13–14.
McGoldrick P, Joyce PI, Fisher EMC, Greensmith L. Rodent models of amyotrophic lateral sclerosis. Biochim Biophys Acta 2013;1832:1421–1436.
Ogata T, Yamazaki H, Teshima T, et al. Early administration of IL-6RA does not prevent radiation-induced lung injury in mice. Radiat Oncol 2010;5:26.
Ogata T, Yamazaki H, Teshima T, Tsuchiya T, Nishimoto N, Matsuura N. Anti-IL-6 receptor antibody does not ameliorate radiation pneumonia in mice. Exp Ther Med 2012;4:273–276.
Sakurai T, Yamada Y, Sawada Y, et al. PRIMe update: innovative content for plant metabolomics and integration of gene expression and metabolite accumulation. Plant Cell Physiol 2013;54:e5.
Ando K, Takahashi F, Kato M, et al. Tocilizumab, a proposed therapy for the cachexia of Interleukin6-expressing lung cancer. PLoS One 2014;9:e102436.
Matthews VB, Allen TL, Risis S, et al. Interleukin-6-deficient mice develop hepatic inflammation and systemic insulin resistance. Diabetologia 2010;53:2431–2441.
Rossi J-F, Lu Z-Y, Jourdan M, Klein B. Interleukin-6 as a therapeutic target. Clin Cancer Res 2015;21:1248–1257.
Tanaka T, Kishimoto T. The biology and medical implications of interleukin-6. Cancer Immunol Res 2014;2:288–294.
Thiolat A, Semerano L, Pers YM, et al. Interleukin-6 receptor blockade enhances CD39+ regulatory T cell development in rheumatoid arthritis and in experimental arthritis. Arthritis Rheumatol 2014;66:273–283.
Hohki S, Ohguro N, Haruta H, et al. Blockade of interleukin-6 signaling suppresses experimental autoimmune uveoretinitis by the inhibition of inflammatory Th17 responses. Exp Eye Res 2010;91:162–170.
Fujimoto M, Serada S, Mihara M, et al. Interleukin-6 blockade suppresses autoimmune arthritis in mice by the inhibition of inflammatory Th17 responses. Arthritis Rheum 2008;58:3710–3719.
Iwanami K, Matsumoto I, Tanaka-Watanabe Y, et al. Crucial role of the interleukin-6/interleukin-17 cytokine axis in the induction of arthritis by glucose-6-phosphate isomerase. Arthritis Rheum 2008;58:754–763.
Moreau C, Devos D, Brunaud-Danel V, et al. Elevated IL-6 and TNF-alpha levels in patients with ALS: inflammation or hypoxia? Neurology 2005;65:1958–1960.
Babu GN, Kumar A, Chandra R, Puri SK, Kalita J, Misra UK. Elevated inflammatory markers in a group of amyotrophic lateral sclerosis patients from northern India. Neurochem Res 2008;33:1145–1149.
Su XW, Simmons Z, Mitchell RM, Kong L, Stephens HE, Connor JR. Biomarker-based predictive models for prognosis in amyotrophic lateral sclerosis. JAMA Neurol 2013;70:1505–1511.
Ehrhart J, Smith AJ, Kuzmin-Nichols N, et al. Humoral factors in ALS patients during disease progression. J. Neuroinflammation 2015;12:127.
Gaber T, Hahne M, Strehl C, et al. Disentangling the effects of tocilizumab on neutrophil survival and function. Immunol Res 2016;64:665–676.
Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta 2011;1813:878–888.
Selvakumaran M, Liebermann D, Hoffman B. The proto-oncogene c-myc blocks myeloid differentiation independently of its target gene ornithine decarboxylase. Blood 1996;88:1248–1255.
Schmitt F, Hussain G, Dupuis L, Loeffler J-P, Henriques A. A plural role for lipids in motor neuron diseases: energy, signaling and structure. Front Cell Neurosci 2014;8.
Dupuis L, Corcia P, Fergani A, et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008;70:1004–1009.
Murai A, Miyahara T, Tanaka T, Kaneko T, Sako Y, Kameyama M. Abnormalities of lipoprotein and carbohydrate metabolism in degenerative diseases of the nervous system-motor neuron disease and spinocerebellar degeneration. Tohoku J Exp Med 1983;139:365–376.
Ikeda K, Hirayama T, Takazawa T, Kawabe K, Iwasaki Y. Relationships between disease progression and serum levels of lipid, urate, creatinine and ferritin in Japanese patients with amyotrophic lateral sclerosis: a cross-sectional study. Intern Med 2012;51:1501–1508.
Patin F, Corcia P, Madji Hounoum B, et al. Biological follow-up in amyotrophic lateral sclerosis: decrease in creatinine levels and increase in ferritin levels predict poor prognosis. Eur J Neurol 2015;22:1385–1390.
Rafiq MK, Lee E, Bradburn M, McDermott CJ, Shaw PJ. Effect of lipid profile on prognosis in the patients with amyotrophic lateral sclerosis: Insights from the olesoxime clinical trial. Amyotroph Lateral Scler Front Degener 2015;16:478–484.
Glund S, Krook A. Role of interleukin-6 signalling in glucose and lipid metabolism. Acta Physiol 2008;192:37–48.
Coppack SW. Pro-inflammatory cytokines and adipose tissue. Proc Nutr Soc 2001;60:349–356.
Wallenius V, Wallenius K, Ahrén B, et al. Interleukin-6-deficient mice develop mature-onset obesity. Nat Med 2002;8:75–79.
Petersen EW, Carey AL, Sacchetti M, et al. Acute IL-6 treatment increases fatty acid turnover in elderly humans in vivo and in tissue culture in vitro. Am J Physiol Endocrinol Metab 2005;288:E155–E162.
Al-Khalili L, Bouzakri K, Glund S, Lönnqvist F, Koistinen HA, Krook A. Signaling specificity of interleukin-6 action on glucose and lipid metabolism in skeletal muscle. Mol Endocrinol 2006;20:3364–3375.
Carey AL, Steinberg GR, Macaulay SL, et al. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes 2006;55:2688–2697.
Blasco H, Patin F, Madji Hounoum B, et al. Metabolomics in amyotrophic lateral sclerosis: how far can it take us? Eur J Neurol 2016;23:447-454.
Lynch CJ, Adams SH. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat Rev Endocrinol 2014;10:723–736.
Batch BC, Hyland K, Svetkey LP. Branch chain amino acids: biomarkers of health and disease. Curr Opin Clin Nutr Metab Care 2014;17:86–89.
Kainulainen H, Hulmi JJ, Kujala UM. Potential role of branched-chain amino acid catabolism in regulating fat oxidation. Exerc Sport Sci Rev 2013;41:194–200.
Wan Z, Ritchie I, Beaudoin M-S, Castellani L, Chan CB, Wright DC. IL-6 indirectly modulates the induction of glyceroneogenic enzymes in adipose tissue during exercise. PLoS One 2012;7:e41719.
Feingold KR, Moser A, Shigenaga JK, Grunfeld C. Inflammation inhibits the expression of phosphoenolpyruvate carboxykinase in liver and adipose tissue. Innate Immun 2012;18:231–240.
Dupuis L, Spreux-Varoquaux O, Bensimon G, et al. Platelet serotonin level predicts survival in amyotrophic lateral sclerosis. PLoS One 2010;5:e13346.
Sandyk R. Serotonergic mechanisms in amyotrophic lateral sclerosis. Int J Neurosci 2006;116:775–826.
Hallen A, Jamie JF, Cooper AJL. Lysine metabolism in mammalian brain: an update on the importance of recent discoveries. Amino Acids 2013;45:1249–1272.
Litzenburger UM, Opitz CA, Sahm F, et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 2014;5:1038–1051.
Acknowledgments
The authors would especially like to thank the Fondation Brou de Laurières and the french “Association pour la Recherche sur la Sclérose Latérale Amyotrophique et autres maladies du motoneurone” (ARSLA) for their support of this project. The authors also thank Chugai Pharmaceutical Co. for providing graciously MR16-1 antibody.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1224 kb)
Table S1
List of all metabolites analyzed (DOCX 18 kb)
Table S2
List of all genes analyzed (DOCX 19 kb)
Table S3
Cytokine measurements (DOCX 27 kb)
Table S4
Univariate findings from metabolomic data (DOCX 22 kb)
Table S5
Multivariate models, orthogonal partial last square discriminant analysis (DOCX 18 kb) (JPG 353 kb)
Fig. S1
Serine and glycerol plasma concentration fold-change. Boxplots are given with whiskers from minimum to maximum (PDF 19 kb) (JPG 279 kb)
Fig. S2
Body weight data of wild-type mice. Error bars represent standard error of the mean (DOCX 27 kb) (JPG 279 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Patin, F., Baranek, T., Vourc’h, P. et al. Combined Metabolomics and Transcriptomics Approaches to Assess the IL-6 Blockade as a Therapeutic of ALS: Deleterious Alteration of Lipid Metabolism. Neurotherapeutics 13, 905–917 (2016). https://doi.org/10.1007/s13311-016-0461-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-016-0461-3