
Abstract
Fragile X syndrome (FXS), an inherited intellectual disability often associated with autism, is caused by the loss of expression of the fragile X mental retardation protein. Tremendous progress in basic, preclinical, and translational clinical research has elucidated a variety of molecular-, cellular-, and system-level defects in FXS. This has led to the development of several promising therapeutic strategies, some of which have been tested in larger-scale controlled clinical trials. Here, we will summarize recent advances in understanding molecular functions of fragile X mental retardation protein beyond the well-known role as an mRNA-binding protein, and will describe current developments and emerging limitations in the use of the FXS mouse model as a preclinical tool to identify therapeutic targets. We will review the results of recent clinical trials conducted in FXS that were based on some of the preclinical findings, and discuss how the observed outcomes and obstacles will inform future therapy development in FXS and other autism spectrum disorders.
Similar content being viewed by others


Fragile X syndrome (FXS) is one of the first single gene disorders manifesting features of autism spectrum disorder (ASD) in which extensive study of the neurobiology and synaptic mechanisms of disease in cellular and animal models has been possible. The enormous progress in basic and preclinical and clinical translational work in FXS in the last several decades has allowed FXS to emerge as an important model to illustrate successes and hurdles in the development of future targeted treatments for autism and related developmental disorders. FXS is the most common known genetic cause of intellectual disability and ASD, with an estimated frequency of about 1 in 4000–5000 [1]. The disorder affects all ethnic groups worldwide.
Genetics and Phenotype of FXS
FXS is one of the fragile X-associated disorders (FXDs), all of which arise from a trinucleotide repeat (CGG) expansion mutation in the promoter region of FMR1. The CGG sequence is transcribed into the 5’ untranslated region of FMR1 mRNA and thus length of the repeat sequence does not affect the sequence of the protein product of FMR1 [fragile X mental retardation protein (FMRP)] [2]. Small expansions in the gene (55–200 CGG repeats), termed the “premutation”, occur in about 1 in 430–468 males and 1 in 151–209 females in the USA [3, 4], and is associated with risk for fragile X-associated tremor/ataxia syndrome and fragile X-associated primary ovarian insufficiency. Although the premutation is transcribed and translated to give FMRP, toxicity in fragile X-associated tremor/ataxia syndrome and fragile X-associated primary ovarian insufficiency is thought to occur through a mechanism related to elevated cellular FMR1 mRNA levels and resultant CGG repeat-mediated RNA toxicity [2]. Large expansions in FMR1 (>200 CGG repeats), termed the “full mutation,” cause FXS, which results from methylation and transcriptional silencing of FMR1 with consequent loss or significant reduction in expression of FMRP [2]. Expansion mutations in FMR1 tend to increase in size as they are inherited through the generations, so fragile X-associated disorders affect families in multiple generations, with more individuals affected with FXS in later generations.
Males with FXS typically present with developmental delay, particularly language delay, and ultimately display intellectual disability that can range from mild to severe [5]. Hypotonia is often seen early in life and evolves into coordination and praxis problems in older children and adults. Physical features include macro-orchidism in most men, and variable presence of craniofacial characteristics, including prominent ears, macrocephaly, long face, prominent jaw and forehead, midfacial hypoplasia, and high arched palate. Loose connective tissue leads to hyperextensible joints, flat feet, and soft redundant skin on the palms [5]. Females with a full mutation are more variably and usually more mildly affected than males because of production of FMRP in cells that express the normal X chromosome with the nonmutated FMR1 allele. The severity of cognitive impairment in females with a full mutation is inversely related to the activation ratio for the normal FMR1 allele and levels of expressed FMRP [6]. Males with size (full and premutation) or methylation (partially unmethylated full mutation) mosaicism may be more mildly affected, with severity related to the percent of unmethylated FMR1 alleles and FMRP levels.
A number of medical problems appear to be more prevalent in FXS than in the general population, based on prior small studies from individual or small groups of centers, and confirmed in a large dataset collected from a natural history study being conducted through the Fragile X Clinic and Research Consortium, a consortium of fragile X clinics in the USA [7]. These problems include seizures, strabismus, frequent otitis media, gastroesophageal reflux, and sleep apnea and other sleep disorders (Table 1).
Cognitive, Behavioral and Language Features of FXS
The majority of males with FXS will meet criteria for mild-to-severe intellectual disability [8]. The average IQ in men with FXS is 40–50, with a mental age of about of 5–6 years. Females with FXS are often less affected than males with about 25 % having cognitive impairment and others frequently being diagnosed with learning disabilities [9]. Average IQ in women is about 80, with a range from severe impairment to normal or even superior ability. There is a relatively consistent pattern of intellectual weaknesses (generally milder in severity in females) and strengths distinct to both males and females with FXS [10]. Relative weaknesses include visuospatial skills, working memory, processing of sequential information, and attention [10], while there are relative strengths in simultaneous processing and long-term memory.
Multiple studies have shown a decrease in full-scale IQ scores with age as children with FXS become older [11–13]. Standard scores on the Vineland Adaptive Behavior Scale for overall adaptive behavior as well as subdomains have also been shown to decline with age during childhood, in males more so than in females with FXS [14]. Decline in standard scores for intelligence and adaptive function is not the results of loss of skills or regression but rather failure to keep pace with the normal rate of intellectual development. Scores are felt to be fairly stable in adulthood.
The behavioral phenotype in males with FXS covers a wide spectrum, although there are certain behaviors that are very commonly seen. These behaviors fall into the broad categories of attention/hyperactivity, hyperarousal, anxiety, and aggression. As in other areas, females with FXS have the same spectrum of behavioral difficulties but with milder symptomology.
Attention disorders, including impulsivity, hyperactivity and inattention, are extremely common in FXS and result in a comorbid attention deficit hyperactivity disorder diagnosis in as many as 73 % of males and 30–63 % of females [10, 15–18]. Impulsivity in boys with FXS does not seem to improve with age, which means that this behavior has the potential to present long-term challenges to the individual and their caregivers [19].
Anxiety disorders have been reported in 86.2 % of males with FXS and 76.9 % of females with FXS [20]. Although individuals with FXS manifest high rates of generalized anxiety and of virtually all subtypes, the anxiety disorders most frequently found in the literature are specific phobias and social anxiety [10, 18, 20].
Hyperarousal, an over-reaction to sensory input, can be triggered in FXS by a wide range of situations, including noises, new environments, crowds, interpersonal distance, eye contact, and new people [21]. The effects of this hyperarousal are widespread, and include high levels of motor activity (e.g., running, jumping), stereotypic motor movements (e.g., hand-flapping), gaze aversion, and perseverative behaviors [21–23].
Aggression and self-injurious behavior are often linked to hyperarousal or anxiety. These problem behaviors are especially detrimental to family functioning as they can significantly impede an individual’s ability to participate in daily activities. Self-injurious behavior typically appears fairly early in development; onset for most individuals (both male and female) occurs at 1–3 years of age [24]. Aggression typically occurs later, mostly during the pubescent and postpubescent period [25].
Language development is globally delayed for most individuals with FXS. It is not uncommon for individuals with FXS to remain prelinguistic communicators until much later in life than is seen in typical development [26]. However, the majority of males and females with FXS will obtain spoken language at some point, and they will continue to gain language skills throughout their lifespan, albeit at a slower pace than typically developing individuals [27, 28].
In general, individuals with FXS have stronger receptive than expressive language skills. While receptive language skills are usually below chronological age expectations, they are commensurate with nonverbal mental age [29], and receptive vocabulary, a major strength, can be well above mental age. However, individuals with comorbid FXS and autism have receptive language skills below their nonverbal mental age, and as autism severity increases so do the deficits in receptive vocabulary and grammar [30, 31]. Expressive language is delayed, with nonverbal mental age being a strong predictor of expressive vocabulary [29].
Pragmatic language (communication in social interactions) is delayed very early in development. Young children with FXS have been shown to have difficulty with joint attention, reciprocating positive facial expressions, eye gaze, and turn-taking [28, 32]. Deficits in pragmatic skills continue to surface as individuals develop. Studies indicate that males with FXS demonstrate high rates of tangential (off-topic) language, overly literal interpretation of language, as well as decreased topic initiation and maintenance [23]. Repetitive language (also called perseverative) is a hallmark of FXS and one that causes significant social difficulty.
Autism and FXS
About a half to two-thirds of males and about 20 % of females with FXS meet criteria for ASD [33]. These two disorders share multiple behaviors (Table 2), including pragmatic deficits, language delays, reduced eye contact, difficulty with regulation of attention and activity level, and additional problematic behaviors (e.g., self-injurious behaviors and aggression).
Characteristics that tend to differ between the FXS behavioral phenotype from the phenotype of idiopathic ASD in general include a higher rate of intellectual disability, more severe motor coordination deficits, worse expressive than receptive language, generally higher interest in socialization (although limited by anxiety), and better imitation skills in FXS relative to idiopathic ASD (Table 2).
Current Supportive Treatment of FXS
Current treatment of FXS is supportive with therapy, educational strategies that take into account cognitive and behavioral strengths and weaknesses in FXS, treatment of medical problems, behavioral modification, and psychopharmacology for behaviors creating dysfunction [33].
Given the global developmental delay most individuals with FXS display, it is likely that they will qualify for multiple services at a young age. Occupational therapy, physical therapy, and speech therapy should all be accessed at the maximum level allowed so as to maximize early growth. Therapy techniques that have been validated for autism are often helpful for FXS but must be modified based on what is known about the FXS phenotype.
Individuals with FXS have been shown to display better behavioral and adaptive functioning when their environment and supportive programming is well matched to their needs [37], and therefore a highly individualized behavioral, therapeutic, and educational intervention plan is needed both for home and school or work environments.
Psychopharmacologic treatment with medications such as stimulants for attention and hyperactivity, selective serotonin reuptake inhibitors for anxiety, alpha-agonists for hyperactivity and overarousal, and antipsychotics for irritable and aggressive behaviors appears to be helpful by assessment in a clinical setting in approximately 50–70 % of patients [34]. Response is not complete, however, and data from a national survey on FXS showed that approximately 10–20 % of respondents thought that medication was not helpful at all for behavior problems, whereas only approximately 40 % felt the medication was helping a lot [38]. Thus, there is a clear unmet need in FXS for better treatments for behavior and for any treatments that target cognitive deficits; thus, treatments that modify the underlying disorder would be an extremely important advance.
Molecular Functions of FMRP: Targets and Molecular Mechanisms
FMRP Domain Function and Human Missense Mutations Provide Insight into Canonical and Noncanonical Mechanisms
FMRP is an mRNA binding protein that regulates the translation of mRNAs encoding proteins that play important roles in neuronal development and synaptic function [39]. An understanding of molecular mechanisms of FMRP domains and target mRNA binding has provided important insight into mechanisms of translational control that go awry in FXS. These mechanisms have been reviewed extensively elsewhere [40]. Here, we will thus focus on recent discoveries and noncanonical mechanisms of FMRP-mediated regulation. FMRP contains three canonical RNA binding domains, two ribonucleoprotein K homology domains (KH1, KH2), and an arginine-glycine–glycine repeat-containing RGG box. An isoleucine 304 to asparagine missense mutation (Ile304Asn) in the KH2 domain, associated with severe clinical symptoms of FXS, has been shown to behave like a null mutation with reduced RNA binding and polyribosome association in a mouse model [41]. More recently, a patient diagnosed with FXS was found to have a glycine 266 to glutamic acid (Gly266Glu) missense mutation in the KH1 domain, which also disrupted RNA binding and polyribosome association [42]. Moreover, the Gly266Glu mutant was unable to rescue the excess α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor internalization in Fmr1 knockout (Fmr1 KO) neurons, which is a key cellular phenotype attributed to loss of translational control by FMRP [43]. The C-terminal RGG box has been shown to bind to G-quadruplex structures using multiple approaches [44–47], although the cellular function of this interaction is unknown. As G-quadruplex structures can facilitate mRNA localization to neuronal processes [48], and FMRP-target mRNAs have impaired activity-regulated mRNA localization in Fmr1 KO neurons [49], this suggests a possible role for the RGG box in mediating mRNA localization. Future work is needed to understand possible mechanistic inter-relationships between the RGG box and KH domains for regulation of FMRP-target mRNA translation.
The role of the amino terminus of FMRP in RNA binding has been reported in several studies, despite the lack of a well-conserved RNA binding domain [40]. Recent crystal structures of the amino terminus uncovered the existence of a novel KH0 domain (residues 126–202) [50]. Of potential interest, KH0 overlaps with the minimal domain (residues 180–217) required for FMRP binding to the small noncoding RNA, BC1 RNA [51]. The role of FMRP–BC1 interactions in translational regulation in neurons has been characterized [52, 53]. More work on the RNA targets and species recognized by KH0 may further clarify its role in mRNA regulation. The KH0 domain has recently been implicated in functions outside of FMRP’s established role in regulating mRNA translation. Of interest, a patient with developmental delay, but not showing the clinical phenotype of FXS, has been identified with an R138Q missense mutation in KH0 [54]. This mutation does not impair canonical functions of FMRP involved in mRNA translation control but does impair presynaptic function and the protein–protein interaction between FMRP and BK channels [54].
FMRP contains nuclear localization and export sequences that facilitate nucleocytoplasmic shuttling [55]. FMRP binds mRNA in the nucleus [56], and has been shown to regulate RNA editing [57, 58]. Two FMRP isoforms (ISO6, ISO12) contain a unique C-terminus, which is absent in common isoforms, for example ISO1, ISO7, and contains a sequence that localizes FMRP to nuclear Cajal bodies [59]. A recent study has shown that the tandem Agenet (Tudor) domain mediates FMRP binding to chromatin involved in the DNA damage response [60]. It will be interesting to further explore possible noncanonical nuclear mechanisms of FMRP.
Mechanisms of FMRP-Mediated Regulation of mRNA Translation in Neurons
Perhaps the most extensively studied function for FMRP has been to repress mRNA translation [39]. Consistent with this role are numerous reports of excess mRNA translation and protein synthesis in animal models of FXS and cells from humans with FXS [61–66]. Recent studies suggest that FMRP-mediated repression involves its association with polyribosomes and ribosome stalling or direct interaction with the 80S ribosomal subunit [39, 67, 68]. Regardless of whether FMRP acts to stall ribosomes by binding indiscriminately along coding sequences [67] or directly to the ribosome [68], these models are unlikely to explain how FMRP selectively binds only 4 % of mRNA in brain [69]. The search for cis-elements is an important direction to pursue, and the role of short sequence motifs in FMRP targets has been reported. The identified motifs included a 7-nucleotide sequence containing a conserved GAC core in one study [70], and 2 shorter motifs ACUK (K = G/U) and WGGA (W = A/U) in another study [71]. Recent bioinformatics analysis of these 2 target mRNA data sets suggests enrichment of GACR and WGGA but not ACUK motifs in FMRP target mRNAs [72], the latter of which would contribute to G-quadruplex structures. Further work is needed to understand the role of cis-elements and secondary structures to confer translational regulation of FMRP target mRNAs. It will be interesting to see if different mRNA binding modalities of FMRP may act together to regulate mRNA translation of specific targets.
FMRP has also been shown to be a positive regulator of mRNA translation in cultured neurons and in the brain [73–76]. In these examples, mRNA targets were observed to have decreased polyribosome association and protein expression in Fmr1 KO mice. More work is needed to characterize the molecular mechanisms involved in the role of FMRP to stimulate translation.
For FMRP to play a key role in protein synthesis-dependent synaptic plasticity underlying learning and memory, it must be capable of reversible regulation of mRNA translation. Few studies have addressed the important question of how FMRP-mediated repression is removed. In one model, activation of group 1 metabotropic glutamate receptors results in the dissociation of cytoplasmic FMR1-interacting protein, a noncanonical eIF4E-BP, from the translationally repressed messenger ribonucleoprotein particle complex [53]. Post-translational modifications of FMRP play key roles in regulating translational repression. Multiple studies have shown that phosphorylation of FMRP at serine 499 in mouse (500 in human) results in translational repression [77–81]. Dephosphorylation of FMRP by PP2A in response to mGlu1/5 activation removes translational repression [82]. FMRP can also be ubiquitinated in response to mGlu1/5 activation [83], and this mechanism can occur locally within dendrites and at synapses [84]. These studies suggest that dephosphorylation and ubiquitination of FMRP are linked mechanistically to the removal of translational repression and receptor-induced protein synthesis.
FMRP Interactions with the microRNA Pathway
FMRP has been shown by several studies to associate with mature microRNAs (miRNAs) and Argonaute proteins, which are part of the RNA-induced silencing complex (RISC) [79, 85–89], and also to interact with other components of the miRNA pathway, including Dicer and miRNA precursors [90]. More recently, interactions between FMRP and RISC/miRNAs have been shown to regulate dendritic protein synthesis and spine morphology [79, 85]. It will be interesting if future studies show that FMRP interactions with G-quadruplexes provide a common mechanism to regulate miRNA seed site accessibility [91]. The tight association of phosphorylated FMRP with RISC/miRNAs may promote translational repression, whereas dephosphorylation of FMRP in response to activation of mGlu1/5 receptor results in release of miRISC and concomitant activation of translation [79]. A role for FMRP phosphorylation to regulate miRNA-mediated repression was recently observed in a non-neuronal culture system [92], which further supports the proposed model. FMRP may also play a role in miRNA stability or turnover, and a recent report suggests dysregulation of several miRNAs in the hippocampus from Fmr1 KO mice [93]. Of interest, FMRP was recently shown to have differential effects on mRNA translation, acting either positively or negatively, depending on the type of molecular interactions between FMRP and the RNA helicase, Mov10, with miRNA recognition elements and hairpin secondary structure [94]. Such interactions could result in either facilitation of RISC-mediated silencing or its inhibition. Taken together these studies suggest a mechanistic cooperation between FMRP and miRNAs that may influence translation in a bidirectional manner. Moreover, these studies suggest that dysregulation of miRNAs may underlie impairments in regulation of protein synthesis in FXS.
FMRP-mediated Regulation of Voltage-gated Ion Channels
Fmr1 KO neurons display increased neocortical network activity [95–98]. Recent evidence suggests that this hyperexcitability in the absence of FMRP may be caused by dysfunctions in voltage-gated ion channels [98, 99]. In vitro screening assays have identified several voltage-gated potassium, sodium, and calcium channels as potential FMRP mRNA targets [44, 67, 100, 101]. Two of them, the voltage-gated potassium channels Kv3.1b and Kv4.2, were validated in independent studies as FMRP mRNA targets [74, 102, 103]. While Kv3.1b was shown to be translationally repressed by FMRP, the exact modalities of FMRP-mediated regulation of Kv4.2 are not fully understood yet, and it is still controversial if FMRP acts as a translational suppressor or activator of Kv4.2 [74, 103]. Kv4.2 is the major mediator of A-type currents in the CA1 region of the hippocampus, and a recent study showed reduced A-type currents and increased backpropagating action potentials in CA1 pyramidal neurons in Fmr1 KO mice [104], suggesting that absence of FMRP decreases Kv4.2 function. Interestingly, apart from translational regulation, FMRP also directly binds to ion channels, which has revealed noncanonical functions. FMRP binds and activates Slack [105, 106], and FMRP’s association with BK channels alters their calcium sensitivity in a protein synthesis-independent manner [98, 107]. A recent study also showed that FMRP interacts with the voltage-gated N-type calcium channel CaV2.2 and modulates its function and expression [108]. Taken together, these studies broaden our view of FMRP as a central regulator of ion channels suggesting novel routes of therapeutic strategies in FXS targeted at these channelopathies.
Preclinical Studies in the FXS Mouse Model
The underlying genetic cause of FXS, a trinucleotide expansion in the 5’ untranslated region of FMR1, which leads to transcriptional silencing, is difficult to replicate in animal models. Instead, Fmr1 KO mice, in which a genetic deletion leads to complete lack of FMRP expression in all cells, are the predominantly used animal model for FXS [109, 110]. Fmr1 KO mice display FXS-like phenotypes (Table 3), and have been widely used to identify and assess potential therapeutic strategies (Table 4). Owing to space limitations we will not discuss the two invertebrate models of FXS, Drosophila and zebrafish [181–184], although these models, as far as they have been studied, show many of the same characteristics and rescue phenomena seen in the mouse model.
Recent Developments in the Characterization of Fmr1 KO Mice as a Preclinical Model for FXS
Fmr1 KO mice have been used to analyze the molecular functions of FMRP, to identify mechanisms underlying FXS and to test potential therapeutic strategies. Major efforts have been made to identify robust and reliable phenotypes in the mouse model that reflect the human disease and pathology and thus can be used to identify and test potential treatment strategies. These phenotypes range from molecular, cellular and network alterations to behavioral and cognitive deficits. However, many of these phenotypes are moderate, difficult to reproduce in different laboratories, and might thus not be reliable tools to evaluate potential therapeutic strategies in preclinical studies. The most frequently used and robust phenotypes are summarized in Table 3. Below, we will discuss recent developments in the phenotypic characterization of the FXS mouse model, which may enable more rigorous and physiologically relevant preclinical testing of therapeutic strategies in the future.
Molecular Defects as Potential Biomarkers
Fragile X is a disease of dysregulated signal transduction and lost control of general and target mRNA translation [39, 185]. Impairments in several signaling pathways and altered expression of many FMRP targets have been detected in the FXS mouse model. Recently, molecular defects that were originally observed in the brains of FXS animal models were also detected in peripheral blood lymphocytes, lymphoblastoid cell lines, and fibroblasts from patients with FXS. Thus, these molecular defects may serve as biomarkers to evaluate the efficiency of disease-targeting therapies in humans. Potential biomarkers for FXS include altered signaling activity of extracellular signal-regulated kinase (ERK)1/2 and phosphoinositide-3-kinase (PI3K) [64–66, 186–188], or altered protein expression or activity of FMRP targets [e.g., amyloid-β precursor protein (AβPP) or matrix metalloproteinase 9 (MMP-9) [162, 189]]. Not all defects in signaling in the mouse model have been reproducible across laboratories. In particular, reports about ERK1/2 and PI3K/mammalian target of rapamycin (mTOR) activation have been conflicting (e.g., [62, 63]). These discrepancies might be explainable by different tissues or tissue preparations used for analysis, but they also show that future work is needed to fully understand ERK1/2 and PI3K signaling defects in FXS in order to be able to comprehensively assess their potential as biomarkers in studies with humans. Nonetheless, impaired ERK1/2 activation and increased AβPP or MMP-9 levels have already been used as outcome measure in open-label clinical trials [189–192]. In the future, analyses of these biomarkers could help to assess if a specific drug treatment targets the underlying molecular mechanisms.
Synaptic, Neuronal, and Network Defects to Test Treatment Strategies
Phenotypes of animal disease models are considered particularly useful if they recapitulate a symptom in human patients. In the case of FXS, increased density and immature appearance of dendritic spines were detected in patients and mouse models [171, 193], and have been widely used to test therapeutic strategies. Pharmacologic and genetic modification of many molecular targets, including neurotransmitter receptors, intracellular signaling molecules and direct targets of FMRP have been shown to rescue this phenotype (Table 4), but it is unclear if correction of altered dendritic spine morphology in the mouse model is a good predictor of the success of a certain therapeutic strategy in patients [194]. More recently, Cruz-Martin et al. [195, 196] have identified defects in activity-induced dendritic spine dynamics in the FXS mouse model in vitro and in vivo, which may be a direct correlate of synaptic plasticity underlying learning and memory. Stimulus-insensitive dendritic spine dynamics and increased turnover of dendritic spines were also observed in two other reports [197, 198]. Of note, defects in dendritic spine dynamics were not rescued, but rather exaggerated, by chronic in vivo administration of the mGlu5 negative allosteric modulator (NAM) 2-methyl-6-(phenylethynyl) pyridine [195]. This is of special interest, because recent clinical trials with mGlu5 NAMs did not show significant behavioral improvements in adolescents and adults with FXS compared with placebo, although cognition and functional outcomes were not tested (Table 5) [194, 210]. In the future, it will be important to identify pharmacologic interventions that restore dynamic alterations in dendritic spine morphology in Fmr1 KO mice in response to extracellular stimuli, as those might be particularly promising therapeutic strategies for patients with FXS.
Recent Developments in the Characterization of Fmr1 KO Mice as a Preclinical Model for FXS
Fmr1 KO mice have been used to analyze the molecular functions of FMRP, to identify mechanisms underlying FXS and to test potential therapeutic strategies. Major efforts have been made to identify robust and reliable phenotypes in the mouse model that reflect the human disease and pathology and thus can be used to identify and test potential treatment strategies. These phenotypes range from molecular, cellular and network alterations to behavioral and cognitive deficits. However, many of these phenotypes are moderate, difficult to reproduce in different laboratories, and might thus not be reliable tools to evaluate potential therapeutic strategies in preclinical studies. The most frequently used and robust phenotypes are summarized in Table 3. Below, we will discuss recent developments in the phenotypic characterization of the FXS mouse model, which may enable more rigorous and physiologically relevant preclinical testing of therapeutic strategies in the future.
Molecular Defects as Potential Biomarkers
Fragile X is a disease of dysregulated signal transduction and lost control of general and target mRNA translation [39, 185]. Impairments in several signaling pathways and altered expression of many FMRP targets have been detected in the FXS mouse model. Recently, molecular defects that were originally observed in the brains of FXS animal models were also detected in peripheral blood lymphocytes, lymphoblastoid cell lines, and fibroblasts from patients with FXS. Thus, these molecular defects may serve as biomarkers to evaluate the efficiency of disease-targeting therapies in humans. Potential biomarkers for FXS include altered signaling activity of extracellular signal-regulated kinase (ERK)1/2 and phosphoinositide-3-kinase (PI3K) [64–66, 186–188], or altered protein expression or activity of FMRP targets [e.g., amyloid-β precursor protein (AβPP) or matrix metalloproteinase 9 (MMP-9) [162, 189]]. Not all defects in signaling in the mouse model have been reproducible across laboratories. In particular, reports about ERK1/2 and PI3K/mammalian target of rapamycin (mTOR) activation have been conflicting (e.g., [62, 63]). These discrepancies might be explainable by different tissues or tissue preparations used for analysis, but they also show that future work is needed to fully understand ERK1/2 and PI3K signaling defects in FXS in order to be able to comprehensively assess their potential as biomarkers in studies with humans. Nonetheless, impaired ERK1/2 activation and increased AβPP or MMP-9 levels have already been used as outcome measure in open-label clinical trials [189–192]. In the future, analyses of these biomarkers could help to assess if a specific drug treatment targets the underlying molecular mechanisms.
Synaptic, Neuronal, and Network Defects to Test Treatment Strategies
Phenotypes of animal disease models are considered particularly useful if they recapitulate a symptom in human patients. In the case of FXS, increased density and immature appearance of dendritic spines were detected in patients and mouse models [171, 193], and have been widely used to test therapeutic strategies. Pharmacologic and genetic modification of many molecular targets, including neurotransmitter receptors, intracellular signaling molecules and direct targets of FMRP have been shown to rescue this phenotype (Table 4), but it is unclear if correction of altered dendritic spine morphology in the mouse model is a good predictor of the success of a certain therapeutic strategy in patients [194]. More recently, Cruz-Martin et al. [195, 196] have identified defects in activity-induced dendritic spine dynamics in the FXS mouse model in vitro and in vivo, which may be a direct correlate of synaptic plasticity underlying learning and memory. Stimulus-insensitive dendritic spine dynamics and increased turnover of dendritic spines were also observed in two other reports [197, 198]. Of note, defects in dendritic spine dynamics were not rescued, but rather exaggerated, by chronic in vivo administration of the mGlu5 negative allosteric modulator (NAM) 2-methyl-6-(phenylethynyl) pyridine [195]. This is of special interest, because recent clinical trials with mGlu5 NAMs did not show significant behavioral improvements in adolescents and adults with FXS compared with placebo, although cognition and functional outcomes were not tested (Table 5) [194, 210]. In the future, it will be important to identify pharmacologic interventions that restore dynamic alterations in dendritic spine morphology in Fmr1 KO mice in response to extracellular stimuli, as those might be particularly promising therapeutic strategies for patients with FXS.
A prominent defect in synaptic plasticity in Fmr1 KO mice is exaggerated and protein synthesis-independent metabotropic glutamate receptor-dependent long-term depression [83, 172]. However, this synaptic deficiency is difficult to translate into a specific defect in patients, and thus might not be ideal to test therapeutic strategies. In contrast, patients with FXS show symptoms of neuronal hyperexcitability, such as increased susceptibility to seizures and hyperarousal. This neuronal hyperexcitability may be reflected by a heightened susceptibility to audiogenic seizures in FXS mice [211], a widely used phenotype to assess therapeutic strategies in preclinical analyses. More recent efforts have focused on identifying measurable defects in neuronal network activity in Fmr1 KO mice that can be used to test therapeutic strategies. For example, prolonged neocortical UP states, a network defect in FXS that reflects the overall neuronal hyperexcitability [95, 96], have been successfully decreased with genetic or pharmacological reduction of mGlu5, genetic removal of Homer 1A, genetic reduction of the PI3K enhancer (PIKE), but not p110β [97, 150, 237, 238]. Of note, a recent study has identified a potential neuronal correlate of hyperarousal in the somatosensory cortex of Fmr1 KO mice that was rescued pharmacologically using a BKCA channel opener [98]. The same BKCA activator has been used previously to rescue behavioral impairments in the FXS mouse model [166], suggesting that neuronal network hyperexcitability is a useful phenotype to evaluate therapeutic strategies in FXS.
Assessing Activity-Dependent Neuronal Defects to Test Therapeutic Strategies in FXS
Many molecular and cellular phenotypes in Fmr1 KO mice are subtle, difficult to reproduce in different laboratories and often discussed controversially. These problems most likely arise from differences in genetic background, handling, and housing of the mice, which may alter the steady-state levels of intracellular signaling, protein synthesis, and dendritic spine morphology. A prominent feature of FMRP-deficient cells is the impairment in stimulus-induced neuronal responses. This has been shown on the molecular level (protein synthesis and signal transduction; e.g., [62, 63]), the cellular level (dendritic spine dynamics [196, 198]), and more recently on the network level (hypersensitivity of neocortical responses to tactile stimuli [98]). Exploring defects of stimulus sensitivity in animal models of FXS as phenotypes to test therapeutic strategies is particularly attractive because of their relevance for synaptic plasticity underlying learning, memory, and behavior. Moreover, these defects may be observed regardless of the “baseline” constitutive phenotype. Of note, impairments in stimulus-induced ERK and Akt signaling and protein synthesis have also been shown in fresh blood lymphocytes and lymphoblastoid cell lines from patients with FXS [64, 187].
Behavioral and Cognitive Phenotypes in the FXS Mouse Model
Behavioral analyses of Fmr1 KO mice are mainly focused on autism-associated defects, for example perseverative behavior, anxiety, and impairments in social interaction (see Table 3; reviewed in [180]). Behavioral phenotypes of FXS mice vary considerably across laboratories, and even defects in the opposite directions have been reported frequently [179]. To improve comparability of results, a future challenge will be to establish rigorous protocols and standards for behavioral testing in FXS that can be applied in various laboratories. Moreover, it will be important to identify more robust and reliable behavioral defects. Here, recent advances in analyzing social behavior and social interactions in FXS mice may hold some promise [119, 161, 212].
FXS is an intellectual disability, and the ultimate goal for the development of therapeutic strategies is to improve cognitive function in patients. Initial analyses to assess defects in hippocampal dependent learning and memory using classic Morris water maze assays revealed only subtle and varying defects in Fmr1 KO mice [213]. Recently, more robust defects in novel object recognition were reported by several laboratories [111, 145, 214], making this paradigm a promising tool to test therapeutic strategies for the improvement of cognition in FXS. Of note, patients with FXS are mostly impaired in higher cognition, behavioral flexibility, and inhibitory control, which depend on the prefrontal cortex. To date, very few studies have reported defects in prefrontal cortex-dependent higher-order cognition in Fmr1 KO mice [215, 237]. A recent study showed that defects in higher cognition in FXS mouse models were rescued by genetic reduction of the PI3K catalytic subunit p110β in the prefrontal cortex [237]. An important future direction will be to use defects in higher cognition in FXS mouse models to assess the efficacy of pharmacological rescue strategies in improving intellectual capabilities in FXS.
Therapeutic Targets and Rescue Strategies in the FXS Mouse Model
Rescue strategies in the FXS mouse model have been based on the analysis of molecular pathomechanisms (e.g., dysregulated mGlu1/5 signaling and protein synthesis), identification of specific FMRP targets that are dysregulated in FXS (e.g., increased protein levels of MMP-9 or striatal-enriched protein tyrosine phosphatase), or have been guided by observations in human patients, and the availability of Food and Drug Administration-approved drugs (N-methyl-D-aspartate antagonists [149]; lovastatin [112]).
Rescue strategies that were tested in the FXS mouse model target virtually every step of cellular signal transmission: neurotransmitter receptors, scaffolding proteins that connect receptors with downstream signaling, intracellular signaling molecules, and regulators of protein synthesis. In some cases, the pharmacologically or genetically modified molecules were confirmed FMRP targets, making these rescue strategies particularly promising. A summary of rescue strategies tested in the mouse model is shown in Table 4. In the following sections, we will discuss a few emerging themes and challenges arising from these studies.
Neurotransmitter Receptors Versus Intracellular Signaling and mRNA Translation
Significant effort has focused on manipulating neurotransmitter-dependent receptors as a therapeutic strategy in FXS. The most widely studied rescue approach targets dysregulated mGlu1/5 signaling in FXS using mGlu5 NAMs [216], which were shown to rescue many phenotypes in both mouse and fly models of FXS. Clinical trials using this approach have, so far, been unsuccessful in showing an improvement of FXS-associated phenotypes in humans, possibly because only behavioral phenotypes were addressed in older patients [194]. Of note, recent studies in the mouse model showed that chronic treatment with mGlu5 NAMs is necessary to improve memory deficits [120, 121, 129], suggesting that prolonged treatment in patients may also be beneficial. Notably, targeting mGlu5 seems to be more efficient in rescuing phenotypes in the FXS mouse model than targeting other G-coupled receptors that were also shown to be dysregulated in FXS [217], such as mGlu1 or muscarinic receptors M1 and M4 [118, 131, 136–138].
Another strategy has been to increase the signaling activity of certain receptors that may have reduced function in FXS, such as gamma-aminobutyric acid (GABA), dopamine and serotonin receptors, or tropomyosin receptor kinase B (Table 4). Here, agonists to GABA and to dopamine receptors are specifically promising because signaling through them was shown to be altered in FXS animal models [144, 169].
In general, neurotransmitter receptors are preferred pharmacological targets; however, in the case of FXS their therapeutic potential maybe limited. A hallmark of FXS animal models and patient cells is the lack of stimulus-induced intracellular signaling and protein synthesis. Dampening the upstream receptor may successfully reduce basal activity but may not restore stimulus sensitivity. Instead, recent studies suggest that strategies targeted at intracellular molecules may be more efficient in restoring the stimulus-induced response of signal transduction and protein synthesis [62, 159]. Extensive efforts have been made to identify signaling molecules or receptor scaffolding molecules that play important roles in the FXS phenotype. Genetic deletion of scaffolding molecules downstream of mGlu1/5 (Homer1a) or general G-coupled proteins (RGS4) have shown rescue of select phenotypes [150, 218]; however, so far it is not clear how they can be used as a pharmacological target in FXS. Furthermore, broad spectrum inhibitors of ERK1/2 and PI3K showed improvement of a few phenotypes in FXS mice but, owing to expected side effects, will not be suitable for treatment in humans [62, 63, 152]. Other efforts have focused on selective phenotypes of FXS, such as, for example, dendritic spine morphology by targeting p21-activating kinase [156, 157], or altered cyclic adenomonophosphate levels in patients with FXS by targeting phosphodiesterase-4 [158, 219].
The absence of FMRP leads to dysregulated protein synthesis, and a general protein synthesis inhibitor was shown to rescue phenotypes in a fly model of FXS [220]. In line with this observation, genetic or pharmacological reduction of central molecules involved in the regulation of general mRNA translation (mTOR [121], S6K1 [111], tuberous sclerosis 2 [160]) and molecules involved in the mRNA translation of select targets (cytoplasmic Poly(A)-binding protein [161]) have been used successfully in the mouse model to rescue many phenotypes. Future studies will have to show if general reduction of mRNA translation is a safe therapeutic strategy in humans.
Manipulating Specific Targets of FMRP
Other approaches have pharmacologically or genetically manipulated select targets of FMRP, such as AβPP, striatal-enriched protein tyrosine phosphatase, MMP-9, and the BKCA channel (Table 4). These strategies led to reversal of several phenotypes in the mouse model. In the future, it will be interesting to further analyze how targeting one out of many FMRP-regulated proteins with restricted and specialized functions in the cell can be therapeutic in FXS. Of particular interest will be strategies that modify FMRP targets with known central functions in intracellular signaling, such as the PI3K enhancer (PIKE), the PI3K catalytic subunit p110β or the glycogen synthase kinase-3β (GSK3β) [62, 122, 155, 221, 237, 238]. Both signaling molecules were identified in recent screens for FMRP targets [67, 71], and p110β has been validated in independent studies as an FMRP target [62, 221]. Moreover, lithium, an antagonist of GSK3β, is already approved for use in humans, and p110β-selective inhibitors have been developed for cancer research. While mouse studies with lithium and a specific GSK3β antagonist already show promising effects on behavior and cognition in FXS mice [122, 123, 134, 155], so far, p110β-selective inhibitors have only been validated in vitro in mice and in vivo in patient-derived cell lines [64, 66].
Challenges and Future Outlook for Preclinical Studies
As outlined above, an extensive number of pharmacologic and genetic rescue strategies have been tested in Fmr1 KO mice and most of them show rescue of a wide range of phenotypes. However, several of these studies also report that specific defects in the mouse model are not rescued by the tested strategy (Table 4). The comparison of defects that were and were not rescued in these different approaches may help to clarify underlying molecular mechanisms, evaluate the potential efficiency to improve core deficits in human patients, and, lastly, suggest potential combinatory therapies using drugs targeting different pathways. Currently, such analyses are complicated by the fact that virtually none of the studies has tested (and/or reported) the same battery of phenotypes, making a direct comparison nearly impossible. Future studies evaluating the effect of different drug treatments in side-by-side experiments will be instrumental to further assess therapeutic strategies.
The identification and development of potential treatment strategies in FXS has been driven substantially by the use of Fmr1 KO mice. These have been a valuable tool to delineate molecular mechanisms underlying FXS and to reveal the cellular functions of FMRP. However, a major problem for preclinical studies in FXS has been that most phenotypes in the mouse model are rather subtle and difficult to reproduce in different laboratories. So, while many different genetic or pharmacological rescue strategies have shown improvements in these phenotypes (see Table 4), the overall effects have been small owing to the subtle nature of the analyzed neuronal and behavioral deficits. This might have contributed to the disappointing results of recent clinical trials that were based on these studies (see following sections) [194]. Thus, a better comprehension of the benefits and limitations of the FXS mouse model is necessary to be able to utilize it as a reliable preclinical tool. One study, for example, revealed differences in autistic-like behaviors of Fmr1 KO mice depending on the genetic background, and argued that some phenotypes might rather represent the specific strain than the absence of FMRP expression [222]. In the future, it will be essential to better understand how genetic background affects phenotypes in the FXS mouse.
Recently, an Fmr1 KO rat model was generated, but, so far, very few studies using this new animal model have been published. One study showed altered speech representation in the auditory cortex of Fmr1 KO rats [223], and another study discovered some autistic-like behaviors but no defects in basic memory tasks [224]. As rats are generally the preferred model to test more complex cognitive function, Fmr1 KO rats hold great promise to evaluate therapeutic strategies for improving intellectual impairments and deficits in higher cognition in FXS.
Another promising novel tool for preclinical studies of brain disorders are human neurons that were generated from patient-derived induced pluripotent stem cells (iPSCs). The recent advances in the use of iPSCs derived from human cells enable the testing of therapeutic strategies “in a dish” [225]. Early studies in FXS patient-derived iPSCs suggest that some FXS-associated defects are replicated in these cells [226–228], although the classic phenotypes reported in the mouse model, for example exaggerated protein synthesis and altered dendritic spine morphology, have yet to be examined in FXS iPSC-derived neurons. In the future, it will be important to identify robust phenotypes in human iPSC-derived neurons from FXS that are suitable to test or screen for therapeutic drugs.
Translation of Targeted Treatments to Humans with FXS
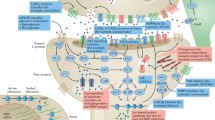
Based on the work with FXS models discussed in the previous sections, treatment targets have been identified for potential clinical development and directed at 1) reduction of excess activity in signal transduction pathways connecting group 1 metabolic glutamate receptors (mGluRs) or other Gq-linked receptors to the dendritic translational machinery, either through (1A) receptor modulation at the cell surface or (1B) through modulation of the intracellular signaling pathway; 2) reduction of excessive activity of proteins normally regulated by FMRP; 3) increasing expression and activation of surface AMPA receptors; 4) modification of activity of GABA and other receptors/proteins that regulate glutamate signaling or translational signaling pathways; 5) using miRNAs to block excessive translation of mRNAs normally regulated by FMRP; and 6) correction of abnormal channel activities normally directly regulated by FMRP (see Table 5, which reviews all targeted treatment trials in FXS with ≥ 8 participants) [229, 230].
Successful preclinical testing in FXS models has led to early proof-of concept clinical trials and subsequent larger trials for some of the proposed targeted treatments (Table 5). CX516, an AMPA activator, did not show efficacy but was used most likely at a subtherapeutic dose, a conclusion that would be supported by a suggestion of efficacy in patients co-treated with antipsychotics, known to potentiate effects of CX516 [199]. Lithium, thought to reduce excess mGluR-dependent activation of translation by attenuating GSK3β activity and possibly phosphatidyl-inositol turnover, resulted in significant improvement in behavioral scales, verbal memory, and abnormal ERK phosphorylation rates in lymphocytes in a 2-month pilot open-label proof-of-concept trial in children and young adults with FXS [200]. A pilot placebo-controlled crossover trial of minocycline, an antibiotic that inhibits overexpressed synaptic MMP-9 in FXS models, conducted in children with FXS demonstrated mild global clinical improvement and reduction of MMP-9 levels in the blood of responders [205].
GABA-B agonist arbaclofen presumably lowers presynaptic glutamate release with resultant reduction of group 1 mGluR signaling (Table 5). In a phase II double-blind placebo-controlled crossover trial [207], arbaclofen showed improvement over placebo in the entire per-protocol group for social withdrawal and parent-nominated problem behaviors. However, a large phase III placebo-controlled trial in adolescents and adults with FXS did not show benefits for arbaclofen over placebo in the primary outcome of social withdrawal. An additional phase III trial in children with FXS showed promise but full analyses are pending. Acamprosate, currently Food and Drug Administration-approved for alcohol withdrawal, with agonist properties at both GABA-A and GABA-B receptors, has shown promise, in an open-label trial, for hyperactivity and social functioning in FXS [209]. Acamprosate and the GABA-A agonist ganaxolone are being tested in small placebo-controlled trials in FXS [203].
Multiple negative modulators of the mGluR5 receptor have been in trials in FXS (Table 5). A single oral dose of fenobam resulted in a significant improvement in abnormal prepulse inhibition compared with untreated control participants with FXS [201]. A phase II double-blind, placebo-controlled, crossover trial of AFQ056 in 30 men with FXS treated for 28 days each with AFQ and placebo suggested improvement in maladaptive behavior in a post hoc analysis in the subgroup with full methylation of FMR1 [206]. Larger multinational trials of AFQ056 and RO4917523, another mGluR5 negative modulator (Roche), have not supported this behavioral outcome but have not addressed cognitive or learning outcomes [203]. Clinical observations from long-term extension studies with both arbaclofen and AFQ056 have suggested there may be long-term cognitive and functional benefits of these drugs that were not captured by formal measures employed in the trials [231].
Challenges and Future Outlook for Clinical Studies
Although many neuronal targets for treating the underlying disorder in FXS have been identified, translational work has begun, and, as can be seen in Table 5, early open-label and some phase II trials have suggested benefits for multiple targeted treatments, there have been problems with demonstrating disease modification in subsequent larger trials because there are still many uncertainties about how to demonstrate treatment effects optimally in a clinical trial setting [229, 232, 233]. Major trial design issues in FXS trials potentially include variable but narrow dosing windows, timing (age) and length of treatment necessary, potential need for cognitive or behavioral interventions to see drug effects on synaptic plasticity in the form of learning, large placebo effects, difficulty predicting a primary outcome in a disease such as FXS with diverse cognitive and behavioral manifestations for a drug that reverses phenotypes in animal models, and lack of validated, sensitive biomarkers, and functional outcome measures in FXS [232]. These trial design issues will need to be resolved to be able to demonstrate disease modification in FXS. It has been recognized that more objective clinician-administered and direct observational measures, measures that address cognition and are validated for populations with intellectual disability, and biomarkers of central nervous system function are needed to reduce placebo effect and establish that the drug is “hitting the target”, and these measures are being increasingly incorporated into more recent trials (Table 5). It is likely that efficacy studies will need to focus on younger study populations, and intensive learning interventions will need to be employed within the trial design in the drug and placebo groups in order to see the enhancement of synaptic plasticity seen in animal models within a time frame compatible with placebo-controlled trials. Thus, the overall approach to process of drug development in FXS needs to change such that efficacy is no longer required in adults before studying children, ways of analyzing global outcomes over multiple domains are developed and accepted by regulatory authorities, and good precedents are established and accepted for embedding form of cognitive training within trial designs. If these problems can be solved, hopefully, in the future, treatment to reverse the underlying disorder will eventually replace or complement supportive treatment.
There is significant overlap in molecular and cellular pathways involving FMRP and those including gene products associated with ASD [6, 230]. This overlap falls roughly into 3 categories: 1) defects in proteins linked to the signaling cascade for regulation of FMRP-regulated translation such as Shank, mTOR, p21-activating kinase, and phosphatase and tensin homolog; 2) defects in proteins regulated directly by FMRP such as postsynaptic density protein 95 and Arc; and 3) defects in proteins involved in the balance of activity in brain glutamate and GABA systems. Indeed, the FMRP/ASD pathway overlap has been recently supported by the findings that 1) in a meta-analysis of exome screening studies, FMRP binds to about a third of all genes associated with ASD [234]; 2) genes that code for FMRP target mRNAs are more likely than other genes with similar expression patterns to contribute to ASD [235]; and 3) common variants in genes involved in postsynaptic regulation of FMRP activity (CAMK4, GRM1, cytoplasmic FMR1-interacting protein) are risk factors for ASD [236]. Treatments directed at all of these pathway-overlap areas are being explored in preclinical or clinical translational work in FXS and, if successful, progress in development of such targeted treatments for FXS may result in treatments to reverse neural defects and clinical manifestations of ASD and intellectual disability.
References
Coffee B, Keith K, Albizua I, et al. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet 2009;85:503-514.
Willemsen R, Levenga J, Oostra BA. CGG repeat in the FMR1 gene: size matters. Clin Genet 2011;80:214-225.
Seltzer MM, Baker MW, Hong J, Maenner M, Greenberg J, Mandel D. Prevalence of CGG expansions of the FMR1 gene in a US population-based sample. Am J Med Genet 2012;159B:589-597.
Tassone F, Iong KP, Tong T-H, et al. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome Med 2012;4:100.
Hagerman RJ, Berry-Kravis E, Kaufmann WE, et al. Advances in the treatment of fragile X syndrome. Pediatrics 2009;123:378-390.
Loesch DZ, Huggins RM, Hagerman RJ. Phenotypic variation and FMRP levels in fragile X. Ment Retard Dev Disabil Res Rev 2004;10:31-41.
Kidd SA LA, Barbouth D, et al. Fragile X syndrome: a review of associated medical problems. Pediatrics 2014;134:995-1005.
Kaufmann WE, Abrams MT, Chen W, Reiss AL. Genotype, molecular phenotype, and cognitive phenotype: Correlations in fragile X syndrome. Am J Med Genet 1999;83:286.
de Vries BB, Wiegers AM, Smits AP, et al. Mental status of females with an FMR1 gene full mutation. Am J Hum Genet 1996;58:1025.
Freund LS, Reiss AL, Abrams MT. Psychiatric Disorders Associated With Fragile X in the Young Female. Pediatrics. 1993;91(2):321.
Dykens EM, Hodapp RM, Leckman JF. Strengths and weaknesses in the intellectual functioning of males with fragile X syndrome. Am J Ment Defic 1987;92:234.
Fisch GS, Carpenter N, Holden JJA, et al. Longitudinal changes in cognitive and adaptive behavior in fragile X females: A prospective multicenter analysis. Am J Med Genet 1999;83:308.
Fisch GS, Simensen R, Tarleton J, et al. Longitudinal study of cognitive abilities and adaptive behavior levels in fragile X males: A prospective multicenter analysis. Am J Med Genet 1996;64:356.
Klaiman C, Quintin E-M, Jo B, et al. Longitudinal profiles of adaptive behavior in fragile X syndrome. Pediatrics 2014;134:315-324.
Baumgardner TL, Reiss AL, Freund LS, Abrams MT. Specification of the neurobehavioral phenotype in males with fragile X syndrome. Pediatrics 1995;95:744.
Hagerman RJ, Falkenstein AR. An association between recurrent otitis media in infancy and later hyperactivity. Clin Pediatr (Phila) 1987;26:253-257.
Hagerman RJ, Jackson C, Amiri K, O’Connor R, Sobesky W, Silverman AC. Girls with fragile X syndrome: physical and neurocognitive status and outcome. Pediatrics 1992;89:395-400.
Mazzocco MM, Pennington BF, Hagerman RJ. The neurocognitive phenotype of female carriers of fragile X: additional evidence for specificity. J Dev Behav Pediatr 1993;14:328-335.
Turk J. Fragile X syndrome and attentional deficits. J Appl Res Intellect Disabil 1998;11:175.
Cordeiro L, Ballinger E, Hagerman R, Hessl D. Clinical assessment of DSM-IV anxiety disorders in fragile X syndrome: prevalence and characterization. J Neurodev Disord 2011;3:57-67.
Cohen IL. A theoretical analysis of the role of hyperarousal in the learning and behavior of fragile X males. Ment Retard Dev Disabil Res Rev 1995;1:286-291.
Bailey DJ, Mesibov GB, Hatton DD, Clark RD, Roberts JE, Mayhew L. Autistic behavior in young boys with fragile x syndrome. J Autism Dev Disord 1998;28:499-508.
Belser RC, Sudhalter V. Conversational characteristics of children with fragile X syndrome: repetitive speech. Am J Ment Retard 2001;106:28-38.
Symons FJ, Byiers BJ, Raspa M, Bishop E, Bailey Jr DB. Self-injurious behavior and fragile X syndrome: findings from the national fragile X survey. Am J Intellect Dev Disabil 2010;115:473-481.
Bailey Jr DB, Raspa M, Bishop E, et al. Health and economic consequences of fragile X syndrome for caregivers. J Dev Behav Pediatr 2012;33:705-712.
Brady N, Skinner D, Roberts J, Hennon E. Communication in young children with fragile X syndrome: A qualitative study of mothers’ perspectives. Am J Speech Lang Pathol 2006;15:353-364.
Roberts JE, Mirrett P, Burchinal M. Receptive and expressive communication development of young males with fragile X syndrome. Am J Ment Retard 2001;106:216-230.
Roberts JE, Mirrett P, Anderson K, Burchinal M, Neebe E. Early communication, symbolic behavior, and social profiles of young males with fragile X syndrome. Am J Speech Lang Pathol 2002;11:295-304.
Abbeduto L, Hagerman RJ. Language and communication in fragile X syndrome. Ment Ret Dev Dis Res Rev 1997;3:313.
Lewis P, Abbeduto L, Murphy M, et al. Cognitive, language and social-cognitive skills of individuals with fragile X syndrome with and without autism. J Intellect Disabil Res 2006;50:532-545.
McDuffie A, Kover S, Abbeduto L, Lewis P, Brown T. Profiles of receptive and expressive language abilities in boys with comorbid fragile X syndrome and autism. Am J Intellect Dev Disabil 2012;117:18.
Murphy MM, Abbeduto L. Indirect genetic effects and the early language development of children with genetic mental retardation syndromes: The role of joint attention. Infants Young Children 2005;18:47-59.
Wang LW, Berry-Kravis E, Hagerman RJ. Fragile X: leading the way for targeted treatments in autism. Neurotherapeutics 2010;7:264-274.
Berry-Kravis E, Sumis A, Hervey C, Mathur S. Clinic-based retrospective analysis of psychopharmacology for behavior in fragile X syndrome. International J of Pediatrics 2012.
Abbeduto L, McDuffie A, Thurman AJ. The fragile X syndrome–autism comorbidity: what do we really know? Front Genet 2014;5:355.
Ozonoff S, Heung K, Byrd R, Hansen R, Hertz‐Picciotto I. The onset of autism: patterns of symptom emergence in the first years of life. Autism Res 2008;1:320-328.
Hessl D, Dyer-Friedman J, Glaser B, et al. The influence of environmental and genetic factors on behavior problems and autistic symptoms in boys and girls with fragile X syndrome. Pediatrics 2001;108:E88.
Bailey DB, Raspa M, Bishop E, Olmsted M, Mallya UG, Berry-Kravis E. Medication utilization for targeted symptoms in children and adults with fragile X syndrome: US survey. J Dev Behav Ped 2012;33:62-69.
Darnell JC, Klann E. The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci 2013;16:1530-1536.
Santoro MR, Bray SM, Warren ST. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol 2012;7:219-245.
Zang JB, Nosyreva ED, Spencer CM, et al. A mouse model of the human Fragile X syndrome I304N mutation. PLoS Genet 2009;5:e1000758.
Myrick LK, Nakamoto-Kinoshita M, Lindor NM, Kirmani S, Cheng X, Warren ST. Fragile X syndrome due to a missense mutation. Eur J Hum Genet 2014;22:1185-1189.
Nakamoto M, Nalavadi V, Epstein MP, Narayanan U, Bassell GJ, Warren ST. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc Natl Acad Sci U S A 2007;104:15537-15542.
Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 2001;107:489-499.
Menon L, Mader SA, Mihailescu MR. Fragile X mental retardation protein interactions with the microtubule associated protein 1B RNA. RNA 2008;14:1644-1655.
Menon L, Mihailescu MR. Interactions of the G quartet forming semaphorin 3F RNA with the RGG box domain of the fragile X protein family. Nucleic Acids Res 2007;35:5379-5392.
Schaeffer C, Bardoni B, Mandel JL, Ehresmann B, Ehresmann C, Moine H. The fragile X mental retardation protein binds specifically to its mRNA via a purine quartet motif. EMBO J 2001;20:4803-4813.
Subramanian M, Rage F, Tabet R, Flatter E, Mandel JL, Moine H. G-quadruplex RNA structure as a signal for neurite mRNA targeting. EMBO Rep 2011;12:697-704.
Dictenberg JB, Swanger SA, Antar LB, Singer RH, Bassell GJ. A direct role for FMRP in activity dependent dendritic mRNA transport links filopodial spine morphogenesis to fragile x syndrome. Dev Cell 2008;14:926-939.
Myrick LK, Hashimoto H, Cheng X, Warren ST. Human FMRP contains an integral tandem Agenet (Tudor) and KH motif in the amino terminal domain. Hum Mol Genet 2015;24:1733-1740.
Zalfa F, Adinolfi S, Napoli I, et al. Fragile X mental retardation protein (FMRP) binds specifically to the brain cytoplasmic RNAs BC1/BC200 via a novel RNA-binding motif. J Biol Chem 2005;280:33403-33410.
Zalfa F, Giorgi M, Primerano B, et al. The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 2003;112:317-327.
Napoli I, Mercaldo V, Boyl PP, et al. The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 2008;134:1042-1054.
Myrick LK, Deng PY, Hashimoto H, et al. Independent role for presynaptic FMRP revealed by an FMR1 missense mutation associated with intellectual disability and seizures. Proc Natl Acad Sci U S A 2015;112:949-956.
Feng Y, Gutekunst CA, Eberhart DE, Yi H, Warren ST. FMRP: Nucleocytoplasmic shuttling and association with somatodendritic polyribosomes. J Neurosci 1997;17:1539-1547.
Kim M, Bellini M, Ceman S. Fragile X mental retardation protein FMRP binds mRNAs in the nucleus. Mol Cell Biol 2009;29:214-228.
Bhogal B, Jepson JE, Savva YA, Pepper AS, Reenan RA, Jongens TA. Modulation of dADAR-dependent RNA editing by the Drosophila fragile X mental retardation protein. Nat Neurosci 2011;14:1517-1524.
Bassell GJ. Fragile balance: RNA editing tunes the synapse. Nat Neurosci 2011;14:1492-1494.
Dury AY, El Fatimy R, Tremblay S, et al. Nuclear fragile X mental retardation protein is localized to Cajal bodies. PLoS Genet 2013;9:e1003890.
Alpatov R, Lesch BJ, Nakamoto-Kinoshita M, et al. A chromatin-dependent role of the fragile X mental retardation protein FMRP in the DNA damage response. Cell 2014;157:869-881.
Muddashetty RS, Kelic S, Gross C, Xu M, Bassell GJ. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci 2007;27:5338-5348.
Gross C, Nakamoto M, Yao X, et al. Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J Neurosci 2010;30:10624-10638.
Osterweil EK, Krueger DD, Reinhold K, Bear MF. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci 2010;30:15616-15627.
Gross C, Bassell GJ. Excess protein synthesis in FXS patient lymphoblastoid cells can be rescued with a p110beta-selective inhibitor. Mol Med 2012;18:336-345.
Hoeffer CA, Sanchez E, Hagerman RJ, et al. Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav 2012;11:332-341.
Kumari D, Bhattacharya A, Nadel J, et al. Identification of fragile X syndrome specific molecular markers in human fibroblasts: a useful model to test the efficacy of therapeutic drugs. Hum Mutat 2014;35:1485-1494.
Darnell JC, Van Driesche SJ, Zhang C, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011;146:247-261.
Chen E, Sharma MR, Shi X, Agrawal RK, Joseph S. Fragile X mental retardation protein regulates translation by binding directly to the ribosome. Mol Cell 2014;54:407-417.
Ashley CT, Wilkinson KD, Reines D, Warren ST. FMR1 protein: conserved RNP family domains and selective RNA binding. Science 1993;262:563-566.
Ray D, Kazan H, Cook KB, et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature 2013;499:172-177.
Ascano M, Jr., Mukherjee N, Bandaru P, et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 2012;492:382-386.
Suhl JA, Chopra P, Anderson BR, Bassell GJ, Warren ST. Analysis of FMRP mRNA target datasets reveals highly associated mRNAs mediated by G-quadruplex structures formed via clustered WGGA sequences. Hum Mol Genet 2014;23:5479-5491.
Bechara EG, Didiot MC, Melko M, et al. A novel function for fragile X mental retardation protein in translational activation. PLoS Biol 2009;7:e16.
Gross C, Yao X, Pong DL, Jeromin A, Bassell GJ. Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J Neurosci 2011;31:5693-5698.
Kwan KY, Lam MM, Johnson MB, et al. Species-dependent posttranscriptional regulation of NOS1 by FMRP in the developing cerebral cortex. Cell 2012;149:899-911.
Fahling M, Mrowka R, Steege A, et al. Translational regulation of the human achaete-scute homologue-1 by fragile X mental retardation protein. J Biol Chem 2009;284:4255-4266.
Ceman S, O’Donnell WT, Reed M, Patton S, Pohl J, Warren ST. Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum Mol Genet 2003;12:3295-3305.
Narayanan U, Nalavadi V, Nakamoto M, et al. S6K1 phosphorylates and regulates fragile X mental retardation protein (FMRP) with the neuronal protein synthesis-dependent mammalian target of rapamycin (mTOR) signaling cascade. J Biol Chem 2008;283:18478-18482.
Muddashetty RS, Nalavadi VC, Gross C, et al. Reversible inhibition of PSD-95 mRNA translation by miR-125a, FMRP phosphorylation, and mGluR signaling. Mol Cell 2011;42:673-688.
Niere F, Wilkerson JR, Huber KM. Evidence for a fragile X mental retardation protein-mediated translational switch in metabotropic glutamate receptor-triggered Arc translation and long-term depression. J Neurosci 2012;32:5924-5936.
Coffee RL, Jr., Williamson AJ, Adkins CM, Gray MC, Page TL, Broadie K. In vivo neuronal function of the fragile X mental retardation protein is regulated by phosphorylation. Hum Mol Genet 2012;21:900-915.
Narayanan U, Nalavadi V, Nakamoto M, et al. FMRP phosphorylation reveals an immediate-early signaling pathway triggered by group I mGluR and mediated by PP2A. J Neurosci 2007;27:14349-14357.
Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 2006;51:441-454.
Nalavadi VC, Muddashetty RS, Gross C, Bassell GJ. Dephosphorylation-induced ubiquitination and degradation of FMRP in dendrites: a role in immediate early mGluR-stimulated translation. J Neurosci 2012;32:2582-2587.
Edbauer D, Neilson JR, Foster KA, et al. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron 2010;65:373-384.
Jin P, Alisch RS, Warren ST. RNA and microRNAs in fragile X mental retardation. Nat Cell Biol 2004;6:1048-1053.
Jin P, Zarnescu DC, Ceman S, et al. Biochemical and genetic interaction between the fragile X mental retardation protein and the microRNA pathway. Nat Neurosci 2004;7:113-117.
Xu XL, Li Y, Wang F, Gao FB. The steady-state level of the nervous-system-specific microRNA-124a is regulated by dFMR1 in Drosophila. J Neurosci 2008;28:11883-11889.
Yang Y, Xu S, Xia L, et al. The bantam microRNA is associated with Drosophila fragile X mental retardation protein and regulates the fate of germline stem cells. PLoS Genet 2009;5:e1000444.
Cheever A, Ceman S. Translation regulation of mRNAs by the fragile X family of proteins through the microRNA pathway. RNA Biol 2009;6:175-178.
Stefanovic S, Bassell GJ, Mihailescu MR. G quadruplex RNA structures in PSD-95 mRNA: potential regulators of miR-125a seed binding site accessibility. RNA 2015;21:48-60.
Li Y, Tang W, Zhang LR, Zhang CY. FMRP regulates miR196a-mediated repression of HOXB8 via interaction with the AGO2 MID domain. Mol Biosyst 2014;10:1757-1764.
Liu T, Wan RP, Tang LJ, et al. A microRNA profile in Fmr1 knockout mice reveals microRNA expression alterations with possible roles in fragile X syndrome. Mol Neurobiol 2014 Jun 7 [Epub ahead of print].
Kenny PJ, Zhou H, Kim M, et al. MOV10 and FMRP regulate AGO2 association with microRNA recognition elements. Cell Rep 2014;9:1729-1741.
Goncalves JT, Anstey JE, Golshani P, Portera-Cailliau C. Circuit level defects in the developing neocortex of Fragile X mice. Nat Neurosci 2013;16:903-909.
Gibson JR, Bartley AF, Hays SA, Huber KM. Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J Neurophysiol 2008;100:2615-2626.
Hays SA, Huber KM, Gibson JR. Altered neocortical rhythmic activity states in Fmr1 KO mice are due to enhanced mGluR5 signaling and involve changes in excitatory circuitry. J Neurosci 2011;31:14223-14234.
Zhang Y, Bonnan A, Bony G, et al. Dendritic channelopathies contribute to neocortical and sensory hyperexcitability in Fmr1(-/y) mice. Nat Neurosci 2014;17:1701-1709.
Brager DH, Johnston D. Channelopathies and dendritic dysfunction in fragile X syndrome. Brain Res Bull 2014;103:11-17.
Brown V, Jin P, Ceman S, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 2001;107:477-487.
Chen L, Yun SW, Seto J, Liu W, Toth M. The fragile X mental retardation protein binds and regulates a novel class of mRNAs containing U rich target sequences. Neuroscience 2003;120:1005-1017.
Strumbos JG, Brown MR, Kronengold J, Polley DB, Kaczmarek LK. Fragile X mental retardation protein is required for rapid experience-dependent regulation of the potassium channel Kv3.1b. J Neurosci 2010;30:10263-10271.
Lee HY, Ge WP, Huang W, et al. Bidirectional regulation of dendritic voltage-gated potassium channels by the fragile X mental retardation protein. Neuron 2011;72:630-642.
Routh BN, Johnston D, Brager DH. Loss of functional A-type potassium channels in the dendrites of CA1 pyramidal neurons from a mouse model of fragile X syndrome. J Neurosci 2013;33:19442-19450.
Brown MR, Kronengold J, Gazula VR, et al. Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat Neurosci 2010;13:819-821.
Zhang Y, Brown MR, Hyland C, et al. Regulation of neuronal excitability by interaction of fragile X mental retardation protein with slack potassium channels. J Neurosci 2012;32:15318-15327.
Deng PY, Rotman Z, Blundon JA, et al. FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 2013;77:696-711.
Ferron L, Nieto-Rostro M, Cassidy JS, Dolphin AC. Fragile X mental retardation protein controls synaptic vesicle exocytosis by modulating N-type calcium channel density. Nat Commun 2014;5:3628.
Anon. Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch–Belgian Fragile X Consortium. Cell 1994;78:23-33.
Mientjes EJ, Nieuwenhuizen I, Kirkpatrick L, et al. The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo. Neurobiol Dis 2006;21:549-555.
Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron 2012;76:325-337.
Osterweil EK, Chuang SC, Chubykin AA, et al. Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 2013;77:243-250.
Liu ZH, Huang T, Smith CB. Lithium reverses increased rates of cerebral protein synthesis in a mouse model of fragile X syndrome. Neurobiol Dis 2012;45:1145-1152.
Henderson C, Wijetunge L, Kinoshita MN, et al. Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci Transl Med 2012;4:152ra128.
Lim CS, Hoang ET, Viar KE, Stornetta RL, Scott MM, Zhu JJ. Pharmacological rescue of Ras signaling, GluA1-dependent synaptic plasticity, and learning deficits in a fragile X model. Genes Dev 2014;28:273-289.
Guo W, Murthy AC, Zhang L, et al. Inhibition of GSK3beta improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum Mol Genet 2012;21:681-691.
Dolen G, Osterweil E, Rao BS, et al. Correction of fragile X syndrome in mice. Neuron 2007;56:955-962.
Thomas AM, Bui N, Graham D, Perkins JR, Yuva-Paylor LA, Paylor R. Genetic reduction of group 1 metabotropic glutamate receptors alters select behaviors in a mouse model for fragile X syndrome. Behav Brain Res 2011;223:310-321.
de Esch CE, van den Berg WE, Buijsen RA, et al. Fragile X mice have robust mGluR5-dependent alterations of social behaviour in the Automated Tube Test. Neurobiol Dis 2015;75:31-39.
Michalon A, Sidorov M, Ballard TM, et al. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 2012;74:49-56.
Busquets-Garcia A, Gomis-Gonzalez M, Guegan T, et al. Targeting the endocannabinoid system in the treatment of fragile X syndrome. Nat Med 2013;19:603-607.
Min WW, Yuskaitis CJ, Yan Q, et al. Elevated glycogen synthase kinase-3 activity in fragile X mice: key metabolic regulator with evidence for treatment potential. Neuropharmacology 2009;56:463-472.
Yuskaitis CJ, Mines MA, King MK, Sweatt JD, Miller CA, Jope RS. Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of fragile X syndrome. Biochem Pharmacol 2010;79:632-646.
Wang GX, Smith SJ, Mourrain P. Fmr1 KO and fenobam treatment differentially impact distinct synapse populations of mouse neocortex. Neuron 2014;84:1273-1286.
Levenga J, Hayashi S, de Vrij FM, et al. AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome. Neurobiol Dis 2011;42:311-317.
de Vrij FM, Levenga J, van der Linde HC, et al. Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis 2008;31:127-132.
Pop AS, Levenga J, de Esch CE, et al. Rescue of dendritic spine phenotype in Fmr1 KO mice with the mGluR5 antagonist AFQ056/Mavoglurant. Psychopharmacology (Berl) 2014;231:1227-1235.
Suvrathan A, Hoeffer CA, Wong H, Klann E, Chattarji S. Characterization and reversal of synaptic defects in the amygdala in a mouse model of fragile X syndrome. Proc Natl Acad Sci 2010;107:11591-11596.
Michalon A, Bruns A, Risterucci C, et al. Chronic metabotropic glutamate receptor 5 inhibition corrects local alterations of brain activity and improves cognitive performance in fragile X mice. Biol Psychiatry 2014;75:189-197.
Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 2005;49:1053-1066.
Thomas AM, Bui N, Perkins JR, Yuva-Paylor LA, Paylor R. Group I metabotropic glutamate receptor antagonists alter select behaviors in a mouse model for fragile X syndrome. Psychopharmacology (Berl) 2012;219:47-58.
Gantois I, Pop AS, de Esch CE, et al. Chronic administration of AFQ056/Mavoglurant restores social behaviour in Fmr1 knockout mice. Behav Brain Res 2013;239:72-79.
Vinueza Veloz MF, Buijsen RA, Willemsen R, et al. The effect of an mGluR5 inhibitor on procedural memory and avoidance discrimination impairments in Fmr1 KO mice. Genes Brain Behav 2012;11:325-331.
Franklin AV, King MK, Palomo V, Martinez A, McMahon LL, Jope RS. Glycogen synthase kinase-3 inhibitors reverse deficits in long-term potentiation and cognition in fragile X mice. Biol Psychiatry 2014;75:198-206.
Volk LJ, Daly CA, Huber KM. Differential roles for group 1 mGluR subtypes in induction and expression of chemically induced hippocampal long-term depression. J Neurophysiol 2006;95:2427-2438.
Veeraragavan S, Graham D, Bui N, Yuva-Paylor LA, Wess J, Paylor R. Genetic reduction of muscarinic M4 receptor modulates analgesic response and acoustic startle response in a mouse model of fragile X syndrome (FXS). Behav Brain Res 2012;228:1-8.
Veeraragavan S, Bui N, Perkins JR, Yuva-Paylor LA, Carpenter RL, Paylor R. Modulation of behavioral phenotypes by a muscarinic M1 antagonist in a mouse model of fragile X syndrome. Psychopharmacology (Berl) 2011;217:143-151.
Veeraragavan S, Bui N, Perkins JR, Yuva-Paylor LA, Paylor R. The modulation of fragile X behaviors by the muscarinic M4 antagonist, tropicamide. Behav Neurosci 2011;125:783-790.
Choi CH, Schoenfeld BP, Bell AJ, et al. Pharmacological reversal of synaptic plasticity deficits in the mouse model of fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res 2011;1380:106-119.
Olmos-Serrano JL, Paluszkiewicz SM, Martin BS, Kaufmann WE, Corbin JG, Huntsman MM. Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J Neurosci 2010;30:9929-9938.
Heulens I, D’Hulst C, Braat S, Rooms L, Kooy RF. Involvement and therapeutic potential of the GABAergic system in the fragile X syndrome. ScientificWorldJournal 2010;10:2198-2206.
Olmos-Serrano JL, Corbin JG, Burns MP. The GABA(A) receptor agonist THIP ameliorates specific behavioral deficits in the mouse model of fragile X syndrome. Dev Neurosci 2011;33:395-403.
Pacey LK, Heximer SP, Hampson DR. Increased GABA(B) receptor-mediated signaling reduces the susceptibility of fragile X knockout mice to audiogenic seizures. Mol Pharmacol 2009;76:18-24.
Wang H, Wu LJ, Kim SS, et al. FMRP acts as a key messenger for dopamine modulation in the forebrain. Neuron 2008;59:634-647.
Ventura R, Pascucci T, Catania MV, Musumeci SA, Puglisi-Allegra S. Object recognition impairment in Fmr1 knockout mice is reversed by amphetamine: involvement of dopamine in the medial prefrontal cortex. Behav Pharmacol 2004;15:433-442.
Costa L, Spatuzza M, D’Antoni S, et al. Activation of 5-HT7 serotonin receptors reverses metabotropic glutamate receptor-mediated synaptic plasticity in wild-type and Fmr1 knockout mice, a model of Fragile X syndrome. Biol Psychiatry 2012;72:924-933.
Tian M, Zeng Y, Hu Y, et al. 7, 8-Dihydroxyflavone induces synapse expression of AMPA GluA1 and ameliorates cognitive and spine abnormalities in a mouse model of fragile X syndrome. Neuropharmacology 2015;89:43-53.
Lauterborn JC, Rex CS, Kramar E, et al. Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome. J Neurosci 2007;27:10685-10694.
Wei H, Dobkin C, Sheikh AM, Malik M, Brown WT, Li X. The therapeutic effect of memantine through the stimulation of synapse formation and dendritic spine maturation in autism and fragile X syndrome. PLoS One 2012;7:e36981.
Ronesi JA, Collins KA, Hays SA, et al. Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat Neurosci 2012;15:431-440.
Pacey LK, Doss L, Cifelli C, der Kooy DV, Heximer SP, Hampson DR. Genetic deletion of regulator of G-protein signaling 4 (RGS4) rescues a subset of fragile X related phenotypes in the FMR1 knockout mouse. Mol Cell Neurosci 2011;46:563-572.
Swanger SA, Yao X, Gross C, Bassell GJ. Automated 4D analysis of dendritic spine morphology: applications to stimulus-induced spine remodeling and pharmacological rescue in a disease model. Mol Brain 2011;4:38.
Liu Z-H, Chuang D-M, Smith CB. Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int J Neuropsychopharmacol 2011;14:618-630.
Chen X, Sun W, Pan Y, et al. Lithium ameliorates open-field and elevated plus maze behaviors, and brain phospho-glycogen synthase kinase 3-beta expression in fragile X syndrome model mice. Neurosciences 2013;18:356-362.
Mines MA, Yuskaitis CJ, King MK, Beurel E, Jope RS. GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS One 2010;5:e9706.
Hayashi ML, Rao BS, Seo JS, et al. Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc Natl Acad Sci U S A 2007;104:11489-11494.
Dolan BM, Duron SG, Campbell DA, et al. Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc Natl Acad Sci U S A 2013;110:5671-5676.
Choi CH, Schoenfeld BP, Weisz ED, et al. PDE-4 inhibition rescues aberrant synaptic plasticity in Drosophila and mouse models of fragile X syndrome. J Neurosci 2015;35:396-408.
Boda B, Mendez P, Boury-Jamot B, Magara F, Muller D. Reversal of activity-mediated spine dynamics and learning impairment in a mouse model of fragile X syndrome. Eur J Neurosci 2014;39:1130-1137.
Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 2011;480:63-68.
Udagawa T, Farny NG, Jakovcevski M, et al. Genetic and acute CPEB1 depletion ameliorate fragile X pathophysiology. Nat Med 2013;19:1473-1477.
Westmark CJ, Westmark PR, O’Riordan KJ, et al. Reversal of fragile X phenotypes by manipulation of AbetaPP/Abeta levels in Fmr1KO mice. PLoS One 2011;6:e26549.
Sidhu H, Dansie LE, Hickmott PW, Ethell DW, Ethell IM. Genetic removal of matrix metalloproteinase 9 rescues the symptoms of fragile X syndrome in a mouse model. J Neurosci 2014;34:9867-9879.
Bilousova TV, Dansie L, Ngo M, et al. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet 2009;46:94-102.
Goebel-Goody SM, Wilson-Wallis ED, Royston S, Tagliatela SM, Naegele JR, Lombroso PJ. Genetic manipulation of STEP reverses behavioral abnormalities in a fragile X syndrome mouse model. Genes Brain Behav 2012;11:586-600.
Hebert B, Pietropaolo S, Meme S, et al. Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by a BKCa channel opener molecule. Orphanet J Rare Dis 2014;9:124.
Choi CH, McBride SM, Schoenfeld BP, et al. Age-dependent cognitive impairment in a Drosophila fragile X model and its pharmacological rescue. Biogerontology 2010;11:347-362.
Chang S, Bray SM, Li Z, et al. Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat Chem Biol 2008;4:256-263.
Gatto CL, Pereira D, Broadie K. GABAergic circuit dysfunction in the Drosophila fragile X syndrome model. Neurobiol Dis 2014;65:142-159.
Siller SS, Broadie K. Neural circuit architecture defects in a Drosophila model of fragile X syndrome are alleviated by minocycline treatment and genetic removal of matrix metalloproteinase. Dis Model Mech 2011;4:673-685.
Irwin SA, Patel B, Idupulapati M, et al. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet 2001;98:161-167.
Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A 2002;99:7746-7750.
Nosyreva ED, Huber KM. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol 2006;95:3291-3295.
Nowicki ST, Tassone F, Ono MY, et al. The Prader-Willi phenotype of fragile X syndrome. J Dev Behav Pediatr 2007;28:133-138.
Hagerman PJ, Stafstrom CE. Origins of epilepsy in fragile x syndrome. Epilepsy Curr 2009;9:108-112.
Hessl D, Berry-Kravis E, Cordeiro L, et al. Prepulse inhibition in fragile X syndrome: feasibility, reliability, and implications for treatment. Am J Med Genet B Neuropsychiatr Genet 2009;150B:545-553.
King MK, Jope RS. Lithium treatment alleviates impaired cognition in a mouse model of fragile X syndrome. Genes Brain Behav 2013;12:723-731.
Gandhi RM, Kogan CS, Messier C. 2-Methyl-6-(phenylethynyl) pyridine (MPEP) reverses maze learning and PSD-95 deficits in Fmr1 knock-out mice. Front Cell Neurosci 2014;8:70.
Bernardet M, Crusio WE. Fmr1 KO mice as a possible model of autistic features. ScientificWorldJournal 2006;6:1164-1176.
Kazdoba TM, Leach PT, Silverman JL, Crawley JN. Modeling fragile X syndrome in the Fmr1 knockout mouse. Intractable Rare Dis Res 2014;3:118-133.
Zhang YQ, Bailey AM, Matthies HJ, et al. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 2001;107:591-603.
McBride SM, Choi CH, Wang Y, et al. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron 2005;45:753-764.
Ng MC, Yang YL, Lu KT. Behavioral and synaptic circuit features in a zebrafish model of fragile X syndrome. PLoS One 2013;8:e51456.
Kim L, He L, Maaswinkel H, Zhu L, Sirotkin H, Weng W. Anxiety, hyperactivity and stereotypy in a zebrafish model of fragile X syndrome and autism spectrum disorder. Prog Neuropsychopharmacol Biol Psychiatry 2014;55:40-49.
Gross C, Berry-Kravis EM, Bassell GJ. Therapeutic strategies in fragile X syndrome: dysregulated mGluR signaling and beyond. Neuropsychopharmacology 2012;37:178-195.
Kim SH, Markham JA, Weiler IJ, Greenough WT. Aberrant early-phase ERK inactivation impedes neuronal function in fragile X syndrome. Proc Natl Acad Sci U S A 2008;105:4429-4434.
Weng N, Weiler IJ, Sumis A, Berry-Kravis E, Greenough WT. Early-phase ERK activation as a biomarker for metabolic status in fragile X syndrome. Am J Med Genet B Neuropsychiatr Genet 2008;147B:1253-1257.
Kovacs T, Bansagi B, Kelemen O, Keri S. Neuregulin 1-induced AKT and ERK phosphorylation in patients with fragile X syndrome (FXS) and intellectual disability associated with obstetric complications. J Mol Neurosci 2014;54:119-124.
Dziembowska M, Pretto DI, Janusz A, et al. High MMP-9 activity levels in fragile X syndrome are lowered by minocycline. Am J Med Genet A 2013;161A:1897-1903.
Erickson CA, Ray B, Maloney B, et al. Impact of acamprosate on plasma amyloid-β precursor protein in youth: A pilot analysis in fragile X syndrome-associated and idiopathic autism spectrum disorder suggests a pharmacodynamic protein marker. J Psychiatr Res 2014;59:220-228.
Erickson CA, Weng N, Weiler IJ, et al. Open-label riluzole in fragile X syndrome. Brain Res 2011;1380:264-270.
Berry-Kravis E, Sumis A, Hervey C, et al. Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J Dev Behav Pediatr 2008;29:293-302.
Irwin SA, Idupulapati M, Gilbert ME, et al. Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am J Med Genet 2002;111:140-146.
Scharf SH, Jaeschke G, Wettstein JG, Lindemann L. Metabotropic glutamate receptor 5 as drug target for fragile X syndrome. Curr Opin Pharmacol 2015;20C:124-134.
Cruz-Martin A, Crespo M, Portera-Cailliau C. Delayed stabilization of dendritic spines in fragile X mice. J Neurosci 2010;30:7793-7803.
Cruz-Martin A, Crespo M, Portera-Cailliau C. Glutamate induces the elongation of early dendritic protrusions via mGluRs in wild type mice, but not in fragile X mice. PLoS One 2012;7:e32446.
Smith LN, Jedynak JP, Fontenot MR, et al. Fragile X mental retardation protein regulates synaptic and behavioral plasticity to repeated cocaine administration. Neuron 2014;82:645-658.
Pan F, Aldridge GM, Greenough WT, Gan WB. Dendritic spine instability and insensitivity to modulation by sensory experience in a mouse model of fragile X syndrome. Proc Natl Acad Sci U S A 2010;107:17768-17773.
Berry-Kravis E, Krause SE, Block SS, et al. Effect of CX516, an AMPA-modulating compound, on cognition and behavior in fragile X syndrome: a controlled trial. J Child Adolesc Psychopharmacol 2006;16:525-540.
Berry-Kravis E, Sumis A, Hervey C, et al. Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J Dev Behav Pediatr 2008;29:293-302.
Berry-Kravis E, Hessl D, Coffey S, et al. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet 2009;46:266-271.
Kesler SR, Lightbody AA, Reiss AL. Cholinergic dysfunction in fragile X syndrome and potential intervention: a preliminary 1H MRS study. Am J Med Genet Part A 2009;149:403-407.
2015 [cited January 21, 2015]. Available from: www.clinicaltrials.gov.
Paribello C, Tao L, Folino A, et al. Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol 2010;10:91.
Leigh MJS, Nguyen DV, Mu Y, et al. A randomized double-blind, placebo-controlled trial of minocycline in children and adolescents with fragile x syndrome. J Dev Behav Pediatr 2013;34:147-155.
Jacquemont S, Curie A, des Portes V, et al. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med 2011;3:64ra1.
Berry-Kravis EM, Hessl D, Rathmell B, et al. Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci Transl Med 2012;4:152ra127.
Berry-Kravis E, Visootsak J, Hagerman R, et al. Arbaclofen in fragile X syndrome: results of phase 3 trials. Ann Neurol 2014;76:174.
Erickson CA, Wink LK, Ray B, et al. Impact of acamprosate on behavior and brain-derived neurotrophic factor: an open-label study in youth with fragile X syndrome. Psychopharmacology (Berl) 2013;228:75-84.
Mullard A. Fragile X disappointments upset autism ambitions. Nat Rev Drug Discov 2015;14:151-153.
Yan QJ, Asafo-Adjei PK, Arnold HM, Brown RE, Bauchwitz RP. A phenotypic and molecular characterization of the fmr1-tm1Cgr fragile X mouse. Genes Brain Behav 2004;3:337-359.
Oddi D, Subashi E, Middei S, et al. Early social enrichment rescues adult behavioral and brain abnormalities in a mouse model of fragile X syndrome. Neuropsychopharmacology 2015;40:1113-1122.
Paradee W, Melikian HE, Rasmussen DL, Kenneson A, Conn PJ, Warren ST. Fragile X mouse: strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience 1999;94:185-192.
Seese RR, Wang K, Yao YQ, Lynch G, Gall CM. Spaced training rescues memory and ERK1/2 signaling in fragile X syndrome model mice. Proc Natl Acad Sci U S A 2014;111:16907-16912.
Krueger DD, Osterweil EK, Chen SP, Tye LD, Bear MF. Cognitive dysfunction and prefrontal synaptic abnormalities in a mouse model of fragile X syndrome. Proc Natl Acad Sci U S A 2011;108:2587-2592.
Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci 2004;27:370-377.
Volk LJ, Pfeiffer BE, Gibson JR, Huber KM. Multiple Gq-coupled receptors converge on a common protein synthesis-dependent long-term depression that is affected in fragile X syndrome mental retardation. J Neurosci 2007;27:11624-11634.
Pacey LK, Doss L, Cifelli C, van der Kooy D, Heximer SP, Hampson DR. Genetic deletion of regulator of G-protein signaling 4 (RGS4) rescues a subset of fragile X related phenotypes in the FMR1 knockout mouse. Mol Cell Neurosci 2011;46:563-572.
Kelley DJ, Bhattacharyya A, Lahvis GP, Yin JC, Malter J, Davidson RJ. The cyclic AMP phenotype of fragile X and autism. Neurosci Biobehav Rev 2008;32:1533-1543.
Bolduc FV, Bell K, Cox H, Broadie KS, Tully T. Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nat Neurosci 2008;11:1143-1145.
Sharma A, Hoeffer CA, Takayasu Y, et al. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci 2010;30:694-702.
Spencer CM, Alekseyenko O, Hamilton SM, et al. Modifying behavioral phenotypes in Fmr1KO mice: genetic background differences reveal autistic-like responses. Autism Res 2011;4:40-56.
Engineer CT, Centanni TM, Im KW, Rahebi KC, Buell EP, Kilgard MP. Degraded speech sound processing in a rat model of fragile X syndrome. Brain Res 2014;1564:72-84.
Hamilton SM, Green JR, Veeraragavan S, et al. Fmr1 and Nlgn3 knockout rats: novel tools for investigating autism spectrum disorders. Behav Neurosci 2014;128:103-109.
Chailangkarn T, Acab A, Muotri AR. Modeling neurodevelopmental disorders using human neurons. Curr Opin Neurobiol 2012;22:785-790.
Liu J, Koscielska KA, Cao Z, et al. Signaling defects in iPSC-derived fragile X premutation neurons. Hum Mol Genet 2012;21:3795-3805.
Sheridan SD, Theriault KM, Reis SA, et al. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One 2011;6:e26203.
de Esch CE, Ghazvini M, Loos F, et al. Epigenetic characterization of the FMR1 promoter in induced pluripotent stem cells from human fibroblasts carrying an unmethylated full mutation. Stem Cell Rep 2014;3:548-555.
Berry-Kravis E. Mechanism-based treatments in neurodevelopmental disorders: fragile X syndrome. Pediatr Neurol 2014;50:297-302.
Berry-Kravis E, Knox A, Hervey C. Targeted treatments for fragile X syndrome. J Neurodev Disord 2011;3:193-210.
De Sonia A, Visootsak J, Smith M, et al. FXCRC analysis of arbaclofen responses in fragile X syndrome. In: Proceedings of the 14th International Fragile X Conference, Orange County, CA, 2004.
Berry-Kravis E, Hessl D, Abbeduto L, et al. Outcome measures for clinical trials in fragile X syndrome. J Dev Behav Pediatr 2013;34:508-522.
Jacquemont S, Berry-Kravis E, Hagerman R, et al. The challenges of clinical trials in fragile X syndrome. Psychopharmacology (Berl) 2014;231:1237-1250.
Iossifov I, Ronemus M, Levy D, et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012;74:285-299.
Steinberg J, Webber C. The roles of FMRP-regulated genes in autism spectrum disorder: single- and multiple-hit genetic etiologies. Am J Hum Genet 2013;93:825-839.
Waltes R, Duketis E, Knapp M, et al. Common variants in genes of the postsynaptic FMRP signalling pathway are risk factors for autism spectrum disorders. Hum Genet 2014;133:781-792.
Gross C, Raj N, Molinaro G, Allen Amanda G, Whyte Alonzo J, Gibson Jay R, et al. Selective Role of the Catalytic PI3K Subunit p110β in Impaired Higher Order Cognition in Fragile X Syndrome. Cell reports. 2015
Gross C, Chang C-W, Kelly Seth M, Bhattacharya A, McBride Sean MJ, Danielson Scott W, et al. Increased Expression of the PI3K Enhancer PIKE Mediates Deficits in Synaptic Plasticity and Behavior in Fragile X Syndrome. Cell reports. 2015
Acknowledgments
This work was supported by National Institutes of Health grants 1U54HD082013 (to GJB), 1R21MH105353 (to GJB and CG), and 1R21MH103748 (to CG), and an Autism Speaks Pilot Grant #8293 (to CG). We thank Dr. Tori Schaefer for helpful discussions.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 260 kb)
Rights and permissions
About this article

Cite this article
Gross, C., Hoffmann, A., Bassell, G.J. et al. Therapeutic Strategies in Fragile X Syndrome: From Bench to Bedside and Back. Neurotherapeutics 12, 584–608 (2015). https://doi.org/10.1007/s13311-015-0355-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-015-0355-9