
Abstract
Spinal muscular atrophies (SMAs) are a group of inherited disorders characterized by motor neuron loss in the spinal cord and lower brainstem, muscle weakness, and atrophy. The clinical and genetic phenotypes incorporate a wide spectrum that is differentiated based on age of onset, pattern of muscle involvement, and inheritance pattern. Over the past several years, rapid advances in genetic technology have accelerated the identification of causative genes and provided important advances in understanding the molecular and biological basis of SMA and insights into the selective vulnerability of the motor neuron. Common pathophysiological themes include defects in RNA metabolism and splicing, axonal transport, and motor neuron development and connectivity. Together these have revealed potential novel treatment strategies, and extensive efforts are being undertaken towards expedited therapeutics. While a number of promising therapies for SMA are emerging, defining therapeutic windows and developing sensitive and relevant biomarkers are critical to facilitate potential success in clinical trials. This review incorporates an overview of the clinical manifestations and genetics of SMA, and describes recent advances in the understanding of mechanisms of disease pathogenesis and development of novel treatment strategies.
Similar content being viewed by others

Introduction
The spinal muscular atrophies (SMAs) are a group of inherited disorders characterized by motor neuron loss in the spinal cord and lower brainstem, muscle weakness, and atrophy. The clinical phenotype incorporates a wide spectrum that is differentiated based on age of onset, pattern of muscle involvement, and inheritance pattern. Broad categories include proximal SMA and distal SMA (DSMA, also known as hereditary motor neuropathy or dHMN), demonstrating considerable genetic and clinical heterogeneity. SMA often refers to the most common form, caused by mutations of SMN1 [1], termed SMA5q or survival of motor neuron (SMN)-related SMA. SMA remains the leading genetic cause of infant death, and without a disease-modifying treatment.
Recently, there have been important advances in understanding the genetic and molecular basis of SMA. Next-generation sequencing technology has accelerated gene discovery, with 13 SMA genes identified since 2011. In total, 33 causative genes have been identified to date. Common pathophysiological themes include defects in RNA metabolism and splicing, axonal transport, and motor neuron development and connectivity.
There is currently great promise to develop a successful, disease-modifying treatment for SMN-related SMA, and extensive efforts are being undertaken towards this aim. Promising therapeutic strategies in development include small-molecule SMN enhancers, antisense oligonucleotides to correct SMN2 splicing, neuroprotectants, stem cell and gene therapies, and regulators of muscle function. This review will focus on recent genetic discoveries in SMA, cellular mechanisms underlying motor neuron degeneration, understanding disease progression, and initiatives to address “clinical trial readiness” and the development of novel treatment strategies.
Clinical Presentations and Genetics
Proximal SMA
SMN1-related SMA
The most common form of SMA is caused by homozygous disruption of SMN1 on chromosome 5q and results in insufficient levels of SMN protein in motor neurons. It is one of the most common autosomal recessive diseases, with an incidence of 1 in 6000–10,000 live births and a carrier frequency of 1 in 40–60 adults [2]. The disease typically presents in infancy or childhood, leading to severe physical disability. The weakness is usually symmetrical, more proximal than distal, the legs are more affected than the arms, and there is relative sparing of the diaphragm, and extraocular and facial muscles. Despite relative sparing of the diaphragm respiratory insufficiency is an important complication of SMA5q. Deep tendon reflexes are generally absent or diminished. There is a broad spectrum of clinical severity, with phenotypes divided into types 1–4 [3], determined principally by maximal motor milestone attained and age of onset. Infants with SMA type 1, or Werdnig–Hoffman disease, do not achieve independent sitting, with onset before 6 months of age, and respiratory failure usually leads to death within the first 2 years without respiratory support. SMA type 2 displays weakness before the age of 18 months. Patients achieve independent sitting, but are not able to stand or walk independently, and life expectancy is often into adulthood. Individuals with SMA type III (also known as Kugelberg–Welander disease) attain the ability to walk unaided and usually manifest after 18 months of age. There is marked variability in the clinical course, with some patients requiring wheelchair assistance in childhood and others walking in adulthood. Life expectancy is normal. SMA type 4 has onset in adulthood.
Non-SMN1-related SMA
Less than 5 % of infantile SMA is non-SMN-related, termed infantile SMA variants or SMA “plus” syndromes, in which additional clinical features may be evident, including arthrogryposis, abnormalities of extraocular movements, brainstem signs, or cardiomyopathy. These are characterized by congenital hypotonia, progressive postnatal weakness and areflexia with anterior horn cell degeneration. The differential diagnosis includes X-linked infantile SMA with arthrogryposis (XL-SMA) [4, 5], SMA due to mitochondrial dysfunction [6–8], SMA with pontocerebellar hypoplasia (SMA-PCH/PCH1) [9–13], and SMA with respiratory distress (SMARD) (Table 1) [14–16]. SMARD1 (or HMN type VI) is probably the second most commonly encountered pediatric from of SMA due to mutations in IGHMBP2 [14]. SMARD1 typically presents with very early respiratory failure due to diaphragmatic paralysis and weakness, which may be diffuse or predominantly upper limb and distal muscles. The phenotype of SMARD1 has recently broadened and includes mild weakness without severe signs of respiratory involvement [17].
While the majority of proximal SMA cases are related to recessive SMN1 mutations, the genetic heterogeneity of proximal SMA beyond infancy has also been recognized for several decades [2, 18, 19], with autosomal dominant SMA accounting for <2 % of cases. This includes lower extremity-predominant SMA types 1 and 2, caused by heterozygous mutations in DYNC1H1 and BICD2, respectively [20, 21]. Muscle weakness and atrophy predominantly affect the proximal lower limbs, although upper limb and distal lower limb involvement may occur [22, 23]. Some patients may demonstrate mild upper motor signs, foot deformities, or lower limb contractures [23, 24]. Sensation, bulbar, and cognitive functions are preserved. Lower extremity-predominant SMA may be static or have very slow progression throughout life. Late-onset autosomal dominant proximal SMA may also be associated with dominant mutations in vesicle-associated membrane-associated protein, protein B, allelic with amyotrophic lateral sclerosis (ALS) type 8 [25]. Although LMNA mutations are more commonly associated with muscle disease (particularly Emery–Dreifuss muscular dystrophy), the phenotypic spectrum also includes adult-onset autosomal dominant SMA followed by cardiomyopathy [26]. Significantly, the phenotype of tauopathies has recently broadened to include lower motor neuron disease, with autosomal dominant mutations in MAPT producing proximal weakness of the upper limbs, and respiratory insufficiency without dementia, pyramidal, or bulbar involvement [27].
Proximal SMA may also be associated with recessive mutations in PLEKHG5 [28]. Although classified as DSMA4, the clinical characteristics are proximal muscle weakness resulting in difficulty walking and climbing stairs with onset by the age of 3 years.
Importantly, the most common adult-onset SMA is bulbospinal muscular atrophy, also known as Kennedy’s disease, related to increased CAG repeats in the androgen receptor [29]. This X-linked recessive neurodegenerative disorder is characterized by widespread and prominent fasciculations, muscle weakness and atrophy, dysarthria, and dysphagia. In addition, patients may have endocrine manifestations, including gynecomastia, reduced fertility, and erectile dysfunction, related to androgen insensitivity, and diabetes mellitus.
DSMA or HMN
In contrast to proximal SMA, DSMA also known as dHMN or HMN, is characterized by a slowly progressive symmetrical and predominantly distal limb weakness and atrophy. Since 2001, mutations in 19 genes for dHMN have been identified, and it has emerged that they are clinically and genetically heterogeneous, with various phenotypes related to individual genes (Table 1). Harding originally classified dHMN into 7 categories, based on inheritance pattern, age of onset, severity, and distinguishing clinical features (Fig. 1) [30]. Autosomal dominant dHMN types I and II present with distal leg and subsequent distal arm weakness in childhood and adulthood, respectively, and are associated with mutations in HSPB1 and HSPB8 [31–33], HSPB3 [34], GARS [35], FBXO38 [36], and DYNC1H1 [37]. Linkage to 11q13 has been demonstrated in the autosomal recessive types III and IV (synonymous with DSMA type 3). HMN type V is distinguished by onset of weakness in the hand muscles, and may be associated with dominant mutations in BSCL2 [38], GARS [39], or REEP1 [40]. HMN type VII is distinguished by vocal cord paresis and may be due to dominant mutations in SLC5A7 (CHT) [41], or DCTN1 [42]. The genetic heterogeneity of HMN is further highlighted with the identification of recessive mutations in DNAJB2 (HSJ1) causing a nonspecific presentation of lower limb predominant slowly progressive weakness with young adult onset, known as DSMA type 5 [43].

Clinical features of spinal muscular atrophies (SMAs). (a) Infant with SMA type 1 with severe weakness, bell-shaped chest, and respiratory insufficiency. (b) Thoracolumbar radiograph of patient with SMA type 2 demonstrating thoracolumbar scoliosis. (c–g) A 36-year-old patient with distal hereditary motor neuropathy type 5C and mutation in BSCL2 demonstrating (c) distal wasting of the legs, (d–f) early and marked weakness and atrophy in the hands with finger contractures, and (g) pes cavus. (h) Chest radiograph of infant with SMA with respiratory distress type 1 demonstrating eventration of the right hemidiaphragm
The clinical spectrum of dHMN/SMA continues to expand and may also include congenital onset and X-linked or mitochondrial inheritance. There may be minor sensory involvement and/or pyramidal signs, and many of these disorders are allelic with axonal Charcot-Marie-Tooth disease (HSPB1, HSPB8, BSCL2, GARS, TRPV4), juvenile forms of ALS (SETX) [44, 45], and hereditary spastic paraplegia (BSCL2, HSPB1). In addition, autosomal recessive dHMN with pyramidal signs, linked to 9p21.1-p12, has been described originating from the Jerash region of Jordan (dHMN-J) [46]. X-linked recessive dHMN/DSMA may be associated with mutations in ATP7A (X-linked dHMN, allelic with Menkes disease) [47, 48] and LAS1L (SMARD2) [16]. Recently, mutations in TRPV4 have been associated with congenital distal SMA and scapuloperoneal SMA [49, 50]. Associated distinguishing clinical features may include vocal cord paralysis, scoliosis, contractures, or proximal upper limb weakness. Challenging the dogma of varied and multisystem involvement in mitochondrial disease, mutations in MT-ATP6 and MT-ATP8 have recently been shown to cause less severe phenotypes, including recurrent attacks of symmetrical limb paralysis and a later-onset distal motor neuropathy, mimicking periodic paralysis due to channelopathies [51, 52].
Diagnostic Evaluation and Management
For any patient presenting with clinical symptoms consistent with proximal SMA, testing for homozygous deletion of SMN1 should be undertaken. This test has 95 % sensitivity and nearly 100 % specificity in confirming the diagnosis of SMN-associated SMA or SMA5q [1, 53]. A negative SMN1 prompts review of clinical features, measurement of creatinine kinase, and neurophysiological studies with repetitive stimulation to help distinguish between motor neuron disease, myopathy, and neuromuscular junction disorders. Compound muscle action potentials (CMAPs) are typically reduced in SMA while motor conduction velocities and distal latencies are normal, or only modestly reduced when the CMAP amplitude is substantially reduced. The identification of sensory involvement is not expected in SMA and suggests Charcot-Marie-Tooth disease, wherein patients may have minimal sensory symptoms or signs. Electromyography shows chronic denervation with motor units of increased amplitude and duration, though in infants with severe SMA there may be normal-sized residual voluntary motor units.
In patients without homozygous SMN1 deletions and proximal SMA, measurement of SMN1 copy number will guide further investigations. A single SMN1 copy may suggest compound heterozygozity, with a deletion on 1 allele and a point mutation on the other, and SMN1 sequencing is indicated [53, 54]. When 2 SMN1 copies are demonstrated, then other motor neuron disorders such as SMARD, Kennedy’s disease, distal SMA, and ALS should be considered. Additional investigations including magnetic resonance imaging of brain and spinal cord, and metabolic and genetic studies may be undertaken. Diagnostic algorithms based on phenotype guide genetic testing in DSMA, although the yield currently remains low [55]. If neurophysiological studies reveal characteristic patterns associated with diseases in muscle, nerve, or neuromuscular junction then muscle or nerve biopsy and edrophonium test may be undertaken.
Practice guidelines for patients with SMN-related SMA have been established, with consensus on pulmonary, gastrointestinal, and orthopedics/rehabilitation to provide consistent management [56]. These incorporate a multidisciplinary and supportive approach. The medical practice and goals of therapy vary according to the patient’s level of function and philosophy of the patient and family. The appropriate level of interventional support to prolong life, particularly in SMA type 1, is controversial, and discussions with the family to explore and define potential quality of life and palliative care issues are important. Patients with SMA may have impaired cough and poor clearance of lower airway secretions, hypoventilation, and recurrent infections related to weakness. Respiratory management includes administration of routine immunizations, employing airway clearance techniques and cough assistance as necessary. Additionally, nocturnal noninvasive ventilation has been routinely introduced for sleep-disordered breathing in patients with SMA types 2 and 3. Inadequate oral intake and malnutrition are managed proactively to avoid potential complications. Treatment strategies may include nutritional supplementation, modifying food consistency, optimizing oral intake, positioning, and seating alterations. Contractures and significant scoliosis from muscle weakness are universal in SMA types 1 and 2, and may also occur in SMA type 3. Provision of equipment to assist with mobility, self-care and function, orthotics, and scoliosis surgery are important interventions.
Insights into SMN1-related SMA Pathogenesis
Almost 2 decades after the identification of SMN1 as a SMA-determining gene, substantial progress has been made in unraveling the molecular, cellular, and physiological processes of disease. Common pathophysiological themes underlying the various forms of SMA include defects in RNA metabolism and splicing, axonal transport, and motor neuron development and connectivity. Taken together, these themes resonate more generally amongst the motor neuron disease. Of further relevance, SMN is a genetic risk factor for ALS [57–59].
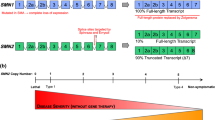
The development of the most common type of SMA relates to insufficient levels of SMN protein expression in motor neurons by homozygous deletion/mutation of SMN1 [1]. Humans possess a variable copy number of SMN2 (0–8 copies) from which SMN is solely derived in patients with SMA [53, 60]. SMN2 is almost identical to SMN1, except that a single translationally silent C to T nucleotide transition causes exon 7 skipping in the splicing of the majority of SMN2 transcripts, producing a truncated and unstable form of the SMN protein [61]. This nucleotide change disrupts an exon splice enhancer sequence and creates an exonic splicing silencer element that binds the splicing repressor heterogenous ribonuclear protein (hnRNP) A1 [62, 63]. A small fraction of SMN2 transcripts are spliced to include exon 7 and produce full-length SMN. The number of SMN2 copies and resultant amount of full-length SMN protein produced in patients with SMA (10–40 % of normal SMN protein levels) correlates with SMA disease severity. Consequently, SMN2 copy number broadly predicts SMA phenotype with the majority of SMA type 1 patients having 1 or 2 copies, type 2 patients usually have 3 copies, and most type 3 patients have 3 or 4 copies. Zero copies of SMN2 is embryonically lethal or associated with SMA type 0 [64–66].
The ubiquitous SMN protein has numerous and diverse functions in cells. The best characterized “housekeeping” function of SMN is in the nucleus and cytoplasm as part of a large macromolecular complex together with other proteins known as gemins [67]. The SMN complex is important in the production, recycling, and maintenance of small nuclear ribonucleoproteins of the Sm class, which are involved in the splicing of pre-mRNA into mRNA [68–70]. Recent studies have provided insights into the selective vulnerability of lower motor neurons to degeneration in SMA, including defective splicing of a subset of lower motor neuron-specific genes [71–73]. In addition, a negative feedback loop specific to motor neurons has been demonstrated, in which SMN depletion decreases exon 7 inclusion, further decreasing the splicing of its own mRNA [74]. SMN also has a role in axonal transport through its ability to regulate actin dynamics, forming a complex with β-actin [75, 76]. SMN also interacts with profilin (ALS18) to influence indirectly actin filament stability [77, 78]. Animal models with SMN depletion are deficient in β-actin mRNA and protein, and numerous disturbances of motor neuron axonal growth and development have been identified [75, 79, 80]. Significantly, plastin 3 has recently been identified as a protective modifier of SMA in females, coding for an actin-modifying protein [81]. Abnormalities in the neuromuscular junction also contribute to SMA pathogenesis, with rapid and progressive dysfunction occurring around the time of clinical disease onset [82–86].
While an important research focus is the direct effect of low SMN on motor neurons, it is not the sole site of pathology. Spinal motoneuronal function is modified by direct and indirect sensory afferent input, such as the spinal reflex circuit [87], and recent SMA animal models indicate that these interactions are important in SMA pathogenesis, shifting the pathophysiological paradigm to one of motor circuit dysfunction. Reduced SMN produced early abnormalities in proprioceptive synaptic input onto motor neurons that paralleled clinical alterations in animal motor behavior [88, 89]. In this model, these abnormalities occurred while motor neuron function was relatively maintained and this suggested deafferentation of motor neurons may be an early event in SMA pathogenesis. In turn, alterations in synaptic inputs induced functional changes in spinal motor neurons, with a compensatory hyperexcitability that may be related to alterations in the activity of ion channels at the cell membrane. Neurophysiological findings in patients with SMA provide support to these observations, with alterations in spinal H reflexes, spinal circuitry ,and ion channel function in motor nerves identified [90–92]. Of therapeutic relevance, increasing the excitability of motor circuits through the pharmacological inhibition of K+ channels ameliorated SMA in animal models [93]. Further evidence of the wider impact of SMN deficiency in disease pathogenesis comes from abnormalities in Schwann cells, skeletal muscle, heart, bone, pancreas, liver, hippocampus, thalamus, and the vascular system [94–101]. Controversy remains over the relative contribution of organ systems other than the motor neuron in SMN1-related SMA pathogenesis. It may be debated that the motor neuron is the only significant tissue, as specific elimination of SMN1 in motor neurons recapitulates all features of SMA and neuronal SMN restoration is necessary and sufficient for therapy [84, 102–104]. In spite of this, SMN upregulation within both the central and peripheral nervous systems may be necessary for optimal therapeutic outcomes [84, 94]. The detrimental effects of low SMN on neuromuscular circuitry raises a significant challenge of solving where SMN targeted therapy will be required and what will be best delivery mode in patients with SMA.
The concept that the pathogenesis of SMA is associated with ubiquitously expressed proteins involved in diverse cellular pathways is reinforced by mechanisms underlying neurodegeneration in non-SMN SMA. Mutations in SMA-related genes are associated with defects in DNA/RNA metabolism and protein synthesis, axonal guidance and transport, protein misfolding and degradation pathways, ion channel function, and energy production (Fig. 2, Table 2). These diverse functional pathways suggest that motor neuron degeneration may be a final common outcome with a number of upstream causes. Future studies incorporating next-generation sequencing and functional models will provide further insights into pathomechanisms underlying motor neuropathy. Furthermore, the overlap among various motor neuron diseases may make similar treatment strategies between these disorders possible. A recent mouse model of ALS demonstrated improvement of neuromuscular function and motor neuron survival with upregulation of SMN overexpression, encouraging further investigation of the potential SMN as a modifier of ALS [105].

Proposed mechanisms underlying spinal muscular atrophies (SMAs). Most of these genes associated with SMA encode for ubiquitously expressed proteins with diverse cellular functions: protein translation and synthesis (glycyl-tRNA synthetase, Bernardinelli-Seip congenital lipodystrophy 2), RNA/DNA metabolism (senataxin, immunoglobulin μ-binding protein 2), axonal guidance and trafficking [heat shock protein (HSP)27, dynactin 1, pleckstrin homology domain containing, family G (with RhoGef domain) member 5], cellular protection (HSP22, HSP27), and apoptosis (HSP27). snRNP = small nuclear ribonucleic particles; SMN = survival motor neuron
Challenges is SMN1-related SMA Therapeutics
These pathophysiological insights have revealed potential novel treatment strategies and extensive efforts are being made towards expedited therapeutics, with clinical trials already in progress. Promising therapeutic strategies in development include small-molecule SMN enhancers, antisense oligonucleotides to correct SMN2 splicing, neuroprotectants, stem cell and gene therapies, and regulators of muscle function (Sumner et al, this issue). To date, the 26 clinical trials that have investigated the effect of 12 potential treatments in patients with SMA have failed to show benefit owing to a number of factors, yet have enabled expertise in trial design to be developed [106, 107]. Experience has suggested that younger patients might be more responsive to treatment [107, 108]. Older patients may be refractive to treatment because they have been living with their disease for longer periods of time or because critical tissues have been irreversibly damaged. Defining therapeutic windows and developing sensitive and relevant biomarkers are critical to facilitating potential success in clinical trials.
There remains a lack of consensus over the extent to which SMA is progressively degenerative and over the rate of clinical progression in SMA. Natural history studies and understanding pathophysiology reveal part of the problem and suggest SMA manifests in 2 phases, with an initial rapid decline followed by the development of a relative plateau phase such that the rate of motor neuron loss in SMA is nonlinear [109, 110]. Some studies suggest gradually progressive degeneration throughout life [111–114], while others suggest that gradual declines are due to the effects of physical growth placing greater demands on the motor system [115]. In further contrast, others describe a static course over a period of up to 2.5 years [116, 117]. The slow rate of progression poses a major challenge to clinical trials in SMA because most trials need to be completed within 1–2 years. Even within SMA subtypes there is significant variation in severity and progression rate throughout the lifespan. Such heterogeneity among clinical trial participants may potentially obscure treatment effects. Further, the combination of clinical decline and plateau phases with simultaneous motor development, myelination, and physical growth complicates the assessment of motor function. In addition, secondary complications such as scoliosis and contractures further obscure the reliability of clinical observations.
Neurophysiological studies have provided further insights into the timing of SMA pathogenesis, with motor unit number estimation (MUNE) and CMAP used to track disease progression [109, 111, 118]. An age-dependent decline in MUNE and CMAP amplitude occurs in both SMA 1 and 2, and significant progressive denervation may be present before the onset of symptoms [111, 119]. The capacity for prolonged motor neuron survival has also been demonstrated by establishing relative stability of CMAP and MUNE values in cross-sectional and longitudinal studies [112, 120]; however, this may not always correlate with decrements in motor function over the same period. In further contrast, while CMAP may remain stable over time, MUNE has been shown to increase, suggesting new motor unit development as a compensatory process [118]. Axonal excitability studies also support a mixed pathology comprising features of axonal degeneration and regeneration [121].
While caution in translating data from preclinical studies into humans remains a challenge, these provide further support for an initial rapid onset of disease, or “up-front course”, coinciding with fetal and early postnatal neurodevelopment. Disruption of neuronal growth, axon branching, and neuromuscular connectivity was observed in zebrafish [122]. While developmental processes were maintained in mouse models, denervation was evident during embryogenesis and early in the postnatal course, around the time of disease onset [86, 123]. In SMA mouse models the temporal requirement for SMN protein encompasses the early postnatal course, with depletion of SMN in adults having minimal effect, coinciding with relative maturity of the neuromuscular junction [82, 124].
Taken together these studies suggest a spectrum in the rate of motor neuron denervation, survival, and potential compensation in SMA that remains a challenge in defining therapeutic windows that may prevent, stabilize, or reverse motor neuron degeneration in humans. It may be expected that early or presymptomatic therapies will provide optimal benefit, particularly in SMA type 1, such that advancing early diagnosis (newborn screening) will be essential. In milder types of SMA, a more extended period for intervention may also possible. National and international SMA registries are expected to accelerate the recruitment process of patients with SMA into new clinical trials, thereby facilitating potential early interventions. Clinical trial design (defining who, when, and where to treat, and expected outcomes) will be critical in developing future treatments. Furthermore, success depends on meeting the efficacy requirements of regulatory agencies, such that outcomes must be both realistic and meaningful.
The search for biomarkers in SMA is one of several research priorities. A number of clinical functional outcome measures have been used in SMA trials; however, current outcome measures may be unable to identify sensitively subtle changes to demonstrate a significant effect. In addition, while all of the scales demonstrate good reliability, the validity of measurement of motor performance in children with different severities of SMA is in question and the relationship to pathophysiology unclear. Ongoing Rasch analyses of multiple motor function scales are expected to create more robust scales [125]. MUNE, CMAP, electrical impedance myography, and axonal excitability have potential as alternative outcome measures that may also enable individual characterization of disease severity and reflect underlying pathophysiology. Aligned with therapeutic approaches designed to increase SMN levels, efforts to develop candidate biomarkers have also concentrated on measurement of SMN protein expression. While this can reliably be measured in peripheral blood and relates to SMA type, SMN protein levels do not predict severity of motor function, and it remains to be determined if this reflects what is happening in motor systems [126, 127]. Development of non-SMN molecular biomarkers using proteomic, metabolomics, and transcriptomic approaches holds promise for SMA, even though these measures may not be able to distinguish primary (initiating) and secondary (responsive) changes in gene expression. The recent BforSMA study identified a new set of 27 validated plasma protein SMA biomarkers significantly associated with motor function and other measures of SMA disease activity, and a commercial SMA-MAP biomarker panel was generated [128, 129]. Further studies will be required to investigate sensitivity to change with disease progression, and assess potential impact on clinical trial design.
Conclusion
SMA is a devastating genetic neuromuscular disorder, leading to significant infant and childhood mortality and morbidity. The most common mutation is homozygous disruption of SMN1 and causative genes implicate altered RNA processing, axonal transport, and protein degradation. Significant advances in patient care and knowledge of the genetics and biology of SMA over the last 2 decades have revealed promising strategies for therapeutics development, with clinical trials already in progress. Extensive efforts are being undertaken towards translating these to reach the ultimate goal of finding an effective treatment in the clinic. If there is hope of identifying a treatment for this disease, a further understanding of the site and timing of disease progression, and potential adaptations, in humans, as well as developing very sensitive and relevant biomarkers over this period of time, is required.
References
Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995;80:155-165.
Pearn J. Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J Med Genet 1978;15:409-413.
Russman BS. Spinal muscular atrophy: clinical classification and disease heterogeneity. J Child Neurol 2007;22:946-951.
Dressman D, Ahearn ME, Yariz KO, et al. X-linked infantile spinal muscular atrophy: clinical definition and molecular mapping. Genet Med 2007;9:52-60.
Ramser J, Ahearn ME, Lenski C, et al. Rare missense and synonymous variants in UBE1 are associated with X-linked infantile spinal muscular atrophy. Am J Hum Genet 2008;82:188-193.
Papadopoulou LC, Sue CM, Davidson MM, et al. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat Genet 1999;23:333-337.
Tarnopolsky MA, Bourgeois JM, Fu MH, et al. Novel SCO2 mutation (G1521A) presenting as a spinal muscular atrophy type I phenotype. Am J Med Genet A 2004;125A:310-314.
Salviati L, Sacconi S, Rasalan MM, et al. Cytochrome c oxidase deficiency due to a novel SCO2 mutation mimics Werdnig-Hoffmann disease. Arch Neurol 2002;59:862–865.
Barth PG. Pontocerebellar hypoplasias. An overview of a group of inherited neurodegenerative disorders with fetal onset. Brain Dev 1993;15:411–422.
Renbaum P, Kellerman E, Jaron R, et al. Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am J Hum Genet 2009;85:281-289.
Wan J, Yourshaw M, Mamsa H, et al. Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat Genet 2012;44:704-708.
Namavar Y, Barth PG, Kasher PR, et al. Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain 2011;134:143-156.
Simonati A, Cassandrini D, Bazan D, Santorelli FM. TSEN54 mutation in a child with pontocerebellar hypoplasia type 1. Acta Neuropathol 2011;121:671-673.
Grohmann K, Schuelke M, Diers A, et al. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat Genet 2001;29:75-77.
Grohmann K, Varon R, Stolz P, et al. Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Ann Neurol 2003;54:719-724.
Butterfield RJ, Stevenson TJ, Xing L, et al. Congenital lethal motor neuron disease with a novel defect in ribosome biogenesis. Neurology 2014;82:1322-1330.
Messina MF, Messina S, Gaeta M, et al. Infantile spinal muscular atrophy with respiratory distress type I (SMARD 1): an atypical phenotype and review of the literature. Eur J Paediatr Neurol 2012;16:90-94.
Hausmanowa-Petrusewicz I, Zaremba J, Borkowska J. Chronic proximal spinal muscular atrophy of childhood and adolescence: problems of classification and genetic counselling. J Med Genet 1985;22:350-353.
Rudnik-Schoneborn S, Wirth B, Zerres K. Evidence of autosomal dominant mutations in childhood-onset proximal spinal muscular atrophy. Am J Hum Genet 1994;55:112-119.
Harms MB, Ori-McKenney KM, Scoto M, et al. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology 2012;78:1714-1720.
Neveling K, Martinez-Carrera LA, Holker I, et al. Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy. Am J Hum Genet 2013;92:946-954.
Harms MB, Allred P, Gardner R, Jr., et al. Dominant spinal muscular atrophy with lower extremity predominance: linkage to 14q32. Neurology 2010;75:539-546.
Oates EC, Rossor AM, Hafezparast M, et al. Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia. Am J Hum Genet 2013;92:965-973.
Synofzik M, Martinez-Carrera LA, Lindig T, Schols L, Wirth B. Dominant spinal muscular atrophy due to BICD2: a novel mutation refines the phenotype. J Neurol Neurosurg Psychiatry 2014;85:590-592.
Nishimura AL, Mitne-Neto M, Silva HC, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 2004;75:822-831.
Rudnik-Schoneborn S, Botzenhart E, Eggermann T, et al. Mutations of the LMNA gene can mimic autosomal dominant proximal spinal muscular atrophy. Neurogenetics 2007;8:137-142.
Di Fonzo A, Ronchi D, Gallia F, et al. Lower motor neuron disease with respiratory failure caused by a novel MAPT mutation. Neurology 2014;82:1990-1998.
Maystadt I, Rezsohazy R, Barkats M, et al. The nuclear factor kappaB-activator gene PLEKHG5 is mutated in a form of autosomal recessive lower motor neuron disease with childhood onset. Am J Hum Genet 2007;81:67-76.
La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991;352:77-79.
Harding AE. Inherited neuronal atrophy and degeneration predominantly of lower motor neurons. In: Peripheral neuropathy. Dyck, PJ, Thomas, PK, Griffin, JW (eds). W. B. Saunders Company, Philadelphia, 1993, pp. 1051-1064.
Evgrafov OV, Mersiyanova I, Irobi J, et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet 2004;36:602-606.
Mandich P, Grandis M, Varese A, et al. Severe neuropathy after diphtheria-tetanus-pertussis vaccination in a child carrying a novel frame-shift mutation in the small heat-shock protein 27 gene. J Child Neurol 2010;25:107-109.
Irobi J, Van Impe K, Seeman P, et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat Genet 2004;36:597-601.
Kolb SJ, Snyder PJ, Poi EJ, et al. Mutant small heat shock protein B3 causes motor neuropathy: utility of a candidate gene approach. Neurology 2010;74:502-506.
Dubourg O, Azzedine H, Yaou RB, et al. The G526R glycyl-tRNA synthetase gene mutation in distal hereditary motor neuropathy type V. Neurology 2006;66:1721-1726.
Sumner Charlotte J, d’Ydewalle C, Wooley J, et al. A dominant mutation in FBXO38 causes distal spinal muscular atrophy with calf predominance. Am J Hum Genet 2013;93:976-983.
Weedon MN, Hastings R, Caswell R, et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet 2011;89:308-312.
Windpassinger C, Auer-Grumbach M, Irobi J, et al. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat Genet 2004;36:271-276.
Antonellis A, Ellsworth RE, Sambuughin N, et al. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet 2003;72:1293-1299.
Beetz C, Pieber Thomas R, Hertel N, et al. Exome sequencing identifies a REEP1 mutation involved in distal hereditary motor neuropathy type V. Am J Hum Geneti 2012;91:139-145.
Barwick KE, Wright J, Al-Turki S, et al. Defective presynaptic choline transport underlies hereditary motor neuropathy. Am J Hum Genet 2012;91:1103-1107.
Puls I, Jonnakuty C, LaMonte BH, et al. Mutant dynactin in motor neuron disease. Nat Genet 2003;33:455-456.
Blumen SC, Astord S, Robin V, et al. A rare recessive distal hereditary motor neuropathy with HSJ1 chaperone mutation. Ann Neurol 2012;71:509-519.
De Jonghe P, Auer-Grumbach M, Irobi J, et al. Autosomal dominant juvenile amyotrophic lateral sclerosis and distal hereditary motor neuronopathy with pyramidal tract signs: synonyms for the same disorder? Brain 2002;125:1320-1325.
Chen YZ, Bennett CL, Huynh HM, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 2004;74:1128-1135.
Christodoulou K, Zamba E, Tsingis M, et al. A novel form of distal hereditary motor neuronopathy maps to chromosome 9p21.1-p12. Ann Neurol 2000;48:877-884.
Takata RI, Speck Martins CE, Passosbueno MR, et al. A new locus for recessive distal spinal muscular atrophy at Xq13.1-q21. J Med Genet 2004;41:224-229.
Kennerson ML, Nicholson GA, Kaler SG et al. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am J Hum Genet 2010;86:343-352.
Auer-Grumbach M, Olschewski A, Papic L, et al. Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C. Nat Genet 2010;42:160-164.
Deng HX, Klein CJ, Yan J, et al. Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat Genet 2010;42:165-169.
Aure K, Dubourg O, Jardel C, et al. Episodic weakness due to mitochondrial DNA MT-ATP6/8 mutations. Neurology 2013;81:1810-1818.
Pitceathly RD, Murphy SM, Cottenie E, et al. Genetic dysfunction of MT-ATP6 causes axonal Charcot-Marie-Tooth disease. Neurology 2012;79:1145-1154.
Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum Mutat 2000;15:228-237.
Farrar MA, Johnston HM, Grattan-Smith P, Turner A, Kiernan MC. Spinal muscular atrophy: molecular mechanisms. Curr Mol Med 2009;9:851-862.
Rossor AM, Kalmar B, Greensmith L, Reilly MM. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry 2012;83:6-14.
Wang CH, Finkel RS, Bertini ES, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol 2007;22:1027-1049.
Moulard B, Salachas F, Chassande B, et al. Association between centromeric deletions of the SMN gene and sporadic adult-onset lower motor neuron disease. Ann Neurol 1998;43:640-644.
Veldink JH, Kalmijn S, Van der Hout AH, et al. SMN genotypes producing less SMN protein increase susceptibility to and severity of sporadic ALS. Neurology 2005;65:820-825.
Veldink JH, van den Berg LH, Cobben JM, et al. Homozygous deletion of the survival motor neuron 2 gene is a prognostic factor in sporadic ALS. Neurology 2001;56:749-752.
Mailman MD, Heinz JW, Papp AC, et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med 2002;4:20-26.
Monani UR, Lorson CL, Parsons DW, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 1999;8:1177-1183.
Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet 2002;30:377-384.
Kashima T, Rao N, David CJ, Manley JL. hnRNP A1 functions with specificity in repression of SMN2 exon 7 splicing. Hum Mol Genet 2007;16:3149-3159.
Lefebvre S, Burlet P, Liu Q, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 1997;16:265-269.
Feldkotter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 2002;70:358-368.
Prior TW, Swoboda KJ, Denman Scott H, Hejmanowski AQ. Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am J Med Genet 2004;130A:307-310.
Liu Q, Fischer U, Wang F, Dreyfuss G. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell 1997;90:1013-1021.
Fischer U, Liu Q, Dreyfuss G. The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell 1997;90:1023-1029.
Pellizzoni L, Yong J, Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science 2002;298:1775-1779.
Meister G, Buhler D, Pillai R, Lottspeich F, Fischer U. A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nat Cell Biol 2001;3:945-949.
Lotti F, Imlach WL, Saieva L, et al. An SMN-dependent U12 splicing event essential for motor circuit function. Cell 2012;151:440-454.
Baumer D, Lee S, Nicholson G, et al. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet 2009;5:e1000773.
Zhang Z, Lotti F, Dittmar K, et al. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 2008;133:585-600.
Ruggiu M, McGovern VL, Lotti F, et al. A role for SMN exon 7 splicing in the selective vulnerability of motor neurons in spinal muscular atrophy. Mol Cell Biol 2012;32:126-138.
Rossoll W, Jablonka S, Andreassi C, et al. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol 2003;163:801-812.
Zhang HL, Pan F, Hong D, Shenoy SM, Singer RH, Bassell GJ. Active transport of the survival motor neuron protein and the role of exon-7 in cytoplasmic localization. J Neurosci 2003;23:6627-6637.
Nolle A, Zeug A, van Bergeijk J, et al. The spinal muscular atrophy disease protein SMN is linked to the Rho-kinase pathway via profilin. Hum Mol Genet 2011;20:4865-4878.
Bowerman M, Anderson CL, Beauvais A, Boyl PP, Witke W, Kothary R. SMN, profilin IIa and plastin 3: a link between the deregulation of actin dynamics and SMA pathogenesis. Mol Cell Neurosci 2009;42:66-74.
McWhorter ML, Monani UR, Burghes AH, Beattie CE. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol 2003;162:919-931.
Jablonka S, Beck M, Lechner BD, Mayer C, Sendtner M. Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J Cell Biol 2007;179:139-149.
Oprea GE, Krober S, McWhorter ML, et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 2008;320:524-527.
Kariya S, Obis T, Garone C, et al. Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation. J Clin Inves 2014;124:785-800.
Kong L, Wang X, Choe DW, et al. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci 2009;29:842-851.
Martinez TL, Kong L, Wang X, et al. Survival motor neuron protein in motor neurons determines synaptic integrity in spinal muscular atrophy. J Neurosci 2012;32:8703-8715.
Kariya S, Park GH, Maeno-Hikichi Y, et al. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet 2008;17:2552-2569.
Murray LM, Lee S, Baumer D, Parson SH, Talbot K, Gillingwater TH. Pre-symptomatic development of lower motor neuron connectivity in a mouse model of severe spinal muscular atrophy. Hum Mol Genet 2010;19:420-433.
Pierrot-Deseilligny E, Burke D. The circuitry of the human spinal cord: spinal and corticospinal mechanisms of movement. Cambridge University Press, Cambridge, 2012.
Ling KK, Lin MY, Zingg B, Feng Z, Ko CP. Synaptic defects in the spinal and neuromuscular circuitry in a mouse model of spinal muscular atrophy. PLoS One 2010;5:e15457.
Mentis GZ, Blivis D, Liu W, et al. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 2011;69:453-467.
Iannaccone ST, Browne RH, Samaha FJ, Buncher CR. Prospective study of spinal muscular atrophy before age 6 years. DCN/SMA Group. Pediatr Neurol 1993;9:187-193.
Renault F, Raimbault J, Praud JP, Laget P. [Electromyographic study of 50 cases of Werdnig-Hoffmann disease]. Rev Electroencephalogr Neurophysiol Clin 1983;13:301-305 (in French).
Farrar MA, Vucic S, Johnston HM, Kiernan MC. Corticomotoneuronal integrity and adaptation in spinal muscular atrophy. Arch Neurol 2011;69:467-473.
Imlach WL, Beck ES, Choi BJ, Lotti F, Pellizzoni L, McCabe BD. SMN is required for sensory-motor circuit function in Drosophila. Cell 2012;151:427-439.
Hua Y, Sahashi K, Rigo F, et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011;478:123-126.
Menke LA, Poll-The BT, Clur SA, et al. Congenital heart defects in spinal muscular atrophy type I: a clinical report of two siblings and a review of the literature. Am J Med Genet A 2008;146A:740-744.
Mutsaers CA, Wishart TM, Lamont DJ, et al. Reversible molecular pathology of skeletal muscle in spinal muscular atrophy. Hum Mol Genet 2011;20:4334-4344.
Wishart TM, Huang JP, Murray LM, et al. SMN deficiency disrupts brain development in a mouse model of severe spinal muscular atrophy. Hum Mol Genet 2010;19:4216-4228.
Bowerman M, Swoboda KJ, Michalski JP, et al. Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann Neurol 2012;72:256-268.
Vitte JM, Davoult B, Roblot N, et al. Deletion of murine Smn exon 7 directed to liver leads to severe defect of liver development associated with iron overload. Am J Pathol 2004;165:1731-1741.
Shanmugarajan S, Tsuruga E, Swoboda KJ, Maria BL, Ries WL, Reddy SV. Bone loss in survival motor neuron (Smn(-/-) SMN2) genetic mouse model of spinal muscular atrophy. J Pathol 2009;219:52-60.
Shanmugarajan S, Swoboda KJ, Iannaccone ST, Ries WL, Maria BL, Reddy SV. Congenital bone fractures in spinal muscular atrophy: functional role for SMN protein in bone remodeling. J Child Neurol 2007;22:967-973.
Gogliotti RG, Quinlan KA, Barlow CB, Heier CR, Heckman CJ, Didonato CJ. Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory-motor defects are a consequence, not a cause, of motor neuron dysfunction. J Neurosci 2012;32:3818-3829.
Park GH, Maeno-Hikichi Y, Awano T, Landmesser LT, Monani UR. Reduced survival of motor neuron (SMN) protein in motor neuronal progenitors functions cell autonomously to cause spinal muscular atrophy in model mice expressing the human centromeric (SMN2) gene. J Neurosci 2010;30:12005-12019.
Gavrilina TO, McGovern VL, Workman E, et al. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum Mol Genet 2008;17:1063-1075.
Turner BJ, Alfazema N, Sheean RK et al. Overexpression of survival motor neuron improves neuromuscular function and motor neuron survival in mutant SOD1 mice. Neurobiol Aging 2014;35:906-915.
Bosboom WM, Vrancken AF, van den Berg LH, Wokke JH, Iannaccone ST. Drug treatment for spinal muscular atrophy type I. Cochrane Database Syst Rev 2009: CD006281.
Wadman RI, Bosboom WM, van der Pol WL, et al. Drug treatment for spinal muscular atrophy types II and III. Cochrane Database Syst Rev 2012;4:CD006282.
Swoboda KJ, Scott CB, Crawford TO, et al. SMA CARNI-VAL trial part I: double-blind, randomized, placebo-controlled trial of L-carnitine and valproic acid in spinal muscular atrophy. PLoS One 2010;5:e12140.
Farrar MA, Vucic S, Johnston HM, du Sart D, Kiernan MC. Pathophysiological insights derived by natural history and motor function of spinal muscular atrophy. J Pediatr 2013;162:155-159.
Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis 1996;3:97-110.
Swoboda KJ, Prior TW, Scott CB, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol 2005;57:704-712.
Kaufmann P, McDermott MP, Darras BT, et al. Prospective cohort study of spinal muscular atrophy types 2 and 3. Neurology 2012;79:1889-1897.
Merlini L, Bertini E, Minetti C, et al. Motor function-muscle strength relationship in spinal muscular atrophy. Muscle Nerve 2004;29:548-552.
Vuillerot C, Payan C, Iwaz J, Ecochard R, Berard C. Responsiveness of the motor function measure in patients with spinal muscular atrophy. Arch Phys Med Rehabil 2013;94:1555-1561.
Dunaway S, Montes J, Ryan PA, Montgomery M, Sproule DM, De Vivo DC. Spinal muscular atrophy type III: Trying to understand subtle functional change over time: A case report. J Child Neurol 2012;27:779-785.
Kaufmann P, McDermott MP, Darras BT, et al. Observational study of spinal muscular atrophy type 2 and 3: functional outcomes over 1 year. Arch Neurol 2011;68:779-786.
Piepers S, van den Berg LH, Brugman F, et al. A natural history study of late onset spinal muscular atrophy types 3b and 4. J Neurol 2008;255:1400-1404.
Kang PB, Gooch CL, McDermott MP, et al. The motor neuron response to SMN1 deficiency in spinal muscular atrophy. Muscle Nerve 2014;49:636-644.
Bromberg MB, Swoboda KJ, Lawson VH. Counting motor units in chronic motor neuropathies. Exp Neurol 2003;184:53-57.
Bromberg MB, Swoboda KJ, Lawson VH. Counting motor units in chronic motor neuropathies. Exp Neurol 2003;184(Suppl. 1):S53-S57.
Farrar MA, Vucic S, Lin CS, et al. Dysfunction of axonal membrane conductances in adolescents and young adults with spinal muscular atrophy. Brain 2011;134:3185-3197.
Hao le T, Duy PQ, Jontes JD, Wolman M, Granato M, Beattie CE. Temporal requirement for SMN in motoneuron development. Hum Mol Genet 2013;22:2612-2625.
McGovern VL, Gavrilina TO, Beattie CE, Burghes AH. Embryonic motor axon development in the severe SMA mouse. Hum Mol Genet 2008;17:2900-2909.
Le TT, McGovern VL, Alwine IE, et al. Temporal requirement for high SMN expression in SMA mice. Hum Mol Genet 2011;20:3578-3591.
Cano SJ, Mayhew A, Glanzman AM, et al. Rasch analysis of clinical outcome measures in spinal muscular atrophy. Muscle Nerve 2014;49:422-430.
Sumner CJ, Kolb SJ, Harmison GG, et al. SMN mRNA and protein levels in peripheral blood: biomarkers for SMA clinical trials. Neurology 2006;66:1067-1073.
Crawford TO, Paushkin SV, Kobayashi DT, et al. Evaluation of SMN protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PLoS One 2012;7:e33572.
Finkel RS, Crawford TO, Swoboda KJ, et al. Candidate proteins, metabolites and transcripts in the Biomarkers for Spinal Muscular Atrophy (BforSMA) clinical study. PLoS One 2012;7:e35462.
Kobayashi DT, Shi J, Stephen L, et al. SMA-MAP: a plasma protein panel for spinal muscular atrophy. PLoS One 2013;8:e60113.
Acknowledgments
This work was supported by funding to Forefront, a collaborative research group dedicated to the study of motor neuron disease, from the National Health and Medical Research Council of Australia program grant (#1037746), and grant support from the Thyne Reid Foundation.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 269 kb)
Rights and permissions
About this article

Cite this article
Farrar, M.A., Kiernan, M.C. The Genetics of Spinal Muscular Atrophy: Progress and Challenges. Neurotherapeutics 12, 290–302 (2015). https://doi.org/10.1007/s13311-014-0314-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-014-0314-x