
Abstract
Tree age affects wood formation and yield. However, the underlying mechanisms are poorly understood, particularly at the molecular level. In this study, we investigated the transcriptomic changes of the uppermost main stems of Larix kaempferi in an entire rotation period using the RNA-Seq method. In total, ∼151 million reads were obtained from the stems of 1-, 2-, 5-, 10-, 25-, and 50-year-old L. kaempferi trees. Combining these with the published Illumina sequencing reads, 299,637 assembled transcripts were generated, of which 161,232 were annotated. Time series expression profiling identified 12,927 transcripts as differentially expressed genes (DEGs); function enrichment analysis of these DEGs showed that 459 gene ontology terms in the biological process category were enriched. These terms were associated with the processes of wood formation, such as cell differentiation, growth and death, and its hormonal regulation. Based on the expression patterns of L. kaempferi homologues of genes associated with ethylene, calcium, and cell wall expansion and synthesis, the regulatory network of tracheid growth was outlined. Altogether, the comparative transcriptomic analysis reported here demonstrated the molecular aspects of aging effects on L. kaempferi wood formation. The identification of genes involved in the regulatory network of tracheid growth provides a means of investigating the regulation of wood formation in gymnosperm trees and also offers potential targets for genetic manipulation to improve the properties of xylem fibers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The seasonal activity of meristem cells in the shoot apex and stem results in the growth in height and girth of a tree. The growth rate changes with age (Lai et al. 2014), indicating that the activity of meristem cells is affected by aging. The growth and development of a tree involve all the activities of a cell, such as division, expansion, cell wall formation, metabolic processes, responses to stimuli, and death, some of which are known to be regulated by aging. For example, cambium cell division in spring restarts earlier in younger than in older trees (Begum et al. 2010; Li et al. 2013b, 2016; Mellerowicz et al. 1995; Rossi et al. 2008), and the sensitivity of cambium cells to auxin declines as a tree ages (Little and Sundberg 1991; Savidge 1983). Notably, cell length and diameter change radially from the pith to the bark and axially from the base to the crown along the main stem in Picea glauca (Mvolo et al. 2015; Yemele et al. 2015) and other plant species (Anfodillo et al. 2006; Olson et al. 2014), suggesting that the process of cell growth changes with tree age or height. Indeed, an increase in the duration of the cell expansion phase has been detected from the crown to the base along the main stem in Picea abies (Anfodillo et al. 2012). However, information about the molecular basis of aging or size effects on meristem cell activity and cell growth is limited.
A linear growth mode has been proposed based on the central roles of ethylene in cotton fiber growth, where calcium activates ethylene biosynthesis and ethylene positively regulates cotton fiber growth (Qin and Zhu 2011). More information about the molecular mechanisms by which ethylene controls wood formation has been reported (Andersson-Gunneras et al. 2003; Love et al. 2009; Vahala et al. 2013). These data indicate that the linear cell growth mode may be common to cotton fibers and xylem cells and facilitate the study of the regulatory network of xylem cell growth. In view of the importance of meristem cell activity and cell growth for wood yield and quality, investigating how age affects the processes of wood formation in trees is of great theoretical and practical importance.
The molecular aspects of the effects of age on wood formation in trees have been studied, and different classes of genes controlling wood formation have been revealed (Li et al. 2010, 2011; Xu et al. 2016; Zhang et al. 2016). Especially, using cryosectioning and RNA-Seq methods, Xu et al. (2016) compared the transcriptome of vascular cambium tissue in Cunninghamia lanceolata at juvenile, transitional, and mature stages to study the effects of age on cambium activity. Within-tree transcriptome and proteome profiling in wood-forming tissues based on changes of cell size and other wood properties have also been studied (Cato et al. 2006; Garces et al. 2014; Kumar et al. 2009; Lorenz and Dean 2002; Paiva et al. 2008; Ranik et al. 2006). However, the regulatory network of age effects on the processes of wood formation remains poorly understood, and systematic investigation at the genome level is still needed.
In a tree’s lifetime, it receives diverse external and endogenous stimuli, and its responses to these stimuli are affected by age. For example, cambium cell division in locally heated portions of the Cryptomeria japonica stem restarts earlier in younger than in older trees (Begum et al. 2010), indicating that age affects the responses of the lateral meristem to temperature signals. Also, age-dependent responses to environmental temperature have been found in Cedrus atlantica, where the negative effects of warming on growth are stronger in older than in younger trees (Linares et al. 2013). In Larix gmelinii, the age-dependent responses of radial growth to climate have also been revealed (Wang et al. 2009). These results are important, considering the effects of increasing temperature on the production of forest plantation in the context of global change. However, the molecular and regulatory mechanisms underlying these ecological and physiological events remain obscure. As for endogenous stimuli, hormones play important roles in tree growth and development, and some appear to be especially involved in aging because changes in their concentrations are associated with developmental stages (Wendling et al. 2014). While there is ample knowledge available about the hormonal regulation of wood formation (Sorce et al. 2013; Ye and Zhong 2015; Zhang et al. 2014), information about how this is affected by aging is still limited.
In studies of the molecular mechanisms of tree aging, many age-related genes have been identified (Busov et al. 2004; Carlsbecker et al. 2004; Diego et al. 2004; Fernández-Ocaña et al. 2010; García-López et al. 2014; Hutchison et al. 1990; Li et al. 2010, 2011; Xu et al. 2016; Zhang et al. 2016), such as miR156 (Wang et al. 2011b) and Flowering Locus T (Böhlenius et al. 2006; Hsu et al. 2006, 2011), and some have been used to change the agronomic traits of plants (Flachowsky et al. 2012; Kotoda et al. 2006; Li et al. 2013a; Yamagishi et al. 2014). Larix kaempferi is a forest tree of important ecological and economic value, widely grown in the Northern Hemisphere, but its genome has not been sequenced. The availability of transcriptome data for L. kaempferi would enable the global analysis of transcriptome reprogramming during aging and wood formation (Li et al. 2014b; Zhang et al. 2012). Here, comparative transcriptomic analysis of the uppermost main stems of 1-, 2-, 5-, 10-, 25-, and 50-year-old L. kaempferi trees was performed to (1) depict the molecular aspects of aging effects on wood formation and (2) propose the regulatory network of tracheid growth based on the expression patterns of genes involved in the linear cell growth mode (Qin and Zhu 2011).
Materials and methods
Sample preparation and transcriptome sequencing
The uppermost main stems produced in the current year were collected from 1-, 2-, 5-, 10-, 25-, and 50-year-old L. kaempferi trees in July 2011. The trees were located in Dagujia seed orchard (42°22′ N, 124°51′ E), Liaoning Province, in northeast China, and from young (1-, 2-, 5-, and 10-year-old), middle-aged (25-year-old), and mature (50-year-old) stands. After removal of branches and needles, the stems from at least three trees from each age category were pooled, frozen in liquid nitrogen, and stored at −80°C until RNA extraction. At the same time, small blocks of vascular tissue were excised from the bottom of each stem and fixed in formalin–alcohol–acetic acid for anatomical observation. The same RNA samples and methods were used for transcriptome library construction and sequencing for each age category as in our previous study (Li et al. 2014b).
Anatomical observation and statistical analysis of secondary vascular tissue
Anatomical examination and statistical analysis were conducted to assess cambium activity and wood formation. The small blocks fixed in formalin–alcohol–acetic acid were dehydrated in an alcohol series and embedded in Spurr’s resin (SPI, USA). Cross-sections 5 μm thick were cut on a microtome (Leitz 1512, Wetzlar, Germany), stained with Toluidine Blue O, and observed under an Axioskop 2 Plus microscope (Zeiss, Gottingen, Germany) equipped with a computer-assisted digital camera. One section from each stem was used for analysis. Fifteen radial files per section were measured. Radial cell layers in the cambium region, and differentiating xylem and phloem, were counted to evaluate cambium activity and wood formation. In addition, the basal diameter and the width of mature xylem were measured. Data are shown as the mean ± SD. Statistical analysis was performed with SPSS 16.0 using ANOVA.
Transcriptome assembly and annotation
The raw Illumina RNA sequencing reads were preprocessed by discarding reads with adaptors, >5% unknown nucleotides, low-quality (quality score <20), or <20 bp. Combining them with the Illumina RNA sequencing reads obtained in our previous study (Li et al. 2014b), we performed de novo assembly using Trinity software (Grabherr et al. 2011) using the default parameters.
To identify the L. kaempferi protein-coding genes, blastx (the basic local alignment tool for searching protein databases using a translated nucleotide query; Altschul et al. 1990) was used to search our assembled sequences against four sets of protein sequences from Arabidopsis thaliana (http://plants.ensembl.org/Arabidopsis_thaliana/Info/Index), P. abies (http://congenie.org/), Populus trichocarpa (http://www.phytozome.net/poplar.php), and Vitis vinifera (http://plants.ensembl.org/Vitis_vinifera/Info/Index/) with an e value of 1e−5. Then, we assigned the Gene Ontology (GO) terms associated with the top hits in the four protein databases to the annotated transcripts.
Identification of DEGs and GO enrichment analysis
Six sets of sequencing reads were mapped to the assembled reference transcripts using Bowtie (Langmead and Salzberg 2012). RNA-Seq by Expectation Maximization (Li and Dewey 2011), an accurate method of transcript quantification from RNA-Seq data, was used to estimate the transcript abundance. The expression of genes was normalized with edgeR (empirical analysis of digital gene expression data in R; Nikolayeva and Robinson 2014). Fragments per kilobase of transcript per million fragments was used to measure the normalized expression value.
Five pairwise comparisons were performed in chronological order to identify the differentially expressed genes (DEGs): (I) 2 vs. 1 year old; (II) 5 vs. 2 years old; (III) 10 vs. 5 years old; (IV) 25 vs. 10 years old; and (V) 50 vs. 25 years old. After pairwise comparison, the DEGs were obtained with stringent cutoffs: a false discovery rate (FDR)-corrected P value cutoff of 0.001 and a minimum expression fold change of 4. The FDR correction is designed to control the expected proportion of incorrectly rejected null hypotheses and is used in multiple-hypothesis testing to reduce type 1 errors.
Genes expressed in at least four of the age categories and with an average normalized expression value >0.5 were selected for correlation analysis. The Pearson correlation coefficient (PCC) between the expression patterns of the DEGs and one homologue of ethylene-responsive transcription factor 1A (LaERF1A) was analyzed, and a PCC value ≥0.8 was considered to indicate correlation and have the same expression pattern as LaERF1A.
In each comparison, to identify the biological processes in which the DEGs participated, GO enrichment analysis was performed separately for the upregulated and downregulated DEGs. Using all the genes as the background, we identified significantly enriched biological processes in one specific group of DEGs with GOatools (https://github.com/tanghaibao/goatools). GOatools uses Fisher’s exact test to identify significant GO terms in one dataset compared with the background. Then, the P value was corrected for multiple testing with the Bonferroni procedure (Hochberg 1988). The biological processes with corrected P values ≤0.05 were considered to be enriched in the group.
Quantitative RT-PCR
To confirm the expression patterns of DEGs, we used the same materials as those in RNA-Seq to perform quantitative reverse transcription polymerase chain reaction (qRT-PCR) as previously described (Li et al. 2014c). We randomly selected eight DEGs for qRT-PCR analysis. The PCC between the expression patterns from RNA-Seq and qRT-PCR was calculated, and a value ≥0.8 was considered to indicate correlation. The primers for qRT-PCR are listed in Supplementary Table S4.
Results
Wood formation in L. kaempferi at six age categories
To determine the effects of age on cambium activity and wood production, we compared wood formation in the uppermost main stems of L. kaempferi at six age categories. Anatomical examination and statistical analysis showed that xylem width and radial cell layers in the cambium region and differentiating xylem and phloem increased before 10 years of age, and then decreased (P < 0.05; Fig. S1); the same pattern was shown in the basal diameter of the sampled stems (P < 0.05; Fig. 1b). In addition, more expanding cells were observed in sections from 5- and 10-year-old trees, in which cell walls were thickening (Fig. 1a).

Overview of transcriptome analysis of Larix kaempferi aging. a Photomicrographs of cambium regions in cross-section in the active stage of 1-, 2-, 5-, 10-, 25-, and 50-year-old trees. b, c Statistical analysis of basal diameter of the uppermost main stems produced in the current year and cell layers in cambium regions and differentiating xylem and phloem during aging. d Analytical flowchart of RNA-Seq analysis. Cell layers = cambium cell layers + recently formed xylem cell layers + recently formed phloem cell layers. The P value was calculated between samples in chronological order. Double-headed arrows indicate cell layers to be counted. Ca cambium region, Ph phloem, Xy xylem. Scale bar, 50 μm
Transcriptome assembly and annotation
A total of 151,413,654 reads were produced from the six age categories (Table 1), with a 101-bp read length. Together with the published Illumina RNA sequencing reads from a pooled sample of L. kaempferi (Li et al. 2014b), all reads were assembled into 299,637 transcripts (Table 2). Then, the six sets of reads were aligned to the assembled reference transcripts, and the mapping ratios ranged from 80.6 to 86.7% with an average of 85.1% (Table 1). All RNA-Seq data in this study have been deposited in the NCBI SRA database (Table 1).
The 299,637 assembled transcripts were first annotated with blastx against the proteins of four species: A. thaliana, P. abies, P. trichocarpa, and V. vinifera (e value < 1e−5). Altogether, 161,232 (53.8%) of the 299,637 transcripts had significant matches, at least one hit in these four species, and the number of the matched transcripts to P. abies was more than that to V. vinifera, P. trichocarpa, and A. thaliana (Table 2). Then, GO annotation was performed for the 161,232 annotated transcripts in terms of “biological process,” “molecular function,” and “cellular component” (Table 2).
GO enrichment analysis of DEGs
To identify DEGs, five pairwise comparisons were performed: (I) 2 vs. 1 year old; (II) 5 vs. 2 years old; (III) 10 vs. 5 years old; (IV) 25 vs. 10 years old; and (V) 50 vs. 25 years old. In total, 12,927 transcripts were differentially expressed, among which 11,926 were annotated and used for further analysis. The largest number of DEGs (5692) was found in group V, while the fewest (2917) were identified in group IV (Fig. 2a).

Numbers of differentially expressed genes (DEGs) during Larix kaempferi aging (a) and Gene Ontology (GO) terms after enrichment analysis of DEGs (b)
To reveal the biological processes involved in wood formation during tree aging, we performed GO enrichment analysis of the DEGs. In total, 459 GO terms in the “biological process” category were enriched in the five groups of DEGs. The greatest number of GO terms (347) was enriched in group V, while the fewest (94) were enriched in group IV (Fig. 2b). In groups II, III, and V, the most enriched GO terms were from downregulated genes, with 73 (64.04%), 106 (100%), and 344 (99.14%) terms, respectively. However, in groups I and IV, the enriched GO terms of the upregulated genes accounted for 136 (99.27%) and 85 (90.43%) terms, respectively (Fig. 2b). Notably, in group V, the downregulated genes accounted for 30.2% of DEGs (3910/12,927), from which the enriched GO terms accounted for 74.9% (344/459).
Based on the annotations of 459 enriched GO terms, many terms related to the different processes of wood formation were found, such as “cell growth,” “cell death,” “cell morphogenesis,” “cell differentiation,” and “plant-type cell wall organization or biogenesis” (Fig. 3 and Table S2). As to hormonal control of wood formation, GO terms related to transport, response, biosynthetic process, metabolic process, regulation or signaling pathway of auxin, abscisic acid, ethylene, salicylic acid, jasmonic acid, gibberellin, brassinosteroid, and karrikin were found (Fig. 4 and Table S2).

Distribution of Gene Ontology (GO) terms associated with wood formation after enrichment analysis of DEGs during Larix kaempferi aging. The enriched GO terms are highlighted in pink and blue for up- and downregulation, respectively. Five pairwise comparisons: (I) 2- vs. 1-year, (II) 5- vs. 2-year, (III) 10- vs. 5-year, (IV) 25- vs. 10-year, and (V) 50- vs. 25-year-old trees

Distribution of Gene Ontology (GO) terms associated with hormone regulation of wood formation after enrichment analysis of DEGs during Larix kaempferi aging. The enriched GO terms are highlighted in pink and blue for up- and downregulation, respectively. Five pairwise comparisons: (I) 2- vs. 1-year, (II) 5- vs. 2-year, (III) 10- vs. 5-year, (IV) 25- vs. 10-year, and (V) 50- vs. 25-year-old trees
Expression patterns of genes related to cell growth
To study the regulatory network of tracheid growth, we analyzed the expression patterns of L. kaempferi homologues of genes associated with ethylene, calcium, and cell wall expansion and synthesis. Among 12,927 DEGs, 138 transcripts were annotated as ethylene-responsive transcription factors (ERFs) and 27 as dehydration-responsive element-binding proteins (DREBs; Table S3). Here, we present the expression pattern of one ERF homologue in L. kaempferi (LaERF1A); it had higher transcript levels than the other 164 APETALA2/ERF (AP2/ERF) superfamily members. LaERF1A transcripts were expressed strongly at 25 years, weakly at 50 years, and they increased from 2 to 10 years (Fig. 5a). In total, 145 members of the AP2/ERF superfamily were selected for expression pattern analysis, and 135 had the same expression patterns as LaERF1A. In addition, we detected the expression of 1-aminocyclopropane-1-carboxylic acid (ACC) synthase (ACS) homologue in L. kaempferi (LaACS) and found that LaACS had the same expression pattern as LaERF1A (Fig. 5a).

a–e Expression patterns of genes in relation to the linear cell growth mode determined by RNA-Seq. The expression pattern of a DEG with the highest average expression value was shown
In total, 100 transcripts were calcium-related (Table S3), of which 78 were used for expression pattern analysis. We found that 35 had the same expression pattern as LaERF1A, and they were all L. kaempferi homologues of calcium-binding proteins (LaCaM, LaCML, and LaKIC; Fig. 5b).
Among 12,927 DEGs, 18 transcripts were annotated as expansins and four as extensins (Table S3), and the expression analysis showed that two expansins (LaEXLA1 and LaEXPB17) and all four extensins (LaLRX3) had the same expression pattern as LaERF1A (Fig. 5c).
We found many genes coding for key enzymes involved in the synthesis of cellulose and hemicellulose. In total, 20 transcripts were annotated as cellulose synthases and 47 as xyloglucan endotransglucosylases/hydrolases, including 15 brassinosteroid-regulated proteins (BRU1; Table S3); among these, two cellulose synthase genes (LaCSLD3) and 14 xyloglucan endotransglucosylase/hydrolase genes (LaXTH5 and LaBRU1) had the same expression patterns as LaERF1A (Fig. 5d).
In total, 26, 23, 10, and 4 transcripts were annotated as MYB, WRKY, Trihelix, and GATA transcription factors from DEGs, respectively (Table S3), and five MYB, nine WRKY, two Trihelix, and three GATA transcription factor members had the same expression patterns as LaERF1A (Fig. 5e).
Among 19 DEGs related to cell growth (Fig. 5), eight were selected for qRT-PCR analysis (Fig. 6). The results showed a close correlation in the expression patterns determined by RNA-Seq and qRT-PCR (PCC ≥ 0.8), indicating the reliability of the RNA-Seq data.

a–c Confirmation of RNA-Seq results by qRT-PCR. Pearson correlation coefficient (PCC) between the expression patterns determined by RNA-Seq and qRT-PCR is shown
Discussion
Tree aging involves extensive and complex transcriptome reprogramming
Based on five pairwise comparisons and GO enrichment analysis, a total of 12,927 DEGs and 459 biological processes were identified. The greatest numbers of DEGs and GO terms were identified in group V, i.e., in the comparison between middle-aged (25-year-old) and mature (50-year-old) trees, showing that this stage involves more biological processes in which larger numbers of genes take part (Fig. 2 and Table S2).
Many aspects of tree growth and development were found in 459 enriched biological processes. For example, the GO term “cell wall biogenesis” was enriched in group II and was assigned to genes with upregulated expression (Fig. 3). This was consistent with the results of anatomical examination (Fig. 1). Here, the enriched biological processes associated with wood formation and its regulation were emphasized.
Age affects the cellular process of wood formation
Wood formation involves cambium cell division, cell expansion, cell wall formation, and cell death (Plomion et al. 2001). These were reflected by the assignment of the assembled transcripts to the related GO terms, such as “cell cycle,” “cell growth,” “cell differentiation,” “cell death,” and “cell wall biogenesis” (Table S1). Notably, aging effects on the cellular process of wood formation were also supported by our GO enrichment analysis. For example, the GO term “cell differentiation” was enriched in group I, in which it was assigned to genes with upregulated expression (Fig. 3). Based on anatomical examination and statistical analysis, cambium cell differentiation indeed increased in this group, indicating that these genes play roles in this process.
Programmed cell death is the last step in xylem development, and much is known about its molecular processes (Escamez and Tuominen 2014; Han et al. 2012). When wood is forming actively and more xylem cells are developing, more cells are dying at the same time. During wood formation, not only cambium cell division but also xylem cell death are affected by aging, and this was also supported by our GO enrichment analysis. The GO term “cell death” was enriched in genes with downregulated expression in group V (Fig. 3), reflecting a positive correlation between xylem development and cell death-related gene expression. Notably, the GO term “regulation of programmed cell death” was enriched in genes with upregulated expression in group II and downregulated expression in group V (Fig. 3), indicating that programmed cell death in wood formation is positively regulated by these genes. Further analysis of these genes will help better understanding of programmed cell death in wood formation.
As we sampled in late summer, the formation of latewood was occurring in L. kaempferi trees. Although the typical anatomy of dormancy was not present, cambium cell division activity was becoming weaker (Fig. 1). This finding also suggested that within the rotation period, the timing of the end of cambium cell division is affected by aging. So in these six categories, cell differentiation and growth were in progress to form the latewood, while the numbers of expanding cells were different. This result indicated that the duration of the cell expansion phase is affected by aging or height (Anfodillo et al. 2012). Aging effects on the timing of the onset or end of cambium cell division have been found in trees (Li et al. 2013b, 2016; Rossi et al. 2008), but more cellular evidence, especially successive cellular assessment of wood formation in a whole growth period, is still needed to clarify the effects of age on the timing or the duration of each cellular process of wood formation in detail.
Age affects the hormonal regulation of wood formation
The environmental conditions under which plants live are complex and changing. To adapt to various environmental conditions, plants have developed many mechanisms to cope with and coordinate their growth and development. Plants respond differently to environmental temperature (Begum et al. 2010; Li et al. 2013b; Rossi et al. 2008) and hormonal signals (Little and Sundberg 1991; Savidge 1983) at different stages of their life span. However, knowledge of the underlying mechanisms is still limited.
Here, using GO enrichment analysis of DEGs, we identified many biological processes associated with responses to endogenous hormonal signals and their biosynthesis, transport, and metabolism (Fig. 4). These hormone-related GO terms were intensely distributed in the late stage of the rotation period, and most (75.8%) were enriched in genes with downregulated expression in group V (Fig. 4). These results indicated that changes in the responses to endogenous stimuli occur at an early stage in the life span, and even in a time window of 50 years, these changes are detectable through comparative transcriptomic analysis.
Auxin is an important hormone that controls many aspects of tree growth and development, such as cambium activity, wood formation (Sorce et al. 2013; Ye and Zhong 2015; Zhang et al. 2014), and the rooting of cuttings (Rosier, et al. 2004). Cambium activity and wood formation in 25- and 50-year-old trees were weaker than those in 10-year-old trees (Fig. 1). Here, the GO terms “auxin polar transport” and “response to auxin” were enriched in genes with downregulated expression in groups IV and V, respectively (Fig. 4), providing not only more evidence on the control of wood formation by auxin but also insights into the control of wood formation with age.
In addition to auxin, other hormones, such as ethylene and brassinosteroid, also control wood formation (Sorce et al. 2013; Ye and Zhong 2015; Zhang et al. 2014), and we also identified biological processes associated with them. For example, the GO terms “response to abscisic acid,” “response to ethylene,” “response to salicylic acid,” “response to jasmonic acid,” “response to karrikin,” and “response to brassinosteroid” were enriched in group V (Fig. 4). These data not only provide new evidence on the involvement of hormones in tree aging and wood formation but also offer a means of investigating the hormonal regulation of the specific process of wood formation.
Regulatory network of tracheid growth
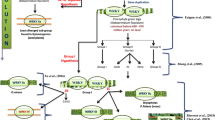
Ethylene acts as a positive regulator of cambium cell division and expansion (Sorce et al. 2013) and cotton fibers (Shi et al. 2006). Both ERF and DREB belong to the AP2/ERF superfamily. The members of this superfamily function downstream of ethylene signaling pathways and play roles in wood development in poplar (Vahala et al. 2013) and vascular cell division in Arabidopsis (Etchells et al. 2012). Conversion of S-adenosyl-l-met to ACC, which is one of the two key steps in ethylene biosynthesis, is catalyzed by ACS. More importantly, calcium activates ethylene biosynthesis through the phosphorylation of ACS by calcium-dependent protein kinase (Wang et al. 2011a). In gymnosperms, tracheid growth and its control remain poorly understood (Carvalho et al. 2013). Here, we found that the expression profiles of the L. kaempferi homologues of genes involved in the linear cell growth mode outlined the regulatory network of tracheid growth. The results show that some genes related to calcium and wall formation had the same expression patterns as LaERFs, suggesting that they might belong to the same regulatory network controlling wood formation.
When a plant cell grows, the primary cell wall loosens and active synthesis of new cell wall occurs. We assessed the expression of expansin, extensin, cellulose synthase, and xyloglucan endotransglucosylase/hydrolase and found that they had the same expression patterns as LaERFs, suggesting that they might function downstream of ethylene and be targets of LaERFs. One of the key enzymes involved in the synthesis of hemicellulose was annotated as brassinosteroid-regulated protein (LaBRU1; Zurek and Clouse 1994) and had the same expression pattern as LaERF1A (Figs. 5 and 6), suggesting that this gene might be controlled by both brassinosteroid and ethylene. Therefore, coupled with ethylene, the promotion of cell growth by brassinosteroid might be involved in the synthesis of hemicellulose. Our results thus provide more information about the roles of brassinosteroid in controlling cell growth during wood formation.
During cotton fiber growth, many transcription factors are differentially expressed, including MYB and WRKY (Liu et al. 2012). Especially, a decrease in the number of MYB binding sites in the promoter region of GaACO3 (ACC oxidase) results in the inactivation of ACO gene transcription and a short-fiber phenotype in Gossypium arboreum ovules (Li et al. 2014a), showing that MYB participates in the linear growth mode by controlling ethylene production. Here, we found that LaMYB44 had the same expression pattern as LaERFs, supporting its involvement in this mode. In addition, overexpression of PttACO1 stimulates cambial cell division in poplar (Love et al. 2009). So these data provide more information about the function of MYB in wood formation besides controlling secondary wall biosynthesis (Demura and Fukuda 2007; Ye and Zhong 2015). As to WRKY and other transcription factors, more experiments are needed to determine how they are involved in cell growth.
Based on our anatomical examination and expression analysis of genes involved in the linear cell growth mode (Qin and Zhu 2011), we propose a regulatory network model for tracheid growth in L. kaempferi (Fig. 7), in which calcium and MYB promote ethylene production, and ethylene stimulates cell wall expansion and synthesis. Large-scale screening of transcription factor–promoter interactions in conifers has been established, and members of the AP2 transcription factor family positively regulate the promoters of genes involved in cell wall synthesis and remodeling (Duval et al. 2014), giving support to this proposed model.

Regulatory network model proposed for tracheid growth. Calcium and MYB promote ethylene production, and ethylene stimulates cell wall expansion and synthesis. SAM S-adenosyl-l-met, ACC 1-aminocyclopropane-1-carboxylic acid, ACS ACC synthase, ACO ACC oxidase, CaM calmodulin, CML calmodulin-like protein, KIC calcium-binding protein, ERF ethylene response factor
Altogether, in this work, we present an integrative analysis of transcriptome reprogramming during tree aging throughout an entire rotation period and emphasize the molecular aspects of aging effects on wood formation. The findings contribute to our understanding of the molecular basis of tree aging. The identification of genes involved in the regulatory network of tracheid growth provides a means of investigating the regulation of wood formation in gymnosperm trees and offers potential targets for genetic manipulation to improve the properties of xylem fibers.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Andersson-Gunneras S, Hellgren JM, Bjorklund S, Regan S, Moritz T, Sundberg B (2003) Asymmetric expression of a poplar ACC oxidase controls ethylene production during gravitational induction of tension wood. Plant J 34:339–349
Anfodillo T, Carraro V, Carrer M, Fior C, Rossi S (2006) Convergent tapering of xylem conduits in different woody species. New Phytol 169:279–290
Anfodillo T, Deslauriers A, Menardi R, Tedoldi L, Petit G, Rossi S (2012) Widening of xylem conduits in a conifer tree depends on the longer time of cell expansion downwards along the stem. J Exp Bot 63:837–845
Begum S, Nakaba S, Oribe Y, Kubo T, Funada R (2010) Cambial sensitivity to rising temperatures by natural condition and artificial heating from late winter to early spring in the evergreen conifer Cryptomeria japonica. Trees 24:43–52
Böhlenius H, Huang T, Charbonnel-Campaa L, Brunner AM, Jansson S, Strauss SH, Nilsson O (2006) CO/FT regulatory module controls timing of flowering and seasonal growth cessation in trees. Science 312:1040–1043
Busov VB, Johannes E, Whetten RW, Sederoff RR, Spiker SL, Lanz-Garcia C, Goldfarb B (2004) An auxin-inducible gene from loblolly pine (Pinus taeda L.) is differentially expressed in mature and juvenile-phase shoots and encodes a putative transmembrane protein. Planta 218:916–927
Carlsbecker A, Tandre K, Johanson U, Englund M, Engström P (2004) The MADS-box gene DAL1 is a potential mediator of the juvenile-to-adult transition in Norway spruce (Picea abies). Plant J 40:546–557
Carvalho A, Paiva J, Louzada J, Lima-Brito J (2013) The transcriptomics of secondary growth and wood formation in conifers. Mol Biol Int 2013:1–12
Cato S, McMillan L, Donaldson L, Richardson T, Echt C, Gardner R (2006) Wood formation from the base to the crown in Pinus radiata: gradients of tracheid wall thickness, wood density, radial growth rate and gene expression. Plant Mol Biol 60:565–581
Demura T, Fukuda H (2007) Transcriptional regulation in wood formation. Trends Plant Sci 12:64–70
Diego LB, Berdasco M, Fraga MF, Canal MJ, Rodriguez R, Castresana C (2004) A Pinus radiata AAA-ATPase, the expression of which increases with tree ageing. J Exp Bot 55:1597–1599
Duval I, Lachance D, Giguere I, Bomal C, Morency MJ, Pelletier G, Boyle B, MacKay JJ, Seguin A (2014) Large-scale screening of transcription factor-promoter interactions in spruce reveals a transcriptional network involved in vascular development. J Exp Bot 65:2319–2333
Escamez S, Tuominen H (2014) Programmes of cell death and autolysis in tracheary elements: when a suicidal cell arranges its own corpse removal. J Exp Bot 65:1313–1321
Etchells JP, Provost CM, Turner SR (2012) Plant vascular cell division is maintained by an interaction between PXY and ethylene signalling. Plos Genet 8:e1002997
Fernández-Ocaña A, Carmen García-López M, Jiménez-Ruiz J, Saniger L, Macías D, Navarro F, Oya R, Belaj A, Rosa R, Corpas FJ, Bautista Barroso J, Luque F (2010) Identification of a gene involved in the juvenile-to-adult transition (JAT) in cultivated olive trees. Tree Genet Genomes 6:891–903
Flachowsky H, Szankowski I, Waidmann S, Peil A, Trankner C, Hanke MV (2012) The MdTFL1 gene of apple (Malus x domestica Borkh.) reduces vegetative growth and generation time. Tree Physiol 32:1288–1301
Garces M, Le Provost G, Lalanne C, Claverol S, Barre A, Plomion C, Herrera R (2014) Proteomic analysis during ontogenesis of secondary xylem in maritime pine. Tree Physiol 34:1263–1277
García-López MC, Vidoy I, Jiménez-Ruiz J, Muñoz-Mérida A, Fernández-Ocaña A, de la Rosa R, Barroso JB, Navarro F, Trelles O, Beuzón CR, Barceló A, Valpuesta V, Luque F (2014) Genetic changes involved in the juvenile-to-adult transition in the shoot apex of Olea europaea L. occur years before the first flowering. Tree Genet Genomes 10:585–603
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Han J, Lin W, Oda Y, Cui K, Fukuda H, He X (2012) The proteasome is responsible for caspase-3-like activity during xylem development. Plant J 72:129–141
Hochberg Y (1988) A sharper Bonferroni procedure for multiple tests of significance. Biometrika 75:800–802
Hsu CY, Liu Y, Luthe DS, Yuceer C (2006) Poplar FT2 shortens the juvenile phase and promotes seasonal flowering. Plant Cell 18:1846–1861
Hsu CY, Adams JP, Kim H, No K, Ma C, Strauss SH, Drnevich J, Vandervelde L, Ellis JD, Rice BM, Wickett N, Gunter LE, Tuskan GA, Brunner AM, Page GP, Barakat A, Carlson JE, DePamphilis CW, Luthe DS, Yuceer C (2011) FLOWERING LOCUS T duplication coordinates reproductive and vegetative growth in perennial poplar. Proc Natl Acad Sci U S A 108:10756–10761
Hutchison KW, Sherman CD, Weber J, Smith SS, Singer PB, Greenwood MS (1990) Maturation in larch. II. Effects of ege on photosynthesis and gene expression in developing foliage. Plant Physiol 94:1308–1315
Kotoda N, Iwanami H, Takahashi S, Abe K (2006) Antisense expression of MdTFL1, a TFL1-like gene, reduces the juvenile phase in apple. J Am Soc Hortic Sci 131:74–81
Kumar M, Saranpää P, Barnett JR, Wilkinson MJ (2009) Juvenile-mature wood transition in pine: correlation between wood properties and candidate gene expression profiles. Euphytica 166:341–355
Lai M, Sun X, Chen D, Xie Y, Zhang S (2014) Age-related trends in genetic parameters for Larix kaempferi and their implications for early selection. BMC Genet 15(Suppl 1):S10
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323
Li X, Wu HX, Southerton SG (2010) Seasonal reorganization of the xylem transcriptome at different tree ages reveals novel insights into wood formation in Pinus radiata. New Phytol 187:764–776
Li X, Wu HX, Southerton SG (2011) Transcriptome profiling of wood maturation in Pinus radiata identifies differentially expressed genes with implications in juvenile and mature wood variation. Gene 487:62–71
Li X, Bian H, Song D, Ma S, Han N, Wang J, Zhu M (2013a) Flowering time control in ornamental gloxinia (Sinningia speciosa) by manipulation of miR159 expression. Ann Bot-London 111:791–799
Li X, Liang E, Gricar J, Prislan P, Rossi S, Cufar K (2013b) Age dependence of xylogenesis and its climatic sensitivity in Smith fir on the south-eastern Tibetan Plateau. Tree Physiol 33:48–56
Li F, Fan G, Wang K, Sun F, Yuan Y, Song G, Li Q, Ma Z, Lu C, Zou C, Chen W, Liang X, Shang H, Liu W, Shi C, Xiao G, Gou C, Ye W, Xu X, Zhang X, Wei H, Li Z, Zhang G, Wang J, Liu K, Kohel RJ, Percy RG, Yu JZ, Zhu YX, Wang J, Yu S (2014a) Genome sequence of the cultivated cotton Gossypium arboreum. Nat Genet 46:567–572
Li W, Han S, Qi L, Zhang S (2014b) Transcriptome resources and genome-wide marker development for Japanese larch (Larix kaempferi). Front Agr Sci Eng 1:77–84
Li WF, Zhang SG, Han SY, Wu T, Zhang JH, Qi LW (2014c) The post-transcriptional regulation of LaSCL6 by miR171 during maintenance of embryogenic potential in Larix kaempferi (Lamb.) Carr. Tree Genet Genomes 10:223–229
Li X, Camarero JJ, Case B, Liang E, Rossi S (2016) The onset of xylogenesis is not related to distance from the crown in Smith fir trees from the southeastern Tibetan Plateau. Can J For Res 46:885–889
Linares JC, Taïqui L, Sangüesa-Barreda G, Seco JI, Camarero JJ (2013) Age-related drought sensitivity of Atlas cedar (Cedrus atlantica) in the Moroccan Middle Atlas forests. Dendrochronologia 31:88–96
Little CHA, Sundberg B (1991) Tracheid production in response to indole-3-acetic acid varies with internode age in Pinus sylvestris stems. Trees 5:101–106
Liu K, Sun J, Yao L, Yuan Y (2012) Transcriptome analysis reveals critical genes and key pathways for early cotton fiber elongation in Ligon lintless-1 mutant. Genomics 100:42–50
Lorenz WW, Dean JF (2002) SAGE profiling and demonstration of differential gene expression along the axial developmental gradient of lignifying xylem in loblolly pine (Pinus taeda). Tree Physiol 22:301–310
Love J, Bjorklund S, Vahala J, Hertzberg M, Kangasjarvi J, Sundberg B (2009) Ethylene is an endogenous stimulator of cell division in the cambial meristem of Populus. Proc Natl Acad Sci U S A 106:5984–5989
Mellerowicz EJ, Riding RT, Greenwood MS (1995) Nuclear and cytoplasmic changes associated with maturation in the vascular cambium of Larix laricina. Tree Physiol 15:443–449
Mvolo C, Koubaa A, Beaulieu J, Cloutier A, Mazerolle M (2015) Variation in wood quality in white spruce (Picea glauca (Moench) Voss). Part I. Defining the juvenile–mature wood transition based on tracheid length. Forests 6:183–202
Nikolayeva O, Robinson MD (2014) EdgeR for differential RNA-seq and ChIP-seq analysis: an application to stem cell biology. Methods Mol Biol 1150:45–79
Olson ME, Anfodillo T, Rosell JA, Petit G, Crivellaro A, Isnard S, León-Gómez C, Alvarado-Cárdenas LO, Castorena M (2014) Universal hydraulics of the flowering plants: vessel diameter scales with stem length across angiosperm lineages, habits and climates. Ecol Lett 17:988–997
Paiva JAP, Garcés M, Alves A, Garnier-Géré P, Rodrigues JC, Lalanne C, Porcon S, Le Provost G, Da Silva PD, Brach J, Frigerio J, Claverol S, Barré A, Fevereiro P, Plomion C (2008) Molecular and phenotypic profiling from the base to the crown in maritime pine wood-forming tissue. New Phytol 178:283–301
Plomion C, Leprovost G, Stokes A (2001) Wood formation in trees. Plant Physiol 127:1513–1523
Qin YM, Zhu YX (2011) How cotton fibers elongate: a tale of linear cell-growth mode. Curr Opin Plant Biol 14:106–111
Ranik M, Creux NM, Myburg AA (2006) Within-tree transcriptome profiling in wood-forming tissues of a fast-growing Eucalyptus tree. Tree Physiol 26:365–375
Rosier CL, Frampton J, Goldfarb B, Blazich FA, Wise FC (2004) Growth stage, auxin type, and concentration influence rooting of stem cuttings of Fraser fir. Hortscience 6:1397–1402
Rossi S, Deslauriers A, Anfodillo T, Carrer M (2008) Age-dependent xylogenesis in timberline conifers. New Phytol 177:199–208
Savidge RA (1983) The role of plant hormones in higher plant cellular differentiation. II. Experiments with the vascular cambium, and sclereid and tracheid differentiation in the pine, Pinus contorta. Histochem J 15:447–466
Shi YH, Zhu SW, Mao XZ, Feng JX, Qin YM, Zhang L, Cheng J, Wei LP, Wang ZY, Zhu YX (2006) Transcriptome profiling, molecular biological, and physiological studies reveal a major role for ethylene in cotton fiber cell elongation. Plant Cell 18:651–664
Sorce C, Giovannelli A, Sebastiani L, Anfodillo T (2013) Hormonal signals involved in the regulation of cambial activity, xylogenesis and vessel patterning in trees. Plant Cell Rep 32:885–898
Vahala J, Felten J, Love J, Gorzsas A, Gerber L, Lamminmaki A, Kangasjarvi J, Sundberg B (2013) A genome-wide screen for ethylene-induced ethylene response factors (ERFs) in hybrid aspen stem identifies ERF genes that modify stem growth and wood properties. New Phytol 200:511–522
Wang X, Zhang Y, McRae DJ (2009) Spatial and age-dependent tree-ring growth responses of Larix gmelinii to climate in northeastern China. Trees 23:875–885
Wang H, Mei W, Qin Y, Zhu Y (2011a) 1-Aminocyclopropane-1-carboxylic acid synthase 2 is phosphorylated by calcium-dependent protein kinase 1 during cotton fiber elongation. Acta Biochim Biophys Sin 43:654–661
Wang J, Park MY, Wang L, Koo Y, Chen X, Weigel D, Poethig RS (2011b) MiRNA control of vegetative phase change in trees. Plos Genet 7:e1002012
Wendling I, Trueman SJ, Xavier A (2014) Maturation and related aspects in clonal forestry—part I: concepts, regulation and consequences of phase change. New For 45:449–471
Xu H, Cao D, Feng J, Wu H, Lin J and Wang Y (2016) Transcriptional regulation of vascular cambium activity during the transition from juvenile to mature stages in Cunninghamia lanceolata. J Plant Physiol 200:7–17
Yamagishi N, Kishigami R, Yoshikawa N (2014) Reduced generation time of apple seedlings to within a year by means of a plant virus vector: a new plant-breeding technique with no transmission of genetic modification to the next generation. Plant Biotechnol J 12:60–68
Ye ZH, Zhong R (2015) Molecular control of wood formation in trees. J Exp Bot 66:4119–4131
Yemele M, Beaulieu J, Defo M, Koubaa A, Cloutier A, Mvolo CS (2015) Prediction of tracheid length and diameter in white spruce (Picea glauca). IAWA J 36:186–207
Zhang Y, Zhang S, Han S, Li X, Qi L (2012) Transcriptome profiling and in silico analysis of somatic embryos in Japanese larch (Larix leptolepis). Plant Cell Rep 31:1637–1657
Zhang J, Nieminen K, Serra JAA, Helariutta Y (2014) The formation of wood and its control. Curr Opin Plant Biol 17:56–63
Zhang Y, Han X, Sang J, He X, Liu M, Qiao G, Zhuo R, He G, Hu J (2016) Transcriptome analysis of immature xylem in the Chinese fir at different developmental phases. Peer J 4:e2097
Zurek DM, Clouse SD (1994) Molecular cloning and characterization of a brassinosteroid-regulated gene from elongating soybean (Glycine max L.) epicotyls. Plant Physiol 104:161–170
Acknowledgements
The authors thank Dr. IC Bruce (Peking University) for critical reading of the manuscript, Ms. Fen Zhou (Beijing University of Chinese Medicine) for statistical analysis, Dr. Tao Wu for sample collection, and the reviewers for their constructive comments on earlier versions of this manuscript. This work was supported by the Basic Research Fund of RIF (grant no. RIF2014-07), the National Natural Science Foundation of China (31200464 and 31330017), and the National High Technology Research and Development Program of China (2013AA102704).
Author contributions
Wan-Feng Li conceived, designed, and carried out the study, and wrote the manuscript. Wen-Hua Yang revised the manuscript. Shou-Gong Zhang, Su-Ying Han, and Li-Wang Qi provided suggestions on the experimental design and analyses. All authors read and approved the manuscript.
Data archiving statement
All RNA-Seq data in this study have been deposited in the NCBI SRA database with the accession number SRR2167459 for the 1-year, SRR2167460 for the 2-year, SRR2167462 for the 5-year, SRR2167463 for the 10-year, SRR2167481 for the 25-year, and SRR2167482 for the 50-year-old sample.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by F. Gugerli
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Figure S1
Xylem width of the uppermost main stems produced in the current year from 1-, 2-, 5-, 10-, 25-, and 50-year-old L. kaempferi trees. (XLS 48 kb)
Supplementary Table S1
Summary of Gene Ontology (GO) annotation in ‘biological process’ (XLS 340 kb)
Supplementary Table S2
Distribution of Gene Ontology (GO) terms after enrichment analysis of differentially expressed genes (DEGs) during Larix kaempferi aging (XLS 108 kb)
Supplementary Table S3
Summary of differentially expressed genes (DEGs) during Larix kaempferi aging (XLS 1655 kb)
Supplementary Table S4
Primers for quantitative reverse transcription polymerase chain reaction (qRT-PCR) (XLS 8 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Li, WF., Yang, WH., Zhang, SG. et al. Transcriptome analysis provides insights into wood formation during larch tree aging. Tree Genetics & Genomes 13, 19 (2017). https://doi.org/10.1007/s11295-017-1106-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-017-1106-3