
Abstract
Coffea canephora is subject to enormous competitive challenges from other crops, especially for farmer sustainability and consumer requirements. Coffee breeding programs have to focus on specific traits linked to these two key targets, such as quality character, largely depending on the bean’s biochemical composition and field yield. Two segregating populations A and B, from crosses between a hybrid (Congolese × Guinean) FRT58 parental clone and a Congolese FRT51 genotype and between two Congolese parents FRT67 and FRT51, respectively, were used to characterize the quantitative trait loci (QTL) involved in agronomic and biochemical traits. A consensus genetic map was established using 249 SSRs covering 1,201 cM. Three QTL detection models per population with MapQTL (model I) and MCQTL (model II) followed by a connected population approach with MCQTL (model III) were compared based on their efficiency, precision for QTL detection, and their genetic effect assessment (additive, dominance, and parental-favorable allele). The analysis detected a total of 143 QTLs, 60 of which were shared between the three models; 28 found with two models; and two, 13, and 40 specific from models I, II, and III, respectively. The last model III based on connected populations is much more efficient in detecting QTLs with low variance explained and led to the genetic characterization of favorable allele. Thanks to this comparison of three QTL detection models on our quantitative genetic study, we will give a new insight for coffee breeding programs dedicated to managing complex agronomic or qualitative traits.
Similar content being viewed by others


Introduction
Coffee is the world’s favorite beverage and the most widely traded tropical agricultural commodity worldwide with more than 8.6 million tonnes of green beans produced in 2012 (IOC 2012). The genus Coffea includes about 125 species (Davis et al. 2006; 2011); however, only two species are cultivated: Coffea canephora (Robusta) and Coffea arabica (Arabica). Robusta represents approximately 38 % of world coffee production and is used for most soluble coffee, which is increasingly consumed throughout the world (IOC 2012). This species is cultivated at low and medium altitudes in the intertropical regions of Africa, America, and Asia.
C. canephora is strictly an outcrossing diploid species and presents a wide natural distribution area which extends west to east from Guinea to Uganda and north to south from Guinea to Angola. Seven genetic and geographic C. canephora groups were identified: the Guinean from Ivory Coast and Guinea, the Congolese from Congo, the Conilons from Gabon, group B from the Central African Republic, group C from Cameroun, group R from Democratic Republic of Congo, and group UW from Uganda (Berthaud 1986; Cubry et al. 2008; Dussert et al. 1999; Gomez et al. 2009; Musoli et al. 2009; Sumirat et al. 2012). This wide genetic diversity of Robusta is an important resource for breeding programs.
Currently, coffee growing is in strong competition with other raw materials such as palm oil or rubber which offer better profits to farmers. One of the main issues for breeding is to develop new varieties with higher yields to sustain coffee farming. Coffee yield is measured by weight of fresh cherries and can be predicted by other yield-related traits such as the number of productive branches, the number of nodes per branch, and the number of cherries per node. Coffee quality is also a major selection criterion for coffee improvement. Robusta is known as a lower quality coffee than Arabica due to its higher caffeine content and bitterness. By focusing on the biochemical compound content related to cup quality (caffeine, trigonelline, chlorogenic acids, lipids, proteins, and sucrose), Robusta quality can be enhanced (Leroy et al. 2006). Considering the complexity of traits such as yield potential and coffee quality, using molecular markers associated with quantitative trait loci (QTL) is expected to improve the efficiency of Robusta breeding.
Recently, an approach by crossing designs composed of bi-parental populations that are connected by common parents was developed to increase genetic diversity under QTL studies (Blanc et al. 2006). The alleles of different parents can be compared within a single model using MCQTL software (Jourjon et al. 2005). Studies on maize (Blanc et al. 2006; Blanc et al. 2007) and on ryegrass (Pauly et al. 2012) reported a higher number of QTLs detected with this connected model than in single-population analyses. The multi-parent approach also made QTL positioning more precise for flowering time QTLs in three connected populations of Medicago truncatula (Pierre et al. 2008). However, it has been reported in different studies (Billotte etal. 2010; Espinoza et al. 2012; Pierre et al. 2008) that some QTLs detected in single-population analyses were not found in the multi-population analyses. The small population size or the dilution of low genetic effect on the whole design explained the loss of QTLs in the connected model (Melchinger et al. 1998; Muranty 1996).
Some QTL studies on coffee have already been conducted, helping to identify QTLs for the self-incompatibility S-locus (Lashermes et al. 1996; Coulibaly et al. 2002), pollen viability restoration (Coulibaly et al. 2003), fructification time (Akaffou et al. 2003), morphological traits (N'Diaye et al. 2007), somatic embryogenesis capacity (Priyono et al. 2010), and flowering time (Priyono 2013). Only two studies reported QTLs related to yield and quality traits for coffee (Ky et al. 2013; Leroy et al. 2011). These last two studies involved bi-parental populations and used single marker analysis (ANOVA) or MapQTL as the QTL detection model (Van Ooijen 2009). Therefore, the use of multi-population connected analysis would seem to be a promising tool for C. canephora breeding, combining the advantage of wide diversity and powerful QTL detection.
The purposes of this study were (1) to compare three QTL detection methods: the model per population of MapQTL and MCQTL and the multi-population connected analysis of MCQTL, (2) to identify QTLs for yield and quality traits for genetic improvement of Robusta, and (3) to learn about our study for new insights into Robusta breeding using marker-assisted selection.
Materials and methods
Plant material
Two progenies were created by crossing three parental clones of C. canephora. The progenies were obtained by controlled pollination using the procedure described by Capot (1964). The two parents FRT67 and FRT51 belong to the Congolese genetic group described by Gomez et al. (2009) while the parent FRT58 is a hybrid between Congolese and Guinean genetic groups obtained in a field plantation.
The two progenies derived from the crosses were planted in an experimental coffee station located in Ecuador near the city of Quevedo. The two plots were planted at an altitude of 65 m.a.s.l. in flat and sandy loamy soil. The annual rainfall is equivalent to 2,200 mm with 4 months of dry season.
Trees were planted at a density of 1,666 trees/ha (2.0 × 3.0 m) and maintained on one single stem topped at 2.0 m. The fertilization program was based on soil analyses, and a complete NPK formula (200–50–150) was applied in three annual applications. No pesticides were applied to the coffee trees; only herbicides (glyphosate or paraquat combined with Diuron) were used to control the weeds in the two plots with six annual treatments.
The experimental design adopted was a fully randomized single tree plot including the parents for which two trees were vegetatively propagated and planted randomly. A progeny of the local Robusta was used as a border for each plot and as pollinators regularly distributed in the plots. The number of trees is 191 for progeny A and 178 for progeny B, and none of them were lost during the 7 years of trials.
Coffee cherries were harvested and weighed on a tree-by-tree base, and seven to eight annual rounds were organized during the harvesting period to collect the mature cherries. Green coffee was processed by sun drying the whole cherries to reach 12 % humidity in the beans and by mechanical hulling to remove the husk.
Observation of phenotypic traits
The coffee trees were individually observed for 7 years (2006 to 2012) for different characteristics including (Table 1):
-
Eight field traits such as the number of primary branches (Br), internode length (In), stem diameter (St), number of cherries per node (Cn), number of nodes on primaries (No), yield (Yi), weight of 100 beans (Bw), and percentage of peaberries (Pe).
-
Six biochemicals and one sensory traits using Near-infrared (NIR) assessment for caffeine bean content (Ca), trigonelline (Tr), lipid (Li), protein (Pr), sucrose (Su), chlorogenic acid (Ch), and bitterness (Bi).
The frequency of the trait observations and the equipment and procedures used to perform the phenotypic observations are summarized in Table 1.
Near-infrared analysis
The green coffee samples were maintained for 15 days at 11 °C and 65 % relative humidity (RH). This phase of storage is necessary to standardize their humidity rate for near-infrared analyses. The biochemical composition and the bitterness sensory trait were predicted by near-infrared spectroscopy using calibration models previously developed for Robusta green (Huck et al. 2005). Spectral data in reflectance mode for each coffee sample was recorded at ambient temperature using the Thermo Electrons FT-NIR Antaris II spectrometer. Spectra were collected using a cup of 12-cm diameter, at a resolution of 8 cm−1, and 80 scans were necessary to complete one round. For each sample, the predictive values were calculated as an average out of four replications.
Statistical analysis
For each trait according to Table 1, the means and the variances of progenies were compared using, respectively, the Welch t test and the Bartlett test. Pearson correlation coefficients were calculated by pairs of variables for each progeny. Statistical analyses were performed using R (R Development Core Team 2012).
SSR genotyping
Total DNA of the two population progenies (A and B) were purified from adult leaves using Dneasy®96 Plant Kit (QIAGEN) following the manufacturer’s protocol. A total of 249 SSRs were used to build the two genetic maps and the consensus map (Table S1) stored in MoccaDB (Plechakova et al. 2009). The markers were selected on a high-density coffee reference map (submitted for publication in Science) according to an average density distribution of 10 to 20 cM and to the highest level of polymorphism detected in the three parents. The SSRs segregating F2 type (a/b × c/d or e/f × e/g) were chosen in priority due to their higher levels of genetic information. The SSR genotyping was performed according to Lefebvre-Pautigny et al. (2010).
Genetic map construction
The two genetic linkage maps A (FRT58 × FRT51) and B (FRT67 × FRT51) were built using JoinMap® 4.0 (Van Ooijen 2006). Log of odds (LOD) scores >3 were used to determine the different linkage groups. The order of the markers was determined with regression mapping function by using the pairwise data of the segregating loci that showed a recombination frequency lower than 0.45 and a LOD >1 (Van Ooijen 2006). The ripple value of 1 and the jump threshold of 5 were selected.
Due to the allogamous status of Robusta, the segregating populations analyzed were cross-pollinated (CP) type resulting from progenies between heterozygous parents with no information available on linkage phases. According to the pseudo-test cross strategy (Grattapaglia and Sederoff 1994), parental maps were built separately using backcross and F2 segregating types. The homologous parental linkage groups were then merged, and integrated maps resulting from the two progenies were constructed based on mean recombination frequencies and LOD scores using the function “combine groups for map integration” in the JoinMap software. The Kosambi mapping function was used to convert recombination frequencies into map distances. Finally, the linkage maps of the two crosses were integrated into a unique consensus linkage map of the multi-cross design using the “combine maps” function in JoinMap software. The genetic maps were drawn using MapChart version 2.2 software (Voorrips 2002). Marker segregation distortion was tested against the expected Mendelian segregation ratios using the chi-square test.
QTL analysis
An initial QTL analysis of each F1 segregating population was performed with MapQTL V.6.0 (Van Ooijen 2009). The interval mapping method was used to test for the presence of a putative QTL with a step size of 2 cM (Jansen and Stam 1994). Log of odds (LOD) thresholds for genome-wide detection analysis (P value of 0.05) was experimentally determined for each trait using the permutation test with 1,000 permutations (Doerge and Churchill 1996). When regions displayed an interval mapping test greater than the threshold, the markers with the highest test values were taken as co-factors for a multiple QTL mapping analysis (MQM) with a step size of 2 cM (Jansen and Stam 1994). Allelic effects were estimated as A f = (μ ac + μ ad)−(μ bc + μ bd) for female additivity, A m = (μ ac + μ bc)−(μ ad + μ bd) for male additivity, and D = (μ ac + μ bd)−(μ ad + μ bc) for dominance where μ ac, μ ad, μ bc, and μ bd are estimated phenotypic means associated to each of the four possible genotypic classes ac, ad, bc, and bd, deriving from a CP segregating cross (Ben Sadok et al. 2013). Each significant QTL was characterized by the closest associated marker, the LOD-1 confidence region, the percentage of phenotypic variation explained (R 2), and the effects of the alleles for each parent. In order to be consistent with the MCQTL analysis, the additive and dominance effects given by MapQTL were divided by four.
A second method for detecting QTL for each F1 population was performed using MCQTL v.5.2.4 (Jourjon et al. 2005) with the linear marker regression model (Haley and Knott 1992). Either the additive allelic effect alone (additive model) or the additive and dominance allelic effects (dominance model), as described in Kawamura et al. (2011), were used as the QTL model. Co-factors were selected by the forward method and multiple QTLs were detected by the iQTLm method (Charcosset et al. 2000) with a window of 10 cM around the putative QTL. The statistical significance of QTLs was assessed using the MCQTL test, which is equal to -log (P value (F test)), as described in MCQTL version 5 (http://carlit.toulouse.inra.fr/MCQTL).The threshold value for genome-wide detection analysis (P value of 0.05) was obtained for each trait with 10,000 permutations (Doerge and Churchill 1996). Model parameters were estimated for each QTL (position, confidence region, and R 2). Finally, a QTL connected analysis for both two F1 populations together was performed with MCQTL v.5.2.4 (Jourjon et al. 2005). The QTL model was a connected model assuming that the QTL allelic effects were identical within the two F1 populations for the common parent FRT51. All the other features of this QTL connected analysis were identical to the per F1 population MCQTL analysis. Each QTL effect was tested by Student t tests. The variances of the estimated QTL effects were computed using the diagonal of the inverse of the incidence matrix provided by MCQTL. A MCQTL plug-in, developed by the University of Poitiers, was used to automatically compute the t tests and produce an Excel file of the results. By definition, we named a QTL as “additive” if the maximum value from the parents of the estimated additive allelic effects was larger than the dominance ones.
Shared QTL between the three detection models were defined trait by trait as QTL which have confidence regions that cover each other and similar genetic effects. We mapped the QTLs detected using models per population on the consensus map before comparing the QTL confidence regions using the QTLproj function of Biomercator v.4.2 software (Sosnowski et al. 2012). A heat map of the QTLs detected with the three models was created using R (R Development Core Team 2012). When QTLs were detected by the connected model, the confidence regions and the R 2 value defined by this model were used to represent the QTLs on the map. For the QTLs detected only by models I and/or II, the QTL with the tightest confidence regions was kept to project the QTL on the map.
Results
Phenotypic data analysis
Statistics for all traits recorded throughout the years for each progeny are given in Table S2 and the significant correlations between quantitative traits are shown in Table 2. Significant mean differences between the two crosses were detected for all traits except for the length of internodes (In) and the bean lipid content (Li).
Progeny B (FRT67 × FRT51) had a higher average yield (Yi) over 7 years. This cross also had a higher number of cherries per node (Cn). This result was consistent with the positive correlation observed between the Yi and Cn in both progenies A and B (0.38 and 0.29, Table 2). Three main secondary yield components such as number of primary branches, number of nodes on primaries, and stem diameter (Br, No, St) were significantly correlated, with Pearson’s coefficient values ranking from 0.21 to 0.31. Cross B (FRT67 × FRT51) presented a higher 100-bean weight (Bw) and a lower percentage of peaberries (Pe) in comparison with progeny A (FRT58 × FRT51).
For the coffee bean biochemical composition, higher caffeine (Ca), chlorogenic acids (Ch), and protein content (Pr) were observed in progeny A (FRT58 × FRT51). These three traits were significantly correlated in both crosses (0.32 to 0.87) with bitterness (Bi), whereas negative correlations were observed between bitterness and lipids (Li) and sucrose (Su) contents (−0.34 to –0.49).
Correlations detected among chemical traits, shared in both progenies, were more frequent than the correlations observed between field characteristics (Table 2). Moreover, variance traits were much higher among field traits than bean chemical characteristics (Table S2). Significant variance differences were detected from the two progenies analyzed.
Genetic linkage maps
Both SSR maps of progenies A (FRT58 × FRT51) and B (FRT67 × FRT51) covered 1,128 cM (199 markers) and 1,135 cM (152 markers), respectively, with an average distance between markers of 6.4 and 8.4 cM (Table 3 and Figure S1). The map of cross A consisted of 11 linkage groups as expected, whereas the linkage groups (LGs) J and H of map B were split into two due to a lack of polymorphic markers on these two map areas (Figure S1). The maximum distance between adjacent markers was 37 cM on LG B for FRT58 × FRT51 and 38 cM on LG C for FRT67 × FRT51 (Figure S1). Among the 199 loci mapped on genetic map A, 94 and 105 showed, respectively, F2 and a backcross segregation types (Table 3). Map B consisted of 72 and 80 loci with, respectively, F2 and a backcross segregation types. However, large parts of some linkage groups such as LGs A and C showed segregation from only one parent with a lack of informative markers from the FRT51 parent. Significant segregation distortions (P value < 0.01) were observed on several loci in both maps A (6 %) and B (3 %) (Figure S1). Strong segregation distortions (P value < 0.001) were identified at the top of LG B for FRT58 × FRT51 and around the locus AY2434 of LG C on both maps.
The consensus map covered 1,201 cM with a total of 249 markers and an average distance between markers of 5.5 cM (Table 3 and Figure S1). The maximum distance between markers was 33 cM on LG B (Figure S1). Among the 249 loci charted on the consensus genetic map, 102 loci (41 %) were shared with both maps and 97 (39 %) and 50 (20 %) loci had originated from genetic maps FRT58 × FRT51 and FRT67 × FRT51, respectively (Table 3). Both genetic maps A and B exhibited a high degree of co-linearity among the different linkage groups in relationship to the consensus map (Figure S1).
QTL detection
Model per population using MapQTL—model I
Very similar LOD threshold values (data not shown) were found using permutation tests for all traits, no matter what cross was analyzed. An average value of 4.1 was then selected to validate QTL detection. Both single-population analyses served to identify 56 QTLs for progeny A (FRT58 × FRT51) and 30 QTLs for progeny B (FRT67 × FRT51) (Table S3). These two populations were found to have six QTLs (6, 19, 59, 62, 90, and 105: Table S3) in common; thus, a total of 80 QTLs were identified with this first QTL detection model. All these QTLs showed additive effects superior to the dominance effects.
A set of 50 QTLs for NIR traits was detected. Among the 11 QTLs detected for bean caffeine content (Ca), four major QTLs (1, 2, 59, and 60: Table S3) were detected consistently throughout the 2 years of observation on LGs A and E with R 2 values ranging from 18.2 to 22.7 %. The FRT58 parent appeared as the favorable parent for the two major QTLs on LG A whereas FRT67 was favorable for one QTL and FRT51 for the other QTL on the LG E.
For chlorogenic acid content (Ch) in the bean, eight QTLs were found. All the QTLs detected for Ch (5, 6, 25, 55, 65, 66, 107, and 108: Table S3) co-localized with QTLs already detected for caffeine. This result was consistent with the highly significant correlation between these two traits in the two progenies (r = 0.80 and r = 0.87: Table 2). The percentage of variance explained by the QTLs detected for Ch varied between 11.2 and 23.1 %.
For the bean lipid content (Li), seven QTLs were detected on LGs B, C, E, G, and I (19, 21, 45, 62, 63, 93, and 113: Table S3). The favorable parents were FRT58 and FRT67 except for two QTLs detected on LG B for which the male additive effect (FRT51) was predominant. On the two LGs B and E, the QTLs detected from FRT67 × FRT51 were detected in both 2007 and 2010 with R 2 values ranging from 16.4 to 24.8 %.
Seven QTLs for sucrose (Su) content were detected in both progenies (3, 4, 64, 115, 125, 126, and 127: Table S3). They were located on four LGs (A, E, I, and J) and explained 9.3 to 16.4 % of the phenotypic variance. The female additive effect (FRT58 or FRT67) was predominant for all these QTLs, promoting a higher sucrose content of the bean and thus a better cup quality. Among the seven QTLs detected for Su, four QTLs were detected both in 2007 and 2010.
Five QTLs (22, 76, 94, 95, and 124: Table S3) were identified for bean protein (Pr) content on the four LGs B, F, G, and J with R 2 values from 14.9 to 19.6 %. Three QTLs showed a major male additive effect (FRT51) whereas the other two QTLs had a predominant female (FRT58) additive effect.
For trigonelline (Tr), three QTLs (78, 97, and 138: Table S3) were detected on LGs F, G, and K. The female FRT58 and FRT67 parents carried the favorable allele for these QTLs for which the R 2 values ranged from 10.2 to 14.5 %.
The bitterness (Bi) appeared to be linked to the bean biochemical composition. Indeed, there was a co-localization of all nine QTLs (13, 35, 37, 69, 70, 100, 102, 111, and 119: Table S3) on LGs A, B, E, G, H and I, and specific biochemical bean composition QTLs, such as Ca, Ch, Su, and Li. In addition, genotypes with higher bitterness, caffeine, and chlorogenic acids content in the bean showed lower sucrose and lipid levels. These significant correlations (Table 2) were consistent with the genetic effects detected in our study; for example in LG A, the FRT58 parent showed a higher content of Su (A f = 0.137 and A f = 0.157 in 2007 and 2010, respectively: Table S3), a lower content of Ca (A f = −0.122 and A f = −0.125 in 2007 and in 2010, respectively) and Ch (A f = −0.210 and A f = −0.250 in 2007 and in 2010, respectively), and a better cup quality with a lower bitterness level (A f = −0.106 in 2010). The R 2 values of the QTLs for Bi ranged from 9.2 to 27.3 %. The QTLs on LGs B, E, and G were detected both in 2007 and 2010.
Finally, four regions with major co-localizations of QTLs for NIR traits were underscored at the end of LG E (nine QTLs), at the end of LG A (seven QTLs), at the top of LG B (seven QTLs), and on LG G (five QTLs). The high number of QTLs detected in these regions was partly explained by the stability of the QTLs over the years (2007 and 2010) and the significant correlations between the NIR traits (Table 2). All QTLs detected for the NIR traits had from minor to major effects with the R 2 varying from 8.3 to 27.3 % and showed additive effects superior to dominance effects, originating predominately from FRT58 (48 %) and secondarily from FRT67 (31 %) and FRT51 (21 %).
A set of 30 QTLs for field traits was detected. Eleven QTLs (10, 11, 67, 81, 82, 83, 84, 98, 99, 131, and 132, Table S3) were found for bean characteristics such as the 100-bean weight (Bw) and five QTLs (9, 117, 118, 129, and 130) for percentage of peaberries (Pe). Among these 16 QTLs, 14 QTLs were stable over the years, and thus detected in 2007 and 2010. They were distributed into six LGs (A, E, F, G, I, and J) and their R 2 reached up to 20.5 %. The FRT58 parent carried the favorable alleles for a higher 100-bean weight for six QTLs. The alleles controlling a lower percentage of peaberries were carried by the female FRT58 or FRT67 parents.
Eight QTLs were detected for traits linked to yield predictors (Table S3). Two QTLs (14 and 39) were identified for the internode length (In) in LGs A and B, three QTLs (72, 141 and 142) for stem diameter (St) in LGs E and K, and three QTLs (42, 86 and 87) for the number of primary branches (Br) in LGs B and F. Their R 2 values ranged from 9.5 to 22.5 %. Female additive effects were predominant for both QTLs detected for In. The QTLs identified for the St on LG K were detected both in 2006 and 2007. FRT58 was the favorable parent for a large stem. The two QTLs controlling Br detected from the two progenies in LG F were not located in the same region, and the origin of their additive predominant effect was different.
Six yield QTLs (15, 44, 90, 103, 134, and 143: Table S3) were detected in 4 years (2007, 2008, 2009 and 2011) on six LGs A, B, F, G, J, and K. These QTLs were not consistent over the years, and their R 2 ranged from 9.6 to 14.9 %. Three QTLs (15, 44, and 90) on LGs A, B, and F were co-localized with the QTLs detected for yield predictor traits such as internode length (In) and number of primary branches (Br), and for bean characteristics such as bean weight (Bw).
Finally, two regions with major co-localizations of QTLs for field traits were found at the end of LG A (five QTLs) and at the end of LG J (five QTLs). All QTLs detected for the field traits showed minor to major effects with the R 2 varying from 7.3 to 22.5 % and showed additive effects superior to dominance effects, originating predominately from FRT58 (58 %) and secondary from FRT67 (23 %) and FRT51 (19 %).
Model per population using MCQTL—model II
Estimated as 3.6 in the permutation tests, the QTL threshold values were always very similar, whatever the cross, trait, or QTL model (additive and dominance). A total of 97 QTLs were identified using this second model, with 52 QTLs and 34 QTLs detected from FRT58 × FRT51 and FRT67 × FRT51, respectively, and 11 QTLs shared between the two progenies (QTL 19, 29, 59, 62, 63, 65, 90, 98, 105, 108, and 123: Table S3). Among these 97 QTLs, 77 were detected using both additive and dominance models, 13 QTLs were only detected with the additive model, and seven QTLs were only identified by the dominance model. The majority of the QTLs showed additive effects superior to dominance effects. But only 25 % of the dominance effects detected were significant at 5 %. However, dominance effects were prevalent for two QTLs on LGs B and C (28 and 49: Table S3) linked to the variation of the number of nodes on primaries (No). Finally, using this model II, 58 QTLs were detected for NIR traits and 39 QTLs for field traits (Table S3).
As for the previous QTL model I, QTLs for NIR traits were detected on the four main QTL locations, at the end of LG E (nine QTLs), at the top of LG B (seven QTLs), at the end of LG A (six QTLs), and on the LG G (four QTLs). Two regions with major co-localizations of QTLs for field traits were noticed at the end of LG A (four QTLs) and at the end of LG J (five QTLs) as highlighted by model I. However, six QTLs (27, 28, 29, 49, 116, and 140) linked to yield predictors such as number of cherries per node and number of nodes on primaries (Cn and No) were only detected by model II.
The majority of the QTLs were shared between the two models per population. However, some differences were observed, thanks to the use of different QTL detection methods.
In fact, the model per population using MCQTL (model II) served to reveal the following:
-
1.
All QTLs detected in the model per population using MapQTL (model I) except for six QTLs (3, 9, 15, 55, 60, and 112: Table S3 and Fig. 1a),
Fig.1 
a Comparison of the three QTL detection models. Number of QTLs for all the traits detected specifically in single analyses with MapQTL (model I), single analyses with MCQTL (model II), multi-population connected analyses (model III), or in common with different models. b Distribution of R 2 values of the 114 QTLs detected with the model III dissociating the QTLs only detected with this model and the QTLs shared between the other two models
-
2.
Twenty-three additional QTLs (12, 17, 18, 27, 28, 29, 34, 41, 49, 56, 58, 61, 68, 85, 101, 110, 114, 116, 120, 123, 136, 137, and 140: Table S3 and Fig. 1a) undetected by model I.

Connected model using MCQTL—model III
The threshold value of 3.6 estimated for this model was very similar no matter which character or QTL model (additive or dominance) was analyzed. A total of 114 QTLs were detected, including 74 QTLs shared between the additive and dominance models. Twenty-five QTLs only identified with the additive model and 15 QTLs only highlighted with the dominance model (Table S3). The majority of these QTLs showed additive effects superior to dominance effects. In addition, only 35 % of the dominance effects detected were significant at 5 %. However, dominance effects were predominant for 10 QTLs on LGs A (8 and 9), C (45 and 46), F (75), J (121, 128, 131, and 134), and K (139). Using this model III, 68 QTLs were detected for NIR traits and 46 QTLs for field traits (Table S3).
As for the two previous QTL models, QTLs for NIR traits were detected with QTL locations at the top of LG B (10 QTLs), on the end of LG E (eight QTLs), at the end of LG A (six QTLs), and on LG G (five QTLs). However, two new areas were detected by model III, at the top of LG H (five QTLs: 105, 106, 108, 109, and 111) and on LG J (five QTLs: 121, 123, 124, 128 and 133). For the field traits, two regions with major QTL locations were found at the end of LG A (seven QTLs) and at the end of LG J (five QTLs) as for the two previous models I and II.
The connected model using MCQTL (model III) detected the following:
-
1.
All QTLs detected in the model per population using MapQTL (model I) and MCQTL (model II) except for 29 QTLs (Fig. 1a). Among the 29 QTLs not detected by model III, two QTLs (3 and 55) were only detected by model I, 13 QTLs (12, 17, 27, 28, 34, 49, 56, 58, 85, 101, 110, 116, and 136) were only detected by model II, and 14 QTLs (44, 50, 52, 64, 69, 76, 82, 84, 95, 107, 127, 135, 138, and 143) were common to both model I and model II. Furthermore, most of these QTLs had a test value just below the threshold of the multi-population connected model, suggesting a dilution effect in the connected model III.
-
2.
Forty additional QTLs detected neither by model I nor by model II. These 40 QTLs generally had R 2 values among the lowest detected in the connected model III (Fig. 1b). This increase of QTL detection, with R 2 values generally less than 10 %, was certainly due to the power of the connected model.
-
3.
Additional information on the comparison of the genetic effects of the different parents in this connected model.
Overview of QTL analyses
A total of 143 QTLs (Fig. 1a) were detected in our study from which 60 were common to the three models. Moreover, 28 QTLs were shared between two models and 55 were specific to a given model with 40 found exclusively in the connected analysis (model III). Figure 2 shows the result of the overview of QTL detection charted on the C.canephora genetic map. QTLs involved in bean biochemical composition were distributed across all LGs except LG H, but six major genomic regions (LGs A, B, E, G, H, and J) with high R 2 values (>12 %) were highlighted. These six QTL hotspots were also involved in the bitterness trait. This result suggests either pleiotropic effect from single gene or the presence of different genes in the same genome area. For field traits, the QTLs were located across all the LGs with co-locations on LGs A, B, F, and J. However they generally had medium-range R 2 values (<12 %) except on LG F for its 100-bean weight. None of the yield QTLs showed higher R 2 values than 10 %, and these QTLs were not consistently detected over the 7 years of the analysis. But secondary yield component traits such as the 100-bean weight showed reliable detection in the 2 years of data recording with high R 2 values.

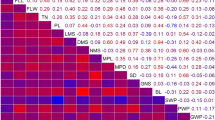
Heat map of the detected QTLs with the three QTL detection models. The chromosomes and the traits are represented in columns and in rows, respectively. The QTLs detected by the model connected were represented on the map with the characteristics (confidence region and R 2 value) of this model by an opaque rectangle. When the QTLs were detected by the per population models only, the QTL with the tightest confidence region was kept to be represented by a translucent rectangle. A color state is used to indicate the percentage of variance explained by the QTL (R 2)
Discussion
Genetic map construction
For cross-pollinated populations such as Robusta coffee, the type of marker segregation varies across the loci. Up to four different alleles may be segregating. For this reason, we used co-dominant markers (SSRs) to facilitate both the map construction and QTL detection (Van Ooijen 2006; Van Ooijen 2009; Jourjon et al. 2005). This type of approach was proved to be successful in other plant species (Clement et al. 2003; Souza et al. 2013). In our study, the two bi-parental maps were similar for both genetic map sizes and number of loci mapped. In the end, the consensus map generated included 249 loci covering 1,201 cM. Furthermore, nearly 40 % of the markers from the consensus map were shared between the two mappings from which QTL analyses could be compared.
Similar Robusta genetic maps have been already published (Paillard et al. 1996; Lashermes et al. 2001; Lefebvre-Pautigny et al. 2010; Leroy et al. 2011) for various genetic studies including syntenic and QTL analyses. Since 2010, the coffee community has adopted the same genetic framework by numbering the 11 linkage groups from A to K, to be able to compare the various genetic studies (submitted for publication in Science) more easily.
Comparison of the three QTL detection models
The analyses per population using MapQTL and MCQTL detected nearly the same set of core QTLs. However, the power of MCQTL was found superior, with an increase of 21 % for the number of QTLs detected. The two software applications do not have the same multiple QTL model. They both begin with a forward search of co-factors; then, MapQTL conducts a MQM mapping (Jansen 1993; Jansen and Stam 1994), while MCQTL uses an iQTLm algorithm (Charcosset et al. 2000). This could explain part of the difference between the results since the iQTLm algorithm analyzes more multiple QTL models than MQM. Moreover, they do not use the same modeling of the putative QTL. MapQTL uses a mixture model while MCQTL uses a regression model. The mixture model is slightly more powerful with major QTL with sparse mapping and small sample sizes, but the two models were proved to be asymptotically equivalent (Rebai et al. 1995) in other cases. In our analyses, the largest interval between two adjacent markers is 33 cM, QTL effects were small or medium (maximum of explained variance 27.9 %), and the sample size was large which led to the near equivalence of the two models. A large part of the power superiority of MCQTL is probably due to the use of an additive QTL effect model, while MapQTL only proposes an additive and dominance. Rebai and Goffinet (1993) showed that it is a powerful strategy to assume that QTLs are additive driven even if dominance effects could exist. In our analyses, 48 % (11/23) of the supplementary QTL detected by MCQTL and not with MaqQTL were detected with the additive model alone.
The advantages of QTL mapping with connected populations have regularly been discussed. They concern the possibility of ranking and studying more than two parental alleles (Lariepe et al. 2012), the reduction of the confidence region that leads to an increase in precision (Espinoza et al. 2012).
For example, in our study, significant different parental allele effects for the chlorogenic acid content in beans (QTL 6) were detected from the three parental FRT clones. These results allowed the breeder to select among the most relevant parental alleles to be used in the new breeding cycle for the trait targeted. The decrease in the confidence region length was small (10 % on average) for connected populations when comparing the shared QTLs from single-population analyses with MCQTL. However, looking closer at the confidence regions, three QTL (QTL 13, 90, and 125) appeared to have a much larger confidence region with the multi-population analyses. They were resolved in two close QTLs by a multi-QTL search beginning with two co-factors on the linkage group. When these three QTLs were discarded, the decrease in the confidence region length jumped to a level of 20 %. The gain in power of the multi-population QTL mapping was clear in our study with an increase of 43 and of 18 % for the number of QTLs detected in comparison to the per population analyses with MapQTL and MCQTL, respectively. However, the risk of dilution of small QTL effects that was highlighted by simulations (Muranty 1996) and observed in an oil palm factorial design with five parents (Billotte et al. 2010) was also present in our design despite the two large connected populations which should have limited this risk. Only 11.3 % (11/97) of the QTLs detected by the model MCQTL per population were shared between the two progenies. This observation could threaten to produce a strong dilution effect in the connected model. However, 72 % (70/97) of the QTLs detected by this model per population were found by the connected model. And, as expected, QTLs shared between the per population and the multi-population analyses had higher average percentages of explained variance and smaller confidence regions than those detected by the per population analyses alone.
QTL detection on coffee in the literature
Previous QTL studies related to biochemical traits for coffee have already been performed. In 2013, Ky et al., using an interspecific cross (Coffea pseudozanguebariae × Coffea liberica var. Dewevrei) identified two QTLs involved in the caffeine bean content on a genetic map with AFLP and RFLP markers. However, the lack of common markers with our genetic maps built only with SSR markers disallows making a comparison between the two studies.
However, seven QTLs related to yield and quality (LGs A, B, I, J, and K) detected in our study also appear to be mapped in areas where Robusta QTLs had already been identified by Leroy et al. (2011). Indeed, common markers between these two studies offer the possibility to compare these two analyses. These QTLs are involved in traits of bean biochemical composition such as the caffeine (QTL 112) and chlorogenic acid content (QTL 5, 6, 25, and 128), bitterness (QTL 119), and yield (QTL 143). The co-localization of QTLs for the same traits identified under different environments and with different genetic backgrounds support the interest in a candidate gene approach. The availability of a complete genome sequence of Robusta in the near future will allow us to identity genes more precisely in relation to the QTLs detected in these studies.
Marker-assisted selection on quality and yield for Robusta
Up to now, all Robusta QTL analyses linked to key economic traits such as the quality and field yield have been performed on Guinean and Congolese genotypes (Leroy et al. 2011). The first breeding programs used these two origins for reciprocal recurrent selection (Leroy et al. 1993). Results of these intergroup hybrid trials indicated high yield and vigor, demonstrating the efficiency of this approach. But recent molecular studies based on SSR markers demonstrated that the Robusta genetic diversity is much more important with seven groups including Guinean and Congolese origins (Dussert et al. 1999; Cubry et al. 2008; Gomez et al. 2009; Musoli et al. 2009). Taking this new classification into consideration, breeders will be able to use the genetic diversity available to create mating schemes for determining the heterotic performance of hybrids by assessing marker-based parental groups. This type of experimental design could be of interest for field traits such as yield and disease resistance (Xie et al. 2014). However, quality traits could also be managed by this type of study since the biochemical composition throughout the genetic groups is highly variable, especially for caffeine and chlorogenic acids (Ky et al. 2001; Campa et al. 2004). Indeed, these biochemical compounds have been already described as important for cup quality (Tessema et al. 2011).
The introgression of QTLs using the marker-assisted selection will require a further step of validation in the different genetic backgrounds in order to keep the most robust and reproducible ones. Once the linked markers to these QTL have been identified, they can be used to predict a quantitative trait and could consequently be at the origin of a MAS program (Collard and Mackill 2008). It will be quicker and more efficient to genotype a population than to phenotype it for complex traits such as cup quality or disease resistance which require mature plants from bean harvest and sensory analyses or intensive pathological studies. Another key advantage of a MAS program is that the screening phase can be performed at the seed level in order to transplant only coffee trees with the desirable characters in the field.
Ideally in the end, MAS can also be used to pyramid genes from multiple genetic parental populations, as demonstrated in cereals for disease resistance (Werner et al. 2005). The subsequent step in coffee breeding will be to focus on validating QTL assessment throughout the various genetic groups characterized in Robusta. This step is certainly important for any efficient breeding program wishing to take advantage of the wide genetic diversity available.
Abbreviations
- Bi:
-
Bitterness
- Br:
-
Number of primary branches
- Bw:
-
100-bean weight
- Ca:
-
Caffeine content in beans
- Ch:
-
Chlorogenic acid content in beans
- Cn:
-
Number of cherries per node
- CP:
-
Cross-pollinated
- In:
-
Internode length
- LG:
-
Linkage group
- Li:
-
Lipid content in beans
- LOD:
-
Log of odds
- MAS:
-
Marker-assisted selection
- NIR:
-
Near-infrared
- No:
-
Number of nodes on primaries
- Pe:
-
Percentage of peaberries
- Pr:
-
Protein content in beans
- QTL:
-
Quantitative trait loci
- RH:
-
Relative humidity
- SSR:
-
Simple sequence repeat
- St:
-
Stem diameter
- Su:
-
Sucrose content in beans
- Tr:
-
Trigonelline content in beans
- Yi:
-
Yield
References
Akaffou DS, Ky CL, Barre P, Hamon S, Louarn J, Noirot M (2003) Identification and mapping of a major gene (Ft1) involved in fructification time in the interspecific cross Coffea pseudozanguebariae x C. liberica var. Dewevrei: impact on caffeine content and seed weight. Theor Appl Genet 106:1486–1490
Ben Sadok I, Celton JM, Essalouh L, El Aabidine AZ, Garcia G, Martinez S, Grati-Kamoun N, Rebai A, Costes E, Khadari B (2013) QTL mapping of flowering and fruiting traits in olive. PLoS One 8:e62831
Berthaud J (1986) Les ressources génétiques pour l’amélioration des caféiers africains diploïdes. Editions de l'ORSTOM, Paris
Billotte N, Jourjon MF, Marseillac N, Berger A, Flori A, Asmady H, Adon B, Singh R, Nouy B, Potier F, Cheah SC, Rohde W, Ritter E, Courtois B, Charrier A, Mangin B (2010) QTL detection by multi-parent linkage mapping in oil palm (Elaeis guineensis Jacq.). Theor Appl Genet 120:1673–1687
Blanc G, Charcosset A, Mangin B, Gallais A, Moreau L (2006) Connected populations for detecting quantitative trait loci and testing for epistasis: an application in maize. Theor Appl Genet 113:206–224
Blanc G, Charcosset A, Veyrieras JB, allais A, reau L (2007) Marker-assisted selection efficiency in multiple connected populations: a simulation study based on the results of a QTL detection experiment in maize. Euphytica
Campa C, Ballester JF, Doulbeau S, Dussert S, Hamon S, Noirot M (2004) Trigonelline and sucrose diversity in wild Coffea species. Food Chemistry 88:39–43
Capot J (1964) La pollinisation artificielle des caféiers allogames et son rôle dans leur amélioration. pp. 75–88
Charcosset A, Mangin B, Moreau L, Combes L, Jourjon MF, Gallais A (2000) Heterosis in maize investigated using connected RIL populations. Quant Genet Breed Methods: the way ahead 96:89–98
Clement D, Risterucci AM, Motamayor JC, N’Goran J, Lanaud C (2003) Mapping QTL for yield components, vigor, and resistance to Phytophthora palmivora in Theobroma cacao L. Genome 46:204–212
Collard BCY, Mackill DJ (2008) Marker-assisted selection: an approach for precision plant breeding in the 21st century. Philos Trans R Soc B 363:557–572
Coulibaly I, Noirot M, Lorieux M, Charrier A, Hamon S, Louarn J (2002) Introgression of self-compatibility from Coffea heterocalyx to the cultivated species Coffea canephora. Theor Appl Genet 105:994–999
Coulibaly I, Louarn J, Lorieux M, Charrier A, Hamon S, Noirot M (2003) Pollen viability restoration in a Coffea canephora P. and C. heterocalyx Stoffelen backcross. QTL identification for marker-assisted selection. Theor Appl Genet 106:311–316
Cubry P, Musoli P, Legnate H, Pot D, De BF, Poncet V, Anthony F, Dufour M, Leroy T (2008) Diversity in coffee assessed with SSR markers: structure of the genus Coffea and perspectives for breeding. Genome 51:50–63
Davis AP, Govaerts R, Bridson DM, Stoffelen P (2006) An annotated taxonomic conspectus of the genus Coffea (Rubiaceae). Bot J Linn Soc 152:465–512
Davis AP, Tosh J, Ruch N, Fay MF (2011) Growing coffee: Psilanthus (Rubiaceae) subsumed on the basis of molecular and morphological data; implications for the size, morphology, distribution and evolutionary history of Coffea. Bot J Linn Soc 167:357–377
Development Core Team R (2012) R : a language and environment for statistical computing. R foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0
Doerge RW, Churchill GA (1996) Permutation tests for multiple loci affecting a quantitative character. Genetics 142:285–294
Dussert S, Lashermes P, Anthony F, Montagon C, Trouslot P, Combes MC, Berthaud J, Noirot M, Hamon S (1999) Le caféier, Coffea canephora. pp. 175–181
Espinoza LC, Huguet T, Julier B (2012) Multi-population QTL detection for aerial morphogenetic traits in the model legume Medicago truncatula. Theor Appl Genet 124:739–754
Gomez C, Dussert S, Hamon P, Hamon S, Kochko A, Poncet V (2009) Current genetic differentiation of Coffea canephora Pierre ex A. Froehn in the Guineo-Congolian African zone: cumulative impact of ancient climatic changes and recent human activities. BMC Evol Biol 9:167
Grattapaglia D, Sederoff R (1994) Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: mapping strategy and RAPD markers. Genetics 137:1121–1137
Haley CS, Knott SA (1992) A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Hered (Edinb) 69:315–324
Huck CW, Guggenbichler GK, Bonn GK (2005) Analysis of caffeine, theobromine and theophylline in coffee by near infrared spectroscopy (NIRS) compared to high-performance liquid chromatography (HPLC) coupled to mass spectrometry. Anal Chim Acta 538:195–203
IOC (2012) Monthly Coffee Market Report - December 2012
Jansen RC (1993) Interval mapping of multiple quantitative trait loci. Genetics 135:205–211
Jansen RC, Stam P (1994) High resolution of quantitative traits into multiple loci via interval mapping. Genetics 136:1447–1455
Jourjon MF, Jasson S, Marcel J, Ngom B, Mangin B (2005) MCQTL: multi-allelic QTL mapping in multi-cross design. Bioinformatics 21:128–130
Kawamura K, Hibrand-Saint OL, Crespel L, Thouroude T, Lalanne D, Foucher F (2011) Quantitative trait loci for flowering time and inflorescence architecture in rose. Theor Appl Genet 122:661–675
Ky CL, Louarn J, Dussert S, Guyot B, Hamon S, Noirot M (2001) Caffeine, trigonelline, chlorogenic acids and sucrose diversity in wild Coffea arabica L. and C. canephora P. accessions. Food Chem 75:223–230
Ky CL, Barre P, Noirot M (2013) Genetic investigations on the caffeine and chlorogenic acid relationship in an interspecific cross between Coffea liberica dewevrei and C. pseudozanguebariae. Tree Genet Genomes 9:1043–1049
Lariepe A, Mangin B, Jasson S, Combes V, Dumas F, Jamin P, Lariagon C, Jolivot D, Madur D, Fievet J, Gallais A, Dubreuil P, Charcosset A, Moreau L (2012) The genetic basis of heterosis: multiparental quantitative trait loci mapping reveals contrasted levels of apparent overdominance among traits of agronomical interest in maize (Zea mays L.). Genetics 190:795–811
Lashermes P, Couturon E, Moreau N, Paillard M, Louarn J (1996) Inheritance and genetic mapping of self-incompatibility in Coffea canephora Pierre. Theor Appl Genet 93:458–462
Lashermes P, Combes MC, Prakash NS, Trouslot P, Lorieux M, Charrier A (2001) Genetic linkage map of Coffea canephora: effect of segregation distortion and analysis of recombination rate in male and female meioses. Genome 44:589–596
Lefebvre-Pautigny F, Wu F, Philippot M, Rigoreau M, Priyono ZM, Frasse P, Bouzayen M, Broun P, Pétiard V, Tanksley S, Crouzillat D (2010) High resolution synteny maps allowing direct comparisons between the coffee and tomato genomes. Tree Genet Genomes 6:565–577
Leroy T, Montagnon C, Charrier A, Eskes AB (1993) Reciprocal recurrent selection applied to Coffea canephora Pierre. Characterization and evaluation of breeding populations and value of intergroup hybrids. Euphytica 67:113–125
Leroy T, Ribeyre F, Bertrand B, Charmetant P, Dufour M, Montagnon C, Marraccini P, Pot D (2006) Genetics of coffee quality. Braz J Plant Physiol 18:229–242
Leroy T, De Bellis F, Legnate H, Kananura E, Gonzales G, Pereira LP, Andrade AC, Charmetant P, Montagnon C, Cubry P, Marraccini P, Pot D, Kochko A (2011) Improving the quality of African robustas: QTLs for yield- and quality-related traits in Coffea canephora. Tree Genet Genomes 7:781–798
Melchinger AE, Utz HF, Schon CC (1998) Quantitative trait locus (QTL) mapping using different testers and independent population samples in maize reveals low power of QTL detection and large bias in estimates of QTL effects. Genetics 149:383–403
Muranty H (1996) Power of tests for quantitative trait loci detection using full-sib families in different schemes. Heredity 76:156–165
Musoli P, Cubry P, Aluka P, Billot C, Dufour M, De BF, Pot D, Bieysse D, Charrier A, Leroy T (2009) Genetic differentiation of wild and cultivated populations: diversity of Coffea canephora Pierre in Uganda. Genome 52:634–646
N’Diaye A, Noirot M, Hamon S, Poncet V (2007) Genetic basis of species differentiation between Coffea liberica Hiern and C. canephora Pierre: analysis of an interspecific cross. Genet Resour Crop Evol 54:1011–1021
Paillard M, Lashermes P, Pétiard V (1996) Construction of a molecular linkage map in coffee. Theor Appl Genet 93:41–47
Pauly L, Flajoulot S, Garon J, Julier B, Beguier V, Barre P (2012) Detection of favorable alleles for plant height and crown rust tolerance in three connected populations of perennial ryegrass (Lolium perenne L.). Theor Appl Genet 124:1139–1153
Pierre JB, Huguet T, Barre P, Huyghe C, Julier B (2008) Detection of QTLs for flowering date in three mapping populations of the model legume species Medicago truncatula. Theor Appl Genet 117:609–620
Plechakova O, Tranchant-Dubreuil C, Benedet F, Couderc M, Tinaut A, Viader V, De BP, Hamon P, Campa C, De KA, Hamon S, Poncet V (2009) MoccaDB—an integrative database for functional, comparative and diversity studies in the Rubiaceae family. BMC Plant Bio 9:123
Priyono ND (2013) Identification of quantitative trait loci (QTLs) determining flowering in the Robusta coffee (Coffea canephora Pierre). J Agric Sci Technol B 3:296–305
Priyono FB, Rigoreau M, Ducos JP, Sumirat U, Mawardi S, Lambot C, Broun P, Pétiard V, Wahyudi T, Crouzillat D (2010) Somatic embryogenesis and vegetative cutting capacity are under distinct genetic control in Coffea canephora Pierre. Plant Cell Rep 29:343–357
Rebai A, Goffinet B (1993) Power of tests for QTL detection using replicated progenies derived from a diallel cross. Theor Appl Genet 86:1014–1022
Rebai A, Goffinet B, Mangin B (1995) Comparing power of different methods for QTL detection. pp. 87–99
Sosnowski O, Charcosset A, Joets J (2012) BioMercator V3: an upgrade of genetic map compilation and quantitative trait loci meta-analysis algorithms. Bioinformatics 28:2082–2083
Souza LM, Gazaffi R, Mantello CC, Silva CC, Garcia D, Le Guen V, Cardoso SE, Garcia AA, Souza AP (2013) QTL mapping of growth-related traits in a full-sib family of rubber tree (Hevea brasiliensis) evaluated in a sub-tropical climate. PLoS One 8(4)
Sumirat U, Bellanger B, L’Anthoëne V, Mawardi S, Nugroho D, Priyono, Wahyundi T, Broun P, Lambot C and Crouzillat D (2012). Genetic diversity assesment in Indonesian Coffea canephora collection using SSR markers. The 24th International Conference on Coffee Science, San José, Costa Rica from 12 to 16 November 2012
Tessema A, Alamerew S, Kufa T, Garedew W (2011) Variability and association of quality and biochemical attributes in some promising Coffea arabica germplasm collections in southwestern Ethiopia. Int J Plant Breeding Genet 5:302–316
Van Ooijen JW (2006) JoinMap ® 4, Software for the calculation of genetic linkage maps in experimental populations. Kyazma B.V, Wageningen, Netherlands
Van Ooijen JW (2009) MapQTL ® 6, Software for the mapping of quantitative trait loci in experimental populations of diploid species. Kyazma B.V, Wageningen, Netherlands
Voorrips R (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. 93:77–78
Werner K, Friedt W, Ordon F (2005) Strategies for pyramiding resistance genes against the barley yellow mosaic virus complex (BaMMV, BaYMV, BaYMV-2). Mol Breed 16:45–55
Xie F, He Z, Esguerra M, Qiu F, Ramanathan V (2014) Determination of heterotic groups for tropical Indica hybrid rice germplasm. Theor Appl Genet 127:407–417
Acknowledgments
The authors are grateful to Julio Torres and his team in the experimental coffee station located in Ecuador involved in trial management, plant sample supply, and the phenotypic observations of the F1 populations. Our thanks are extended to Stéphane Michaux and Blandine Landré for the NIR analyses. We also thank Laurence Pauly and Philippe Barre (INRA, Lusignan) for the helpful advice on QTL detection, the students from the University of Poitiers (Mathilde Forêt, Céline Debuiselle, Amélie Ricros and Pascal Gonidec) for the development of a plug-in of MCQTL, and Christine Tranchant-Dubreuil and Valérie Poncet (IRD, Montpellier) for integrating molecular marker sequences into the MoccaDB database. Finally, we thank Alexandre de Kochko and Valérie Poncet (IRD, Monptellier) for their helpful comments on the manuscript.
Data archiving statement
The SSRs sequences have been submitted to NCBI (http://www.ncbi.nlm.nih.gov/), Sol Genomics (http://solgenomics.net/), and MoccaDB (https://moccadb.mpl.ird.fr/) databases. The consensus genetic map has been submitted in MoccaDB (https://moccadb.mpl.ird.fr/).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by D. Grattapaglia
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Mérot-L’Anthoëne, V., Mangin, B., Lefebvre-Pautigny, F. et al. Comparison of three QTL detection models on biochemical, sensory, and yield characters in Coffea canephora . Tree Genetics & Genomes 10, 1541–1553 (2014). https://doi.org/10.1007/s11295-014-0778-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-014-0778-1