
Abstract
To trace the genetic history of porcine reproductive and respiratory syndrome virus (PRRSV), we determined the complete sequences of ORFs 5 to 7 of four PRRSV isolates. These sequences were analyzed together with published sequences from 146 isolates from various parts of the world using a Bayesian coalescent approach as well as Bayesian inference and maximum-likelihood methods. All of the European-type (EU-type) viruses were classified into one of two groups or unclassified (4 isolates), while all North American–type (NA-type) viruses belonged to one of three major groups or were unclassified (5 isolates). Within each genotype, no apparent periodic and/or geographic influence on the evolution of PRRSVs was observed. The evolutionary rate of PRRSV isolates was estimated to be 1.55 × 10−3 substitutions/site/year, and the time of the most recent common ancestor (TMRCA) was 491.2 years ago. Here, the TMRCA for the EU- and NA-type viruses was 58.7 and 62.6 years ago, respectively. A Bayesian skyline plot revealed that the viruses evolved at an almost constant population size until the late 1970s, when they experienced a population expansion that continued until the late 1980s. The population size then remained constant again until the early 2000s, when a rapid, sharp decline in the effective number of infections occurred.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Porcine reproductive and respiratory syndrome (PRRS) is one of the most notorious diseases in the global swine industry [5]. It is characterized by reproductive failure in sows and respiratory illness in young pigs [21]. The causative agent, PRRS virus (PRRSV), is classified as a member of the order Nidovirales, family Arteriviridae, genus Arterivirus. PRRSV is an enveloped, single-stranded, positive-sense RNA virus [34]. The linear genome of about 15 kb consists of nine open reading frames (ORFs), two of which encode polymerases (ORF 1a and ORF 1b) and seven of which encode structural genes (ORF 2a, 2b, 3, 4, 5, 6, and 7). Among them, ORF 5, 6, and 7 encode the three major structural proteins, glycoprotein 5 (GP5), membrane (M) protein, and nucleocapsid (N) protein, respectively [5]. GP5 is the major virus antigen associated with the development of neutralizing antibodies and protection [41], and it is often used as a phylogenetic marker. The M protein is thought to be associated with the development of strong cellular immunity [2]. Recently, Jiang et al. [16] reported that DNA vaccines co-expressing two major structural proteins (GP5 and M proteins) with two promoters displayed enhanced immunogenicity. Zheng et al. [45] also suggested co-expression of GP4, GP5, and M proteins of vaccine virus, Ankala. The remaining major structural protein, N protein, is the most abundant and most immunogenic protein in the virion [9] and is an important target for virological detection by PCR [15] and serological detection by ELISA [30], as well as being a phylogenetic marker [44].
PRRSV can be divided into two distinct genotypes, type 1 (European, EU) and type 2 (North American, NA), with Lelystad virus and VR-2332, respectively, as their prototype strains [42]. Also, although the disease symptoms are similar following infections with these viruses, they are antigenically and genetically very different [23].
Since the first identification of PRRSV in the United States in 1987 [17] and in Europe in 1990 [24], many global studies have been published on the basis of its gene [e.g., 25, 40, 46] and genome [e.g., 4, 22, 47] sequences. However, the evolutionary phylogenetics of PRRSV remains controversial [10, 11, 14, 26, 31–33].
To further explore the genetic history of the PRRS virus, we determined the complete ORF 5 to 7 sequences from four Korean NA-type isolates and analyzed them together with published sequences for 23 EU-type (one from Korea) and 123 NA-type (three from Korea) isolates from different parts of the world. The first objective of this study was to examine the key mechanisms that drive the evolution of this virus, such as recombination and selection pressure. Subsequently, Bayesian inferences (BI) approaches as well as the maximum-likelihood (ML) method were used to investigate the phylogeny of PRRSVs. Finally, a Bayesian coalescent analysis was conducted to estimate substitution rates, divergence times, and changes in population size.
Materials and methods
Sample and data collection
Using MARC-145 cells, four type-2 PRRSV strains were isolated from three Korean prefectures (Gyeonggi, Gangwon, and Chungnam) in 2007, 2009, and 2010. The PRRSV isolates, CP07-401-9 (passage 9), CP07-626-2 (passage 13), e417-2 (passage 5), and A4699 (passage 9) were used for sequencing ORFs 5 to 7. The complete ORF 5 to 7 sequences (all NA type) were analyzed together with published sequences from four Korean isolates (one EU type and three NA type) and 142 non-Korean PRRSV isolates (22 EU type and 120 NA type). The PRRSVs utilized in the present study are listed in Table S1.
RNA extraction, RT-PCR, and sequencing
The PRRSV isolates in this study were cultured on MARC-145 cells in Dulbecco’s modified Eagle’s medium (5 % fetal bovine serum) for 5 days and harvested by two rounds of freezing and thawing and centrifugation (3,000 rpm for 20 min) when cytopathic changes were observed in about 80 % of the cells. The harvested viruses were used for RNA extraction. Total RNA was extracted from the virus stocks using TRIzol® LS (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. The cDNA was synthesized with random hexamer using a SuperScript® III First-Strand Synthesis Kit (Invitrogen) according to the manufacturer’s instructions. For sequencing ORFs 5 to 7, the following primers were selected from a previous study (numbers correspond to the positions in the North American prototype PRRSV strain [VR-2332] [47]): (a) for the ORF 5 gene, O5P1 (13,671–13,693), 5-TCCTACTGGCAATTTGAATG-3, and O5P2 (14,369-14,390), 5-CCTTTAGAGCATATATCATCAC-3; (b) for the ORF 6 gene, O6P1 (14,271-14,292), 5-GTTTCAGCGGAACAATGGGGTC-3, and O6P2 (14,820-14,840), 5-GCACAGCTGATTGACTGGCTG-3; and (c) for the ORF 7 gene, O7P1 (14,751-14,770), 5-TGGGTGGCAGAAAAGCTGTT-3, and O7P2 (15,219-15,240), 5- GTGTCAATCAGTGCCATTGACC-3. PCR was performed separately with each primer set using a Maxime PCR Premix Kit (Intron, Seongnam, Korea). The PCR amplification conditions were 94 °C for 2 min followed by 40 cycles at 94 °C for 10 s, 58 °C for 30 s, and 72 °C for 30 s, followed by a final elongation step at 72 °C for 7 min. The amplified fragments were purified using a QIAquick Gel Extraction Kit (QIAGEN, Valencia, CA) and sequenced in both directions by Macrogen Company (Seoul, Korea) using the corresponding ORF 5, ORF 6, and ORF 7 PCR primers. The complete ORF 5 to 7 sequences from each isolate determined in this study were submitted to GenBank under accession numbers JN809804–JN809807.
Sequence, recombination, and selection pressure analysis
After sequencing three fragments, each fragment was assembled with overlapping sequences to obtain the complete ORF 5 to 7 sequences. These sequences were initially aligned using CLUSTAL X 1.81 [39]. For each genetic region, as well as the entire ORF 5-through-7 region, the following calculations were made from the dataset using the programs PUZZLE 4.0.2 [36] and Modeltest 3.7 [27]: total sites, conserved sites, variable sites, base frequencies, and transition/transversion ratios. The nucleotide and amino acid sequence identities among the PRRSV isolates were also estimated using BIOEDIT 7.0.9 [13].
To detect putative recombinants, the identification of likely parental sequences and localization of possible recombination breakpoints were performed using the RDP 3.0b41 package [19] with the default parameters. This package consists of six different recombination detection programs: RDP, GENECONV, MaxChi, BOOTSCAN, Chimeara, and SiScan. To exclude the possibility of detecting false-positive recombinants, we considered only putative recombinant sites detected by at least three methods. In all analyses, p < 0.05 was considered to indicate statistical significance.
To evaluate the selective pressure driving PRRSV evolution, nonsynonymous/synonymous substitution ratio (ω = dN/dS) values were estimated using ClustalX 1.81 [39], PAL2NAL [38], and the codeml program in the PAML 3.14.1 package [43].
Phylogeny inference
Phylogenetic analysis was carried out using two different analytical methods, BI and ML. For phylogenetic analysis based on the complete ORF 5 to 7 nucleotide sequences from the 150 PRRSVs, GTR+I+G was selected as the best-fit model using Akaike’s information criterion (AIC) in MODELTEST 3.7 [27]. The BI method was executed in MrBayes 3.1.2 [28] with the following options: nst, 6; rates, gamma; number of generations, 2,000,000; sample frequency, 100; number of chains, 4; burn-in generation, 20,000. To estimate the reliability of nodes, Bayesian posterior probability (BPP) values are shown on the BI tree. ML analysis was conducted using PHYML 3.0 [12], The parameters used for tree construction were as follows: model of nucleotide substitution, GTR; initial tree, BIONJ; nonparametric bootstrap analysis, yes, 500 replicates; proportion of invariable sites, estimated; number of substitution rate categories, 6; gamma shape parameter, estimated by program; optimize tree topology, yes.
Substitutions rates, divergence times, and population size changes
To investigate the evolutionary history of global PRRSVs, the rates of nucleotide substitutions, times of the most recent common ancestor (TMRCA), and changes in population size were co-estimated using the Bayesian Markov chain Monte Carlo (MCMC) approach as implemented in BEAST 1.5.3 [8]. The dataset for these analyses comprised the complete sequences of the ORF 5-to-7 region from 150 PRRSVs circulating worldwide during the last 22 years (1989–2010). Here, GTR+I+G and nst = 6 and rates = gamma were used as the best-fitting model and the likelihood setting, respectively, which were generated with MODELTEST 3.7 [27]. Subsequently, we employed both strict and relaxed (both uncorrelated exponential and uncorrelated lognormal) molecular clocks [7] with five different demographic models (constant size, exponential growth, expansion growth, logistic growth, and Bayesian skyline). Using the Bayesian factor test (log10 Bayes factors >2 in all cases) based on the relative marginal likelihoods of the models [37], the relaxed uncorrelated exponential clock and expansion population size models were selected as showing the best fit for the PRRSV datasets. The changes in effective population size over time were examined using the Bayesian skyline plot (BSP). The datasets were each run for 200,000,000 generations to ensure convergence of all parameters (ESSs>200) with discarded burn-in of 10 %. The resulting convergence was analyzed using Tracer 1.5 (http://beast.bio.ed.ac.uk/Tracer), and the statistical uncertainties were summarized in the 95 % highest probability density (HPD) intervals. Trees were summarized as maximum clade credibility trees using the TreeAnnotator program and visualized using FigTree 1.3.1 [29].
Results
Sequence analysis, selection pressure analysis, and recombination detection
The complete ORF 5 to 7 sequences from four Korean PRRSV isolates were determined. They all belonged to the NA genotype and were the same length (1,494 nucleotides: 603 for ORF 5, 525 for ORF 6, and 372 for ORF 7).
For a total of 150 global PRRSVs (23 EU and 127 NA types), the characteristics of the complete of ORF 5 to 7 region and their individual gene sequences are summarized in Table 1. The overall length of the alignment (including gaps) was 1,535 base pairs, and the deduced amino acid sequences were 502 residues in length. The complete sequences of the ORF 5 to 7 region of the PRRSVs revealed a very low degree of genetic similarity: 619 (40.3 %) of the nucleotides and 102 (20.3 %) of the amino acids were conserved. Pairwise comparisons demonstrated that the nucleotide sequence identity among those sequences ranged from 61.4 % to 100 %, corresponding to 59.6 %–100 %, similarity, respectively, at the amino acid level. Of the three individual genes, ORF 5 was the most variable (average sequence identity of 85.4 % and 83.2 % for nucleotides and amino acids, respectively), whereas ORF 6 was the most conserved (average sequence similarity of 89.8% and 93.2% for nucleotides and amino acids, respectively). The transition/transversion ratio estimated from sequences of the entire ORF 5 to 7 region was 3.71. Here, the transition/transversion ratio of the ORF 5 gene was the highest (3.80), whereas ORF 7 had the lowest ratio (3.39).
The complete ORF 5 to 7 sequences of the EU-type viruses were 1,509 nucleotides and 502 amino acids (including gaps) in length and revealed considerable genetic variation: 1,016 (67.3 %) of the nucleotides and 347 (69.1 %) of the amino acids were conserved. The nucleotide sequence identities among this type of isolates ranged from 87.2 % to 99.9 %, corresponding to 87.4–100 % identity at the amino acid level. Of the three individual genes, ORF 5 was the most variable (average sequence similarity, 88.8 % for nucleotides and 89.0 % for amino acid), whereas ORF 7 was the most conserved (average sequence identity, 93.3 % for nucleotides and 93.9 % for amino acids). The transition/transversion ratio estimated from the dataset was 6.20. The transition/transversion ratio of the ORF 7 gene was the highest (7.47), whereas that of the ORF 5 region was the lowest (6.15).
The total sites (including insertions) of the NA-type ORF 5 to 7 region had 1,494 base pairs and 500 deduced amino acids. In comparison with EU-type sequences, they had similar configurations: 914 (61.2 %) of the nucleotides and 308 (61.6 %) of the amino acids were conserved. Pairwise comparisons showed that the identities between NA-type isolates ranged from 87.5 % to 100 % for the nucleotide sequences and from 87.1 % to 100 % for the amino acid sequences. ORF 6, rather than ORF 7 in the EU type, was the most conserved (average sequence identity, 97.2 % for nucleotides and 98.2 % for amino acids). The transition/transversion ratio calculated from the entire NA-type virus dataset was 4.51. The transition/transversion ratio of the ORF 6 gene was the highest (5.61), and the ratio of ORF 5 was the lowest (4.17).
The nonsynonymous/synonymous substitution ratio (ω = dN/dS) value of the complete ORF 5 to 7 sequences of 150 global PRRSVs was 0.2229: 0.2372 for EU-type isolates and 0.2914 for NA-type viruses. Accordingly, our analysis showed that purifying selection acted on the ORF 5-to-7 sequences of both EU- and NA-type PRRSVs. Additionally, no recombination events was detected among the viruses.
Phylogenetic analysis
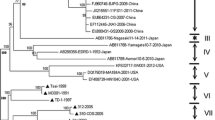
Ambiguous positions were excluded from the nucleotide alignment so that 1,514 nucleotide sites were used for the phylogenetic analysis, BI, and ML methods, which yielded consistent findings in terms of the phylogeny of PRRSV. The topology of the maximum clade credibility tree (Fig. 1) indicated that all of the global PRRSVs are divided into two different clades corresponding to the EU and NA genotypes (all posterior probabilities = 1.00 and bootstrap values = 100 %). Here, all of the EU-type viruses were divided into one of two groups (all posterior probabilities = 1.00) or were unclassified (4 isolates: JF276431, EU076704, JF304781, and GU047344). Group 1 contained a total of 14 isolates, with four from the United States, three from Spain, two from the Netherlands, and one each from Korea, China, Thailand, Germany, and Portugal during 1991–2007. All five isolates in Group 2 originated from China in 2009. Within the NA clade, all members belonged to one of three major groups (all posterior probabilities = 1.00) or were unclassified (5 isolates: AF325691, AF494042, AY545985, AY424271, and AY262352). Group 1 consisted of 90 Chinese viruses (70.9 % of all NA-type viruses) collected between 2002 and 2009. Group 2 was composed of six Chinese viruses from 1996 to 2008. Group 3 contained 26 isolates, with 12 from China, seven from Korea, four from the United States, and one each from Japan, Thailand, and Canada from 1989 to 2010.

Bayesian maximum clade credibility phylogenetic tree obtained from complete ORF 5 to 7 region sequences of 150 PRRSVs from around the world. The dataset (1,514 sites) was also analyzed phylogenetically by the BI and ML methods, and an identical topology was produced. The robustness of the phylogenetic analysis is presented above the nodes: numbers at the left represent Bayesian posterior probabilities (≥0.80), and those at the right represent ML bootstrap values (≥70 %). Divergence times (in years) are positioned below the nodes; the 95 % HPD intervals are indicated in brackets. Groups are marked by a “G”
Substitution rates, divergence times, and population size changes
The sequences of the ORF 5 to 7 region of PRRSVs collected during the past 22 years (1989–2010) were also analyzed using a Bayesian coalescent approach. The evolutionary rate of PRRSV isolates was estimated to be 1.55 × 10−3 (95 % HPD = 9.06 × 10−4–2.23 × 10−3) substitutions/site/year, projecting the TMRCA back to 491.2 years ago (95 % HPD = 199.6–864.4). Two distinct genotypes diverged at about the same time; the TMRCA estimated for the current diversity of EU-type viruses was 58.7 years ago (95 % HPD = 40.6–191.9), whereas that for the NA-type isolates was calculated at 62.6 years ago (95 % HPD = 41.5–153.7). In contrast, BSP analysis on the basis of the ORF 5-to-7 sequences of the isolates (Fig. 2) showed that the viruses appear to have evolved at an almost constant population size until the late 1970s, when they experienced a population expansion lasting until the late 1980s. The population size then remained constant again until the early 2000s, when a rapid, sharp decline in the effective number of infections occurred.

Bayesian skyline plot of global PRRSVs sampled between 1989 and 2010. The thicker black line represents the median estimate of the effective number of infections over time, and the thinner blue lines indicate the upper and lower bounds of the 95 % highest posterior density
Discussion
To date, the ORF 5-through-7 region has been regarded as the most important target for PRRSV studies involving PCR detection, immunology, and evolutionary phylogenetics. Here, to explore the genetic history of these viruses further, we determined the complete ORF 5-to-7 sequences from four Korean PRRSV isolates from 2007 to 2010 and then analyzed them together with 146 sequences from different parts of the world that were available in the GenBank database. Pairwise comparisons of the ORF 5 to 7 region confirmed that the virus is highly variable in sequence diversity: between intergenotypic sequences, maximum divergences of 38.6 % for nucleotides, and 40.4 % for amino acids were observed. Intra-dissimilarities of both genotypes were similar to each other. Maximum divergences among the EU-type isolates reached 12.8 % for the nucleotide sequences and 12.6 % for the amino acid sequences, respectively, and those among NA-type viruses were 12.5 % and 12.9 %, respectively. This indication is in agreement with previous studies, e.g., Stadejek et al. [35], An et al. [1], and Yoon et al. [44]. Domingo et al. [6] stated that RNA viruses have high mutation rates of 10−3 to 10−5 nucleotide substitutions per site per replication cycle due to their inaccurate RNA replication. In particular, of the three major structural genes, our analysis showed that ORF 5 was the most variable. This finding supports the results from previous studies [20, 35]. Because PCR detection sensitivity and vaccine development depend on sequence variability [18], continuous monitoring of the sequence variability of ORFs 5 to 7 could be taken into account for updating primers and for studies on immunology as well as epidemiology and evolutionary phylogenetics.
Regarding the selection pressure on PRRSVs, our results based on ORF 5 to 7 sequence data revealed that purifying selection acted on the viruses. That is, the nonsynonymous/synonymous substitution ratio (ω = dN/dS) value of the complete ORF 5 to 7 sequences from 150 global PRRSVs was 0.2229: 0.2372 for EU-type isolates and 0.2914 for NA-type isolates. This point confirms the views of Pesch et al. [25], while it contradicts the predictions of others such as Hanada et al. [14] and Song et al. [33], who demonstrated that the evolution of PRRSV was driven by positive selection. The exchange of different genetic sites between viruses by recombination, however, contributes to PRRSV genetic diversity and evolution, although in the present study, no pattern of recombination events was detected that was consistent across all PRRSVs.
Regarding the phylogenetic relationships among the PRRSVs, our results based on ORF 5 to 7 sequence data revealed two clearly defined clades corresponding to the EU and NA genotypes. Despite the striking genetic distances between two genotypes, their clinical symptoms are notably similar following infections with these viruses.
Next, this study focused on geographic and/or temporal influences on the evolution of PRRSVs. Recently, several phylogeny investigators such as Cha et al. [3] and Zhu et al. [47] documented that geographic separation is a factor influencing the PRRSV evolution, based on ORF 5 and genome sequences, respectively. However, the findings of the present study do not support their viewpoints; no apparent correlation was observed between country and/or time and the evolution of global PRRSVs. Within the EU-type clade, group 1 members were found in eight countries (the United States, Spain, the Netherlands, Korea, China, Thailand, Germany, and Portugal) during 1991–2007, although all isolates of group 2 originated from a single country (China) in 2009. Similar features were observed within the NA clade as well. Although all members of both group 1 and group 2 were collected in China, their sampling times were different: group 1 during 2002–2009 and group 2 during 1996–2008. Moreover, the group 3 viruses of the NA type occurred in six countries (China, Korea, the United States, Japan, Thailand, and Canada) during 1989–2010. These features are also consistent with the results of previous work [25, 44] and may be largely due to the rapid expansion and diversification of PRRSVs over a relatively short period of time and their rapid spread via the frequent international trade in livestock. The mixed population structure can make vaccine strategies more difficult. Thus, continuously screening for changes in the mixed population structure of this virus is needed.
The substitution rates and divergence times of PRRSV have been investigated in recent years from the molecular standpoint. Forsberg et al. [10], on the basis of the ORF 3 sequences of EU isolates, estimated mean evolutionary rates of 5.8 × 10−3 (95 % HPD = 4.8–6.9 × 10−3) and dated the most recent common ancestor to 1979, more than 10 years before the start of the European epidemic. Plagemann [26] analyzed ORF 5, ORF 7, and ORF 1b sequences and then postulated that the TMRCA was approximately 100 years ago. Plagemann suggested that a mutant of LDV (lactate dehydrogenase-elevating virus) infected wild boars in central Europe in 1912 and that this intermediate host spread the virus to North Carolina imports; the virus then evolved independently on the European and American continents in the wild boar populations for about 70 years until independently entering the domestic swine population. Subsequently, two debates have occurred [11, 14] on the evolutionary rate and the divergence time of PRRSV. As a result, their values were estimated to be 4.17–9.8 × 10−2 and about 1980, respectively, in the Hanada et al. [14] investigation using ORF 3-to-5 data from two genotypes, whereas those values were recalculated to be around the year 1880 by Forsberg [11] based on ORF 3 sequences of two genotypes. Most recently, Shi et al. [32] estimated a mean evolutionary rate of 1.46 × 10−3 substitutions/site/year for ORF 5 data and noted that the common ancestors of genotype NA viruses appeared approximately in 1979 (1977–1982). Additionally, Song et al. [33] used ORF 5 sequences of two genotypes and demonstrated that the mean evolutionary rates of PRRSVs were 3.29 × 10−3 and that the virus diverged in 1894. Our findings on this topic do not support the previous suggestions. Here, we used a Bayesian coalescent approach using the most extended ORF 5 to 7 region sequences from 150 EU and NA genotype PRRSVs collected during the past 22 years (1989–2010). The relaxed uncorrelated exponential clock and expansion population size model were selected as the best fit for our PRRSV dataset. As a result, in contrast to previous work, our findings indicate that the PRRSV might be an “ancient virus” in spite of its recent emergence. The average substitution rate was 1.55 × 10−3 (95 % HPD = 9.06 × 10−4–2.23 × 10−3) substitutions/site/year, and the time of the most recent common ancestor was 491.2 years ago (95 % HPD = 199.6–864.4); they segregated approximately 1,519.8 years ago (95 % HPD = 1,146.6–1,811.4). Then, two distinct genotypes diverged approximately at the same time; the TMRCA estimated for the current diversity of EU-type viruses was 58.7 years ago (95 % HPD = 40.6–191.9), and that for the NA-type isolates was 62.6 years ago (95 % HPD 41.5–153.7). This configuration is in concordance with the conclusions of several PRRSV researchers and shows that the two lineages must have evolved separately from a very distant common ancestor prior to their emergence into the pig populations on the two continents [23].
Regarding the effective population size changes of PRRSVs, our BSP analysis on the basis of ORF 5 to 7 sequences of the global PRRSV isolates (Fig. 2) revealed that the viruses appear to have evolved at an almost constant population size until the late 1970s, when they experienced a population expansion that continued until the late 1980s. Their population size then remained constant again until the early 2000s, when a rapid, sharp decline in the effective number of infections occurred. The sharp decrease of the PRRSV effective population size might have been due to vaccinations that effectively controlled PRRSV worldwide for many years.
Here, we further studied the genetic history of the PRRSV on the basis of the most extensive ORF 5 to 7 sequences of the EU and NA genotypes. We first explained the evolutionary and past population dynamics of the global PRRSVs inferred from ORF 5 to 7 sequences using a Bayesian coalescent approach. We also demonstrated evolutionary mechanisms such as recombination and selection pressures, as well as phylogeny of PRRSV for two genotypes. Since its outbreaks two decades ago, PRRSV apparently radiated worldwide in an “explosive” fashion during a relatively short time. To date, despite intensive immunological efforts, the porcine reproductive and respiratory syndrome is still the most significant swine disease worldwide. Accordingly, both national and global strategies are necessary to prevent and control this acute disease. The expanding database and genetic history information of PRRSV sequences obtained from the present study might be useful for the prevention and control of this virus as well as for advancing our knowledge about its outbreak and epidemiology.
References
An TQ, Zhou YJ, Liu GQ, Tian ZJ, Li J, Qiu HJ, Tong GZ (2007) Genetic diversity and phylogenetic analysis of glycoprotein 5 of PRRSV isolates in mainland China from 1996 to 2006: coexistence of two NA-subgenotypes with great diversity. Vet Microbiol 123:43–52
Bautista EM, Suárez P, Molitor TW (1999) T cell responses to the structural polypeptides of porcine reproductive and respiratory syndrome virus. Arch Virol 144:117–134
Cha SH, Choi EJ, Park JH, Yoon SR, Song JY, Kwon JH, Song HJ, Yoon KJ (2006) Molecular characterization of recent Korean porcine reproductive and respiratory syndrome (PRRS) viruses and comparison to other Asian PRRS viruses. Vet Microbiol 117:248–257
Chen N, Cao Z, Yu X, Deng X, Zhao T, Wang L, Liu Q, Li X, Tian K (2011) Emergence of novel European genotype porcine reproductive and respiratory syndrome virus in mainland China. J Gen Virol 92:880–892
Dea S, Gagnon CA, Mardassi H, Pirzadeh B, Rogan D (2000) Current knowledge on the structural proteins of porcine reproductive and respiratory syndrome (PRRS) virus: comparison of the North American and European isolates. Arch Virol 145:659–688
Domingo E, Baranowski E, Escarmis C, Sobrino F (2002) Foot and-mouth disease virus. Comp Immunol Microbiol Infect Dis 25:297–308
Drummond AJ, Ho SY, Phillips MJ, Rambaut A (2006) Relaxed phylogenetics and dating with confidence. PLoS Bio 4:e88. doi:10.1371/journal.pbio.0040088
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Duran JP, Climent I, Sarraseca J, Urniza A, Cortés E, Vela C, Casal JI (1997) Baculovirus expression of proteins of porcine reproductive and respiratory syndrome virus strain Olot/91. Involvement of ORF3 and ORF5 proteins in protection. Virus Genes 14:19–29
Forsberg R, Oleksiewicz MB, Petersen AM, Hein J, Bøtner A, Storgaard T (2001) A molecular clock dates the common ancestor of European-type porcine reproductive and respiratory syndrome virus at more than 10 years before the emergence of disease. Virology 289:174–179
Forsberg R (2005) Divergence time of porcine reproductive and respiratory syndrome virus subtypes. Mol Biol Evol 22:2131–2134
Guindon S, Gascuel O (2003) A simple, fast and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Hall TA (1999) BIOEDIT: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hanada K, Suzuki Y, Nakane T, Hirose O, Gojobori T (2005) The origin and evolution of porcine reproductive and respiratory syndrome viruses. Mol Biol Evol 22:1024–1031
Inoue R, Tsukahara T, Sunaba C, Itoh M, Ushida K (2007) Simple and rapid detection of the porcine reproductive and respiratory syndrome virus from pig whole blood using filter paper. J Virol Methods 141:102–106
Jiang Y, Xiao S, Fang L, Yu X, Song Y, Niu C, Chen H (2006) DNA vaccines co-expressing GP5 and M proteins of porcine reproductive and respiratory syndrome virus (PRRSV) display enhanced immunogenicity. Vaccine 24:2869–2879
Keffaber K (1989) Reproductive failure of unknown etiology. Amer Assoc Swine Pract Newsl 1:1–10
Labarque G, Van Reeth K, Nauwynck H, Drexler C, Van Gucht S, Pensaert M (2004) Impact of genetic diversity of European-type porcine reproductive and respiratory syndrome virus strains on vaccine efficacy. Vaccine 22:4183–4190
Martin DP, Williamson C, Posada D (2005) RDP2: recombination detection and analysis from sequence alignments. Bioinformatics 21:260–262
Meng XJ, Paul PS, Halbur PG, Morozov I (1995) Sequence comparison of open reading frames 2 to 5 of low and high virulence United States isolates of porcine reproductive and respiratory syndrome virus. J Gen Virol 76:3181–3188
Meulenberg JM (1998) Porcine reproductive and respiratory syndrome virus (PRRSV) molecular characterization of the agent. Proceedings of fifteenth IPVS congress, Birmingham, In, pp 149–157
Nam E, Park CK, Kim SH, Joo YS, Yeo SG, Lee C (2009) Complete genomic characterization of a European type 1 porcine reproductive and respiratory syndrome virus isolate in Korea. Arch Virol 154:629–638
Nelsen CJ, Murtaugh MP, Faaberg KS (1999) Porcine reproductive and respiratory syndrome virus comparison: divergent evolution on two continents. J Virol 73:270–280
Paton DJ, Brown IH, Edwards S, Wensvoort G (1991) “Blue ear” disease of pigs. Vet Rec 128:617
Pesch S, Meyer C, Ohlinger VF (2005) New insights into the genetic diversity of European porcine reproductive and respiratory syndrome virus (PRRSV). Vet Microbiol 107:31–48
Plagemann PG (2003) Porcine reproductive and respiratory syndrome virus: origin hypothesis. Emerg Infect Dis 9:903–908
Posada D, Crandall KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rambaut A (2006) Available at: http://evolve.zoo.ox.ac.uk/beast
Seuberlich T, Tratschin JD, Thür B, Hofmann MA (2002) Nucleocapsid protein-based enzyme-linked immunosorbent assay for detection and differentiation of antibodies against European and North American porcine reproductive and respiratory syndrome virus. Clin Diagn Lab Immunol 9:1183–1191
Shi M, Lam TT, Hon CC, Hui RK, Faaberg KS, Wennblom T, Murtaugh MP, Stadejek T, Leung FC (2010) Molecular epidemiology of PRRSV: a phylogenetic perspective. Virus Res 154:7–17
Shi M, Lam TT, Hon C-C, Murtaugh MP, Davies PR, Hui RK, Li J, Wong LT, Yip CW, Jiang JW, Leung FC-C (2010) A phylogeny-based evolutionary, demographical and geographical dissection of North American type 2 porcine reproductive and respiratory syndrome viruses. J Virol 84:8700–8711
Song J, Shen D, Cui J, Zhao B (2010) Accelerated evolution of PRRSV during recent outbreaks in China. Virus Genes 41:241–245
Stadejek T, Stankevicius A, Storgaard T, Olekssiewicz MB, Belak S, Drew YW, Pejsak Z (2002) Identification of radically different variants of porcine reproductive and respiratory syndrome virus in Eastern Europe: Towards a common ancestor for European and American viruses. J Gen Virol 83:1861–1873
Stadejek T, Oleksiewicz MB, Potapchuk D, Podgorska K (2006) Porcine reproductive and respiratory syndrome virus strains of exceptional diversity in Eastern Europe support the definition of new genetic subtypes. J Gen Virol 87:1835–1841
Strimmer K, von Haeseler A (1996) Quartet puzzling: a quartet maximum likelihood method for reconstructing tree topologies. Mol Biol Evol 13:964–969
Suchard MA, Weiss RE, Sinsheimer JS (2001) Bayesian selection of continuous- time Markov chain evolutionary models. Mol Biol Evol 18:1001–1013
Suyama M, Torrents D, Bork P (2006) PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acid Res 34:W609–W612
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The Clustal-windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 22:4673–4680
Wang C, Wu B, Amer S, Luo J, Zhang H, Guo Y, Dong G, Zhao B, He H (2010) Phylogenetic analysis and molecular characteristics of seven variant Chinese field isolates of PRRSV. BMC Microbiol 20(10):146
Weiland E, Wieczorek-Krohmer M, Kohl D, Conzelmann KK, Weiland F (1999) Monoclonal antibodies to the GP5 of porcine reproductive and respiratory syndrome virus are more effective in virus neutralization than monoclonal antibodies to the GP4. Vet Microbiol 66:171–186
Wensvoort G, Terpstra C, Pol JM, ter Laak EA, Bloemraad M, de Kluyver EP, Kragten C, van Buiten L, den Besten A, Wagenaar F et al (1991) Mystery swine disease in The Netherlands: the isolation of Lelystad virus. Vet Q 13:121–130
Yang Z (1997) PAML: A program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13:555–556
Yoon SH, Song JY, Lee CH, Choi EJ, Cho IS, Kim B (2008) Genetic characterization of the Korean porcine reproductive and respiratory syndrome viruses based on the nucleocapsid protein gene (ORF7) sequences. Arch Virol 153:627–635
Zheng QS, Bi ZX, Li P, Chen DS, Chen PY (2007) Construction and the immunogenicity of the recombinant Modified Vaccinia Virus Ankala co-expressing ORF4, ORF5 and ORF6 genes of porcine reproductive and Respiratory Syndrome Virus NJ-a strain. Wei Sheng Wu Xue Bao 47:345–349
Zhou YJ, Yu H, Tian ZJ, Li GX, Hao XF, Yan LP, Peng JM, An TQ, Xu AT, Wang YX, Wei TC, Zhang SR, Cai XH, Feng L, Li X, Zhang GH, Zhou LJ, Tong GZ (2009) Genetic diversity of the ORF5 gene of porcine reproductive and respiratory syndrome virus isolates in China from 2006 to 2008. Virus Res 144:136–144
Zhu L, Zhang G, Ma J, He X, Xie Q, Bee Y, Gong SZ (2011) Complete genomic characterization of a Chinese isolate of porcine reproductive and respiratory syndrome virus. Vet Microbiol 147:274–282
Acknowledgments
This work was supported by a grant from the Next-Generation BioGreen 21 Program (No. PJ008196), Rural Development Administration, Republic of Korea.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Yoon, S.H., Kim, H., Park, B. et al. Tracing the genetic history of porcine reproductive and respiratory syndrome viruses derived from the complete ORF 5-7 sequences: a Bayesian coalescent approach. Arch Virol 157, 2143–2151 (2012). https://doi.org/10.1007/s00705-012-1408-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-012-1408-6