
Abstract
Several decades of research on human cytomegalovirus (HCMV) and the principal mammalian cytomegaloviruses which to varying degrees act as models of HCMV infection, particularly murine, guinea pig and rhesus CMV, have led to the recognition of the CMVs as interesting models of persistent infection with a large and complex DNA virus, which have been highly informative of the immunology and molecular pathogenesis of the virus–host relationship in the normal host. However, it is appropriate to ask how this relative wealth of knowledge has influenced the understanding and management of clinical disease due to HCMV. This article considers the immunology of cytomegalovirus in the normal human host, and the interrelated issue of the sites of HCMV latency and mechanisms of reactivation in the myeloid cell lineage, and in related in vitro model systems. The way in which this site of latency conditions the immune response, and emerging information on the special features of the adaptive immune response to HCMV during latency are also considered. Examples of HCMV disease associated with acquired immunosuppression, principally in the context of transplantation, but also as a consequence of HIV/AIDS and immune reconstitution inflammatory syndrome, are then discussed, with a particular emphasis on how understanding the immunology of persistent infection may contribute to managing CMV disease now and in future.
Similar content being viewed by others


Introduction
There have now been several decades of research on human cytomegalovirus (HCMV) and the principal mammalian cytomegaloviruses which to varying degrees act as models of HCMV infection, particularly murine, guinea pig and rhesus CMV: this has led to the recognition of the CMVs as interesting models of a persistent infection with a large and complex DNA virus, which have been highly informative of the immunology and molecular pathogenesis of the virus–host relationship in the normal host. However, it is appropriate to ask how this accumulated knowledge has influenced the understanding and management of clinical disease due to HCMV: this is the main question we address in this article. It is worth emphasising at the outset that as is the case with clinical research on all human disease, the flow of information is bidirectional: the informed application of knowledge derived from laboratory and animal model studies to the clinical setting is important, but it is equally important that observations derived from clinical studies on HCMV disease are used to inform the design of laboratory and animal model studies (sometimes encapsulated in the aphorism used to summarise translational research as being “from bench to bedside and bedside to bench”).
Immunology of cytomegalovirus in the normal human host
Innate immunity: NK cells
The role of natural killer cells in controlling HCMV infection is covered in detail in this volume, and their importance is inferred from the now large number of HCMV genes that encode functions enabling virus-infected cells to evade recognition and lysis by NK cells. Deficiency states specific for NK cells would also provide the opportunity to assess their relative contribution to controlling CMV infection in normal subjects in vivo. Immunodeficiency syndromes predominantly characterised by NK cell dysfunction are certainly now described, and interestingly, these tend to have herpesvirus infections as a common phenotypic feature [1]. However, thus far, there are no really “pure” immunodeficiencies, affecting only NK cells, described—which is unsurprising given the extent to which the differentiation pathway, phenotypic markers and lytic machinery of NK cells have features in common with those in T cells and some monocytes. For example, the patient who was the subject of a frequently cited report [2], with a near complete deficiency of phenotypic NK cells and persistent active HCMV infection and disease, was eventually found to be one of a group of patients who have genetic haplo-insufficiency of GATA-2 [3], which is associated with a variety of additional defects in the myeloid lineage pathway [4]. Whilst it seems reasonable to infer that the NK cell deficiency was causally related to the CMV, it can be envisaged that the myeloid lineage defects could also influence CMV latency and reactivation.
Another interesting point, indicating that there is continuing interaction between host NK cells and HCMV infection during persistent infection, is the accumulating evidence that HCMV is associated with a distinctive pattern of expression of phenotypic markers on peripheral blood NK cells which persists during long-term CMV carriage (reviewed in [5]); thus, expansion of NK cells expressing the activating receptor CD94-NKG2C in HCMV seropositive subjects has been reported in two studies [6, 7]. This has been adduced as evidence of “NK cell memory”, although a preferable way to describe such continued expansion of an apparently pathogen-specific subset of NK cells during persistent infection might be to say that HCMV is associated with a stable “imprint” on the expressed NK cell receptor (KIR) repertoire. It is also possible that such “imprinting” may have pathologic associations—it has been reported that these HCMV-induced NK cell expansions may be associated with the development of specific de novo malignancies in liver transplant recipients [8].
Adaptive immunity
T cells
Primary HCMV infection induces robust CD8+ cytotoxic, and CD4+ helper, T cell-mediated immune responses, which are associated with the resolution of acute primary infection: these responses are then maintained at high frequency in long-term memory as the virus establishes persistent infection, with latency and periodic reactivation. Although many of these T cells are specific for epitopes in the pp65 and IE1 HCMV proteins, it is apparent that many other viral proteins can also be T cell targets, and in some individuals, pp65 and IE1 responses are not immunodominant [9, 10].
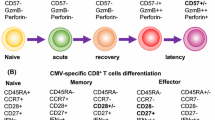
HCMV exerts a major influence on the clonal composition of the human T cell memory pool in normal virus carriers. Analysis of the CD8+ response in individual subjects at the clonal level has shown that it comprises large clonal expansions specific for limited number of allele-specific HCMV peptides: clones are present early in primary infection and stable over many years. These clones comprising the memory response have high TCR affinity for antigen, and high affinity associates with selection from the primary response into long-term memory [11]. Immunophenotyping shows that the bulk of the HCMV CD8+ memory pool is CD28−, CD45RA+: it has been proposed that in general such T “effector memory RA+” (TEMRA) cells are terminally differentiated, but at least for HCMV-specific T cells, this is certainly not the case [12, 13]. Functional analysis shows that they remain precursor memory cells, albeit requiring specific co-stimulatory signals additional to that through the TCR (e.g. through the costimulatory ligand pair CD137/41BB) to differentiate and become activated [14]. These observations from HCMV make the point that models of T cell memory which divide T cells into “central” and “effector” memory cells and imply a unidirectional linear differentiation pathway to effector cells, do not accurately reflect the level of plasticity inherent in memory T cells (see Chang et al. [15] for a recent review on memory T cell differentiation). One limitation of clinical investigation of human cellular immunity is that, in contrast to the mouse, it is usually only peripheral blood lymphocytes that can be readily sampled, and inferences have to be made about the memory pool based on sampling which might be unrepresentative of the situation in lymph nodes and tissues. In fact, one study that did compare the frequencies of EBV- and CMV-specific T cells in blood and lymph nodes reported much lower frequencies in lymph nodes than blood for HCMV, whereas the frequencies were similar for EBV [16]. Another related constraint that deserves mention is that it has recently been recognised that there is a specialised subset of memory T cells, tissue-resident T lymphocytes, which do not recirculate but reside long-term in sites where antigen is present, particularly in skin and mucosa, and likely play an important role in infections involving such tissues, including some herpesviruses [17]. Studies of the role of these tissue-resident T cells in CMV infection are in progress, but will clearly be difficult to conduct in humans for HCMV.
During long-term carriage of the virus, a balance is thus established between the T cell-mediated immune response and viral reactivation: the T cell response controls viral spread following reactivation, but the virus encodes multiple “immunoevasion” genes that interfere with MHC-I antigen processing and with MHC-II processing and NK cell killing, allowing limited viral evasion of the response, which is probably particularly important during initial virus reactivation from its cellular sites of latency [18]. Loss of this balance, whereby the T cell response keeps persistent reactivating virus in check in healthy carriers, is most evident in the immunosuppressed host in whom reactivation of latent virus or primary infection can lead to unchecked viral replication, with consequent disease and mortality. This article focuses on the extent to which current understanding of CD8+ and CD4+ T cell responses to HCMV in normal subjects, and of their perturbation in disease, can usefully inform the clinical management of immunosuppressed patients with CMV disease.
B cells and antibody
The role of humoral immunity in immunity to HCMV has recently been reviewed elsewhere [19] and is not considered further here. Whilst there are protective neutralising antibodies to HCMV which may play a role in limiting primary infection, the conventional view is that protection against CMV, particularly the control of reactivation, is more dependent on T cells. In the murine MCMV model, protection can be mediated completely by T cells in the face of B cell depletion [20]. Nevertheless, vaccination against HCMV will have to be shown to work practically and mechanistically, and most current candidates are likely to do so mainly, or at least in part, by being designed to induce antibody, as for most vaccines. The role of antibody in HCMV is thus an active area of research, with much interest currently focussed on antibody to the pentameric glycoprotein complex on virions, which plays a major role in virus binding to, and entry via, the surface of infected cells, including a wider range of cell types of likely importance in vivo, rather than just fibroblasts in vitro [21].
HCMV latency and reactivation in the myeloid lineage
A model of HCMV latency and reactivation
Any discussion of the immunology of HCMV has to consider the mechanism and site of HCMV latency and their influence on the immune response to the virus. This topic is discussed in depth in another article in this volume and is only briefly summarised here.
Work over the last 10 years, much of it from the Cambridge group, has shown that human CMV is latent in CD34+ cells in the myeloid lineage (with HCMV DNA present in 1 in 103–105 cells) and reactivates as they mature to dendritic cells (DC) [22–24]. Although it is possible, if not likely, there may be other cellular sites of latency, this site of latency within key antigen-presenting cells of the immune system likely influences the scale of the immune response and immunoevasion, which HCMV produces, and also illustrates the particular therapeutic challenge presented by HCMV.
Although it was assumed that there was little or no virus gene expression during latency, recent work has shown that viral gene expression is far from quiescent during latent infection, and there is now much published data detailing expression of specific viral genes during natural or experimental latent infection in CD34 cells or their myeloid derivatives. Transcripts expressed during natural latency are known to include RNAs from the major IE region (UL122–123 CLTs), as well as UL81–82ast (LUNA), UL138, UL111a, UL14428 and US28. More recently, additional latency-associated viral transcripts have also been identified, but the detailed functions of all these viral genes, even where known, cannot be considered here but are described in recent reviews [23, 25, 26].
HCMV, as have many other herpesviruses, has been shown to encode a wealth of microRNAs (miRNAs) with the potential to orchestrate both cell and viral gene regulation. During lytic infection, HCMV expresses approximately 24 miRNAs derived from 13 pre-miRNAs which have been shown to target both viral and cellular RNAs. Viral targets include IE72 as well as a number of viral genes involved in DNA synthesis [27].
Regulation of virus reactivation as CD34+ cells differentiate is associated with, and very probably consequent on, changes in the chromatin structure of “latency breaking” viral genes, such as HCMV IE1, which occurs with myeloid lineage differentiation [28, 29], and appears to be a common mechanism in all herpesviruses (e.g. the BZLF1 promoter of EBV and ICP0 promoter of HSV1).
HCMV latency also results in multiple changes in cellular gene expression to allow long-term carriage of viral genome in the face of potent immune responses and to optimise virus reactivation. The changes in the total plasma membrane proteome of cells expressing UL138, one of the viral genes expressed during latency, have been quantified in an unbiased way by Weekes et al. [30] using the technique of SILAC (stable isotope labelling with amino acids in culture) and mass spectrometry. They showed that transfection of cells with latency-associated UL138 specifically down-regulates the multi-drug transporter MRP1 and that this also occurs in model latency systems (a nearly eightfold reduction compared to all other plasma membrane proteins). Studies in the MRP1 knockout mouse [31] have previously suggested that MRP1 is involved in DC migration to lymph nodes and is also required for myeloid differentiation, and it thus seems plausible that this profound down-regulation of surface MRP occurring in latently infected human dendritic cells may alter their trafficking.
The identification of the above changes in both cellular and viral gene expression specific to latently infected cells raises the prospect of using the gene products as targets for eliminating or purging cells carrying latent virus, for example in a haematopoietic stem cell graft from a CMV-positive donor. Cells in which MRP1 is down-regulated accumulate toxic levels of drugs which are normally dependent on MRP1 for being exported from the cell, and it has been shown that cells carrying latent CMV are selectively killed when treated with low-dose vincristine. To give a further example, latency can be broken by histone deacetylase (HDAC) inhibitors which open the closed chromatin conformation around the IE genes, allowing lytic cycle genes to be expressed and thus rendering the infected cells susceptible to killing by lytic cycle specific antibody or T cells. Such mechanisms of “purging” latently HCMV-infected cells have yet to find clinical application, but it can be envisaged that they may soon do so, particularly in the context of HSCT.
Special features of the adaptive cellular immune response to HCMV during latency
Given that there are specific HCMV genes that are selectively expressed during latent infection, an obvious question is whether they elicit an immune response: the answer is that they do, but one that is significantly different from that observed to HCMV proteins expressed during lytic infection—the area has been reviewed recently by Wills et al. [32], and further details of the immune response during HCMV latency are available there.
The T cell response to lytic cycle virus gene products is dominated by CD8 T cells, but virus-specific CD4 cells specific for epitopes restricted by class II MHC can also be detected (e.g. gB and IE1): they are mostly IFNγ secreting Th1-type CD4+ T cells and indeed provide essential help for the induction of CD8 responses. In contrast, it has proved difficult to generate latent protein-specific CD8 cells by in vitro restimulation; instead, the latency-associated gene products (LUNA, UL138, US28, US111A) elicit a predominant or exclusively CD4+ T cell response—a subpopulation of which secrete the immunomodulatory cytokines IL-10 and TGFβ and express markers of regulatory T (Treg) cells. In general, we find UL138- and LUNA-specific T cells are composed of IFNγ- and IL10-producing cells, and US28- and UL111A-specific T cells are dominated by IL10 production (and are of FoxP3+ CD25+ Treg phenotype) [33].
Furthermore, the “secretome” (the total secreted proteins in the cell culture supernatant) of latently infected cells in experimental latency systems favours recruitment of helper CD4+ T cells (principally through it containing the chemokine CCL-8), but then imparts an immunosuppressive microenvironment for the recruited cells, inhibiting their killing function, principally mediated by increased levels of the immunosuppressive cytokines cellular IL-10 and TGF-β in the secretome derived from the latently infected cells [34].
Spectrum of HCMV disease associated with immunosuppression
There is a fairly broad spectrum of disease which has been associated with HCMV, ranging from clinical syndromes unequivocally causally attributable to HCMV, nearly always in the context of acquired immunosuppression (but sometimes related to genetic immunodeficiency syndromes), to chronic diseases of multifactorial aetiology where a possible causal role for HCMV has been suggested more on the principle of “guilt by association”, rather than any established mechanistic link. Only the former are discussed here—with the exception of congenital HCMV infection which, although it is clearly associated with both maternal (the immunology of pregnancy) and foetal (developmental immunology) aspects of immunology and immunosuppression, has recently been reviewed elsewhere [35].
The focus here is on adaptive immunity given its importance in controlling HCMV infection, and the scale of the problem in clinical medicine presented by HCMV disease in the context of acquired and iatrogenic immunosuppression due to defective T cell function. However (as discussed earlier), there are clearly informative examples of defects in innate immunity being associated with HCMV infection, which indicate the likely importance of NK cells in controlling infection.
In the modern era of medicine, most HCMV disease has occurred in association with the iatrogenic immunosuppression required for transplantation. A fundamental therapeutic dilemma in transplantation is that the effector T cell mechanisms that mediate organ rejection and thus need to be therapeutically inhibited did not evolve to frustrate transplant surgeons but precisely to recognise and eliminate intracellular pathogens, such as viruses, through MHC-dependent mechanisms. In causing organ rejection through recognising foreign MHC molecules, alloreactive effector T cells are thus “only doing their job”, and finding highly selective, or even antigen-specific, immunosuppressive agents has always been a principal challenge to the field. From the earliest inception of organ transplantation, HCMV has presented a significant problem in post-operative patient management, principally due to the relatively high population prevalence of virus carriage and the relative lack of effective therapies for HCMV. HCMV disease presents clinical problems which may differ depending on which organ is being transplanted, but the main conceptual difference in management is between solid organ transplantation and haematopoietic stem cell transplantation (HSCT), which are contrasted below.
HCMV and solid organ transplantation: the kidney
Renal transplantation has been longer established and performed in larger numbers, as a routine treatment for end-stage renal disease than any other solid organ transplant. Whilst any of the three combinations in which either donor or recipient is seropositive (as an index of HCMV carriage) carries an increased risk of HCMV disease, the highest risk is clearly associated with the Donor+ Recipient− combination, in which an organ highly likely to contain latent virus in passenger or tissue-resident monocytes or epithelial cells is transplanted into a recipient with no prior immunity to HCMV: the risk of transmission is about 90 % and of disease ~50 % (without prophylaxis). In general, where there is immunity, the scale of recipient HCMV-specific CD8+ T cell response, as assessed quantitatively with class I MHC tetramers and qualitatively (e.g. with IFNγ production), correlates with protection.
The incidence and severity of HCMV disease in renal transplant recipients appears to be declining: this probably reflects both newer prophylaxis and pre-emptive therapy regimes for HCMV disease, based on the use of newer antivirals such as valganciclovir and quantitative PCR (qPCR) monitoring, and more recent selective immunosuppressive regimes based on the mTOR (mammalian target of rapamycin) inhibitors.
There is a long-running issue of whether there is a causal association of CMV with graft rejection, which remains unresolved, despite a large literature. An obvious confounding element is that HCMV disease may occur consequent on an increase in immunosuppressive therapy given because of rejection due to independent risk factors or that severe HCMV disease may lead to immunosuppressive therapy being reduced with consequent rejection—in neither case is the association directly causal. There is, however, some evidence that the presence of HCMV DNA in the renal graft is associated with a risk of long-term graft dysfunction, whatever the reason.
Of note, HCMV vaccine trials have been carried out in renal transplant recipients, as accessible defined patient groups: examples are both the original Towne strain candidate vaccine [36] (see article by Plotkin in this issue) and a recent phase 2 trial of a recombinant gB-based vaccine [37], given to seronegative and seropositive recipients. In both trials, CMV infection was reduced in the D+R− population, but in the latter trial the incidence of viraemia was no less in R+ recipients. The suggestion from both appears to be that the vaccine may reproduce the effect of primary CMV infection.
Haematopoietic stem cell transplantation (HSCT)
The major conceptual difference when considering the host immune response to infection or other antigens after HSCT is that the recipient host immune system has been ablated and reconstituted with the donor immune system, and immune responses of the recipient thus reflect the prior experience of the donor. Analysis of the generation or reconstitution of HCMV-specific immunity, including CD4+ and CD8+ T cells, following HSCT must thus take account of several important factors. The donor (D) and recipient (R) may be HLA-matched siblings, but this is in a minority of transplants and more usually they are HLA matched but unrelated (allo-SCT). In addition, the HCMV serostatus of the donor and recipient needs to be considered with there being four possible combinations: D+/R+, D−/R−, D+/R− and D−/R+.
It is well established that primary infection or reactivation of latent HCMV can lead to serious morbidity and mortality in HSCT recipients, and it has long been recognised that there is a direct correlation between effective HCMV-specific T cell cytotoxicity and recovery from CMV infection or reactivation [38]. The greatest risk of HCMV disease in HSCT thus occurs in D−/R+ transplantation, because the graft from the seronegative donor does not contain antigen-experienced HCMV-specific T cells and the recipient has latent virus whilst also receiving immunosuppressive treatment to prevent GvHD.
The reconstitution of HCMV-specific T cell immunity following HSCT has been examined in several studies. We have previously undertaken a longitudinal analysis of HCMV-specific CD8+ T cell clones, using clonotypic TCR probes derived from prior clonal analysis in the donor to measure the kinetics of reappearance and size of donor-derived clones in the recipient following allogeneic HSCT [39]. For the combination Donor+/Recipient+, donor antigen-experienced T cell clones readily expanded and persisted; for Donor−/Recipient+, there was a delayed T cell response of the type we have reported in primary infection, using cells of donor origin (obviously no prior analysis was possible in the donor); and for Donor+/Recipient−, donor antigen-experienced T cell clones were undetectable in two of three recipients for up to 9 months. Particularly in the latter two combinations, D−/R+ and D+/R−, prophylaxis or pre-emptive therapy with antivirals is often given to mitigate the risk of CMV disease associated with delayed reconstitution, but this then carries the risk of late CMV disease occurring many months after HSCT when such treatment is stopped. This and the delayed reconstitution seen in the D+/R− setting suggest a requirement for the presence of HCMV antigen, probably derived from intermittently replicating virus, in order to expand HCMV-specific memory T cell precursors in vivo.
These difficulties exemplify why HCMV disease remains a significant clinical problem complicating HSCT and have provided a driver for haemato-oncologists to develop additional treatments, particularly the cell-based therapies described below.
Acquired immunodeficiency: HIV/AIDS and immune reconstitution inflammatory syndromes (IRIS)
The most widespread and important cause of immunosuppression, in global terms and over the last three decades, has been HIV and the acquired immunodeficiency syndrome (HIV/AIDS). Prior to the use of highly active antiretroviral therapy (HAART), HCMV-related diseases were common in patients with HIV/AIDS, although very much correlated with advanced HIV infection and severe immunosuppression. HCMV disease was mainly seen in people with CD4+ T cell counts of <50/μl, and CMV retinitis was the commonest presenting syndrome and seen in about 25 % of this group with such very low CD4 counts. Following the advent of HAART, HIV-infected patients were started on therapy when their CD4 count had declined to 200 μl or if they presented with already advanced disease. HAART resulted in remarkable immune reconstitution and resolution of opportunistic infections: since its introduction the incidence of serious CMV disease has fallen by ~90 %, and for those patients who do require it for presenting CMV disease, CDC guidelines advise stopping antiviral treatment for CMV 3–6 months after HAART brings the CD4 count back to over 100/μl.
However, a new clinical problem then began to be described in HIV patients starting HAART whilst they were still receiving, or had recently stopped, treatment for an opportunistic infection: these were a related series of clinical syndromes named the “immune reconstitution inflammatory syndromes” (IRIS). These are characterised by marked inflammation occurring at tissue sites where there is a significant accumulation of microbial antigen and temporal association with reconstitution of T cell immunity due to HAART. If the inflammation leads to tissue damage at a critical organ site, for example in the CNS or the eye, significant loss of function may occur. The commonest opportunistic infectious diseases associated with IRIS, in global terms, are tuberculosis and cryptococcal disease, often in the central nervous system, but HCMV retinitis is the individual opportunistic infection most frequently associated with IRIS with around 37 % of patients who have had CMV retinitis subsequently having an episode of “immune recovery uveitis” (IRU). The use of intravitreal cidofovir is a major risk factor for IRU (relative odds = 19), and the early introduction of HAART is another, in patients being treated for CMV retinitis (see [40, 41] for reviews).
The IRIS syndromes are a good example of the unexpected—they had not been predicted from experimental models and, although clearly immunologically mediated, understanding of their pathogenesis remains limited—not least because of the difficulty in obtaining adequate biopsy material from the sites of inflammation. The simplest hypothesis to explain the occurrence of HCMV retinitis in association with the very low CD4 counts associated with advanced HIV/AIDS is that a profound lack of HCMV-specific T cell help from Th1-type CD4+ cells results in failure of HCMV-specific CD8T cells to become activated and expand into effector cells—and indeed this may well be a sufficient explanation. IRIS/IRU may then reflect the accumulation of large amounts of microbial/viral antigen in the absence of immune control, followed by tissue inflammation as reconstituted effector T cells interact with it at the site, with release of mediators. A recent study comparing systemic T cell responses in 25 patients with CMV IRU to controls with CMV retinitis but no IRU found that the IRU patients had poor CMV-specific CD4+ T cell responses compared with controls, whereas CD8(+) T cell responses were comparable: patients with IRU had lower numbers of circulating Th17 cells, but no differences in T(reg) cell function [42]. A study directly examining aqueous and vitreous fluids (which might more accurately reflect the pathogenic process in the eye) from patients with IRU and active CMV retinitis reported that IRU can be differentiated from active CMV retinitis by the presence of IL-12 and less IL-6 and the absence of detectable CMV replication, in IRU [43]. The precise immunopathogenic mechanism of CMV IRU is thus undefined and is likely to remain so in the absence of a model, leaving clinical management largely empirical, with local corticosteroids, coupled with restarting or continuing antiviral agents to reduce the amount of HCMV antigen.
Adoptive transfer of HCMV-specific T cells for prevention and treatment of HCMV disease
Perhaps it is in the application of adoptive cellular immunotherapy to CMV infection that clinical investigators managing patients with HCMV, and those working on experimental models using murine CMV, come closest together. Adoptive transfer of HCMV-specific T cells for prevention of HCMV disease in humans was first reported by Riddell and Greenberg in 14 patients—five D+/R+ and nine D+/R− [44]. HCMV-specific CD8+ T cell clones were generated from seropositive allo-BMT donors by stimulation of PBMCs with HCMV-infected autologous fibroblasts. In this trial, HCMV-specific CD8+ T cell numbers declined in the absence of CD4+ T cell help, which could be due to the absence of HCMV antigen in the recipients, as most of the recipients were HCMV negative and no instances of HCMV viraemia occurred. However, more recent work gives direct evidence that HCMV-specific CD4+ T cell help is needed for the induction of effective CD8+ T cell function. This is reflected in recent attempts to refine the generation of HCMV-specific T cells for adoptive immunotherapy, not only to include HCMV-specific CD4+ T cell help but also to remove live HCMV from the culture systems, and reduce the risk of GvHD by directly selecting for HCMV-specific CD4+ and CD8+ T cells using techniques based on peptide–MHC-I multimers or IFN-γ capture, thus reducing the amount of time consuming ex vivo manipulation of cells prior to their infusion into patients [45–47]. The proof of concept for these antigen-specific selection and adoptive transfer experiments was first demonstrated in the MCMV model [48, 49]. In vitro stimulation and expansion prior to infusion, having first been achieved with infected autologous fibroblasts [50], has more recently employed HCMV antigen-pulsed dendritic cells [47] or ex vivo selection and stimulation (with MHC class I tetramers) prior to infusion [45]. The requirement for HCMV-specific CD4+ cells for in vivo expansion is reinforced by recent studies [46, 51].
Thus far, all reports have been of relatively small clinical investigational studies: it is clear that if the reconstitution of HCMV-specific T cell responses by adoptive cell therapy is to become routine, the techniques used will need to be rapid, induce high frequencies of HCMV-specific T cells which contain both CD4+ and CD8+ T cell populations, and in order to cover as wide a range of patient, MHC-I and II haplotypes as possible should not just rely on the very well characterised pp65 and IE1 peptides specific for the HLA-A201 and B7 alleles. Indeed, it can be argued that it is unlikely adoptive T cell therapy will become routine for the treatment of HCMV unless there is some fundamental technical advance driven by its wider application in other diseases, and possibly a failure in the pipeline of effective new antivirals against HCMV.
In this context, the recent use of T cells with engineered chimeric antigen receptors to achieve sustained remissions in leukaemia [52] may well be pertinent. In this work, autologous T cells were transduced with chimeric antigen receptor directed against a relevant antigen on leukaemic cells (CD19), in a lentiviral vector, and infused in patients with relapsed or refractory ALL. The chimeric antigen receptor (CAR) was constructed from the Fv fragment of anti-CD19, and the TCR (CD3) zeta and 41BB (CD137) signalling domains. High remission rates were achieved (even if patients had previously relapsed after HSCT), with ~80 % survival at 2 years. The CAR T cells underwent expansion in vivo, which persisted over 2 years—although interestingly less in the case of CAR with the CD28 (rather than 41BB) signalling domain. Of note, 30 % of patients receiving these CAR T cells experienced severe cytokine release syndrome—which was anticipated in the protocol and treated with anti-IL6R (tocilizumab).
Such use of CAR T cells, by using a generic antigen receptor, makes the technique potentially more widely applicable in other diseases where there is a defined cell membrane antigen marking a cell population to be targeted and eliminated, and these could include intracellular infections such as CMV. Obviously, this would require selection of a virus-specified antigen to target which was ubiquitously expressed on infected cells.
“Lectures held by MCMV relevant to cytoimmunotherapy of clinical CMV disease” (from [54] in “Cytomegaloviruses: From Molecular Pathogenesis to Intervention”, ed. Matthias J. Reddehase) |
Lecture 1 viral epitope specificity of protection |
Lecture 2 loss of antiviral potency in CTLL (with in vitro expansion) |
Lecture 3 protection by immunotherapy does not care about immunodominance |
Lecture 4 escape variants of CTL epitopes are no issue of serious concern in immunotherapy |
Lecture 5 viral immunoevasion proteins limit AT efficacy but do not prevent protection |
Lecture forecast: analysis of allo-HCT (vs syngeneic HCT) in murine CMV model |
This article has not referenced the large body of work on murine CMV, and although usually informative, it is by no means always the case that findings in the MCMV model can be automatically extrapolated to HCMV and the clinical setting. In this respect, it is appropriate to conclude this section by referring to the work of the Mainz group on adoptive T cell therapy, whose work stands out in being characterised by careful and insightful design of their murine model to ensure it recapitulates the human setting of HCMV disease in the context of HSCT as faithfully as is possible [53]. Above is a table based on a review of their recent work, in which they summarise the lessons learned (or “Lectures”) from the model [54]. Given their record, those working on adoptive T cell therapy for HCMV would do well to heed their conclusions from the model, and we look forward to the new insights they will gain from future studies of the even more clinically relevant model of allo-SCT in the mouse.
How understanding immunology contributes to managing CMV disease now and in future
HCMV is a large complex persistent DNA virus, from a virus family which has been co-evolving with humans for millions of years, and we have only a very partial understanding of the innate and adaptive immune systems and the full complexity of their interactions with intracellular pathogens. Our understanding of the immunology of HCMV is limited, but accruing at a steady rate—some questions are at least partly answered but many remain. Some such outstanding questions, in what is inevitably a personal list, include:
Does the myeloid lineage/dendritic cell site of latency and reactivation of HCMV, within cells whose principal function is antigen presentation, account for the proportion of its genome devoted to “immunoevasion”?
Is the principal role of “immunoevasins” to gain a window of opportunity to reactivate from the HCMV site of latency, and possibly facilitate “strain superinfection” (as shown in the Rhesus CMV model [55]).
Is the DC site of latency, with persistent antigen production, responsible for maintaining the oligoclonal T cell expansions characteristic of HCMV, and larger than for other virus pathogens?
Should we not question the use of descriptive terms, or assumptions, about the T cell immune response to HCMV which imply a role in pathogenesis, where none may exist? Examples include “CTL exhaustion” to describe differentiated effector T cells which are fully functional when given appropriate signals, and large oligoclonal expansions of T cells described as evidence of memory “inflation”, a term which has pathological implications, when such expansions may be within normal ranges. Finally, a causal association of HCMV with “immunosenescence” and age-related functional deterioration in the immune response has been postulated. We and others have argued elsewhere that such a causal association is unsupported by strong evidence and remains unproven. Space does not permit these arguments to be rehearsed here, but we believe that they remain valid [10].
In conclusion, if we attempt to answer the question posed at the outset “How Understanding Immunology Contributes to Managing CMV Disease in Immunosuppressed Patients: Past and Future” it can be said that such understanding helps enable us to:
-
Stratify the risk of CMV disease in patients with immunodeficiency—acquired or iatrogenic.
-
Devise preventive strategies and select management options for solid organ transplantation and HSCT.
-
Understand and influence the factors that control the establishment and maintenance of effective T cell immunity to HCMV.
-
Undertake logical approaches to cell therapy: adoptive transfer of T cells is so far experimental and trials are needed. Developing widely applicable cell therapies may only be driven by lack of new safe antivirals and more generically applicable technologies for targeting effector cells.
-
Consider possible novel approaches to purging latently infected cells (DC) from HSCT.
-
Apply knowledge to vaccine development.
All these questions, answered or not, of course reflect the biological complexity of this fascinating virus, both normal human passenger and potentially severe pathogen. No doubt still more questions remain, all worth attempting to answer because clinical practice which is based on scientific understanding is always superior to that based only on empiricism!
References
Orange JS (2013) Natural killer cell deficiency. J Allergy Clin Immunol 132(3):515–525. doi:10.1016/j.jaci.2013.07.020
Biron CA, Byron KS, Sullivan JL (1989) Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med 320(26):1731–1735
Mace EM, Hsu AP, Monaco-Shawver L, Makedonas G, Rosen JB, Dropulic L, Cohen JI, Frenkel EP, Bagwell JC, Sullivan JL, Biron CA, Spalding C, Zerbe CS, Uzel G, Holland SM, Orange JS (2013) Mutations in GATA2 cause human NK cell deficiency with specific loss of the CD56(bright) subset. Blood 121(14):2669–2677. doi:10.1182/blood-2012-09-453969
Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, Lakey JH, Rahman T, Wang XN, McGovern N, Pagan S, Cookson S, McDonald D, Chua I, Wallis J, Cant A, Wright M, Keavney B, Chinnery PF, Loughlin J, Hambleton S, Santibanez-Koref M, Collin M (2011) Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood 118(10):2656–2658. doi:10.1182/blood-2011-06-360313
Della Chiesa M, Marcenaro E, Sivori S, Carlomagno S, Pesce S, Moretta A (2014) Human NK cell response to pathogens. Semin Immunol 26(2):152–160. doi:10.1016/j.smim.2014.02.001
Beziat V, Liu LL, Malmberg JA, Ivarsson MA, Sohlberg E, Bjorklund AT, Retiere C, Sverremark-Ekstrom E, Traherne J, Ljungman P, Schaffer M, Price DA, Trowsdale J, Michaelsson J, Ljunggren HG, Malmberg KJ (2013) NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood 121(14):2678–2688. doi:10.1182/blood-2012-10-459545
Hendricks DW, Balfour HH Jr, Dunmire SK, Schmeling DO, Hogquist KA, Lanier LL (2014) Cutting edge: NKG2C(hi)CD57+ NK cells respond specifically to acute infection with cytomegalovirus and not Epstein–Barr virus. J Immunol 192(10):4492–4496. doi:10.4049/jimmunol.1303211
Achour A, Baychelier F, Besson C, Arnoux A, Marty M, Hannoun L, Samuel D, Debre P, Vieillard V (2014) Expansion of CMV-mediated NKG2C+ NK cells associates with the development of specific de novo malignancies in liver-transplanted patients. J Immunol 192(1):503–511. doi:10.4049/jimmunol.1301951
Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, Sleath PR, Grabstein KH, Hosken NA, Kern F, Nelson JA, Picker LJ (2005) Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med 202(5):673–685
Wills MR, Mason GM, Sissons JGP (2013) Adaptive cellular immunity to human cytomegalovirus. I. In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention. Caister Academic Press, Norfolk, pp 142–172
Day EK, Carmichael AJ, Ten Berge IJ, Waller EC, Sissons JG, Wills MR (2007) Rapid CD8+ T cell repertoire focusing and selection of high-affinity clones into memory following primary infection with a persistent human virus: human cytomegalovirus. J Immunol 179(5):3203–3213
Wills MR, Okecha G, Weekes MP, Gandhi MK, Sissons PJ, Carmichael AJ (2002) Identification of naive or antigen-experienced human CD8(+) T cells by expression of costimulation and chemokine receptors: analysis of the human cytomegalovirus-specific CD8(+) T cell response. J Immunol 168(11):5455–5464
Jackson SE, Mason GM, Okecha G, Sissons JG, Wills MR (2014) Diverse specificities, phenotypes, and antiviral activities of cytomegalovirus-specific CD8+ T cells. J Virol 88(18):10894–10908. doi:10.1128/JVI.01477-14
Waller EC, Day E, Sissons JG, Wills MR (2008) Dynamics of T cell memory in human cytomegalovirus infection. Med Microbiol Immunol 197(2):83–96. doi:10.1007/s00430-008-0082-5
Chang JT, Wherry EJ, Goldrath AW (2014) Molecular regulation of effector and memory T cell differentiation. Nat Immunol 15(12):1104–1115. doi:10.1038/ni.3031
Klarenbeek PL, Remmerswaal EB, ten Berge IJ, Doorenspleet ME, van Schaik BD, Esveldt RE, Koch SD, ten Brinke A, van Kampen AH, Bemelman FJ, Tak PP, Baas F, de Vries N, van Lier RA (2012) Deep sequencing of antiviral T-cell responses to HCMV and EBV in humans reveals a stable repertoire that is maintained for many years. PLoS Pathog 8(9):e1002889. doi:10.1371/journal.ppat.1002889
Ariotti S, Haanen JB, Schumacher TN (2012) Behavior and function of tissue-resident memory T cells. Adv Immunol 114:203–216. doi:10.1016/B978-0-12-396548-6.00008-1
Jackson SE, Mason GM, Wills MR (2011) Human cytomegalovirus immunity and immune evasion. Virus Res 157(2):151–160. doi:10.1016/j.virusres.2010.10.031
Mach M, Wiegers A, Spindler N, Winkler T (2013) Protective Humoral Immunity. In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention. Caister Academic Press, Norfolk, pp 215–231
Polic B, Hengel H, Krmpotic A, Trgovcich J, Pavic I, Luccaronin P, Jonjic S, Koszinowski UH (1998) Hierarchical and redundant lymphocyte subset control precludes cytomegalovirus replication during latent infection. J Exp Med 188(6):1047–1054
Macagno A, Bernasconi NL, Vanzetta F, Dander E, Sarasini A, Revello MG, Gerna G, Sallusto F, Lanzavecchia A (2010) Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J Virol 84(2):1005–1013. doi:10.1128/JVI.01809-09
Reeves MB, MacAry PA, Lehner PJ, Sissons JG, Sinclair JH (2005) Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc Natl Acad Sci USA 102(11):4140–4145
Sinclair JH, Reeves MB (2013) Human cytomegalovirus manipulation of latently infected cells. Viruses 5(11):2803–2824. doi:10.3390/v5112803
Reeves MB, Sinclair JH (2010) Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J Gen Virol 91(Pt 3):599–604. doi:10.1099/vir.0.015602-0
Sinclair J, Reeves M (2014) The intimate relationship between human cytomegalovirus and the dendritic cell lineage. Front Microbiol 5:389. doi:10.3389/fmicb.2014.00389
Poole E, Wills M, Sinclair J (2014) Human cytomegalovirus latency: targeting differences in the latently infected cell with a view to clearing latent infection. New J Sci Article ID 313761
Hook L, Hancock M, Landais I, Grabski R, Britt W, Nelson JA (2014) Cytomegalovirus microRNAs. Curr Opin Virol 7:40–46. doi:10.1016/j.coviro.2014.03.015
Reeves MB, Lehner PJ, Sissons JG, Sinclair JH (2005) An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J Gen Virol 86(Pt 11):2949–2954. doi:10.1099/vir.0.81161-0
Sinclair J, Sissons P (2006) Latency and reactivation of human cytomegalovirus. J Gen Virol 87(Pt 7):1763–1779
Weekes MP, Tan SY, Poole E, Talbot S, Antrobus R, Smith DL, Montag C, Gygi SP, Sinclair JH, Lehner PJ (2013) Latency-associated degradation of the MRP1 drug transporter during latent human cytomegalovirus infection. Science 340(6129):199–202. doi:10.1126/science.1235047
Robbiani DF, Finch RA, Jager D, Muller WA, Sartorelli AC, Randolph GJ (2000) The leukotriene C(4) transporter MRP1 regulates CCL19 (MIP-3beta, ELC)-dependent mobilization of dendritic cells to lymph nodes. Cell 103(5):757–768
Wills MR, Poole E, Lau B, Krishna B, Sinclair JH (2015) The immunology of human cytomegalovirus latency: could latent infection be cleared by novel immunotherapeutic strategies? Cell Mol Immunol 12(2):128–138. doi:10.1038/cmi.2014.75
Mason GM, Jackson S, Okecha G, Poole E, Sissons JG, Sinclair J, Wills MR (2013) Human cytomegalovirus latency-associated proteins elicit immune-suppressive IL-10 producing CD4(+) T cells. PLoS Pathog 9(10):e1003635. doi:10.1371/journal.ppat.1003635
Mason GM, Poole E, Sissons JG, Wills MR, Sinclair JH (2012) Human cytomegalovirus latency alters the cellular secretome, inducing cluster of differentiation (CD)4+ T-cell migration and suppression of effector function. Proc Natl Acad Sci USA 109(36):14538–14543. doi:10.1073/pnas.1204836109
Reddehase MJ (ed) (2013) Cytomegaloviruses: from molecular pathogenesis to intervention. Caister Academic Press, London. ISBN:978-1-908230-18-8 (with the assistance of Niels A.W. Lemmermann)
Plotkin SA, Farquhar J, Horberger E (1976) Clinical trials of immunization with the Towne 125 strain of human cytomegalovirus. J Infect Dis 134(5):470–475
Griffiths PD, Stanton A, McCarrell E, Smith C, Osman M, Harber M, Davenport A, Jones G, Wheeler DC, O’Beirne J, Thorburn D, Patch D, Atkinson CE, Pichon S, Sweny P, Lanzman M, Woodford E, Rothwell E, Old N, Kinyanjui R, Haque T, Atabani S, Luck S, Prideaux S, Milne RS, Emery VC, Burroughs AK (2011) Cytomegalovirus glycoprotein-B vaccine with MF59 adjuvant in transplant recipients: a phase 2 randomised placebo-controlled trial. Lancet 377(9773):1256–1263. doi:10.1016/S0140-6736(11)60136-0
Quinnan GV Jr, Kirmani N, Rook AH, Manischewitz JF, Jackson L, Moreschi G, Santos GW, Saral R, Burns WH (1982) Cytotoxic t cells in cytomegalovirus infection: HLA-restricted T-lymphocyte and non-T-lymphocyte cytotoxic responses correlate with recovery from cytomegalovirus infection in bone-marrow-transplant recipients. N Engl J Med 307(1):7–13
Gandhi MK, Wills MR, Okecha G, Day EK, Hicks R, Marcus RE, Sissons JG, Carmichael AJ (2003) Late diversification in the clonal composition of human cytomegalovirus-specific CD8+ T cells following allogeneic hemopoietic stem cell transplantation. Blood 102(9):3427–3438. doi:10.1182/blood-2002-12-3689
Jabs DA (2011) Cytomegalovirus retinitis and the acquired immunodeficiency syndrome–bench to bedside LXVII Edward Jackson Memorial Lecture. Am J Ophthalmol 151(2):198–216. doi:10.1016/j.ajo.2010.10.018 e191
Urban B, Bakunowicz-Lazarczyk A, Michalczuk M (2014) Immune recovery uveitis: pathogenesis, clinical symptoms, and treatment. Mediat Inflamm 2014:971417. doi:10.1155/2014/971417
Hartigan-O’Connor DJ, Jacobson MA, Tan QX, Sinclair E (2011) Development of cytomegalovirus (CMV) immune recovery uveitis is associated with Th17 cell depletion and poor systemic CMV-specific T cell responses. Clin Infect Dis 52(3):409–417. doi:10.1093/cid/ciq112
Schrier RD, Song MK, Smith IL, Karavellas MP, Bartsch DU, Torriani FJ, Garcia CR, Freeman WR (2006) Intraocular viral and immune pathogenesis of immune recovery uveitis in patients with healed cytomegalovirus retinitis. Retina 26(2):165–169
Riddell SR, Watanabe KS, Goodrich JM, Li CR, Agha ME, Greenberg PD (1992) Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science 257(5067):238–241
Cobbold M, Khan N, Pourgheysari B, Tauro S, McDonald D, Osman H, Assenmacher M, Billingham L, Steward C, Crawley C, Olavarria E, Goldman J, Chakraverty R, Mahendra P, Craddock C, Moss PA (2005) Adoptive transfer of cytomegalovirus-specific CTL to stem cell transplant patients after selection by HLA-peptide tetramers. J Exp Med 202(3):379–386
Einsele H, Roosnek E, Rufer N, Sinzger C, Riegler S, Loffler J, Grigoleit U, Moris A, Rammensee HG, Kanz L, Kleihauer A, Frank F, Jahn G, Hebart H (2002) Infusion of cytomegalovirus (CMV)-specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood 99(11):3916–3922
Peggs KS, Verfuerth S, Pizzey A, Chow SL, Thomson K, Mackinnon S (2009) Cytomegalovirus-specific T cell immunotherapy promotes restoration of durable functional antiviral immunity following allogeneic stem cell transplantation. Clin Infect Dis 49(12):1851–1860
Pahl-Seibert MF, Juelch M, Podlech J, Thomas D, Deegen P, Reddehase MJ, Holtappels R (2005) Highly protective in vivo function of cytomegalovirus IE1 epitope-specific memory CD8 T cells purified by T-cell receptor-based cell sorting. J Virol 79(9):5400–5413
Reddehase MJ (2002) Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat Rev Immunol 2(11):831–844
Riddell SR, Watanabe KS, Goodrich JM, Li CR, Agha ME, Greenberg PD (1992) Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science 257 (5067):238–241; ISSN: 0036-8075
Einsele H, Kapp M, Grigoleit GU (2008) CMV-specific T cell therapy. Blood Cells Mol Dis 40(1):71–75
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA (2014) Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371(16):1507–1517. doi:10.1056/NEJMoa1407222
Ebert S, Podlech J, Gillert-Marien D, Gergely KM, Buttner JK, Fink A, Freitag K, Thomas D, Reddehase MJ, Holtappels R (2012) Parameters determining the efficacy of adoptive CD8 T-cell therapy of cytomegalovirus infection. Med Microbiol Immunol 201(4):527–539. doi:10.1007/s00430-012-0258-x
Holtappels R, Ebert S, Podlech J, Fink A, Böhm V, Lemmermann NAW, Freitag K, Renzaho A, Thomas D, Reddehase MJ (2013) Murine model for cytoimmunotherapy of CMV disease after hematopoietic cell transplantation. In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention. Caister Academic Press, Norfolk, pp 353–381
Früh K, Malouli D, Oxford KL, Barry PA (2013) Non-human-primate models of cytomegalovirus infection, prevention, and therapy. In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention. Caister Academic Press, Norfolk, pp 463–496
Acknowledgments
The author wishes to acknowledge the generosity of close colleagues in Cambridge who have allowed him to talk about their work, in particular John Sinclair, Matt Reeves, Paul Lehner and Michael Weekes, and all the members of their groups who contributed to the work on HCMV, the long-standing Programme Grant support from the Medical Research Council and Fellowship support of the Wellcome Trust. He apologises for the limited citations of the extensive clinical and experimental literature that has contributed so much to understanding CMV biology and disease.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Special Issue on Cytomegalovirus.
Rights and permissions
About this article

Cite this article
Sissons, J., Wills, M.R. How understanding immunology contributes to managing CMV disease in immunosuppressed patients: now and in future. Med Microbiol Immunol 204, 307–316 (2015). https://doi.org/10.1007/s00430-015-0415-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00430-015-0415-0