
Abstract
The Antarctic ecotype of Colobanthus quitensis is a vascular plant highly adapted to the harsh environmental conditions of Maritime Antarctica which is now facing with the rapid local warming experienced in the Antarctic Peninsula during the last decades. Thus, the identification of the molecular mechanisms leading to the adaptation to this warming trend is a new target for modern cell physiology. The selection of suitable reference genes for quantification of key stress-responsive genes through quantitative Reverse Transcriptase-Polymerase Chain Reaction (qRT-PCR) is important to ensure accurate and reliable results. In this study, we evaluated the expression stability of eleven candidate genes in C. quitensis under different abiotic stress conditions using geNorm and RefFinder tools. The statistical analysis showed that the appropriate reference genes varied depending on the experimental conditions, even if EF1α and PP2Acs ranked as the most stable reference genes when all stress conditions were considered. To further validate the stability of the selected reference genes, the expression patterns of C. quitensis catalase gene (CqCAT) was analyzed. The reference genes validated in this study will be useful for improving the accuracy of qRT-PCR analysis for gene expression studies of the Antarctic ecotype of C. quitensis and could be extended to other ecotypes adapted to low temperatures.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The rise in average Earth’s temperatures is one of the main consequences of the increase of atmospheric concentrations of greenhouse gases. The areas along the Antarctic Peninsula are warming faster than many other parts of the world (Turner et al. 2005; Mulvaney et al. 2012). This makes particularly interesting to investigate the genetic and molecular traits of the local plants to shed some light on the molecular basis of adaptation to low temperatures and their ability to cope with climatic changes. During evolution, the climatic conditions of Maritime Antarctic ecosystems allowed spreading and survival of only two highly adapted vascular plants species: the Antarctic hairgrass (Deschampsia antarctica Desv., Poaceae) and the Antarctic pearlwort (Colobanthus quitensis (Kunth) Bartl., Caryophillaceae) (Longton 1979; Smith 2003), which are also endemic of South and Central America. In particular, C. quitensis is present in the Andes of the central Chile as well as in Mexico, while D. antarctica occurs from Antarctica to South Central Chile through the Andes Mountains, neighboring Argentina (Moore 1970). The identification of the mechanisms underlying their stress and cold adaptations is a challenge for modern cell biology and cell physiology. It has been reported that during evolution and following glaciation events the Antarctic ecotype of C. quitensis adapted both physiologically and morphologically to cold environment (Alberdi et al. 2002; Pérez-Torres et al. 2004; Bravo et al. 2007; Bascuñán-Godoy et al., 2012). For instance, it has been demonstrated that the photosynthetic machinery evolved to cope with low temperature-induced photoinhibition modifying chloroplasts in order to restore the energy balance (Krause 1994). Moreover, the global warming has also influenced the life of organisms in the Antarctic Peninsula. In particular, it has been proved that the size and number of populations of C. quitensis as well as germination ability increased, suggesting an improvement in its reproductive performances as result of longer and warmer growing seasons (Day et al. 1999; Vera et al. 2013; Sanhueza et al. 2017). Furthermore, it has been reported that higher temperature also influences in situ freezing resistance (Sierra-Almeida et al. 2018) and photosynthesis rates (Xiong et al. 2000; Sáez et al. 2018). Recently, ecophysiological traits of Antarctic vascular plants have been unveiled figuring out that response to climate change relies on many factors including metabolic reprogramming as well as morphological modifications (Cavieres et al. 2016). Interestingly, it has been also demonstrated that C. quitensis populations show anatomical and physiological adaptation to simulated global warming along a latitudinal gradient (Acuña-Rodríguez et al. 2017). Moreover, many studies have been also proving that the interaction with microorganisms may help C. quitensis colonize the unique Antarctic environment (Torres-Díaz et al. 2016; Nibert et al. 2018; Ballesteros et al. 2020). Despite the growing number of physiological studies addressing the ability of C. quitensis to adapt to the Antarctic environment and its progressive warming, little is known about the molecular characterization of genes involved in its adaptation to these climate conditions. Changes in photosynthesis and stress response genes were recently reported to be related to extreme environments adaptation (Cho et al. 2018), but more studies are needed to disclose the transcriptomic reprogramming induced by the rapid local warming experienced in the Antarctic Peninsula during the last decades.
Recently, we performed de novo assembly of C. quitensis transcriptome (Arthofer et al. 2015). With the aim to study the influence of temperature on C. quitensis transcriptomic reprogramming, small plexiglass greenhouses open on the top (Open Top Chambers, OTC) have been installed in December 2012 near the Arctowski base in King George Island, Maritime Antarctic (62°14′S; 58°48′W), in a field where C. quitensis grows and expands together with D. antarctica. Plants growing inside OTC experienced a mean temperature increase of around 4 °C at midday, mimicking regional warming for remote areas such as polar habitats (Bokhorst et al. 2013). It is worthwhile mentioning that the OTC may influence plant growth also affecting the light quality, air humidity and water content. After one year, C. quitensis individuals grown inside OTC (condition IN) and control plants grown in close proximity outside OTC (condition OUT) were harvested. RNA sequencing (RNA-seq) analysis of both samples resulted in a high number of genes differentially expressed among the two experimental conditions (Bertini et al. in preparation). This approach is of great sensitivity for gene expression profiling, representing indeed the gold standard for studying the influence of different experimental conditions on transcriptome remodeling. Nevertheless, RNA-seq, and high-throughput transcriptomic data in general, are often validated using a different approach and quantitative Reverse Transcriptase-Polymerase Chain Reaction (qRT-PCR) is the most extensively used (Chuaqui et al. 2002; Tsukagoshi et al. 2015). Indeed, qRT-PCR has become extremely useful in many research fields contributing to understand signaling and metabolic pathways involved in several biological processes, including the molecular adaptation of different C. quitensis haplotypes coming from multiple colonization events of the Maritime Antarctic (Biersma et al. 2020). This method is rapid, sensitive and specific; moreover, it has good reliability to study gene expression, determining changes of the transcripts in different experimental conditions (Gachon et al. 2004; Wong and Medrano 2005; Nolan et al. 2006). Despite its powerful capacity, qRT-PCR presents some pitfalls such as the use of unreliable reference genes for the normalization steps (Huggett et al. 2005). Consequently, qRT-PCR could be underused due to the lack of characterized reference genes that enable robust normalization, especially in less known plant species.
Reference genes are usually housekeeping genes (HKGs) that are stably and constitutively expressed in all plant tissues regardless of growth, developmental stage or environmental conditions, therefore having an expression level unaffected by experimental conditions (Radonic et al. 2004; Huggett et al. 2005). Generally, HKGs encode for proteins involved in basic metabolism or in the cytoskeleton architecture, such as Elongation Factor 1-α (EF1-α), Glyceraldehyde 3-Phosphate Dehydrogenase (GAPDH), ubiquitin, α-actin or β-tubulin. Several papers reported the identification of HKGs that can be used for validation and gene expression analyses in different plant species (Czechowski et al. 2005; Paolacci et al. 2009; Dai et al. 2018; Ruduś and Kępczyński 2018; Santos et al. 2018; Zhang et al. 2018). However, there are no universal normalizing genes that can be used for all plant species because many of the genes examined showed a variable expression depending on the experimental conditions and the species tested (Thellin et al. 2009; Artico et al. 2010; Kozera and Rapacz 2013). Thus, it is of utmost importance to evaluate the stability of reference genes under certain experimental conditions in order to select the most appropriate ones to be used for normalization of gene expression assessment. Therefore, validation studies are fundamental to select new reference genes that are constitutively expressed regardless of experimental condition prior to their use in qRT-PCR analysis.
Several algorithms are now freely available that allow the evaluation of the best candidate reference genes related to a given set of experimental conditions, such as: the comparative Δ cycle threshold (ΔCt) method (Silver et al. 2006), geNorm (Vandesompele et al. 2002), NormFinder (Andersen et al. 2004), BestKeeper (Pfaffl et al. 2004) and qBase (Hellemans et al. 2007). In addition, RefFinder (Kim et al. 2010) that integrates geNorm, NormFinder, BestKeeper and the comparative ΔCt method is very useful to calculate the rank of the selected candidate reference genes. It is important to underline that some of these algorithms have been developed using animal or human systems, so their use in plant science needs to be integrated with more specific analysis.
In this paper, we report the validation of eight specific reference genes suited for quantitative analysis of C. quitensis grown under different abiotic stresses (i.e. nitrogen supply, salinity and OTC). In particular, we selected Translation Elongation Factor 1-α and 1-β (EF1α and EF1β, respectively), Glucose 6-Phosphate Dehydrogenase (G6PDH), Cyclophilin (CYP), Phospholipase A2 (PLPA2), catalytic subunit of Protein Phosphatase 2A (PP2Acs), Phosphoglycerate Kinase (PGK) and F-box family protein (FBX). In addition, to test the reliability of the selected reference genes, the expression level of the gene coding for catalase from C. quitensis (CqCAT) was examined. Ultimately, our data provide a useful set of reference genes that can be used in qRT-PCR to analyze gene expression in C. quitensis under different experimental conditions in order to disclose how plants react and adapt to environmental stressors, including abiotic stress as well as selective pressure due to multiple colonization events.
Materials and methods
In vivo plant growth under control and warmer conditions
Field activity was carried out in the area close to Henryk Arctowski Antarctic Research station, King George Island, Maritime Antarctica (62° 14′S, 58° 48′W). Permits for entrance and plant collection in the Antarctic Specially Protected Area (ASPA) 128 were provided by The Chilean Antarctic Institute (INACH) and by the Italian National Agency for New Technologies, Energy and Sustainable Economic Development—Technical Antarctic Unit (ENEA-UTA). Colobanthus quitensis plants were grown in the field for 15 months inside OTC (December 2012–March 2014), which determined an increase of about 4 °C in the internal temperature during midday. Leaf samples from individuals grown either in warm conditions (inside OTC, samples IN) or in natural conditions (outside OTC, samples OUT) were harvested and pooled. Freshly collected leaves were soaked in RNAlater® solution (2:10, w/v) (Sigma-Aldrich, Saint Louis, Missouri, USA), and stored at -80 °C until RNA isolation.
Plant growth and treatments
Entire plants of C. quitensis with underground portion were extracted near to the Henryk Arctowski Polish Antarctic station in King George Island, and transported to Universidad de La Frontera, Temuco, Chile. Plants were propagated and maintained in a greenhouse, using plastic pot with soil/peat/vermiculite mixture (3:1:1) and fertilized with a Phostrogen® (Bayer-garden, UK). Before treatment application, plants were exposed to nutrient drainage for one month, suppressing addition of fertilization product and supplied only with distilled water. For the experiment, plants were separated into individuals and their roots were rinsed with distilled water. Homogeneous size plants (aerial and subterranean portions) were selected and transferred to plastic pots containing vermiculite as substrate. Ammonium fertilization assay was applied after establishment as described in Rabert et al. (2017). Briefly, plants were watered by saturating the substrate with a modified Hoagland nutrient solution at pH 5.6 every 72 h for the next five months. The following treatments were applied: 0, 8 and 32 mmol L−1 of N-NH4+ (samples N0, N1, N2, respectively). The treatment without ammonium application (0 mmol/L mmol L−1 of N) was prepared as followed: 2 mmol L−1 KH2PO4, 1 mmol L−1 K2SO4, 2.05 mmol L−1 KCl, 3 mmol L−1 Ca(H2PO4)2H2O, 2 mmol L−1 MgSO47H2O, 1 mmol L−1 CaSO4, 0.025 mmol L−1 H3BO3, 0.002 mmol L−1 MnSO4, 0.002 mmol L−1 ZnSO4, 0.05 μmol L−1 CuSO45H2O, 0.05 μmol L−1 H2MoO4, and 0.02 mmol L−1 Fe-EDTA. For ammonium treatments, the solution without N-NH4+ was used as basal solution and ammonium application was supplied as (NH4)2H2PO4 and CO(NH2)2 at required concentrations. At the end of treatments, plants were collected and stored at − 80 °C. Meanwhile, for salinity assay, after establishment, plants were irrigated for the next two months every 48 h at field capacity of substrate with a distilled water or modified Hoagland nutrient solution at pH 5.6 consisting in: 2 mmol L−1 (NH4)2H2PO4, 2 mmol L−1 CO(NH2)2, 2 mmol L−1 K2HPO4, 1 mmol L−1 K2SO4, 2.05 mmol L−1 KCl, 3 mmol L−1 Ca(H2PO4)2H2O, 2 mmol L−1 MgSO47H2O, 1 mmol L−1 CaSO4, 0.025 mmol L−1 H3BO3, 0.002 mmol L−1 MnSO4, 0.002 mmol L−1 ZnSO4, 0.05 μmol L−1 CuSO4 5H2O, 0.05 μmol L−1 H2MoO4, and 0.02 mmol L−1 Fe-EDTA. The following treatments were applied: 0, 250, 500 and 750 mmol L−1 of NaCl (samples S0, S1, S2, S3, respectively). The application of salinity treatments was gradual. In the first month of treatment, every two days, the irrigation was alternated between distilled water and Hoagland solution plus NaCl at required concentrations. In the second month of treatment, the Hoagland solution plus NaCl was applied every two days. At the end of this period, plants were collected and stored at − 80 °C until use.
Total RNA isolation and complementary DNA synthesis
Total RNA was extracted using the Nucleospin® RNA Plant kit (Macherey–Nagel, Düren, Germany), starting from 100 mg of finely grinded leaf samples, according to the manufacturer’s instructions. RNA concentration was estimated by reading spectrophotometric absorbance at 260 nm, whereas the OD260/OD280 nm and OD260/OD230 nm absorption ratios were calculated to evaluate RNA quality and purity (spectrophotometer UV-30 SCAN, ONDA). RNA integrity was also verified by agarose gel electrophoresis (Online Resource 1), whereas the absence of DNA contamination was tested using 100 ng of total RNA as template in a PCR reaction using EF1α specific primers for amplification.
Complementary DNA (cDNA) was synthesized using the ImProm-II™ Reverse Transcription System (Promega, Madison, WI, USA) starting from 1 μg of RNA as template and using the oligo-dT primer for first strand synthesis.
Candidate reference genes selection and primers design
Sequence information of the candidate reference genes were obtained from the Shotgun Transcriptome Assembly of C. quitensis leaves (National Center of Biotechnology Information (NCBI) Sequence Read Archive (SRA), accession SRX814890). In particular, the following accession numbers are related to samples IN (SRR1720758, SRR1720760, SRR1720762) while the subsequent accession numbers are related to samples OUT (SRR1720763, SRR1720765, SRR1720767). Eleven candidate reference genes were selected for analysis of their expression profile, based on the stability of their RPKM (Reads Per Kilobase per Million mapped reads) values obtained from the transcriptomic analysis of both IN and OUT samples (Table 1 and Online Resource 2): Translation Elongation Factor 1-α and 1-β (EF1α and EF1β, respectively), Polypyrimidine Tract Binding protein (PTB), Glucose 6-Phosphate Dehydrogenase (G6PDH), Cyclophilin (CYP), Porphobilinogen Deaminase (PBGD), TATA-box Binding Protein (TBP), catalytic subunit of Protein Phosphatase 2A (PP2Acs), Phosphoglycerate Kinase (PGK), F-box family protein (FBX) and Phospholipase A2 (PLPA2). The raw data relative to the RPKM values of all the contigs obtained from the Shotgun Transcriptome Assembly of C. quitensis leaves are supplied in Online Resource 3. Blast hits can be accessed in Dryad: https://doi.org/10.5061/dryad.jp1mk. For primer pairs design, we searched for the contigs coding for each of the selected candidate genes inside the transcriptome data (Table 1) and we aligned them using the freely available ClustalW bioinformatics tool (https://embnet.vital-it.ch/software/ClustalW.html). Then, primers were designed inside the most conserved region using Primer3 software (http://bioinfo.ut.ee/primer3-0.4.0/). The specificity of each selected primer pair was observed via standard PCR on synthesized cDNA using the BIOTAQ DNA polymerase (Bioline, London, UK), and each amplification product was verified by 1.5% agarose gel electrophoresis.
Quantitative reverse transcriptase-PCR analysis
Quantitative Reverse Transcriptase-PCR (qRT-PCR) reactions were performed in 96-well plates on a Bio-Rad CFX96 Real-Time PCR thermal cycler (Bio-Rad, Hercules, California, USA), using the SYBR Green detection system. The reaction mixture (10 μL) contained 1 μL of four-fold diluted cDNA, 5 μL Sso Advanced SYBR Green Supermix (Bio-Rad, Hercules, California, USA) and 0.5 μM of each gene specific primer, except for FBX (0.3 μM) and PTB (0.4 μM) primers. The cycling conditions were the following: initial denaturation step at 95 °C for 3 min, followed by 44 cycles at 95 °C for 10 s and primer annealing at different temperature (Table 2) for 30 s. Next, the melting curves ranging from 70 to 95 °C (with constant increase of 0.5 °C every 5 s) were evaluated in order to check the PCR specificity. Each assay included no-template controls (NTCs) for each primer pair. Primers efficiency (E) was calculated by generating standard curves for each oligonucleotide pair with at least five serial fourfold dilution points and the slope of the amplification curve was used to calculate E = 10(−1/slope). Output data were processed using the CFX ManagerTM Software (Bio-Rad, Hercules, California, USA). All qRT-PCR reactions were run in three technical and biological replicates.
Data analysis for expression stability of candidate reference genes
geNorm and RefFinder algorithms were used to calculate and rank the stability of the candidate reference genes. The geNorm software (Vandesompele et al. 2002) determines the expression stability of control genes based on non-normalized expression levels. A stability parameter M is calculated as the average pairwise variation between all tested reference genes, with a recommended cut-off value of 1.5. The genes are ranked according to their M value and those with the lowest M value are the most stable ones. RefFinder (Kim et al. 2010) is a freely available software platform, which integrates four different algorithms, namely the above mentioned geNorm, NormFinder (Andersen et al. 2004), BestKeeper (Pfaffl et al. 2004) and the comparative ∆Ct method (Silver et al. 2006), to analyze the stability of reference genes comprehensively. It uses the cycle threshold (Ct) value as input data and ranks the reference genes according to the four mentioned algorithms independently. Moreover, it assigns a weight to each reference gene and calculates the geometric mean (Geomean) of their weights to obtain the recommended comprehensive ranking order, identifying the best reference gene or pair of genes.
The NormFinder software (Andersen et al. 2004) uses a model-based approach to estimate expression variation. The software automatically calculates a stability value for all reference genes by organizing samples in groups. This approach considers as best genes those having the least intra- and inter-group variation in expression, so that the genes with the lowest value are the best normalizers.
The BestKeeper software (Pfaffl et al. 2004) evaluates the stability of reference genes calculating their standard deviation (SD) using CP (crossing point) or Ct values as input in different experimental conditions. The genes with a SD [± CP] value below 1.0 are considered to be stably expressed.
The ∆Ct method (Silver et al. 2006) chooses the best reference gene by comparing the variation of the cycle threshold values between different pairs of reference genes. If the ∆Ct value between two genes remains constant when analyzed in several experimental samples, this means that both genes are stably expressed or that they are co-regulated. However, if the ∆Ct oscillates, the introduction of a third, fourth or fifth gene in the comparison provides information on which pairs show a lower variability and thus which gene has a more stable expression among the tested samples.
Validation of reference genes
For the validation of selected reference genes for qRT-PCR analysis under different abiotic stress in C. quitensis, the relative transcript accumulation of catalase gene from C. quitensis (CqCAT) was normalized using the two most stable (EF1α and PP2Acs), and the least stable (PLPA2) reference genes identified in this study. Sequence information and specific primers for CqCAT were obtained as described above. Primers sequence and related information are shown in Table 2. Quantitative analysis for CqCAT gene was performed according to the 2−∆∆Ct method by applying the efficiency correction formula (Pfaffl 2001).
Results
Selection of candidate reference genes and their characterization
In this study, eleven reference genes showing stable expressions in C. quitensis transcriptome data from both inside (IN) and outside (OUT) OTC samples were selected as candidate for normalization of gene expression analysis. The cut-off value for the stability of gene expression between the two conditions was fixed as − 1 < Log2 Fold Change IN/OUT < 1 (Table 1).
The selected genes could be grouped into three categories: (i) genes involved in transcription, i.e. TBP (TATA-box Binding Protein) and PTB (Polypyrimidine Tract-Binding protein); (ii) translation, i.e. EF1α and EF1β (Translation Elongation Factor 1-α and 1-β); (iii) cellular metabolism, i.e. PBGD (Porphobilinogen Deaminase), G6PDH (Glucose-6-Phosphate Dehydrogenase), PLPA2 (Phospholipase A2) and PGK (Phosphoglycerate Kinase). In addition, three traditional housekeeping genes were also selected: CYP (Cyclophylin), PP2Acs (Protein Phosphatase 2A catalytic subunit) and FBX (F-box family protein). Gene names and description, accession ID, primer sequences, annealing temperatures and amplicon length as well as amplification efficiency and regression coefficient (R-squared) are listed in Table 2. Gene-specific amplification was confirmed by the presence of a single peak in the melting curves obtained by qRT-PCR (Online Resource 4). Moreover, no primer dimers were detected during the amplification reactions neither nonspecific products were detected in negative controls. Efficiency values were accepted within a range of 100 ± 10%, accompanied by a linear regression value as close to 1.
Expression profiles of the candidate reference genes
In order to detect the expression profiles of the eleven candidate reference genes of C. quitensis and to study their stability in qRT-PCR, we analyzed several experimental samples subjected to different abiotic stresses (i.e. nitrogen supply, salinity and OTC).
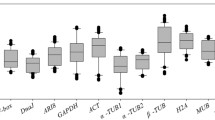
The cycle threshold (Ct) values were determined for each gene through all the experimental conditions and the mean Ct values along with the standard deviation (SD) were determined (Online Resource 5). The Ct values measured in each run allowed us to assess the transcript abundance of the candidate genes. The mean Ct value ranged from 19.3 to 25.96, which is considered a moderate to high level of expression. In particular, EF1α showed the highest expression level with a mean Ct value of 19.3 ± 2.62, whereas G6PDH showed the lowest transcript abundance with a mean Ct value of 25.96 ± 1.30. Moreover, SD of Ct values, which can reveal the expression stability of candidate reference genes, ranged from 1.30 to 2.98 for six genes (G6PDH, FBX, PP2Acs, EF1α, CYP and EF1β), indicating a good stability (Online Resource 5). On the other hand, PGK and PLPA2 showed SD values of 3.25 and 4.24, respectively, indicating a lower level of stability. Furthermore, it is worthwhile mentioning that PBGD, TBP and PTB displayed a different behavior in relation to the stress conditions. As far as PTB is concerned, its expression level was undetectable in samples subjected to nitrogen supply (Ct > 40) at any concentration (N0 through N2), whereas TBP expression was undetectable during both nitrogen supply (N0 through N2) and salinity stress (S0 through S3); finally, PBGD expression was undetectable only at the highest concentration of NaCl (S2 and S3) (Online Resource 5). The different behavior of the last three genes indicates a different impact of the experimental conditions on their expression as already reported in similar studies in other plant species (Chen et al. 2011; Tillett et al. 2011). Since a good reference gene should be expressed at good level and with high stability independently from experimental conditions, we decided to continue our analysis only with eight genes, namely CYP, EF1α, EF1β, G6PDH, PP2Acs, FBX, PLPA2 and PGK. The box plot graph of the Ct values for all the selected candidate reference genes is showed in Fig. 1.

Box-plot graph of the cycle threshold (Ct) values referred to the eleven candidate reference genes across the experimental samples (n = 9). The line across the box shows the median value, lower and upper boxes indicate the 25th percentile to the 75th percentile, the whiskers represent the 95% confidence intervals. Ct values for all the reference genes are given in Online Resource 5
Expression stability of the candidate reference genes
Since the eight candidate reference genes did not show a completely constant expression level across all samples, geNorm and RefFinder tools were used in order to evaluate and rank the stability of the selected genes for qRT-PCR studies.
The geNorm algorithm calculates the stability M value that is used to arrange the gene ranking order from the smallest to the largest value, i.e. from the highest to the lowest stability, with a suggested cut-off range of M ≤ 1.5 (Vandesompele et al. 2002). During nitrogen supply stress, the lowest M values (M < 0.15) were attributed to PGK, FBX, PP2Acs and CYP even though all other genes displayed very low M values (< 0.6), indicating that they can be all considered reliable candidates for normalization studies under this stress condition (Fig. 2 and Online Resource 6).

Expression stability values (M) of eight candidate reference genes calculated by geNorm software in different sample groups. The genes with M ≤ 1.5 are considered significant with stable expression
During salinity stress, EF1β, EF1α and PGK were found to be the most stable genes (M < 0.6). However, M values < 1.5 were also found for PP2Acs, CYP and FBX, indicating that also these genes can be suitable for normalization studies under this stress condition. On the contrary, G6PDH and PLPA2 with M values > 1.5 should be considered the least stable (Fig. 2 and Online Resource 6). As far as OTC stress is concerned, PP2Acs, G6PDH and PGK were the most stable genes showing M value ≤ 0.1. The other genes displayed an M value slightly higher but, in any case < 1.5, so they all can be considered reliable and stable reference genes under this stress condition (Fig. 2 and Online Resource 6). When all stress conditions were taken into account together, the best reference genes were EF1β, PGK and EF1α with M values < 0.9. In addition, PP2Acs, CYP, FBX and G6PDH displayed M value < 1.5, indicating that they are all suitable reference genes, whereas PLPA2 with an M value slightly greater than 1.5 is borderline (Fig. 2 and Online Resource 6).
The geNorm program was also used to assess the optimal number of reference genes required for accurate normalization across the experimental conditions by calculating the pairwise variation (Vn/Vn+1) between two sequential normalization factors NF (NFn and NFn+1). According to this software, a cut-off value of 0.15 is the recommended threshold to determine whether additional reference genes are required for better normalization (Vandesompele et al. 2002). A little variation between Vn/Vn+1 indicates that the number of reference genes identified is sufficient for normalization. The V2/3 values in nitrogen supply and OTC stresses were lower than 0.15 (Fig. 3) suggesting that the top two reference genes were sufficient for accurate normalization. Nevertheless, when salinity and all stress conditions were considered, the lowest pairwise variation value was above 0.15. As recommended in this case, the lowest Vn/Vn+1 value could be considered to determine the number of reference genes adequate for normalization (Vandesompele et al. 2002). Thus, the use of the four most stable genes could be considered a valid normalization strategy for both conditions and the addition of two more genes has a significant effect on the results compared to the other conditions.

Pairwise variation (V) analysis of eight candidate reference genes in different sample groups, calculated by geNorm software. The optimal number of the reference genes required for accurate normalization was determined by pairwise variation (Vn/Vn+1). The dotted line indicates the recommended threshold value of 0.15 below which the inclusion of an additional reference gene is not necessary
In Fig. 4 the expression stabilities of the selected reference genes according to NormFinder, BestKeeper and ΔCt algorithms, as derived by RefFinder, are reported. In particular, the results of NormFinder analysis highlighted that all genes have a good stability during OTC and nitrogen supply stresses displaying expression stability values ranging from 0.289 (PP2Acs, PGK, CYP) to 1.0 (PLPA2) for nitrogen stress and from 0.354 (G6PDH, PP2Acs, CYP, PLPA2, EF1α) to 0.9 (EF1β) for OTC stress (Fig. 4a and Online Resource 7). On the contrary, great variability among the reference genes was found during salinity stress, ranging from 0.126 (EF1α) to 2.918 (G6PDH). When all stress conditions were taken into account together, PP2Acs (0.554) ranked first as the most stable expressed gene, followed by EF1α (0.566) and EF1β (0.633), whereas PLPA2 was the least stable with a stability value of 2.184.

Expression stability analysis of eight candidate reference genes assayed by the software: NormFinder (a), based on the stability values calculated by the software; BestKeeper (b), based on the standard deviation values ± crossing point (SD [± CP]); ΔCt (c), based on the average of standard deviation (SD) values
BestKeeper analysis (Fig. 4b and Online Resource 8) showed a good stability for all reference genes when considering OTC and nitrogen supply stresses, as found with NormFinder analysis. In fact, SD [± CP] values ranged from 0.0 (PP2Acs, PGK, CYP) to 0.67 (PLPA2) during nitrogen stress and from 0.0 (FBX, PGK) to 1 (EF1α) under OTC stress. As regards to salinity stress, G6PDH was the only gene with a SD [± CP] close to 1 (1.11), indicating a good stability under these conditions, whereas all other reference genes displayed values > 1. When considering all stress conditions together, G6PDH (1.03) ranked first while PLPA2 (3.08) was the least stable reference gene as also found using the NormFinder and geNorm algorithms.
According to the previous results, the analysis with ΔCt method showed that all genes have a good stability during OTC and nitrogen supply stresses having average of SD values below 1 (Fig. 4c and Online Resource 9). In particular, values ranged from 0.47 (PP2Acs, PGK, CYP) to 1.06 (PLPA2) during nitrogen stress and from 0.30 (G6PDH, PP2Acs, CYP, PLPA2, EF1α) to 0.91 (EF1β) for OTC stress. As far as salinity stress is concerned, values of SD average varied from 1.33 (CYP) to 2.98 (G6PDH), indicating a moderate stability as reference genes. When all stress conditions were considered together, the stability values ranged from 1.28 (EF1α) and 2.34 (PLPA2). Once again, PLPA2 displayed the highest value correlated to the lowest stability, as also found using the other algorithms.
On the basis of the above results and taking into account all software (Figs. 2 and 4), we can infer that under nitrogen stress, PP2Acs, PGK and CYP could be considered as the most stable genes, while the least stable should be PLPA2. Under salinity stress, the gene stability order was different among the four software, but in general, EF1α, CYP and PP2Acs were the most stable, while G6PDH and PLPA2 were the least stable. Finally, as far as the OTC stress is concerned, the most stable genes were G6PDH and PP2Acs, being EF1β the least stable one. Although the stability order of the candidate reference genes varied as a function of the software used and the experimental conditions, a general consensus can be inferred when the stress conditions were considered all together. Indeed, PP2Acs and EF1α were often among the most stable genes, while PLPA2 and G6PDH were frequently the least stable in all conditions.
These evidences were confirmed by the comprehensive expression stability ranking of the candidate reference genes obtained by RefFinder (Table 3). Results confirmed PP2Acs and EF1α as the most stable genes due to the lowest Geomean of the ranking values. Furthermore, regardless of the order, the software substantiates the lower stability for PLPA2 and G6PDH ranking them as the most unstable gene in almost all sample sets, with the exception of the OTC stress. It is worth highlighting that small discrepancies were obtained between the original geNorm software package and the geNorm output of the RefFinder tool, probably because RefFinder uses the raw Ct values as input data, while the original software makes use of PCR efficiencies (Online Resources 6 and 10).
Catalase gene expression analysis under different abiotic stresses for reference genes validation
In order to validate the reliability of the best reference genes identified, the expression profiling of the C. quitensis catalase (CqCAT) gene was evaluated using the most stable reference gene (EF1α), the combination of the two most stable genes (EF1α + PP2Acs) and the least stable gene (PLPA2) as normalizers. The expression levels of CqCAT were determined by qRT-PCR on C. quitensis leaves harvested from plants subjected to different abiotic stresses, i.e. nitrogen supply, salinity and OTC (Fig. 5).

Relative expression of Colobanthus quitensis catalase gene (CqCAT) using the selected reference genes for transcript normalization following different stress treatments. (a) Nitrogen supply; (b) Salinity; (c) OTC. Bar indicates the standard error (±SE) evaluated from three biological replicates
In this study, when the most stable reference genes (EF1α and EF1α + PP2Acs) were used for accurate normalization of the samples under nitrogen supply stress, CqCAT transcript was found to increase along with the rise of ammonium concentration, whereas using the least stable reference gene (PLPA2), a decrease on CqCAT expression was observed at the highest ammonium concentration (Fig. 5a). A different expression pattern of CqCAT was also highlighted under salinity stress when the best or the least stable reference genes were used for data normalization. In particular, EF1α and EF1α + PP2Acs allowed to detect an increasing CqCAT expression between 0 and 250 mM NaCl and a strong decrease when 500 and 750 mM NaCl concentrations were reached. The expression pattern of CqCAT gene did not significantly change when the four top genes (EF1α, PP2Acs, PGK and EF1β) were used for normalization as suggested by the geNorm pairwise variation results (data not shown). On the other hand, using PLPA2 a very strong increase in CqCAT expression was registered until 500 mM NaCl (Fig. 5b). Finally, when OTC stress was considered, the CqCAT expression profile followed the same trend with all the reference genes (Fig. 5c). This result could be related to the good performance of PLPA2 gene, when the OTC stress was considered per se (Figs. 2 and 4).
Discussion
The recent climate changes affecting the entire planet have a strong impact on the Antarctic environment, with clear signs both on land and sea (Schofield et al. 2010). It has been reported an increase of the Antarctic Peninsula air temperature of about 2.6 °C in the last 50 years that influences and shapes the life and expansion of endemic organisms (Turner et al. 2014). In the Maritime Antarctic, only two endemic flowering plants found the way to survive and propagate in this harsh environment and, nowadays, they must overcome a further challenge represented by the climate change. Colobanthus quitensis represents the only native dicotyledonous plant able to grow in this region withstanding the hostile climate (Smith 2003) and a recent study assessed its very close origin dating its arrival after the last glacial event (Biersma et al. 2020). Due to the great impact that climate change could have all over the world, we wondered whether C. quitensis could be used as bioindicator of regional warming in the Antarctic Peninsula helping discover new molecular biomarkers useful to evaluate biological effects of climate change in Antarctica. The transcriptome of this plant has been recently unveiled (Arthofer et al. 2015; Cho et al. 2018) allowing design specific PCR primers to be used in gene expression profiling studies aimed at disclosing the differentially expressed genes in plants subjected to different stressors. Moreover, studies on the molecular adaptation of different C. quitensis haplotypes coming from multiple colonization events of the Maritime Antarctic might support genetic population studies aimed at dating its origin and age in Antarctica. However, although quantitative Reverse Transcriptase-PCR (qRT-PCR) is the technique of choice for gene expression studies, unfortunately it also has some weaknesses as the need to use reliable normalizing genes to obtain accurate and meaningful expression values (Gutierrez et al. 2008). Furthermore, it should also be taken into account that the transcription levels of reference genes can vary greatly in response to different physiological conditions (plant tissues, organs or developmental stages) as well as under different stress conditions or even depending on the experimental design (Czechowski et al. 2005; Le et al. 2012). Thus, the simply use of housekeeping genes commonly thought to be good reference genes could affect the reliability of results. Therefore, there are no universal reference genes for all seasons, but it is mandatory to select a panel of reliable genes within specific experimental treatments or tissues from the same individual. In a previous work, Cho et al. (2018) selected 18S rRNA and TIM (chloroplast-like Triosephosphate Isomerase, also known as TPI) as reference genes for gene expression analysis in C. quitensis plants challenged with abiotic stress. We searched for these genes in our experimental system, but could not find any contig coding for 18S rRNA, since our transcriptome analysis was performed after ribosomal RNA (rRNA) depletion. Moreover, we found three contigs coding for TIM, but with very unstable Log2 Fold Change (viz. -0.61, -0.82, -2.2). Indeed, genes that are good normalizers for some stresses may not be reliable for others, thus we searched for new reference genes and in this work we tested the reliability of eleven different candidates under diverse experimental conditions. Between them are some traditional housekeeping genes, such as EF1α and EF1β, widely used in several plant species as conventionally considered stably expressed under different treatments and various development stages (Zhou et al. 2016; Wan et al. 2017), and some new genes reported to be highly stable in diverse conditions, such as CYP, PP2Acs, PGK, TBP, PTB and FBX (Expósito-Rodríguez et al. 2008; Hao et al. 2014; Li et al. 2015; Wang et al. 2015; Taylor et al. 2016; Zhou et al. 2016). Moreover, three more genes, i.e. PLPA2, G6PDH and PBGD, which have proved to be suitable reference genes in insects (Meunier et al. 2006), fungi (Yin et al. 2013) and humans (Dundas and Ling 2012), respectively, were added to this study with the aim of testing their reliability in plants as well. At first, the expression profile of the candidate reference genes under various abiotic stress conditions (i.e., nitrogen supply, salinity and OTC) was evaluated by qRT-PCR. This preliminary analysis revealed that the expression of three out of eleven genes (i.e. TBP, PTB and PBGD) varied strongly as a function of the stress condition (Table 3 and Fig. 1), thus they were excluded from the following computational analysis. Although TBP and PTB were reported to rank between the first top reference genes in some plant systems (Hao et al. 2014; Taylor et al. 2016; Expósito-Rodríguez et al. 2017), they were also reported to be strongly unstable in tea plants under metal stress, in Oenanthe javanica under abiotic stress, in Olea europea between different cultivars, in cotton between different tissues (Jiang et al. 2014; Ray and Johnson 2014; Sun et al. 2015; Wang et al. 2017), corroborating our results. As far as PBGD is concerned, its reliability was well known in animal systems (Dundas et al. 2012; Yu et al. 2015), but its use as reference gene was unsuccessful in our model system. The stability ranking of the remaining eight candidate reference genes was evaluated using geNorm software package and RefFinder, a comprehensive software platform which integrates four different statistical algorithms, i.e. geNorm, NormFinder, BestKeeper and ΔCt. All software generally ranked the same genes as the least stable, but showed discrepancy in the ranking of the most stable candidate reference genes (Figs. 2 and 4). In particular, the least stable genes were identified as PLPA2 and G6PDH considering both individual stress, such as nitrogen supply or salinity, and all stress conditions jointly. These genes, which are not frequently used in plants, were selected with the aim of identifying new reference genes suitable for plant systems, but they turned out to be unreliable in our study. However, our findings are in agreement with some previous studies where these genes ranked between the last positions. In particular, PLPA2 was found to be unsuitable as internal control in peach under different treatments and development stages (Tong et al. 2009), and G6PDH was estimated as the least stable gene in soybean exposed to cadmium (Wang et al. 2012). Nevertheless, if OTC stress was considered per se, G6PDH and PLPA2 ranked between the first top genes according to all software, proving to be reliable reference genes in C. quitensis under this condition. As regards the best reference genes, only under nitrogen stress the four algorithms agreed identifying the same ranking positions for all the genes, classifying PP2Acs and PGK as the best reference genes. For salinity and OTC stresses, the four algorithms differed in the choice of the best reference genes, being ΔCt and Normfinder the algorithms with a more similar output. According to RefFinder analysis (Table 3), the comprehensive ranking recommended G6PDH and PP2Acs as the best reference genes under OTC stress and EF1α and CYP under salinity stress. Interestingly, EF1α and CYP were also reported to be the most stable genes in Deschampsia antarctica under salt stress (Lee et al. 2010). When all stress conditions were considered together, the comprehensive ranking indicated EF1α and PP2Acs as the most stable reference genes. Our findings were corroborated by several previous studies that reported the above genes as the most stable under different abiotic stresses. For example, they were recommended as internal control genes in Agrostis stolonifera (bentgrass) and Cenchrus americanus (pearl millet) under multiple abiotic stresses (Chen et al. 2014; Shivhare and Lata 2016). Moreover, EF1α was selected as the best reference gene in tomato under nitrogen and cold stresses (Løvdal and Lillo 2009), in cucumber under osmotic and salt stresses (Migocka and Papierniak 2011), in sugarcane (Guo et al. 2014) and Glycine max (soybean) (Ma et al. 2013) under salinity and drought stresses. Likewise, PP2Acs was found to be the most stable reference gene in pearl millet under salinity and drought stresses (Saha and Blumwald 2014), in Pisum sativum under salinity stress (Die et al. 2010), in soybean exposed to cadmium (Wang et al. 2012).
To further validate the selected reference genes in C. quitensis, the expression of catalase gene (CqCAT) under nitrogen supply, salinity and OTC stresses was evaluated. The choice of CqCAT as a target gene was due to its important role in scavenging the ROS notoriously produced at high levels under the considered abiotic stresses. Moreover, this gene is often exploited as an abiotic stress-responsive marker in many plant species. Indeed, many papers report the behaviour of CAT expression in different model systems under the same abiotic stresses studied in this paper and they can be taken into account to compare its expression trend in our model plant. The qRT-PCR analysis was carried out using the most stable gene (EF1α), the combination of the two most stable genes (EF1α + PP2Acs) and the least stable gene (PLPA2), according with the RefFinder comprehensive ranking as normalizers (Table 3). Catalase is a very important enzyme in protecting cells against ROS, catalyzing the breakdown of hydrogen peroxide to water and oxygen. Its expression was found to be greatly affected in Arabidopsis and rice plants after hormone treatment (Proietti et al. 2013; Bertini et al. 2019a) as well as in Posidonia oceanica under heavy metal stress (Bertini et al. 2019b). A significant increase in CAT activity was also reported in wheat under drought, salinity, nitrogen or heat stresses (Sairam et al. 2002; Polesskaya et al. 2004; Luna et al. 2005; Almeselmani et al. 2006; Esfandiari et al. 2007; Heidari 2009; Gupta et al. 2013) as well as in Cicer arietinum (chickpea) seedlings, in the unicellular alga Chlorella sp., in the aquatic macrophyte Najas graminea, in the mangrove plant Suaeda maritime under salt stress (Eyidogan and Öz 2007; Mallik et al. 2011). Nevertheless, high saline concentrations (200 to 500 mM NaCl) were found to induce a strong decrease in CAT activity in Triticum aestivum (wheat) leaves (Heidari 2009) as well as in leaf tissue of chickpea seedlings (Eyidogan and Öz 2007). According to these findings, we found CqCAT overexpressed following nitrogen supply, OTC and salinity (≤ 250 mM NaCl) treatments; a strong decrease in its expression pattern was registered only under ≥ 500 mM NaCl (Fig. 5). When the most stable reference genes (EF1α and EF1α + PP2Acs) were used for normalization, CqCAT expression trend was found similar along all the treatments. On the other hand, when using the least stable reference gene, PLPA2, the expression pattern of CqCAT was different and generally overestimated from that obtained using the other reference genes, in particular for nitrogen supply and salinity stresses. Under OTC stress, PLPA2 showed a greater stability, as confirmed by the stability ranking derived from all four software kinds when single stress was considered (Figs. 2 and 4).
In summary, we identified two stable reference genes, namely EF1α and PP2Acs, which can be used for accurate transcript normalization in gene expression studies using qRT-PCR in C. quitensis under different individual abiotic stresses. Nevertheless, it is well known that the expression level of genes identified as good reference genes in specific experimental conditions may vary when diverse experimental conditions are considered (Huggett et al. 2005), thus could be necessary to test the reliability of other candidate reference genes in order to identify the most appropriate ones for any specific circumstance.
References
Acuña-Rodríguez IS, Torres-Díaz C, Hereme R, Molina-Montenegro MA (2017) Asymmetric responses to simulated global warming by populations of Colobanthus quitensis along a latitudinal gradient. PeerJ 5:e3718. https://doi.org/10.7717/peerj.3718
Alberdi M, Bravo LA, Gutiérrez AH, Gidekel M, Corcuera LJ (2002) Ecophysiology of Antarctic vascular plants. Physiol Plantarum 115:479–486. https://doi.org/10.1034/j.1399-3054.2002.1150401.x
Almeselmani M, Deshmukh PS, Sairam RK, Kushwaha SR, Singh TP (2006) Protective role of antioxidant enzymes under high temperature stress. Plant Sci 17:382–388. https://doi.org/10.1016/j.plantsci.2006.04.009
Andersen CL, Jensen JL, Orntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64:5245–5250. https://doi.org/10.1158/0008-5472.CAN-04-0496
Arthofer W, Bertini L, Caruso C, Cicconardi F, Delph LF, Fields PD, Ikeda M, Minegishi Y, Proietti S, Ritthammer H, Schlick-Steiner BC, Steiner FM, Wachter GA, Wagner HC, Weingartner LA (2015) Transcriptome sequencing of the Antarctic Colobanthus quitensis (Kunth) Bartl (Caryophillaceae). Mol Ecol Resour 15:1014–1015. https://doi.org/10.1111/1755-0998.12419
Artico S, Nardeli SM, Brilhante O, Grossi-de-Sa MF, Alves-Ferreira M (2010) Identification and evaluation of new reference genes in Gossypium hirsutum for accurate normalization of real-time quantitative RT-PCR data. BMC Plant Biol 10:49. https://doi.org/10.1186/1471-2229-10-49
Ballesteros GI, Torres-Díaz C, Bravo LA, Balboa K, Caruso C, Bertini L, Proietti S, Molina-Montenegro MA (2020) In silico analysis of metatranscriptomic data from the Antarctic vascular plant Colobanthus quitensis: Responses to a global warming scenario through changes in fungal gene expression levels. Fungal Ecol 43:100873. https://doi.org/10.1016/j.funeco.2019.100873
Bascuñán-Godoy L, Sanhueza C, Cuba M, Zuñiga GE, Corcuera LJ, Bravo LA (2012) Cold-acclimation limits low temperature induced photoinhibition by promoting a higher photochemical quantum yield and a more effective PSII restoration in darkness in the Antarctic rather than the Andean ecotype of Colobanthus quitensis Kunt Bartl (Cariophyllaceae). BMC Plant Biol 12:114–128. https://doi.org/10.1186/1471-2229-12-114
Bertini L, Palazzi L, Proietti S, Pollastri S, Arrigoni G, Polverino de Laureto P, Caruso C (2019a) Proteomic analysis of MeJa-induced defense responses in rice against wounding. Int J Mol Sci 20:2525. https://doi.org/10.3390/ijms20102525
Bertini L, Focaracci F, Proietti S, Papetti P, Caruso C (2019b) Phsiological response of Posidonia oceanica to heavy metal pollution along the Tyrrhenian coast. Funct Plant Biol 46:933–941. https://doi.org/10.1071/FP18303
Biersma EM, Cristian Torres-Díaz C, Molina-Montenegro MA, Newsham KK, Vidal MA, Collado GA, Acuña-Rodríguez IS, Ballesteros GI, Figueroa CC, Goodall-Copestake WP, Leppe MA, Cuba-Díaz M, Valladares MA, Pertierra LR, Peter Convey P (2020) Multiple late-Pleistocene colonisation events of the Antarctic pearlwort Colobanthus quitensis (Caryophyllaceae) reveal the recent arrival of native Antarctic vascular flora. J Biogeogr 47:1663–1673. https://doi.org/10.1111/jbi.13843
Bokhorst S, Huiskes A, Aerts R, Convey P, Cooper EJ, Dalen L, Erschbamer B, Gudmundsson J, Hofgaard A, Hollister RD, Johnstone J, Jónsdóttir IS, Lebouvier M, Van de Vijver B, Wahren CH, Dorrepaal E (2013) Variable temperature effects of open top chambers at polar and alpine sites explained by irradiance and snow depth. Glob Change Biol 19:64–74. https://doi.org/10.1111/gcb.12028
Bravo LA, Saavedra-Mella FA, Vera F, Guerra A, Cavieres LA, Ivanov AG, Huner NPA, Corcuera LJ (2007) Effect of cold acclimation on the photosynthetic performance of two ecotypes of Colobanthus quitensis (Kunth.) Bartl. J Exp Bot 58:3581–3590. https://doi.org/10.1093/jxb/em206
Cavieres LA, Sáez P, Sanhueza C, Sierra-Almeida A, Rabert C, Corcuera LJ, Alberdi M, Bravo LA (2016) Ecophysiological traits of Antarctic vascular plants: their importance in the responses to climate change. Plant Ecol 217:343–358. https://doi.org/10.1007/s11258-016-0585-x
Chen L, Zhong H, Kuang J, Li J, Lu W, Chen J (2011) Validation of reference genes for RT-qPCR studies of gene expression in banana fruit under different experimental conditions. Planta 234:377–390. https://doi.org/10.1007/s00425-011-1410-3
Chen Y, Hu B, Tan Z, Liu J, Yang Z, Li Z, Huang B (2014) Selection of reference genes for quantitative real-time PCR normalization in creeping bentgrass involved in four abiotic stresses. Plant Cell Rep 34:1825–1834. https://doi.org/10.1007/s00299-015-1830-9
Cho SM, Lee H, Jo H, Lee H, Kang Y, Park H, Lee J (2018) Comparative transcriptome analysis of field- and chamber-grown samples of Colobanthus quitensis (Kunth) Bartl, an Antarctic flowering plant. Sci Rep 8:11049. https://doi.org/10.1038/s41598-018-29335-4
Chuaqui RF, Bonner RF, Best CJM, Gillespie JW, Flaig MJ, Hewitt SM, Phillips JL, Krizman DB, Tangrea MA, Ahram M, Linehan WM, Knezevic V, Emmert-Buck MR (2002) Post-analysis follow-up and validation of microarray experiments. Nat Genet 32:509–514. https://doi.org/10.1038/ng1034
Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible WR (2005) Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol 139:5–17. https://doi.org/10.1104/pp.105.063743
Dai F, Zhao X, Tang C, Wang Z, Kuang Z, Li Z, Huang J, Luo G (2018) Identification and validation of reference genes for qRT-PCR analysis in mulberry (Morus alba L.). PLoS ONE. https://doi.org/10.1371/journal.pone.0194129
Day TA, Ruhland CT, Grobe CW, Xiong F (1999) Growth and reproduction of antarctic vascular plants in response to warming and UV radiation reductions in the field. Oecologia 119:24–35. https://doi.org/10.1007/s004420050757
Die JV, Román B, Nadal S, González-Verdejo CI (2010) Evaluation of candidate reference genes for expression studies in Pisum sativum under different experimental conditions. Planta 232:145–153. https://doi.org/10.1007/s00425-010-1158-1
Dundas J, Ling M (2012) Reference genes for measuring mRNA expression. Theory Biosci 131:215–223. https://doi.org/10.1007/s12064-012-0152-5
Esfandiari E, Shekari F, Shekari F, Esfandiari M (2007) The effect of salt stress on antioxidant enzymes activity and lipid peroxidation on the wheat seedling. Not Bot Hort Agrobot Cluj 35:48–56. https://doi.org/10.15835/nbha.35.1.251
Expósito-Rodríguez M, Borges AA, Borges-Pérez A, Pérez JA (2008) Selection of internal control genes for quantitative real-time RT-PCR studies during tomato development process. BMC Plant Biol 8:131. https://doi.org/10.1186/1471-2229-8-131
Eyidogan F, Öz MT (2007) Effect of salinity on antioxidant responses of chickpea seedlings. Acta Physiol Plant 29:485–493. https://doi.org/10.1007/s11738-007-0059-9
Gachon CMA, Mingam A, Charrier B (2004) Real-time PCR: what relevance to plant studies? J Exp Bot 55:1445–1454. https://doi.org/10.1093/jxb/erh181
Guo J, Ling H, Wu Q, Xu L, Que Y (2014) The choice of reference genes for assessing gene expression in sugarcane under salinity and drought stresses. Sci Rep 4:7042. https://doi.org/10.1038/srep07042
Gupta NK, Agarwal S, Agarwal VP, Nathawat NS, Gupta S, Singh G (2013) Effect of short-term heat stress on growth, physiology and antioxidative defence system in wheat seedlings. Acta Physiol Plant 35:1837–1842. https://doi.org/10.1007/s11738-013-1221-1
Gutierrez L, Mauriat M, Guénin S, Pelloux J, Lefebvre JF, Louvet R, Rusterucci C, Moritz T, Guerineau F, Bellini C, Van Wuytswinkel O (2008) The lack of a systematic validation of reference genes: a serious pitfall undervalued in reverse transcription-polymerase chain reaction (RT-PCR) analysis in plants. Plant Biotechnol J 6:609–618. https://doi.org/10.1111/j.1467-7652.2008.00346.x
Hao X, Horvath DP, Chao WS, Yang Y, Wang X, Xiao B (2014) Identification and evaluation of reliable reference genes for quantitative Real-Time PCR analysis in tea plant (Camellia sinensis (L.) O. Kuntze). Int J Mol Sci 15:22155–22172. https://doi.org/10.3390/ijms151222155
Heidari M (2009) Antioxidant activity and osmolyte concentration of sorghum (Sorghum bicolor) and wheat (Triticum aestivum) genotypes under salinity stress. Asian J Plant Sci 8:240–244. https://doi.org/10.3923/ajps.2009.240.244
Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J (2007) qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8:R19. https://doi.org/10.1186/gb-2007-8-2-r19
Huggett JDK, Bustin S, Zumla A (2005) Real-time RT-PCR normalisation; strategies and considerations. Genes Immun 6:279–284. https://doi.org/10.1038/sj.gene.6364190
Jiang Q, Wang F, Li MY, Ma J, Tan GF, Xiong AS (2014) Selection of suitable reference genes for qPCR normalization under abiotic stresses in Oenanthe javanica (BI.) DC. PLoS ONE 9:e92262. https://doi.org/10.1371/journal.pone.0092262
Kim M, Gee M, Loh A, Rachatasumrit N (2010) Ref-Finder: a refactoring reconstruction tool based on logic query templates. Proceedings of the eighteenth ACM SIGSOFT international symposium on Foundations of software engineering, Santa Fe, NM, USA, November 7–11. ACM Digital Library, pp. 371–372. Doi:https://doi.org/10.1145/1882291.1882353
Kozera B, Rapacz M (2013) Reference genes in real-time PCR. J Appl Genet 54:391–406
Krause GH (1994) Photoinhibition induced by low temperatures. In: Baker NR, Bowyer JR (eds) Photoinhibition of photosynthesis from molecular mechanism to the field. BiosScientific Publishers Ltd, Oxford, pp 301–348
Le DT, Aldrich DL, Valliyodan B, Watanabe Y, Ha CV, Nishiyama R, Guttikonda SK, Quach TN, Gutierrez-Gonzalez JJ, Tran LSP, Nguyen HT, HT, (2012) Evaluation of candidate reference genes for normalization of quantitative RT-PCR in soybean tissues under various abiotic stress conditions. PLoS ONE 7(9):e46487. https://doi.org/10.1371/journal.pone.0046487
Lee H, Kim JH, Park M, Kim IC, Yim JH, Lee HK (2010) Reference genes validation for qPCR normalization in Deschampsia antarctica during abiotic stresses. Antarct Sci 22:477–484. https://doi.org/10.1017/S0954102010000428
Li X, Zhang D, Li H, Gao B, Yang H, Zhang Y, Wood AJ (2015) Characterization of reference genes for RT-qPCR in the desert moss Syntrichia caninervis in response to abiotic stress and desiccation/rehydration. Front Plant Sci 6:38. https://doi.org/10.3389/2Ffpls.2015.00038
Longton RE (1979) Vegetation Ecology and Classification in the Antarctic Zone. Can J Botany 57:2264–2278. https://doi.org/10.1139/b79-273
Løvdal T, Lillo C (2009) Reference gene selection for quantitative real-time PCR normalization in tomato subjected to nitrogen, cold, and light stress. Anal Biochem 387:238–242. https://doi.org/10.1016/j.ab.2009.01.024
Luna C, Pastori GM, Driscoll S, Foyer CH (2005) Drought controls on H2O2 accumulation, catalase (CAT) activity and CAT gene expression in wheat. J Exp Bot 56:417–423. https://doi.org/10.1093/jxb/eri039
Ma S, Niu H, Liu C, Zhang J, Hou C, Wang D (2013) Expression stabilities of candidate reference genes for RT-qPCR under different stress conditions in soybean. PLoS ONE 8:e75271. https://doi.org/10.1093/jxb/eri039
Mallik S, Nayak M, Sahu BB, Panigrahi AK, Shaw BP (2011) Response of antioxidant enzymes to high NaCl concentration in different salt-tolerant plants. Biol Plant 5:191–195. https://doi.org/10.1007/s10535-011-0029-3
Meunier L, Préfontaine G, Van Munster M, Brousseau R, Masson L (2006) Transcriptional response of Choristoneura fumiferana to sublethal exposure of Cry1Ab protoxin from Bacillus thuringiensis. Insect Mol Biol 15:475–483. https://doi.org/10.1111/j.1365-2583.2006.00659.x
Migocka M, Papierniak A (2011) Identification of suitable reference genes for studying gene expression in cucumber plants subjected to abiotic stress and growth regulators. Mol Breed 28:343–357. https://doi.org/10.1007/s11032-010-9487-0
Moore DM (1970) Studies in Colobanthus quitensis (Kunth.) Bartl. and Deschampsia antarctica Desv. II. Taxonomy, distribution and relationships. BAS Bulletin 23:63–80
Mulvaney R, Abram NJ, Hindmarsh RC, Arrowsmith C, Fleet L, Triest J, Sime LC, Alemany O, Foord S (2012) Recent Antarctic Peninsula warming relative to Holocene climate and ice-shelf history. Nature 489:141–144. https://doi.org/10.1038/nature11391
Nibert ML, Manny AR, Debat HJ, Firth AE, Bertini L, Caruso CA (2018) A barnavirus sequence mined from a transcriptome of the Antarctic pearlwort Colobanthus quitensis. Arch Virol 163:1921–1926. https://doi.org/10.1007/s00705-018-3794-x
Nolan T, Hands RE, Bustin SA (2006) Quantification of mRNA using real-time RT-PCR. Nat Protoc 1:1559–1582. https://doi.org/10.1038/nprot.2006.236
Paolacci AR, Tanzarella OA, Porceddu E, Ciaffi M (2009) Identification and validation of reference genes for quantitative RT-PCR normalization in wheat. BMC Mol Biol 10:11. https://doi.org/10.1186/1471-2199-10-11
Pérez-Torres E, Dinamarca J, Bravo LA, Corcuera LJ (2004) Responses of Colobanthus quitensis (Kunth) Bartl. to high light and low temperature. Polar Biol 27:183–189. https://doi.org/10.1007/s00300-003-0577-x
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time PCR. Nucleic Acids Res. https://doi.org/10.1093/2Fnar/2F29.9.e45
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP (2004) Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol Lett 26:509–515. https://doi.org/10.1023/B:BILE.0000019559.84305.47
Polesskaya OG, Kashirina EI, Alekhina ND (2004) Changes in the activity of antioxidant enzymes in wheat leaves and roots as a function of nitrogen source and supply. Russ J Plant Physiol 51:615–620. https://doi.org/10.1023/B:RUPP.0000040746.66725.77
Proietti S, Bertini L, Timperio AM, Zolla L, Caporale C, Caruso C (2013) Crosstalk between salicylic acid and jasmonate in Arabidopsis investigated by an integrated proteomic and transcriptomic approach. Mol Biosyst 9:1169–1187. https://doi.org/10.1039/c3mb25569g
Rabert C, Reyes-Díaz M, Corcuera LJ, Bravo LA, Alberdi M (2017) Contrasting nitrogen use efficiency of Antarctic vascular plants may explain their population expansion in Antarctica. Polar Biol 40:1569–1580. https://doi.org/10.1007/s00300-017-2079-2
Radonic A, Thulke S, Mackay IM, Landt O, Siegert W, Nitsche A (2004) Guideline to reference gene selection for quantitative real-time PCR. Biochem Biophys Res Commun 23:856–862. https://doi.org/10.1016/j.bbrc.2003.11.177
Ray DL, Johnson JC (2014) Validation of reference genes for gene expression analysis in olive (Olea europaea) mesocarp tissue by quantitative real-time RT-PCR. BMC Res Notes 7:304. https://doi.org/10.1186/1756-0500-7-304
Ruduś I, Kępczyński J (2018) Reference gene selection for molecular studies of dormancy in wild oat (Avena fatua L) caryopses by RT-qPCR method. PLoS ONE. https://doi.org/10.1371/journal.pone.0192343
Sáez PL, Cavieres LA, Galmés J, Gil-Pelegrín E, Peguero-Pina JJ, Sancho-Knapik D, Vivas M, Sanhueza C, Ramírez CF, Rivera BK, Corcuera LJ, Bravo LA (2018) In situ warming in the Antarctic: effects on growth and photosynthesis in Antarctic vascular plants. New Phytol 218:1406–1418. https://doi.org/10.1111/nph.15124
Saha P, Blumwald E (2014) Assessing reference genes for accurate transcript normalization using quantitative Real-Time PCR in pearl millet [Pennisetum glaucum (L.) R. Br.]. PLoS ONE. https://doi.org/10.1371/journal.pone.0106308
Sairam RK, Rao KV, Srivastava GC (2002) Differential response of wheat genotypes to long term salinity stress in relation to oxidative stress, antioxidant activity and osmolyte concentration. Plant Sci 163:1037–1046. https://doi.org/10.1016/S0168-9452(02)00278-9
Sanhueza C, Vallejos V, Cavieres LA, Sáez P, Bravo LA, Corcuera LJ (2017) Growing temperature affects seed germination of the Antarctic plant Colobanthus quitensis (Kunth) Bartl (Caryophyllaceae). Polar Biol 40:449–455. https://doi.org/10.1007/s00300-016-1972-4
Santos FIdCd, Marini N, Santos RSd, Hoffman BSF, Alves-Ferreira M, de Oliveira AC (2018) Selection and testing of reference genes for accurate RT-qPCR in rice seedlings under iron toxicity. PLoS ONE 13:e0193418. https://doi.org/10.1371/journal.pone.0193418
Schofield O, Ducklow HW, Martinson DG, Meredith MP, Moline MA, Fraser WR (2010) How do polar marine ecosystems respond to rapid climate change? Science 328:1520. https://doi.org/10.1126/science.1185779
Shivhare R, Lata C (2016) Selection of suitable reference genes for assessing gene expression in pearl millet under different abiotic stresses and their combinations. Sci Rep 6:23036. https://doi.org/10.1038/srep23036
Sierra-Almeida A, Cavieres LA, Bravo LA (2018) Warmer temperatures affect the in situ freezing resistance of the Antarctic vascular plants. Front Plant Sci 9:1456–1469. https://doi.org/10.3389/2Ffpls.2018.01456
Silver N, Best S, Jiang J, Thein SL (2006) Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol Biol 7:33. https://doi.org/10.1186/1471-2199-7-33
Smith LRI (2003) The enigma of Colobanthus quitensis and Deschampsia antarctica in Antarctica. In: Huiskes AHL, Gieskes WWC, Rozema J, Schorno RML, van der Vies SM, Wolff WJ, Wolf WJ (eds) Antarctic biology in a global context. Backhuys, Leiden, pp 234–2395
Sun R, He Q, Zhang B, Wang Q (2015) Selection and validation of reliable reference genes in Gossypium raimondii. Biotechnol Lett 37:1483–1493. https://doi.org/10.1007/s10529-015-1810-8
Taylor CM, Jost R, Erskine W, Nelson MN (2016) Identifying stable reference genes for qRT-PCR normalisation in gene expression studies of narrow-leafed lupin (Lupinus angustifolius L.). PLoS ONE. https://doi.org/10.1371/journal.pone.0148300
Thellin O, ElMoualij B, Heinen E, Zorzi W (2009) A decade of improvements in quantification of gene expression and internal standard selection. Biotechnol Adv 27:323–333. https://doi.org/10.1016/j.biotechadv.2009.01.010
Tillett RL, Ergül A, Albion RL, Schlauch KA, Cramer GR, Cushman JC (2011) Identification of tissue-specific, abiotic stress responsive gene expression patterns in wine grape (Vitis vinifera L.) based on curation and mining of large-scale EST data sets. BMC Plant Biol. https://doi.org/10.1186/1471-2229-11-86
Tong Z, Gao Z, Wang F, Zhou J, Zhang Z (2009) Selection of reliable reference genes for gene expression studies in peach using real-time PCR. BMC Mol Biol. https://doi.org/10.1186/1471-2199-10-71
Torres-Díaz C, Gallardo-Cerda J, Lavin P, Oses R, Carrasco-Urra F, Atala C, Acuña-Rodríguez IS, Convey P, Molina-Montenegro M (2016) Biological interactions and simulated climate change modulates the ecophysiological performance of Colobanthus quitensis in the Antarctic ecosystem. PLoS ONE. https://doi.org/10.1371/journal.pone.0164844
Tsukagoshi H, Suzuki T, Nishikawa K, Agarie S, Ishiguro S, Higashiyama T (2015) RNA-Seq analysis of the response of the halophyte, Mesembryanthemum crystallinum (Ice Plant) to high salinity. PLoS ONE 10:e0118339. https://doi.org/10.1371/journal.pone.0118339
Turner J, Colwell SR, Marshall GJ, Lachlan-Cope TA, Carleton AM, Jones PD, Lagun V, Reid PA, Iagovkina S (2005) Antarctic climate change during the last 50 years. Int J Climatol 25:279–294. https://doi.org/10.1002/joc.1130
Turner J, Barrand NE, Bracegirdle TJ, Convey P, Hodgson DA, Jarvis M, Jenkins A, Marshall G, Meredith MP, Roscoe H, Shanklin J (2014) Antarctic climate change and the environment: an update. Polar Rec 50:237–259. https://doi.org/10.1017/S0032247413000296
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. https://doi.org/10.1186/gb-2002-3-7-research0034
Vera M, Fernández-Teruel T, Quezada A (2013) Distribution and reproductive capacity of Deschampsia antarctica and Colobanthus quitensis on Byers Peninsula, Livingston Island, South Shetland Islands, Antarctica. Antarct Sci 25:292–302. https://doi.org/10.1017/S0954102012000995
Wan D, Wan Y, Yang Q, Zou B, Ren W, Ding Y, Wang Z, Wang R, Wang K, Hou X (2017) Selection of reference genes for qRT-PCR analysis of gene expression in Stipa grandis during environmental stresses. PLoS ONE 12:e0169465. https://doi.org/10.1371/journal.pone.0169465
Wang Y, Yu K, Poysa V, Shi C, Zhou Y (2012) Selection of reference genes for normalization of qRT-PCR analysis of differentially expressed genes in soybean exposed to cadmium. Mol Biol Rep 39:1585–1594. https://doi.org/10.1007/s11033-011-0897-9
Wang H, Chen S, Jiang J, Zhang F, Chen F (2015) Reference gene selection for cross-species and cross-ploidy level comparisons in Chrysanthemum spp. Sci Rep. https://doi.org/10.1038/srep08094
Wang ML, Li QH, Xin HH, Chen X, Zhu XJ, Li XH (2017) Reliable reference genes for normalization of gene expression data in tea plants (Camellia sinensis) exposed to metal stresses. PLoS ONE. https://doi.org/10.1371/journal.pone.0175863
Wong ML, Medrano JF (2005) Real-time PCR for mRNA quantitation. Biotechniques 39:75–85. https://doi.org/10.2144/05391RV01
Xiong FS, Mueller EC, Day TA (2000) Photosynthetic and respiratory acclimation and growth response of Antarctic vascular plants to contrasting temperature regimes. Am J Bot 87(5):700–710. https://doi.org/10.2307/2656856
Yin Z, Ke X, Huang D, Gao X, Voegele RT, Kang Z, Huang L (2013) Validation of reference genes for gene expression analysis in Valsa mali var. mali using real-time quantitative PCR. World J Microbiol Biotechnol 29:1563–1571. https://doi.org/10.1007/s11274-013-1320-6
Yu S, Yang Q, Yang JH, Du Z, Zhang G (2015) Identification of suitable reference genes for investigating gene expression in human gallbladder carcinoma using reverse transcription quantitative polymerase chain reaction. Mol Med Rep 11:2967–2974. https://doi.org/10.3892/mmr.2014.3008
Zhang Y, Peng X, Liu Y, Li Y, Luo Y, Wang X, Tang H (2018) Evaluation of suitable reference genes for qRT-PCR normalization in strawberry (Fragaria×ananassa) under different experimental conditions. BMC Mol Biol 19:8–17. https://doi.org/10.1186/s12867-018-0109-4
Zhou L, Shi Q, Wang Y, Li K, Zheng B, Miao K (2016) Evaluation of candidate reference genes for quantitative gene expression studies in tree peony. J Am Soc Hortic Sci 141:99–111. https://doi.org/10.21273/JASHS.141.2.99
Acknowledgements
The authors thank the members of Arctowski Research Station for their collaboration with the logistics and with a great hospitality.
Funding
This research was funded by the Ministero dell’Istruzione, Università e Ricerca Scientifica (MIUR) in the framework of the PNRA (National Program for Antarctic Research), grant number 2013/C1.02 and PNRA16_00068 (CC), and by the Ministerio de Educación, Project Conicyt-PIA grant number ART11-02 and by NEXER grant number NXR17-0002 (LAB).
Author information
Authors and Affiliations
Contributions
CC and LB conceived and designed research; CC collected the field samples; LB, SP, FC, FF and CR conducted experiments; LB, SP, FC and FF analyzed data; CC and LAB retrieved funds; CC and LB wrote the manuscript; CC, LB, SP, CR and LAB reviewed the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bertini, L., Proietti, S., Focaracci, F. et al. Identification and validation of new reference genes for accurate quantitative reverse transcriptase-PCR normalization in the Antarctic plant Colobanthus quitensis under abiotic stress conditions. Polar Biol 44, 389–405 (2021). https://doi.org/10.1007/s00300-021-02801-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00300-021-02801-y