
Abstract
Aims/hypothesis
The aims of this study were to determine the role of lipolysis in hypoglycaemia and define the underlying intracellular mechanisms.
Methods
Nine healthy volunteers were randomised to treatment order of three different treatments (crossover design). Treatments were: (1) saline control; (2) hyperinsulinaemic hypoglycaemia (HH; i.v. bolus of 0.1 U/kg insulin); and (3) hyperinsulinaemic euglycaemia (HE; i.v. bolus of 0.1 U/kg insulin and 20% glucose). Inclusion criteria were that volunteers were healthy, aged >18 years, had a BMI between 19 and 26 kg/m2, and provided both written and oral informed consent. Exclusion criteria were the presence of a known chronic disease (including diabetes mellitus, epilepsy, ischaemic heart disease and cardiac arrhythmias) and regular use of prescription medication. The data was collected at the medical research facilities at Aarhus University Hospital, Denmark. The primary outcome was palmitic acid flux. Participants were blinded to intervention order, but caregivers were not.
Results
Adrenaline (epinephrine) and glucagon concentrations were higher during HH than during both HE and control treatments. NEFA levels and lipid oxidation rates (determined by indirect calorimetry) returned to control levels after 105 min. Palmitate flux was increased to control levels during HH (p = NS) and was more than twofold higher than during HE (overall mean difference between HH vs HE, 114 [95% CI 64, 165 μmol/min]; p < 0.001). In subcutaneous adipose tissue biopsies, we found elevated levels of hormone-sensitive lipase (HSL) and perilipin-1 phosphorylation 30 min after insulin injection during HH compared with both control and HE. There were no changes in the levels of adipose triglyceride lipase (ATGL), comparative gene identification-58 (CGI-58) or G0/G1 switch gene 2 (G0S2) proteins. Insulin-stimulated phosphorylation of Akt and mTOR were unaffected by hypoglycaemia. Expression of the G0S2 gene increased during HE and HH compared with control, without changes in ATGL (also known as PNPLA2) or CGI-58 (also known as ABHD5) mRNA levels.
Conclusions/interpretation
These findings suggest that NEFAs become a major fuel source during insulin-induced hypoglycaemia and that lipolysis may be an important component of the counter-regulatory response. These effects appear to be mediated by rapid stimulation of protein kinase A (PKA) and HSL, compatible with activation of the β-adrenergic catecholamine signalling pathway.
Trial registration:
ClinicalTrials.gov NCT01919788
Funding:
The study was funded by Aarhus University, the Novo Nordisk Foundation and the KETO Study Group/Danish Agency for Science Technology and Innovation (grant no. 0603–00479, to NM).
Similar content being viewed by others

Introduction
Hypoglycaemia is a common side effect of insulin treatment, and remains the leading limiting factor in glucose management of insulin-treated diabetes mellitus [1, 2].
Hypoglycaemia is counteracted by a cascade of behavioural, hormonal and metabolic events. In healthy volunteers, this defence mechanism involves a reduction in endogenous insulin secretion, release of the counter-regulatory hormones glucagon, adrenaline (epinephrine), growth hormone (GH) and cortisol, and carbohydrate ingestion [3–5]. Individuals with type 1 diabetes mellitus depend entirely on exogenous insulin and cannot reduce endogenous insulin production during hypoglycaemia. Furthermore, with long-standing disease and recurrent episodes of hypoglycaemia, the counter-regulatory hormonal responses to hypoglycaemia become compromised (due to loss of glucagon response and hypoglycaemia-associated autonomic failure) [2, 6], rendering these individuals particularly vulnerable to hypoglycaemia.
Hormonal responses to hypoglycaemia have profound metabolic effects in healthy individuals, leading to increased endogenous glucose production and decreased glucose disposal. In addition, lipolysis is acutely stimulated by adrenaline [7] and, after a delay, by GH [8] and cortisol [9]. A series of studies in healthy volunteers during insulin-induced hypoglycaemia, have shown that NEFA and 3-hydroxybutyrate (3-OHB) levels and lipid oxidation rates are restored after some hours to above basal levels [10, 11]. Furthermore, these studies demonstrate that increased lipolysis plays a pivotal role in mediating hypoglycaemia-associated insulin resistance [10, 11]. Very few studies have measured the actual rates of lipolysis during hypoglycaemia. In apparent contrast with these findings, one study reported unchanged rates of lipolysis in healthy volunteers, measured using [14C]palmitate after insulin-induced hypoglycaemia with mean glucose nadir of ≈2.8 mmol/l [12]. Another study from the 1960s reported increased NEFA production in sheep following hypoglycaemia, as measured with [9,10-3H2]palmitate [13].
Similarly, the intracellular signalling mechanisms in adipose tissue (AT) that regulate lipolysis in response to hypoglycaemia in humans are unclear. Lipolysis is regulated by adipose triglyceride lipase (ATGL), hormone-sensitive lipase (HSL) and monoacylglycerol lipase (MAGL), which hydrolyse triacylglycerol to form NEFAs and glycerol [14].This process is mediated by protein kinase A (PKA), which phosphorylates HSL and lipid droplet-associated protein (PLIN1). Phosphorylation of PLIN1 leads to the dissociation of comparative gene identification-58 (CGI-58) from PLIN1, and it then binds to and strongly increases the activity of ATGL [15]. The counter-regulatory G0/G1 switch gene 2 (G0S2) protein is a major inhibitor of ATGL [15]. PKA is activated by catecholamines, which act via β-adrenergic receptors to activate adenylyl cyclase and increase intracellular cAMP [15]. The intracellular signalling events in AT that lead to increased lipolysis during hypoglycaemia are largely unknown.
Consequently, our study was designed to test whether hypoglycaemia induces a detectable increase in the rate of lipolysis, as measured with labelled palmitate, and whether this is associated with activation of intracellular signalling in AT. Based on previous observations of a central role for adrenergic mechanisms, we hypothesised that PKA activity and subsequently HSL phosphorylation would increase.
Methods
A randomised, crossover study was conducted involving nine male volunteers over three study days. Randomisation was performed using Research Randomizer (www.randomizer.org). Inclusion criteria were that volunteers were healthy, aged >18 years, had a BMI between 19 kg/m2 and 26 kg/m2, and provided both written and oral informed consent. Exclusion criteria were the presence of a known chronic disease (including diabetes mellitus, epilepsy, ischaemic heart disease and cardiac arrhythmias) and regular use of prescription medication. Each volunteer was randomly allocated for the order of three treatments: (1) control (i.v. bolus of 2 ml NaCl); (2) hyperinsulinaemic hypoglycaemia (HH; i.v. bolus of insulin [0.1 U Actrapid/kg in 2 ml NaCl]; and (3) HE (i.v. bolus of insulin [0.1 U Actrapid/kg in 2 ml NaCl] and 20% glucose). During HE, glucose levels were clamped based on frequent (every 5 min) measurements of plasma glucose. Volunteers were blinded to treatment and reported to the clinical research unit at 07:50 hours on each study day, after a 10 h fast. One i.v. catheter was inserted into the right antecubital vein for infusions and another was inserted, in a retrograde fashion, into the left antecubital vein for measuring venous blood glucose from the forearm (arteriovenous blood glucose measurements were not used in this study). A third i.v. catheter was inserted into a dorsal hand vein on the volunteer’s right hand for sampling arterialised blood with the heated hand technique [16–18]. Each volunteer was placed in a supine position in a thermoneutral room and each study day commenced at 09:00 hours (t = 0 min) with the injection of one of the three interventions. Volunteers were assessed from 09:00 to 10:45 hours (t = 0–105 min).
Each of the study days were separated by a minimum of 21 days.
Abdominal subcutaneous fat biopsy
Abdominal subcutaneous fat was biopsied under sterile conditions 15 min after local anaesthesia (1% lidocaine, 10 ml) was applied. Biopsies were obtained from the right side at t = 30 min and from the left side at t = 75 min. See electronic supplementary material (ESM) Methods for further details.
Western blotting
Western blotting analyses were performed using 4–15% Criterion XT Bis-Tris gels (Bio-Rad, Hercules, CA, USA). Primary antibodies against HSL (no. 4107), p-HSL Ser563, Ser565 and Ser660 (nos 4139, 4137 and 4126; measuring p-HSL Ser552, Ser554 and Ser650 in human AT), mammalian target of rapamycin (mTOR; no. 2972), p-mTOR Ser2448 (no. 5536), PKA substrate (no. 9624), AMP-activated protein kinase α (AMPKα) (no. 2532), p-AMPK Thr172 (no. 2531; all from Cell Signaling, Danvers, MA, USA), ATGL (no. GTX62840; GeneTex, Irvine, CA, USA), CGI-58 (ab183739, Abcam, Cambridge, UK) and G0S2 (sc-133424, Santa Cruz Biotechnology, Santa Cruz, CA, USA) were obtained from the respective supplier. Protein phosphorylation is expressed as a ratio compared with total protein expression as measured on the same membrane. Protein levels are expressed as a ratio to total protein content in the sample as measured by stain-free protein technology [19]. Stain-free protein technology was also used to demonstrate equal loading. See ESM Methods.
mRNA isolation
mRNA was isolated and quantified by quantitative PCR (qPCR) analysis using standard protocols. See ESM Methods.
Biochemical variables
Plasma glucose was measured using the glucose oxidase method (YSI 2300 STAT plus, YSI Life Sciences, Yellow Springs, OH, USA). Serum insulin was measured using ELISA (Dako, Glostrup, Denmark). C-peptide was measured by ELISA (Alpco, Salem, NH, USA). Serum NEFA was measured using a commercial kit (Wako Chemicals, Neuss, Germany). Plasma glucagon was measured using a RIA kit (EMD Millipore, Darmstadt, Germany). Plasma adrenaline and noradrenaline (norepinephrine) were measured by electrochemical detection following HPLC [20]. 3-OHB was measured by hydrophilic interaction liquid chromatography-tandem MS [21].
Palmitic acid flux
Palmitic acid flux was measured with the isotope dilution technique by [9,10-3H]palmitic acid (0.3 μCi/min = 10.500 Bq/min) infusion from t = 30 min to t = 105 min. Blood samples used to measure palmitic acid concentration and specific activity (ratio between isotope and total palmitic acid levels) were obtained at baseline (t = 0 min) and at t = 60, 75, 90 and 105 min. Palmitic acid concentration and specific activity were measured using [2H31]palmitic acid as an internal standard [22]. A quality control standard was included in every assay.
Indirect calorimetry
Primary outcomes
The primary outcomes were palmitic acid flux and changes in the phosphorylation levels of target proteins during HH.
Statistical analysis
Statistical analyses were performed using Stata version 13.0 (StataCorp, College Station, Texas, USA), with a repeated measurement mixed model that included visit number, visit order, time, intervention and the interaction between time and intervention as factors. A value of p < 0.05 was considered statistically significant. Graphs have crude geometric means with SEM. Numerical data are presented as adjusted geometric means and 95% CIs. Western blotting protein measurements are given as the ratio to the mean control value at t = 30 min. See ESM Methods.
Data on the effects of hypoglycaemia on ghrelin concentrations have been published previously [25].
Ethics statement
The study protocol was approved by the local scientific ethics committee (1-10-72-113-13). The study was conducted in accordance to the Helsinki Declaration II and volunteers gave both oral and written informed consent.
Results
Characteristics of participants
Participants had a median age of 22 years (range 18–27 years) and a median BMI of 23.6 (range 21.7–26.1)kg/m2. All nine participants completed all three study days and were included in the analyses. Baseline measurements are shown in Table 1. During HH, volunteers experienced varying degrees of symptoms related to hypoglycaemia and stress hormone release (heart palpitations, sweating, nausea, stomach aches, hot flushes and dizziness) that started at 20–30 min post insulin administration and disappeared after another 20–30 min. Three volunteers reported being hungry approximately 60 min post insulin injection. All volunteers reported extreme fatigue from approximately 30 min post insulin administration onwards throughout the study day. There were no serious adverse events.
Glucose infusion during HE
The glucose infusion rate required to maintain euglycaemia during HE quickly reached a peak level of 13.5 (95% CI 12.5, 14.6) mg/kg/min at t = 15 min, followed by a slow decrease toward zero at t = 105 min (data not shown).
Biochemical variables
Hormone and substrate levels
All baseline measurements of insulin, C-peptide, adrenaline, glucagon, GH, glucose, NEFA, 3-OHB, lactate and noradrenaline were comparable among interventions.
Insulin levels
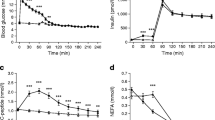
A comparison of HH and HE data revealed similar peak insulin levels at t = 15 min: (in pmol/l) HH, 897 (95% CI 779, 1033); HE, 881 (95% CI 715, 1086); p = 0.87 (Fig. 1a). Insulin levels during HH were comparable with those during control treatment at t = 60, 75 and 90 min.

(a–h) Substrate and hormone concentrations. Empty squares with solid line, control; filled circles with dotted line, HH; filled triangles with dashed line, HE. Symbols indicate p < 0.05: *HH vs HE; †HH vs control; ‡HE vs control; §all
C-peptide levels
During HH, C-peptide concentrations were rapidly reduced (Fig. 1b). A mean nadir of 96 (95% CI 51, 142) pmol/l was observed at t = 60 min. During HE, C-peptide levels were initially reduced, but increased at t = 30 min to exceed control levels (p < 0.01). The peak C-peptide level during HE was 569 (95% CI 502, 636) pmol/l at t = 45 min. During control treatment, mean C-peptide concentrations remained unaltered, with an overall mean of 352 (95% CI 312, 391) pmol/l.
Adrenaline levels
Adrenaline concentrations markedly increased during HH compared with HE and control treatment (Fig. 1c). During HH, a peak adrenaline value of 891 (95% CI 560, 1221) pg/ml was observed at t = 30 min; the concentration at the equivalent time point during HE was 43 (95% CI 8, 79) pg/ml (p < 0.001). Adrenaline levels during HE and control treatment were comparable at all times.
Noradrenaline levels
Noradrenaline concentrations peaked at 387 (95% CI 349, 425) pg/ml during HH at t = 30 min, representing a marked increase compared with both control (223 [95% CI 185, 261] pg/ml; p < 0.001) and HE (259 [95% CI 221, 298] pg/ml; p < 0.001; data not shown). There was no difference in noradrenaline concentrations during control treatment and HE at t = 30 min (p = 0.17). Noradrenaline concentrations decreased during HH to 285 (95% CI 247, 323) pg/ml at t = 75 min; these were comparable to levels during HH at t = 0 min (244 [95% CI 190, 298] pg/ml; p = 0.21).
Glucagon levels
During HH, glucagon levels were higher than during HE from t = 30 min (p < 0.001) up to t = 105 min (p < 0.01), with a peak of 133 (95% CI 119, 147) pg/ml at t = 45 min (Fig. 1d). During HE, glucagon levels were reduced compared with control from t = 30 min (p < 0.01) up to t = 105 min (p = 0.01) and glucagon nadir of 34 (95% CI 28, 39) pg/ml was observed at t = 60 min (p < 0.001). During control treatment, mean glucagon levels remained unaltered at approximately 50 pg/ml.
Glucose levels
During HH, a glucose nadir of 2.0 (95% CI 1.7, 2.4) mmol/l was reached at t = 25 min, whereas the glucose level during HE at the same time point was 5.3 (95% CI 4.9, 5.6) mmol/l (p < 0.001; Fig. 1e).
NEFA levels
During HH and HE, NEFA levels were reduced to an equal degree at t = 15 min: HH, 131 (95% CI 87, 198) μmol/l; HE, 151 (95% CI 97, 234) μmol/l (p = 0.4; Fig. 1f). Thereafter, NEFA levels increased steadily during HH to reach a peak of 584 (95% CI 455, 751) μmol/l at t = 105 min, approximately four times higher than during HE (140 [95% CI 89, 220] μmol/l; p < 0.001). During HE, a NEFA nadir of 66 (95% CI 45, 99) μmol/l was reached at t = 45 min.
3-OHB levels
During HH, 3-OHB levels were comparable with those during HE at t = 45 min (p = 0.44), but were higher at t = 75 min (p = 0.02) and t = 105 min (p < 0.001; Fig. 1g). During control treatment, 3-OHB levels were higher than those during HH at t = 45 min (p < 0.001) and t = 75 min (p < 0.001), but comparable with those during HH at t = 105 min (p = 0.27) and were higher than those during HE at t = 45, 75 and 105 min (p < 0.001).
Lactate levels
A comparison of peak lactate levels during HE (t = 30 min) and during HH (t = 45 min) revealed higher levels during HH, with a mean difference of 0.66 (95% CI 0.32, 1.00) mmol/l (p < 0.001; Fig. 1h). During control treatment, mean lactate levels remained unaltered throughout the study day at approximately 0.77 (95% CI 0.59, 0.94) mmol/l.
Palmitate flux
Palmitate maintained steady-state levels during HE, but not during control treatment and HH. Palmitate flux is therefore shown as a function of time, and not as a single measure (Fig. 2a). Palmitate flux throughout HE was significantly lower than throughout both the control treatment and HH, with a steady-state palmitate flux of 83 (95% CI 44, 123) μmol/min. The overall mean difference between palmitate flux during HH and HE was 114 (95% CI 64, 165 μmol/min) (p < 0.001). Compared with control, there was no difference in palmitate flux (p > 0.05) during HH, but there was a tendency toward higher flux rate at t = 105 min (p = 0.07).

Palmitate flux (a) and lipid oxidation (b) rates. Empty squares with solid line, control; filled circles with dotted line, HH; filled triangles with dashed line, HE. (a) Symbols indicate p < 0.05: *HH vs HE; ‡HE vs control (CTR). (b) **p < 0.01, ***p < 0.001
Lipid oxidation rates
Similar to palmitate fluxes, lipid oxidation rates at 90–105 min were decreased during HE compared with both control treatment and HH (both p < 0.01). Lipid oxidation rates were comparable during both HH and control treatment: HE, 946 (95% CI −1153, 3046) kJ/day; control, 3754 (95% CI 1631, 5876) kJ/day; HH, 3011 (95% CI 870, 5152) kJ/day; p = 0.19; Fig. 2b)
Gene expression, protein modification and total protein levels
PKA phosphorylation of PLIN1
At t = 30 min, PKA-dependent PLIN1 phosphorylation during HH was 3.8-fold (95% CI 1.9-fold, 7.4-fold) greater than during control treatment (p < 0.001) and 14.3-fold (95% CI 8.9-fold, 23.1-fold) greater than during HE (p < 0.001; Fig. 3a). PKA phosphorylation of PLIN1 was reduced to 26% (95% CI 31%, 57%) of control levels during HE (p < 0.01). At t = 75 min, the differences were smaller, but levels were still 2.4-fold (95% CI 1.3-fold, 4.8-fold) greater during HH than during control treatment (p < 0.01) and 3.6-fold (95% CI 2.2-fold, 5.8-fold) greater than during HE (p < 0.001). No significant differences were detected between HE and control treatment at t = 75 min (p = 0.33).

Protein levels at t = 30 min and t = 75 min shown relative to control (CTR) levels at t = 30 min. Western blots are shown below graphs. (a) PKA-dependent PLIN1 phosphorylation. (b, c) Ratio of Ser552 and Ser650 phospho-HSL to total HSL. (d) Total ATGL. Empty squares with solid line, control; filled circles with dotted line, HH; filled triangles with dashed line, HE. Symbols indicate p < 0.05: *HH vs HE; †HH vs CTR; §all. Phos., phosphorylation; RU, relative unit
HSL Ser552 phosphorylation
At t = 30 min, the p-Ser552 HSL/total HSL ratio was 3.6-fold (95% CI 2.6-fold, 5.0-fold) higher for HH than for control treatment (p < 0.001) and 2.9-fold (95% CI 2.1-fold, 4.1-fold) higher for HH than for HE (p < 0.001; Fig. 3b). There was no difference between HE and control treatment (p = 0.22). At t = 75 min, the fold differences in HSL Ser552 phosphorylation between interventions diminished, but were still significant between HH and control treatment (1.9 [95% CI 1.3, 2.6]; p < 0.001) and between HH and HE (2.1 [95% CI 1.5, 2.9]; p ≤ 0.001).
HSL Ser650 phosphorylation
At t = 30 min, the p-Ser650 HSL/total HSL ratio was 2.4-fold (95% CI 1.6-fold, 3.8-fold) higher for HH than for control treatment (p < 0.001) and 3.4-fold (95% CI 2.2-fold, 5.3-fold) higher for HH than for HE (p < 0.001; Fig. 3c). There was no significant difference between HE and control treatment (p = 0.16). At t = 75 min, the differences were lower but the fold increase in phosphorylation was still significantly higher during HH than during control treatment (1.8-fold [95% CI 1.2-fold, 2.8-fold]; p < 0.01) and HE (1.6-fold [95% CI 1.1-fold, 2.6-fold]; p = 0.03). However, no differences were detected between control treatment and HE (p = 0.68).
ATGL
No differences were seen among treatments in the levels of total ATGL at t = 30 min or at t = 75 min (Fig. 3d).
Akt Ser473 phosphorylation
At t = 30 min, the p-Ser473 Akt/total Akt ratio was 22.5-fold (95% CI 13.6-fold, 37.1-fold) higher during HH compared with control (p < 0.001), 9.7-fold (95% CI 5.8-fold, 16.3-fold) higher during HE compared with control (p < 0.001) and 2.3-fold (95% CI 1.4-fold, 4.0-fold) higher during HH compared with HE (p < 0.01; Fig. 4a). At t = 75 min, the ratio was 1.6-fold (95% CI 1.0-fold, 2.7-fold) higher during HH compared with control, but the difference was not significant (p = 0.055). No differences were detected between HH and HE (p = 0.42) and between HE and control (p = 0.30).

Protein and mRNA levels at t = 30 min and t = 75 min shown relative to control (CTR) at t = 30 min. Western blots are shown below graphs. (a) Ratio between Ser473 phospho-Akt and total Akt. (b) Ratio between Ser2448 phospho-mTOR and total mTOR. (c) G0S2 mRNA levels. Empty squares with solid line, CTR; filled circles with dotted line, HH; filled triangles with dashed line, HE. Symbols indicate p < 0.05: †HH vs CTR; ‡HE vs CTR; §all
mTOR Ser2448 phosphorylation
At t = 30 min, the p-Ser2448 mTOR/total mTOR ratio was 2.6-fold (95% CI 2.0-fold, 3.2-fold) higher during HH compared with control (p < 0.001) and 2.4-fold (95% CI 1.9-fold, 2.9-fold) higher during HE compared with control (Fig. 4b). No significant differences were detected between HH and HE (p = 0.1). At t = 75 min, the differences were smaller, but still present: the ratio was 1.7-fold (95% CI 1.4-fold, 2.2-fold) higher during HH compared with control (p < 0.001) and similarly 1.7-fold (1.4-fold, 2.1-fold) higher during HE compared with control (p < 0.001). No differences were detected between HH and HE at this time point (p = 0.8).
G0S2 mRNA
At t = 30 min, the level of G0S2 gene expression was 1.5-fold (95% CI 1.1-fold, 2.1-fold) higher during HH compared with control (p = 0.01) and 1.6-fold (95% CI 1.1-fold, 2.2-fold) higher during HE compared with control (p < 0.01; Fig. 4c). No differences were detected between HH and HE (p = 0.67). At t = 75 min, the level of G0S2 gene expression was 1.9-fold (95% CI 1.3-fold, 2.6-fold) higher during HH compared with control and 1.9-fold (95% CI 1.3-fold, 2.7-fold) higher during HE compared with control. No differences were detected between HH with HE (p = 0.82).
Other proteins and genes
No significant differences in the levels of total CGI-58-protein and in the p-Thr172 AMPK/AMPK ratio were observed at t = 30 min or t = 75 min. Neither did we observe any differences in levels of CGI-58 or ATGL mRNA at these time points (data not shown).
Discussion
Our study was designed to determine the increase in lipolysis rate during acute insulin-induced hypoglycaemia and whether an increase in lipolysis is reflected in activation of pro-lipolytic signalling in AT. In contrast to HE, HH induced a gradual increase in plasma NEFA and 3-OHB concentrations starting from 15 min and 45 min, respectively. Furthermore, we found an approximate threefold increase in whole body palmitate lipolysis at t = 105 min and lipid oxidation rates close to control values at t = 105 min. These findings suggest that activation of lipolysis occurs rapidly and that lipids are eventually restored to the position of major oxidative fuel. In line with these findings, we observed a robust 14-fold increase in AT PKA-mediated PLIN1 phosphorylation and a threefold increase in pro-lipolytic HSL phosphorylation at 30 min post insulin injection during HH compared with HE. Notably, this activation occurred in the presence of unaltered insulin-induced mTOR phosphorylation, unaltered G0S2 mRNA expression and augmented Akt phosphorylation, suggesting that the activation of β-adrenergic signals overrides intact insulin signals during hypoglycaemia. From a clinical point of view, these findings place activation of lipolysis high in the counter-regulatory response hierarchy and have implications for a large number of patients with diabetes and a smaller number of patients with spontaneous or reactive hypoglycaemia. It remains uncertain whether patients with diabetes and/or recurrent hypoglycaemia have intact lipolytic responses to hypoglycaemia and to what extent these responses may be compromised in hypoglycaemia-associated autonomic failure.
Experimental insulin-induced hypoglycaemia can be achieved in a variety of ways. For example, insulin can be continuously infused and glucose concentrations can be manipulated by varying the rate of exogenously administered glucose to accomplish a desired glucose level during a hyperinsulinaemic–hypoglycaemic clamp [7, 10, 11, 20]. This allows the gradual control of changes in glycaemia at close to steady-state conditions, but also involves massive insulin exposure over a long period of time. Alternatively, the hyperinsulinaemic–hypoglycaemic clamp technique can be combined with manipulating hormonal responses and metabolic processes with inhibitors such as adrenergic blockers, somatostatin and the nicotinic acid derivative Acipimox. Our study used a bolus injection of 0.1 IU insulin/kg body mass to provide a clearly defined stimulus to AT and avoid prolonged tissue exposure to insulin. It is likely that, in general, clinically occurring hypoglycaemia in insulin-treated individuals may differ from both bolus insulin administration and continuous i.v. insulin administration in terms of the insulinaemia pattern, rate of glucose excursions, possible autonomic failure, antecedent hypoglycaemia, exercise and food ingestion, all of which may affect the counter-regulatory responses. Nonetheless, we succeeded in establishing three well-controlled experimental situations: one in which glucose levels were close to normal and suppressed lipolysis (HE); a second with hypoglycaemia (glucose nadir of 2 mmol/l), clear-cut stress hormone responses and stimulation of lipolysis (HH); and a third reflecting the basal post-absorptive state (control).
Studies have consistently demonstrated that 2–3 h after the induction of hypoglycaemia, NEFA levels, 3-OHB levels and lipid oxidation rates are restored to above basal levels and that increased lipolysis therefore seems to play a pivotal role in mediating hypoglycaemia-associated counter-regulation and insulin resistance [10, 11]. To our knowledge, only one study has investigated lipolytic rates during hypoglycaemia in humans; this somewhat surprisingly reported unchanged [14C]palmitate lipolytic fluxes [12]. However, it should be noted that only five volunteers were included, only a modest extent of hypoglycaemia was achieved (2.8 mmol/l) and insulin levels were substantially higher during hypoglycaemia than during euglycaemia. It is plausible that the latter factor independently reduced palmitate fluxes. In our study, we observed a clear increase of more than twofold in the palmitate flux at 60–105 min after insulin administration. The glucose nadir and peak adrenaline responses occurred approximately 30 min after insulin administration, whereas glucagon responses were slightly later. We also confirmed that blood NEFA concentrations, palmitate fluxes and lipid oxidation rates were similar to control levels at the end of the study. In this context, it is important to consider that, in principle, measurements of lipolytic fluxes and lipid oxidation rates rely on steady-state or ‘quasi’ steady-state conditions. The possibility that rates of lipolysis and ketogenesis were increasing during HH 60–105 min after insulin administration cannot be ruled out. On the other hand, we found that levels of specific activity reach a plateau from 90 to 105 min and, even in the presence of major changes in the rates of NEFA appearance, it has been shown that steady-state equations yield the best estimates of actual appearance rates [26]. The observation that hypoglycaemia leads to increased rates of lipolysis is supported by a study that used continuous infusions of [9,10-3H2]palmitate in sheep [13].
Lipolysis is regulated by three lipases (ATGL, HSL and MAGL) which hydrolyse triacylglycerol to NEFAs and glycerol and are regulated by complex interplay among a number of pro- and anti-lipolytic intracellular agents [14]. The most prominent effects of HH on AT signalling were a marked stimulation of PKA-dependent phosphorylation on perilipin and similar increases in HSL Ser552 and Ser650 phosphorylation, all indicative of increased β-adrenergic pro-lipolytic signalling. During HE, insulin alone more than halved PKA activity toward perilipin, whereas during HH with concomitant spikes of adrenaline, PKA activity toward perilipin more than doubled compared with control at 30 min (these differences persisted to t = 75 min). Our findings of persistent increases in HSL Ser552 and Ser650 phosphorylation of more than twofold at t = 30 and t = 75 min suggest a strong stimulation of PKA activity toward perilipin and HSL.
It has been demonstrated that ATGL is phosphorylated at Ser406 by AMPK in mice [27] and that ATGL Ser406 is phosphorylated by PKA, leading to increased ATGL activity [28]. It has also been demonstrated that ATGL Ser404 phosphorylation in human AT explants (corresponding to Ser406 in mice) is increased by β-adrenergic stimulation and corresponds to increased lipolysis rates [28]. Thus, it is plausible that ATGL activation by PKA contributes to the enhanced lipolysis during hypoglycaemia observed in our study. There is, however, a lack of a reliable measure of ATGL activity, which could have confirmed this finding.
Furthermore, we found that Akt phosphorylation was rapidly stimulated by insulin and actually increased during hypoglycaemia at t = 30 min. This finding, together with comparable levels of mTOR phosphorylation during HE and HH and similar 50–100% increments in G0S2 mRNA levels, suggests that the anti-lipolytic insulin signal remains intact during HH [14, 29, 30]. It should be noted that insulin and C-peptide levels from t = 30 min onwards were modestly increased during HE (reflecting relative hyperglycaemia), leading to increased endogenous insulin release during the euglycaemic clamp. This increase, however, did not alter insulin signalling in AT, which appears to be primarily triggered by the initial very high spike concentrations of insulin.
It is unlikely that other counter-regulatory hormones significantly contribute to the rapid stimulation of lipolysis during hypoglycaemia. Although both GH and cortisol have lipolytic activity, this only becomes apparent after a latency of more than 1–2 h [31, 32].
Conclusion
Our data show that insulin-induced hypoglycaemia rapidly activates lipolysis and restores rates of lipolysis to close to basal values within 60–105 min, thus mobilising alternative fuel sources and contributing to NEFA-induced insulin resistance. These rapid effects appear to be mediated by the stimulation of PKA activity and pro-lipolytic HSL phosphorylation, compatible with the activation of β-adrenergic catecholamine signalling, to override insulin inhibition of lipolysis in AT.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- AT:
-
Adipose tissue
- ATGL:
-
Adipose triglyceride lipase
- CGI-58:
-
Comparative gene identification-58
- GH:
-
Growth hormone
- G0S2:
-
G0/G1 switch gene 2
- HE:
-
Hyperinsulinaemic euglycaemia
- HH:
-
Hyperinsulinaemic hypoglycaemia
- HSL:
-
Hormone-sensitive lipase
- MAGL:
-
Monoacylglycerol lipase
- mTOR:
-
Mammalian target of rapamycin
- 3-OHB:
-
3-Hydroxybutyrate
- PKA:
-
Protein kinase A
- PLIN1:
-
Lipid droplet-associated protein
- qPCR:
-
Quantitative PCR
References
Cryer PE (2008) Hypoglycemia: still the limiting factor in the glycemic management of diabetes. Endocr Pract 14:750–756
Cryer PE (2013) Mechanisms of hypoglycemia-associated autonomic failure in diabetes. N Engl J Med 369:362–372
Cryer P (1997) Hypoglycemia: pathophysiology, diagnosis and treatment. Oxford University Press, Oxford
Sprague JE, Arbeláez AM (2011) Glucose counterregulatory responses to hypoglycemia. Pediatr Endocrinol Rev 9:463–473
Cryer PE, Davis SN, Shamoon H (2003) Hypoglycemia in diabetes. Diabetes Care 26:1902–1912
Raju B, Cryer PE (2005) Loss of the decrement in intraislet insulin plausibly explains loss of the glucagon response to hypoglycemia in insulin-deficient diabetes: documentation of the intraislet insulin hypothesis in humans. Diabetes 54:757–764
Fanelli CG, De Feo P, Porcellati F et al (1992) Adrenergic mechanisms contribute to the late phase of hypoglycemic glucose counterregulation in humans by stimulating lipolysis. J Clin Invest 89:2005–2013
De Feo P, Perriello G, Torlone E et al (1989) Demonstration of a role for growth hormone in glucose counterregulation. Am J Physiol 256:E835–E843
De Feo P, Perriello G, Torlone E et al (1989) Contribution of cortisol to glucose counterregulation in humans. Am J Physiol 257:E35–E42
Fanelli C, Calderone S, Epifano L et al (1993) Demonstration of a critical role for free fatty acids in mediating counterregulatory stimulation of gluconeogenesis and suppression of glucose utilization in humans. J Clin Invest 92:1617–1622
Lucidi P, Rossetti P, Porcellati F et al (2010) Mechanisms of insulin resistance after insulin-induced hypoglycemia in humans: the role of lipolysis. Diabetes 59:1349–1357
Godfried MH, Romijn JA, Endert E, Sauerwein HP (1994) Metabolic effects of hypoglycemic counterregulation during sustained mild hyperinsulinemia and constant glucose availability in healthy men. Nutrition 10:5–10
West CE, Passey RF (1967) Effect of glucose load and of insulin on the metabolism of glucose and of palmitate in sheep. Biochem J 102:58–64
Nielsen TS, Jessen N, Jorgensen JO, Moller N, Lund S (2014) Dissecting adipose tissue lipolysis: molecular regulation and implications for metabolic disease. J Mol Endocrinol 52:R199–R222
Bolsoni-Lopes A, Alonso-Vale MI (2015) Lipolysis and lipases in white adipose tissue – an update. Arch Endocrinol Metab 59:335–342
Jensen MD, Heiling VJ (1991) Heated hand vein blood is satisfactory for measurements during free fatty acid kinetic studies. Metabolism 40:406–409
Liu D, Moberg E, Kollind M, Lins PE, Adamson U, Macdonald IA (1992) Arterial, arterialized venous, venous and capillary blood glucose measurements in normal man during hyperinsulinaemic euglycaemia and hypoglycaemia. Diabetologia 35:287–290
Nauck MA, Liess H, Siegel EG, Niedmann PD, Creutzfeldt W (1992) Critical evaluation of the ‘heated-hand-technique’ for obtaining ‘arterialized’ venous blood: incomplete arterialization and alterations in glucagon responses. Clin Physiol 12:537–552
Gilda JE, Gomes AV (2013) Stain-Free total protein staining is a superior loading control to beta-actin for Western blots. Anal Biochem 440:186–188
Orskov L, Bak JF, Abildgard et al (1996) Inhibition of muscle glycogen synthase activity and non-oxidative glucose disposal during hypoglycaemia in normal man. Diabetologia 39:226–234
Sorensen LK, Rittig NF, Holmquist EF et al (2013) Simultaneous determination of beta-hydroxybutyrate and beta-hydroxy-beta-methylbutyrate in human whole blood using hydrophilic interaction liquid chromatography electrospray tandem mass spectrometry. Clin Biochem 46:1877–1883
Moller N, Jensen MD, Rizza RA, Andrews JC, Nair KS (2006) Renal amino acid, fat and glucose metabolism in type 1 diabetic and non-diabetic humans: effects of acute insulin withdrawal. Diabetologia 49:1901–1908
Merilainen PT (1987) Metabolic monitor. Int J Clin Monit Comput 4:167–177
Schadewaldt P, Nowotny B, Strassburger K, Kotzka J, Roden M (2013) Indirect calorimetry in humans: a postcalorimetric evaluation procedure for correction of metabolic monitor variability. Am J Clin Nutr 97:763–773
Lauritzen ES, Voss T, Kampmann U et al (2015) Circulating acylghrelin levels are suppressed by insulin and increase in response to hypoglycemia in healthy adult volunteers. Eur J Endocrinol 172:357–362
Miles JM, Ellman MG, McClean KL, Jensen MD (1987) Validation of a new method for determination of free fatty acid turnover. Am J Physiol 252:E431–E438
Ahmadian M, Abbott MJ, Tang T et al (2011) Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab 13:739–748
Pagnon J, Matzaris M, Stark R et al (2012) Identification and functional characterization of protein kinase A phosphorylation sites in the major lipolytic protein, adipose triglyceride lipase. Endocrinology 153:4278–4289
Chakrabarti P, English T, Shi J, Smas CM, Kandror KV (2010) Mammalian target of rapamycin complex 1 suppresses lipolysis, stimulates lipogenesis, and promotes fat storage. Diabetes 59:775–781
Caron A, Richard D, Laplante M (2015) The roles of mTOR complexes in lipid metabolism. Annu Rev Nutr 35:321–348
Djurhuus CB, Gravholt CH, Nielsen S et al (2002) Effects of cortisol on lipolysis and regional interstitial glycerol levels in humans. Am J Physiol Endocrinol Metab 283:E172–E177
Djurhuus CB, Gravholt CH, Nielsen S, Pedersen SB, Moller N, Schmitz O (2004) Additive effects of cortisol and growth hormone on regional and systemic lipolysis in humans. Am J Physiol Endocrinol Metab 286:E488–E494
Acknowledgements
We would like to thank A. Mengel, K. Nyborg Rasmussen, E. Søgaard Hornemann and K. Mathiassen of the Medical Research Laboratories, H. Zibrandtsen of the Research Laboratories for Biochemical Pathology and L. Pedersen of the Department of Endocrinology and Internal Medicine, Aarhus University Hospital, Denmark, for their excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported by Aarhus University, the Novo Nordisk Foundation and the KETO Study Group/Danish Agency for Science Technology and Innovation (grant no. 0603–00479, to NM).
Duality of interest statement
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
NM, MHV, TSV and UK designed the study; TSV recruited the participants; TSV conducted the trial; NJ, NM, MJ, MVS, SBP, TSN and TSV collected the data; TSV conducted the statistical analyses; NM and TSV wrote the manuscript; and NJ, MJ, MHV, MVS, SBP, TSN, UK reviewed and edited the manuscript. NM is the guarantor of this work. All authors approved the final version of the manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 167 kb)
Rights and permissions
About this article

Cite this article
Voss, T.S., Vendelbo, M.H., Kampmann, U. et al. Effects of insulin-induced hypoglycaemia on lipolysis rate, lipid oxidation and adipose tissue signalling in human volunteers: a randomised clinical study. Diabetologia 60, 143–152 (2017). https://doi.org/10.1007/s00125-016-4126-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4126-x