
Abstract
Aims/hypothesis
The aim of this study was to test the hypothesis that type 1 diabetes alters renal amino acid, glucose and fatty acid metabolism.
Materials and methods
We studied five C-peptide-negative, type 1 diabetic subjects during insulin replacement (glucose 5.6 mmol/l) and insulin deprivation (glucose 15.5 mmol/l) and compared them with six non-diabetic subjects. Leucine, phenylalanine, tyrosine, glucose and palmitate tracers were infused after an overnight fast and samples were obtained from the renal vein, femoral vein and femoral artery.
Results
Insulin deprivation significantly increased whole-body fluxes (20–25%) of phenylalanine, tyrosine and leucine, and leucine oxidation (50%). Kidney contributed 5–10% to the whole-body leucine and phenylalanine flux. A net uptake of phenylalanine, conversion of phenylalanine to tyrosine (5 μmol/min) and net release of tyrosine (∼5 μmol/min) occurred across the kidney. Whole-body (three-fold) and leg (two-fold) leucine transamination increased but amino acid metabolism in the kidney did not alter with diabetes or insulin deprivation. Insulin deprivation doubled endogenous glucose production, renal glucose production was unaltered by insulin deprivation and diabetes (ranging between 100 and 140 μmol/min). Renal palmitate exchange was unaltered by insulin deprivation.
Conclusions/interpretation
In conclusion, kidney postabsorptively accounts for 5–10% of whole-body protein turnover, 15–20% of leucine transamination and 10–15% of endogenous glucose production, and actively converts phenylalanine to tyrosine. During insulin deprivation, leg becomes a major site for leucine transamination but insulin deprivation does not affect renal phenylalanine, leucine, palmitate or glucose metabolism. Despite its key metabolic role, insulin deprivation in type 1 diabetic patients does not alter many of these metabolic functions.
Similar content being viewed by others

Introduction
In people with type 1 diabetes, lack of insulin has a profound effect on intermediary metabolism. Insulin deprivation increases production of glucose and lipid intermediates [1], accelerates proteolysis [2, 3] and alters substrate disposal. This results in high plasma concentration levels of NEFA, ketone bodies and increased circulating concentrations of most amino acids [1–3]. Until recently it was assumed that the primary tissues participating in these events were liver (increased glucose production and ketogenesis), adipose tissue (increased lipolysis) and muscle (increased proteolysis) [1–3]. Several reports have suggested that in addition the kidney may be an important component in the regulation of intermediary metabolism, in particular glucose metabolism [4]. This renewed interest in the metabolic role of the kidney was fuelled by a study by Cersosimo et al. [5] who used arteriovenous balance and isotopic dilution techniques to report that in postabsorptive dogs renal glucose utilisation and production each accounted for close to 30% of total glucose turnover in the presence of net balances close to zero. More recent studies in humans have reported that the renal contribution to postabsorptive whole-body glucose production may amount to between 15 and 30%, that glutamine appears to be an important precursor for renal gluconeogenesis and that renal glucose production increases in response to epinephrine and hypoglycaemia and decreases during insulin exposure [6–11]. Other investigators, however, have failed to show any significant renal contribution to overall endogenous glucose production [12].
It is well established that insulin deprivation leads to high protein turnover with disproportionately increased protein degradation [2, 3]. Analysis of regional protein turnover by means of catheterisation has revealed that the increase in protein synthesis almost exclusively occurs in the splanchnic bed, whereas the excessive protein breakdown is most pronounced in muscle [3]. It was additionally found that deamination of leucine to ketoisocaproate (KIC) and reamination of KIC to leucine was increased five-fold during insulin deprivation, but the tissues responsible for this phenomenon were not identified. A recent report used renal vein catheterisation and radioactive amino acid tracers and demonstrated that in non-diabetic postabsorptive humans 25% of leucine oxidation and around 10% of leucine appearance and leucine protein synthesis took place in the kidney [13]. The kidney has also been shown to be a major site of phenylalanine conversion to tyrosine and a contributor of tyrosine to the circulation [14].
Little information is available about the role of the kidney in lipid metabolism. One previous report has found that the main oxidative fuels extracted by the kidney in the postabsorptive state were lactate and free fatty acids [15], but these results could not be confirmed in insulin-depleted diabetic patients [16].
The present study was therefore undertaken to define the role of the kidney in intermediary metabolism with particular reference to alterations of amino acid, fat, and glucose metabolism following acute insulin withdrawal. To answer these questions we studied five type 1 diabetic subjects during insulin replacement and insulin deprivation and compared the results to those observed in six healthy non-diabetic control subjects. Renal glucose, amino acids and palmitate production and uptake, as well as phenylalanine conversion to tyrosine, by the kidney were measured using the renal catherisation and tracer dilution method.
Research design and methods
Subjects
Six type 1 diabetic patients were recruited for the study. Problems with maintenance of correct catheter placement caused one patient to be excluded. Hence five C-peptide-negative, diabetic subjects (two male and three female, age 30.4±2.9 years, diabetes duration 12.4±2.0 years, insulin dose 46.4±5.9 IU/day, BMI between 28 and 19 kg/m2, HbA1c 8.5±0.2%) were each studied twice and six healthy control subjects (three female, three male, age 30.7±2.0 years, BMI between 28 and 19 kg/m2 ) were each studied once. One patient from the diabetic group and one patient from the control group were on stable l-thyroxine replacement therapy because of hypothyroidism. Otherwise none received any medication. Two of the diabetic subjects had mild background retinopathy. None had microalbuminuria, hypertension or signs or symptoms of neuropathy or macroangiopathy. Studies in the diabetic subjects were separated by a minimum of 2 months. Women participating in the study were examined during the luteal phase of the menstrual cycle. Before the study the protocol was submitted to and approved by the Mayo Institutional Review Board and the purpose and potential risks of the study were explained to all subjects and informed, written consent was obtained from all of them.
Analysis of samples
l-[15N]phenylalanine (99 atom% excess) and l-[1-13C,15N]leucine (99 atom% excess) were purchased from Cambridge Isotope Laboratories (Andover, MA, USA) and l-[ring-2H4] tyrosine (99 atom% excess) and l-[15N]tyrosine (99 atom% excess) were purchased from Isotec (Maimisburgh, OH, USA). [6,6-2H2]glucose (2H, 99 atom%) was purchased from Cambridge Isotope Laboratories.
The chemical, isotopic and optical purity of these compounds was confirmed before use. Solutions were prepared under sterile conditions in the pharmacy and were shown to be free of bacteria and pyrogens before administration.
[9,10-3H]palmitate was obtained from NEN Research Products (Wilmington, DE, USA). Cardiogreen (indocyanine) was purchased from Becton Dickinson (Cockeysville, MD, USA) and paraaminohippurate (PAH) was obtained from Merck (West Point, PA, USA).
Study protocol
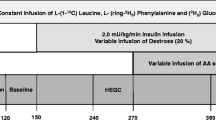
Type 1 diabetic patients were treated with 6-hourly regular insulin for 3 days before the study and their long-acting insulins were discontinued to avoid a carryover effect of insulin during the metabolic study. All subjects ingested a standardised weight-maintaining diet for 3 days before the study. The diabetic patients were studied on two separate occasions—during insulin deprivation and during insulin treatment. On each occasion subjects were admitted to the clinical research centre at 17.00 h the evening before the study and, following ingestion of a standard meal, kept fasted until the end of the study. Diabetic subjects received their last injection of slowly acting insulin 72 h before the study and the last subcutaneous injection of regular insulin 12 h before the study. After catheterisation of an antecubital vein, infusion of either insulin (to keep plasma glucose between 4.5 and 6.5 mmol/l) or saline was commenced at 24.00 h and continued throughout the study. The non-diabetic subjects received saline. Glucose concentrations were monitored throughout the night. Between 07.30 and 08.30 h the following morning cannulas were placed in the femoral vein, one renal vein and the femoral artery, as described previously [6, 13, 16, 17]. Renal vein catheters were inserted under fluoroscopic guidance; the femoral artery line was used for infusion of indocyanine green (0.6 mg/min) to measure blood flow in the leg and for blood sampling. Femoral vein and renal vein catheters were used to collect blood samples. The peripheral antebrachial vein was used for infusion of isotopes, insulin and PAH.
After taking duplicate baseline samples, priming doses followed by continuous infusion of [15N,1-13C]leucine (1 mg/kg and 1 mg kg−1 h−1), [15N]phenylalanine (0.75 mg/kg and 0.75 mg kg−1 h−1), [2H4]tyrosine (0.6 mg/kg and 0.6 mg kg−1 h−1) and [6,6-2H2]glucose (2.4 mg/kg and 2.4 mg kg−1 h−1), together with a priming dose of [13C]sodium bicarbonate (0.2 mg/kg ) and [15N]tyrosine (0.3 mg/kg ) were given at 08.00 h. Infusions were continued for 4 h in all cases. At 11.00 h a 1-h infusion of [3H]palmitate (11.1 kBq/min) was administered for determination of palmitate turnover. Cardiogreen was infused at a rate of 0.6 mg/min from 10.30 h for 90 min. PAH was given as a 3-mmol bolus followed by a constant infusion of 0.1 mmol/min for three sequential clearance periods from 10.30 h onwards [8, 11, 12].
Urine was collected over the entire 240-min study period in two portions (0–120 and 120–240 min). A total minimum water input (i.v. + oral) of 250 ml/h was maintained throughout the 4-h study period. Indirect calorimetry was performed from 10.30 to 11.00 h. Duplicate blood and breath samples were collected from 07.45–08.00 h and every 15 min from 11.00–12.00 h.
Blood glucose was measured by a glucose oxidase method (Beckman Instruments, Fullerton, CA, USA). Lactate, 3-hydroxybutyrate and NEFA were analysed as described previously [3], as was circulating epinephrine [18]. Plasma insulin and growth hormone were measured using a chemiluminescent sandwich assay (Sanofi Diagnostics, Chaska, MN, USA) and glucagon and cortisol were measured with radioimmunoassays (Diagnostic Products, Los Angeles, CA, USA). Indocyanine green concentrations were measured by spectrophotometry and PAH was measured calorimetrically.
Plasma enrichment levels of [15N]phenylalanine, [2H4]tyrosine, [15N]tyrosine, [1-13C,15N]leucine and [1-13C]leucine were determined by gas chromatography/mass spectrometry as described elsewhere [3, 14].
Isotopic enrichment of [6,6-2H2]glucose was measured by gas chromatography/mass spectrometry (Hewlett Packard 5972 GC/MS, Palo Alto, CA, USA) using the trimethylsilyl-O-methyloxime derivate and monitoring fragment ions at m/z 321 and 319, as described previously [19].
Plasma palmitate concentrations and specific activity were determined by HPLC with the use of [2H31]palmitate as internal standard [20]. Palmitate concentrations in the insulin-withdrawn state were sufficiently high that it was necessary to calculate concentration and measure the palmitate content (nmol) in these samples using curvilinear formulae (R 2>0.99 for standard curves).
Calculations
Plasma flows (PFs) from the kidney were calculated as outlined previously [16] and converted to blood flows (BFs) using the equation:
An isotopic plateau was observed from 180–240 min in all cases. This was assessed based on the observation that when isotopic enrichment values (or specific activity) of the tracers utilised in different sites were plotted against time the ensuing slopes were not different from zero. Mean values for any isotope at each plateau were used for all calculations of substrate kinetics.
Whole-body phenylalanine flux (Q Phe) was calculated as follows:
where i Phe is the rate of phenylalanine infusion (μmol kg−1 body weight min−1), i ePhe is the isotopic enrichment of the infusate and P ePhe is [15N]phenylalanine enrichment at isotopic plateau. Similar calculations were performed for [2H4]tyrosine (Q Tyr).
Conversion to tyrosine is the initial step in phenylalanine degradation and whole-body phenylalanine-to-tyrosine (I PT) conversion was calculated using the equation:
where [15N]P Tyr and [15N]P Phe are steady-state isotopic enrichments of tyrosine and phenylalanine, respectively.
Whole-body incorporation of phenylalanine into protein is calculated by subtracting I PT from Q Phe because apart from conversion to tyrosine the only other fate of phenylanine is protein synthesis. The basis of these equations is discussed elsewhere [3, 21].
Regional dynamics of phenylalanine, tyrosine and leucine
Net amino acid balances across organs were calculated by multiplying arteriovenous differences by blood flows. For calculation of across-organ isotope kinetics we used the equations described previously [3, 14].
The rate of appearance of phenylalanine (R a(Phe)), representing local protein breakdown was determined as follows:
where [15N]PheEA and [15N]PheEV are enrichments in artery and vein, Phea is arterial concentration of phenylalanine and BF is the blood flow. Rate of appearance of tyrosine was calculated similarly.
Local conversion of phenylalanine-to-tyrosine (I PT) was calculated by:
Calculations of local phenylalanine incorporation into protein can now be determined after calculation of phenylalanine rate of disappearance (Rd(Phe))
Leucine kinetics, leucine appearance rate from protein and leucine transamination were measured using the equations previously described by Cheng et al. [3, 22]. Leucine oxidation rates were measured using [13C]KIC as the precursor pool [3] and protein synthesis (disappearance) rates were determined as the sum of net balances and calculated breakdown (appearance) rates.
Whole-body rate of appearance (Ra) for glucose was calculated by dividing the rate of infusion of labelled glucose by enrichment.
Net renal glucose exchange was calculated as the product of arteriovenous plasma glucose concentration differences and renal blood flow.
Regional renal production of glucose was in all circumstances calculated using the simple equation [12, 23]:
in which R a is renal glucose production, BF is total renal blood flow, [glucose]art is the arterial glucose concentration and E ven and E art represent enrichment in venous or arterial blood. As pointed out earlier [12] the small size of the fractional extractions (<3%) of glucose across the renal bed means that their contributions to the calculation become negligible. For that reason the parameter was omitted. Negative values for regional glucose production were included in the mean calculations.
Regional renal glucose uptake was determined as the sum of renal glucose production and net renal glucose exchange.
Systemic palmitate R a and regional palmitate exchange were calculated as described previously [24], with the use of the measured tracer purity to determine the correct infusion rate.
Statistics
All values are given as mean±SEM. Paired and unpaired t-tests were used to assess whether any parameter differed significantly between the insulin-treated and insulin-deprived states and between diabetic and non-diabetic subjects, respectively. A p value <0.05 (two-tailed) was considered statistically significant.
Results
Blood flow, circulating substrates and hormones and indirect calorimetry
In all cases renal blood flow was stable at ∼1 l ml/min and leg blood flow at ∼400–500 ml/min (Table 1). Plasma glucose averaged 5.0 and 5.6 mmol/l in the non-diabetic and insulin-treated subjects, respectively. It was elevated to 15.5 mmol/l in the diabetic subjects when they were insulin deprived. Plasma insulin averaged 4.4 and 10.8 mU/l in the non-diabetic and insulin-treated type 1 diabetic subjects and was decreased (1.5 mU/l, p<0.05) in the insulin-deprived type 1 diabetic subjects. No differences in circulating counter-regulatory hormones were detected. There was a non-significant trend towards elevated energy expenditure and a decreased respiratory exchange ratio during insulin deprivation (0.10>p>0.05).
Phenylalanine and tyrosine dynamics
Whole body
Circulating concentrations of phenylalanine and whole-body fluxes of phenylalanine and tyrosine (reflecting protein breakdown) were elevated during insulin deprivation (p<0.05) (Table 2). The increase in phenylalanine flux was accompanied by a high rate of protein synthesis (p<0.05), whereas phenylalanine to tyrosine conversion was unaffected.
Kidney
In the basal state there was a small net uptake of phenylalanine of ∼2 μmol/min and a more pronounced net release of tyrosine of ∼5 μmol/min across the renal bed (Table 2). Renal appearance rates in plasma of [15N]tyrosine originating from [15N]phenylalanine (phenylalanine-to-tyrosine conversion) were close to 5 μmol/min in all cases. Renal phenylalanine and tyrosine dynamics were not altered by insulinopaenia.
Leg
Phenylalanine appearance rates from protein (protein breakdown) were 5.4±0.6 (control), 5.6±1.2 (insulin-treated) and 7.0±1.1(insulin-deprived) μmol/min (p=0.07 for insulin-treated vs insulin-deprived subjects) (Table 2). Local disappearance rates for phenylalanine (protein synthesis) were between 3.3 and 3.8 μmol/min in all cases (p>0.05).
Leucine dynamics
Whole body
Total leucine C-flux representing protein breakdown was increased by 20% (p<0.05) by insulin deprivation (Table 3). In parallel leucine oxidation was raised by 50% (p<0.05) and protein synthesis from leucine by 15% (p<0.05). Furthermore leucine-KIC interconversion was markedly increased in insulin-deprived subjects (KIC to leucine from 40 to 160 μmol kg−1 h−1 and leucine to KIC from 60 to 190 μmol kg−1 h−1, p<0.05).
Kidney
Local leucine appearance from protein (protein breakdown) was between 3.7 and 5.7 μmol/min in all groups (p>0.05) and disappearance rates (protein synthesis) were between 4.3 and 4.9 μmol/min (p>0.05) (Table 3). Renal rates of leucine deamination were also stable (8.1–11.9 μmol/min) as were reamination rates (6.2–10.2 μmol/min) (p>0.05 for all comparisons).
Leg
Rates of protein breakdown and protein synthesis were comparable in all situations (Table 3), ranging from 7.5 to 12.8 and 6.1 to 6.8 μmol/min, respectively (p>0.05). Compared with the insulin-treated state rates of leucine reamination and deamination were both doubled during insulin deprivation (52 vs 26 and 55 vs 25 μmol/min, p<0,05); compared with control subjects p=0.08 and p=0.07 for reamination and deamination during insulinopaenia, respectively.
Glucose dynamics
During insulin deprivation glucose appearance rates increased approximately two-fold (p<0.05) (Table 4). Concomitantly there was a trend towards increased renal glucose uptake (p>0.05), while renal glucose production remained unaffected (p>0.05). Renal glucose production rates remained low with a high variation between subjects. This variation was primarily the result of highly variable dilution of deuterated glucose across the kidney, ranging from 11.8 to −9.5% (mean of five measurements) in each individual. The renal contribution to total body endogenous glucose production during insulin deprivation decreased slightly, but not significantly, from ∼15 to 7%.
NEFA dynamics
Under conditions of insulin deprivation circulating palmitate concentrations and whole-body appearance rates were more than doubled and there was a trend towards increased femoral release of palmitate, although this was not statistically significant (Table 5). Regardless of conditions, kidney exchange of palmitate was close to zero. When deprived of insulin, subjects tended to exhibit increased palmitate concentrations, appearance rates and femoral release and uptake rates (all p values between 0.06 and 0.2). It could be argued that the comparisons between insulin-deprived and insulin-replaced values should be one-sided because we know the direction of change.
Discussion
The current study was conducted to define the role of the kidney in amino acid, fat and glucose metabolism in non-diabetic and insulin-treated type 1 diabetic subjects and to identify renal components that contribute to the metabolic derangement caused by acute insulinopaenia in type 1 diabetic subjects. Our main findings are that in the basal state 5–10% of whole-body protein flux, close to 20% of leucine–KIC interconversion and a major part of phenylalanine-to-tyrosine conversion occur in kidney. During acute insulin withdrawal the leg becomes a major site for leucine transamination, whereas local renal phenylalanine, leucine and glucose kinetics remained unaffected. Of note, the diabetic subjects studied were not insulin deficient for a long time; although fasting plasma glucose levels were above 15 mmol/l, 3-hydroxybutyrate concentrations were only modestly raised at 1.6 mmol/l, and it is conceivable that more prolonged insulin deficiency might have affected the outcome of the study. On the other hand, the insulinopaenia imposed under the present experimental conditions did not increase circulating levels of any of the counterregulatory hormones, this allowed assessment of insulin per se without interference from other metabolic hormones.
It is well recognised that insulin is a protein anabolic hormone. In untreated type 1 diabetes prolonged lack of insulin leads to cachexia, excessive urinary nitrogen loss and decreased lean body mass, all of which revert to normal upon insulin therapy [25–27]. In accordance with previous studies [2, 3] we found that insulin deprivation increases whole-body protein breakdown, as evidenced by increased flux rates of phenylalanine, tyrosine and leucine of around 20%. This effect is to some extent offset by a concomitant increase in protein synthesis, which could relate to increased levels of circulating amino acids. In addition, uncontrolled diabetes is characterised by dramatic three- to five-fold increases of leucine–KIC transamination rates, as also observed in the present study [3]. Theoretically this biochemical reaction could provide a mechanism for the transfer of surplus nitrogen from gluconeogenic precursors such as glutamine. The tissues behind these metabolic aberrations have not been precisely defined in previous studies [3], although it has been shown that both splanchnic and leg tissues contribute to increased protein breakdown. The present study adds new evidence that the leg is a primary site for leucine transamination—the doubling of both leucine deamination and reamination rates observed across the leg accounts for the majority of the three- to four-fold increase of leucine transamination at the whole-body level.
Our observations do not support the premise that the kidney plays any major role during insulin withdrawal on protein metabolism. The finding that 5–10% of whole-body phenylalanine and leucine fluxes occur across the renal bed is in line with a previous study [13], which reported a renal component of 10% in patients with hypertension and cardiac disease. In addition, our data demonstrated that the kidney participates in leucine transamination; in all three groups studied reamination and deamination rates consistently ranged from 6 to 12 μmol/min, corresponding to between 10 and 20% of whole-body transamination under conditions of well-controlled insulin. These findings are in contrast to the previous study [13], which reported renal transamination rates below zero. The discrepancy could relate to the different patient categories studied, because it cannot be excluded that patients with hypertension and cardiac disease may have subtly compromised renal function. Finally, we confirm our recent observation that the kidney is an important site for phenylalanine-to-tyrosine conversion [14], by showing that this also applies to diabetic subjects, in whom we observed renal conversion rates of around 5 μmol/min, regardless of whether insulin was administered. This indicates that the kidney’s key role as a supplier of tyrosine to the systemic circulation during the postabsorptive state is unaffected by insulin deficiency and the associated metabolic derangements. In addition, these data indicate that kidney does not contribute actively to the altered metabolic conditions associated with insulin deficiency.
The renal contribution to endogenous glucose production has been a matter of much controversy [4–12, 28]. Much of the confusion in all likelihood has arisen from the fact that measurements across the renal bed, because of the high renal blood flow of around 1 l/min, are highly susceptible to methodological imprecision as regards analysis of differences in arteriovenous glucose concentrations and dilution of tracers. In general, the observed differences are close to the detection limits of the assays employed. In the present study we found average renal glucose production rates of 100–140 μmol/min, amounting to around 15% of endogenous glucose production. There was, however, huge interindividual variation. This picture is in agreement with the literature, where production rates between 0 and 30% have been reported [28]. Previous studies have reported that high levels of insulin may suppress renal glucose production in healthy control subjects [6, 11]. We failed to observe any effect of acute insulin deficiency on renal glucose production in type 1 diabetic subjects, but as noted above it is possible that more severe hypoinsulinaemia could have promoted renal glucose production. In particular, it should be noticed that under the current conditions insulin withdrawal did not augment circulating epinephrine concentrations and that epinephrine has been reported to stimulate renal glucose production; and blood levels of epinephrine increase in severe diabetic derangement, such as ketoacidosis [4, 6].
Enhanced lipolysis and uncontrolled ketogenesis following insulin deprivation can lead to diabetic ketoacidosis. Our results suggest that moderate hypoinsulinaemia increases appearance rates and circulating concentrations of NEFA and that adipose tissue in the leg contributes by releasing high amounts of NEFA. Only very few studies have examined renal free fatty acid metabolism in humans [15, 16, 29]. One of these studies reported a substantial renal uptake of NEFA [15], whereas another failed to confirm this observation [16]. More recently Jensen et al. reported a total NEFA uptake in the kidney of 26 μmol/min in 60-h-fasted subjects [30]. The present findings suggest that the kidney plays a minor role in NEFA metabolism
In summary, we find that in the postabsorptive state the kidney contribute 5–10% of whole-body protein turnover, 15–20% of leucine transamination, 15% of endogenous glucose production and that the kidney actively converts phenylalanine to tyrosine. Acute insulin withdrawal has no detectable effect on these parameters, albeit the leg becomes a major site for leucine reamination and deamination.
Abbreviations
- KIC:
-
ketoisocaproate
- PAH:
-
paraaminohippurate
References
Miles JM, Rizza RA, Haymond MW, Gerich J (1980) Effects of acute insulin deficiency on glucose and ketone body turnover in man. Evidence for the primacy of overproduction of glucose and ketone bodies in the genesis of diabetic ketoacidosis. Diabetes 29:926–930
Nair KS, Garrow JS, Ford C, Mahler RF, Halliday D (1983) Effect of poor diabetic control and obesity on whole body protein metabolism in man. Diabetologia 25:400–403
Nair KS, Ford GC, Ekberg K, Fernqvist-Forbes E, Wahren J (1995) Protein dynamics in whole body and in splanchnic and leg tissues in type I diabetic patients. J Clin Invest 95:2926–2937
Stumvoll M, Meyer C, Mitrakou A, Nadkarni V, Gerich JE (1997) Renal glucose production and utilization: new aspects in humans. Diabetologia 40:749–757
Cersosimo E, Judd RL, Miles JM (1994) Insulin regulation of renal glucose metabolism in conscious dogs. J Clin Invest 93:2584–2589
Stumvoll M, Chintalapudi U, Perriello G, Welle SL, Gutierrez O, Gerich JE (1995) Uptake and release of glucose by the human kidney. Postabsorptive rates and responses to epinephrine. J Clin Invest 96:2528–2533
Stumvoll M, Meyer C, Perriello G, Kreider M, Welle S, Gerich J (1998) Human kidney and liver gluconeogenesis: evidence for organ substrate selectivity. Am J Physiol 274:E817–E826
Stumvoll M, Meyer C, Kreider M, Perriello G, Gerich J (1998) Effects of glucagon on renal and hepatic glutamine gluconeogenesis in normal postabsorptive humans. Metabolism 47:1227–1232
Meyer C, Stumvoll M, Nadkarni V, Dostou J, Mitrakou A, Gerich J (1998) Abnormal renal and hepatic glucose metabolism in type 2 diabetes mellitus. J Clin Invest 102:619–624
Cersosimo E, Garlick P, Ferretti J (1999) Insulin regulation of renal glucose metabolism in humans. Am J Physiol 276:E78–E84
Cersosimo E, Garlick P, Ferretti J (1999) Renal glucose production during insulin-induced hypoglycemia in humans. Diabetes 48:261–266
Ekberg K, Landau BR, Wajngot A et al (1999) Contributions by kidney and liver to glucose production in the postabsorptive state and after 60 h of fasting. Diabetes 48:292–298
Tessari P, Garibotto G, Inchiostro S et al (1996) Kidney, splanchnic, and leg protein turnover in humans: insight from phenylalanine kinetics. J Clin Invest 98:1481–1492
Moller N, Meek S, Bigelow M, Andrews J, Nair KS (2000) The kidney is an important site for in vivo phenylalanine-to-tyrosine conversion in adult humans: a metabolic role of the kidney. Proc Natl Acad Sci USA 97:1242–1246
Nieth H, Schollmeyer P (1966) Substrate utilization of the human kidney. Nature 209:1244–1245
Wahren J, Felig P (1975) Renal substrate exchange in human diabetes mellitus. Diabetes 24:730–734
Brundin T, Wahren J (1994) Renal oxygen consumption, thermogenesis, and amino acid utilization during i.v. infusion of amino acids in man. Am J Physiol 267:E648–E655
Charlton MR, Adey DB, Nair KS (1996) Evidence for a catabolic role of glucagon during an amino acid load. J Clin Invest 98:90–99
Krury D, Keller U (1991) Trimethylsilyl-O-derivatives for the measurement of [6,6-2H2]-d-glucose-enriched plasma samples by gas chromatography–mass spectrometry. J Chromatogr Appl 572:302–306
Jensen MD, Rogers PJ, Ellman MG, Miles JM (1988) Choice of infusion-sampling mode for tracer studies of free fatty acid metabolism. Am J Physiol 254:E562–E656
Thompson GN, Pacy PJ, Merritt H et al (1989) Rapid measurement of whole body and forearm protein turnover using a [2H5]phenylalanine model. Am J Physiol 256:E631–E639
Cheng KN, Dworzak F, Ford GC, Rennie MJ, Halliday D (1985) Direct determination of leucine metabolism and protein breakdown in humans using l-(1-13C,15N)leucine and forearm model. Eur J Clin Invest 15:345–353
Wolfe RR (1992) Radioactive and stable isotopes tracers in biomedicine. In: Principles and Practice of Kinetic Analysis. Wiley-Liss, New York p. 186
Jensen MD, Cryer PE, Johnson CM, Murray MJ (1996) Effects of epinephrine on regional free fatty acid and energy metabolism in men and women. Am J Physiol 270:E259–E264
Geyelin HR, Harrop G, Murray MF, Corwin E (1922) The use of insulin in juvenile diabetes. J Metab Res 11:767–791
Atchley DW, Loeb RF, Richards DW Jr, Benedict EIM, Driscoll ME (1933) On diabetic acidosis—a detailed study of electrolyte balances following the withdrawal and reestablishment of insulin therapy. J Clin Invest 12:297–326
Walsh CH, Soler NG, James H et al (1976) Studies in whole body potassium and whole body nitrogen in newly diagnosed diabetics. Q J Med 45:295–301
Moller N, Rizza RA, Ford GC, Nair KS (2001) Assessment of postabsorptive renal glucose metabolism in humans with multiple glucose tracers. Diabetes 50:747–751
Hillier TA, Pedula KL (2001) Characteristics of an adult population with newly diagnosed type 2 diabetes. Diabetes Care 24:1522–1527
Jensen MD, Ekberg K, Landau BR (2001) Lipid metabolism during fasting. Am J Physiol—Endocrinol Metab 281:E789–E793
Acknowledgements
We thank the nursing and dietetic staff of Mayo General Clinical Research Center. We are grateful for the skilful technical assistance of C. Ford, M. Persson, J. Kahl, D. Morse and J. Schimke, and for assistance in the conduct of the study by M. Bigelow. The study is supported by National Institutes of Health grants RO1 DK41973 and MO1 RR00585 and Mayo Foundation. K. S. Nair is David Murdock-Dole Professor of Nutrition at the Mayo Clinic.
Duality of interest
The authors declare that they have no conflicts of interest as regards this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moller, N., Jensen, M.D., Rizza, R.A. et al. Renal amino acid, fat and glucose metabolism in type 1 diabetic and non-diabetic humans: effects of acute insulin withdrawal. Diabetologia 49, 1901–1908 (2006). https://doi.org/10.1007/s00125-006-0287-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-006-0287-3