
Abstract
Chemical components of air pollutant exposures that induce oxidative stress and subsequent inflammation may be partly responsible for associations of cardiovascular morbidity and mortality with airborne particulate matter and combustion-related pollutant gasses. However, epidemiologic evidence regarding this is limited. An exposure-assessment approach is to measure the oxidative potential of particle mixtures because it is likely that hundreds of correlated chemicals are involved in overall effects of air pollution on health. Oxidative potential likely depends on particle composition and size distribution, especially ultrafine particle concentration, and on transition metals and certain semi-volatile and volatile organic chemicals. For health effects, measuring systemic oxidative stress in the blood is one feasible approach, but there is no universal biomarker of oxidative stress and there are many potential target molecules (lipids, proteins, DNA, nitric oxide, etc.), which may be more or less suitable for specific study goals. Concurrent with the measurement of oxidative stress, it is important to measure gene and/or protein expression of endogenous antioxidant enzymes because they can modify relations between oxidative stress biomarkers and air pollutants. Conversely, the expression and activities of these enzymes are modified by oxidative stress. This interplay will likely determine the observed effects of air pollutants on systemic inflammatory and thrombotic mediators and related clinical outcomes. Studies are needed to assess the reliability and validity of oxidative stress biomarkers, evaluate differences in associations between oxidative stress biomarkers and various pollutant measurements (mass, chemical components, and oxidative potential), and evaluate impacts of antioxidant responses on these relations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Overview
Cardiovascular hospitalization and mortality have been consistently associated with increased exposure to airborne mass concentrations of particulate matter (PM) < 2.5 μm (PM2.5) and <10 μm in diameter (PM10) (Pope and Dockery 2006; Brook et al. 2010). Although epidemiologic studies have shown associations of cardiovascular morbidity and mortality with ambient CO and NO2 (Brook et al. 2010; Bhaskaran et al. 2009), concentrations of these gasses have been considerably lower relative to levels causing effects in experimental models, and it is possible that these gasses are serving as surrogates for other causal components from fossil fuel combustion. A recent review also reports lack of consistent findings in time series studies for the relation of ozone to daily hospital data for myocardial infarction (Bhaskaran et al. 2009). Together, these findings call for the need to better understand the underlying causal chemical components and pathophysiological mechanisms responsible for the morbidity and mortality associations (Mills et al. 2009).
Oxidative stress responses to reactive oxygen species (ROS) and subsequent inflammation resulting from air pollution exposure may be one of these important mechanisms (Brook et al. 2010; Frampton 2006; Xia et al. 2006). However, epidemiologic evidence in humans that ambient PM or gaseous pollutant exposures increases biomarkers of oxidative stress is limited. The following review assesses the importance of addressing this gap to the field of air pollution research and we offer some possible methods of doing so by focusing on a few selected biomarkers that can be measured in the blood for large population studies. We will not discuss the use of other body fluids to measure oxidative stress that are reviewed elsewhere, for example exhaled breath condensates (Silkoff et al. 2006) and urine (Cooke et al. 2002). We also focus on inhalation and pulmonary deposition as the route of exposure for cardiovascular effects of interest in the paper (we do not discuss dermal, gastrointestinal, or transplacental routes). Finally, the air pollutants of primary focus in the review are those associated with particles.
Oxidative stress, inflammation, and air pollution exposures
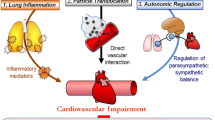
Oxidative stress is a biochemical imbalance in which production of ROS exceeds the natural antioxidant capacity. This imbalance can occur in the body following exposure to pro-oxidant air pollutants (Fig. 1). Given this balance between oxidants and antioxidants, a threshold level for exposure to the pro-oxidant pollutant might be expected to lead to a measurable oxidative stress response. In the presence of oxidative stress, uncontained ROS cause tissue damage and dysfunction by attacking and denaturing structural and functional molecules (lipids, proteins, carbohydrates, DNA, RNA, NO, etc.) and by modulating the activities of redox-sensitive transcription factors (e.g., nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), AP-1, and signal transduction pathways). In general, the first line of defense against oxidative stress is via antioxidant enzymes and proteins involved in phase II detoxification. This response is regulated by the nuclear factor-erythroid 2-related factor 2 (Nrf2)/antioxidant-response element (ARE) pathway. The pathway is activated when signals from oxidative and electrophilic chemicals modify Kelch-like ECH associated protein 1 to release Nrf2, which then moves to the nucleus and activates the expression of genes with ARE that include antioxidant and phase II genes. This is a key pathway in responses to toxic pollutants (Kensler et al. 2007), and is important in mitigating oxidative stress-induced endothelial dysfunction (Mann et al. 2007). When these defense mechanisms are overwhelmed at higher levels of exposure-induced oxidative stress, pro-inflammatory effects follow via the activation of NFκB. Activation of NFκB increases the transcription of chemokines, cytokines and acute phase proteins that contribute to and are predictive of coronary artery disease risk (Pai et al. 2004). Nitrosative stress from reactive nitrogen species (RNS) such as peroxynitrite may also be an important driver of inflammatory responses (Shishehbor et al. 2003). Finally, cytotoxic effects could occur at higher levels of exposure (Ayres et al. 2008).

Stress response pathways. Air pollutant effects may occur in large part through the generation of ROS or reactive nitrogen species (RNS) from organic chemicals either directly from air pollutant components, or through activation of bronchial epithelial cells, macrophages, neutrophils, and endothelial cells (specific details in text). Air pollutant components from fossil fuel combustion including polycyclic aromatic hydrocarbons (PAH), quinones, and transition metals (e.g., Fe) have pro-oxidant properties and may induce oxidative stress through the production of free radicals and subsequent activation of redox-sensitive signaling pathways (e.g., Nrf2, ARE, NF-κB). These pathways may be behind findings indicating that exposure to fine particles trigger inflammatory and cytotoxic responses in the human lung as well as systemic inflammation and thrombosis when oxidant defenses are inadequate. Systemic oxidative stress and inflammation may occur as a result of effects of pollutants on the airways (indirect pathway) or from the direct systemic distribution of pollutant chemicals following pulmonary deposition
Extensive data show linkages of oxidative stress and depressed antioxidant capacity with the risk of cardiovascular disease (Dhalla et al. 2000), hypertension (Vaziri and Rodríguez-Iturbe 2006), and chronic kidney disease (Vaziri 2004). Oxidative stress may play a central role in the respiratory and cardiovascular effects of air pollution through its immunomodulating effects and its ability to initiate the inflammatory process and thrombogenic activity (Dhalla et al. 2000). Experimental data shows that redox active PM components (that are especially enriched in ultrafine PM < 0.1 μm) lead to the production of ROS in various cells in the lungs, blood and vascular tissues. This is followed by oxidative stress, which can then lead to increased airway and systemic inflammation, and adverse cardiovascular responses when antioxidant defenses are overwhelmed (Ayres et al. 2008; Utell et al. 2002). Epidemiologic data directly supporting this experimental evidence is limited, but there is indirect support from studies that have examined the effect modification of responses to air pollutant exposures by variants in oxidative stress-related genes (Alexeeff et al. 2008; Chahine et al. 2007; Islam et al. 2008, 2009; Park et al. 2006; Ren et al. 2010; Schwartz et al. 2005).
There is also some epidemiologic evidence that PM exposure significantly increases biomarkers of oxidative stress in blood, but the data are limited for susceptible populations exposed to urban air pollution. The epidemiologic evidence include panel studies of healthy subjects with few repeated measures (Chuang et al. 2007; Liu et al. 2007; Sørensen et al. 2003, 2005; Vinzents et al. 2005), and studies of workers occupationally exposed to combustion aerosols (Autrup et al. 1999; Kim et al. 2004; Lai et al. 2005; Rossner et al. 2007, 2008a, b; Sharman et al. 2002; Singh et al. 2007). The oxidation of low density lipoprotein (ox-LDL) has been found to be increased among people exposed to environmental tobacco smoke (Panagiotakos et al. 2004), which may carry redox active components similar to those in urban air.
Urban air pollution has been linked to increased ox-LDL in mice (Soares et al. 2009) and impairment of the anti-inflammatory capacity of high density lipoprotein (HDL) in mice (Araujo et al. 2008). This effect may play a role in the pro-atherosclerotic effects of air pollution that have been found using experimental models (Araujo and Nel 2009; Chen and Nadziejko 2005; Sun et al. 2005; Suwa et al. 2002), and have been evidenced in epidemiologic studies showing increased risk of atherosclerotic lesions among subjects living near busy traffic (Hoffmann et al. 2007; Künzli et al. 2005, 2010).
The importance of endogenous antioxidants to air pollutant responses
Evidence for the important role of antioxidant reactions (discussed below) in vivo is that alterations in antioxidant enzyme activities have been associated with cardiovascular (Espinola-Klein et al. 2007; Förstermann 2008; Fukai et al. 2002), renal (Vaziri 2004; Vaziri et al. 2009), and respiratory disease pathologies (Rahman et al. 2006). More recently, meta-analyses of published human studies identified a strong, inverse association of superoxide dismutase (SOD), glutathione peroxidase (GPx-1), and catalase enzyme activities in blood with coronary artery disease risk in people (Flores-Mateo et al. 2009). Such chronic antioxidant enzyme deficiencies raise the susceptibility to and increase the severity of tissue injury in response to oxidative stress. However, acutely, up-regulation of antioxidant enzymes can be a marker of responses to pollutant exposures denoting ROS/electrophile-induced Nrf2 activation.
The antioxidant activity of key proteins can also be used as an indirect measurement of oxidative stress under the assumption that it is a protective response. For example, this approach has been used with heme oxygenase-1 (HO-1) gene and protein expression in leukocytes exposed in vitro to particle mixtures (Li et al. 2002). HO-1 catalyzes the first step in heme degradation, and produces a general response to oxidative stress induced by ROS via the production of biliverdin which is a potent antioxidant molecule (Ryter et al. 2006).
Other key antioxidant enzymes include the glutathione S-transferases (GSTM1, GSTT1, and GSTP1), which are critical for the detoxification of xenobiotics including polycyclic aromatic hydrocarbons (PAH)-epoxides (phase II reactions) through the mercapturic acid pathway (Keen and Jakoby 1978). They also protect cells from protein, lipid and DNA oxidation products by the conjugation of harmful electrophiles to glutathione (GSH) (Bogaards et al. 1997; Hayes and McLellan 1999; Hayes et al. 2005). In human lung epithelial cells and in leukocytes, GSTP1 contributes most GST-derived enzyme activity, whereas GSTM1 is highly expressed in lymphoid cell lines and GSTT1 in erythroid cell lines (Fryer et al. 1986; Wang et al. 2000).
Oxidant air pollutants can also induce NADPH quinone-oxidoreductase (NQ01) expression. NQ01 detoxifies highly reactive quinones to hydroquinones (Monks et al. 1992), but some of these hydroquinones may still be reactive (Ross et al. 2000).
Mitochondrial manganese superoxide dismutase (MnSOD), extracellular Cu,Zn-SOD (extracellular SOD), cytosolic Cu,Zn-SOD, GPx-1, and catalase (CAT) are part of the frontline defense against ROS. SOD converts superoxide radicals to H2O2, whereas CAT and GPX1 catalyze the reduction of H2O2 to water and molecular oxygen. Superoxide anion scavenging activity in erythrocytes is driven largely by cytosolic Cu,ZnSOD since they lack mitochondria. extracellular SOD is the predominant SOD isoenzyme in plasma and its gene is 60% homologous with cytosolic Cu,ZnSOD, but is minimally homologous to mitochondrial MnSOD. It is highly expressed in arterial walls, where it is produced predominantly by smooth muscle cells (Fattman et al. 2003). Diminished expression of extracellular SOD may be important in the progression of coronary artery disease (Fukai et al. 2002) and may play an important role in endothelial dysfunction in patients with this disease (Landmesser et al. 2000). Specifically, endothelial NO synthase produces NO, which traverses the extracellular space to induce smooth muscle relaxation in the vessel wall. Superoxide can react with NO to form the potent oxidant, peroxynitrite, which is likely involved in lipid peroxidation (O’Donnell and Freeman 2001). Gurgueira et al. (2002) found that rats exposed to concentrated ambient particles showed an adaptive increase in Cu,Zn-SOD activity in heart but not lung tissue. However, the standard colorimetric measurement of Cu,Zn-SOD activity does not distinguish between extracellular and cytosolic Cu,Zn-SOD (which predominates in erythrocytes and other cell types) potentially invalidating the measurement of extracellular SOD in plasma. Low hemolysis after blood cell separation could result in an overestimation of SOD as well as GPx in plasma.
Paraoxonase-1 (PON1) is an antioxidant enzyme that contributes to the antioxidant property of HDL by reversing/preventing lipid peroxidation, particularly peroxidation on LDL (Watson et al. 1995), and by limiting production of oxidized arachidonic acid-derived products that lead to the formation of 8-isoprostane (Adkins et al. 1993). The antioxidant properties of PON1 are based on its capacity to hydrolyze lipid peroxidation products (Mackness et al. 1998).
Production of several other enzymes capable of promoting oxidative stress may be induced by pollutant chemicals or particles that interact with cells. For example, myeloperoxidase which is released by activated neutrophils, monocytes, and some macrophages can damage lung (Repine et al. 1997; Gilliland et al. 1999) and arterial tissue (Schindhelm et al. 2009) by producing potent oxidants such as hypochlorous acid. Macrophages produce ROS as well in response to particle exposures. The cytochrome P450 (CYP) family is also important in the generation of ROS by activating inhaled toxicants via reductase-catalyzed quinone reduction (phase I reactions) (Kumagai and Shimojo 2002; Fig. 1). Microsomal epoxide hydrolase 1 as such acts in association with CYP enzymes by metabolizing PAH to highly reactive epoxide intermediates, which lead to ROS generation (Decker et al. 2009). The above data regarding cell-based pro-oxidant responses has led to the suggestion that inflammatory cell cultures may serve as indicators of the oxidative potential of particle mixtures via in vitro test systems that measure ROS production (discussed below; Ayres et al. 2008).
In addition to endogenous antioxidant enzymes and ROS scavenger molecules, exogenous dietary antioxidants may also protect people from the adverse effects of oxidative stress by up-regulating endogenous antioxidant defenses (or acting as cofactors), and by their own antioxidant properties (Romieu et al. 2008). However, their importance relative to the endogenous system itself is controversial (Limón-Pacheco and Gonsebatt 2009; Viña et al. 2007; Wenger 2007).
Finally, several drugs used in the treatment of cardiovascular diseases, especially statins (cholesterol-lowering medications), may inhibit the progression of atherosclerosis by their anti-inflammatory and anti-oxidant properties (Adameova et al. 2009). This may be relevant to findings in several cohort panel studies that show weaker associations between circulating biomarkers of inflammation and air pollution among subjects using statins (Delfino et al. 2009; Dubowsky et al. 2006; O’Neill et al. 2007; Ruckerl et al. 2006).
Oxidative stress and post-translational modification of proteins
ROS and RNS can lead to many different types of post-translational modifications of proteins including electrophilic modifications, protein carbonlyation, S-glutathionylation, oxidation of methionine residues, nitration, and hydroxylation of amino acids, oxidation of sulfhydryl groups, etc. (Gianazza et al. 2007). These effects have functional implications including inactivation of proteins, and downstream effects such as epigenetic changes. For example, S-glutathionylation occurs under oxidative conditions when a disulfide bond is made between a cysteinyl residue of a protein and oxidized glutathione disulfide (GSSG), or with GSH via one of the GSTs, and is reversed by thiol-disulfide oxidoreductase enzymes. This is important in redox signal transduction, in the regulation of c-Jun, JNK, and TRAF2 (apoptotic and proliferative effects), redox-sensitive thiol proteins, and proteins involved in the folding and maturing of transmembrane and secretory proteins in the endoplasmic reticulum (ER; Mieyal et al. 2008; Townsend 2007). S-glutathionylation may be a regulator of an accumulation of misfolded or unfolded proteins secondary to an increased redox state in the ER (ER stress) resulting in an adaptive response (unfolded protein response), which if prolonged leads to apoptosis (Townsend 2007). Indirect evidence that this biological pathway can be influenced by air pollutant-induced oxidative stress comes from several experimental studies that have found exposure to urban PM or diesel exhaust particles leads to upregulation of unfolded protein responses in cells from in vitro cell cultures and from lung and liver tissue of exposed mice (Gong et al. 2007; Laing et al. 2010; Watterson et al. 2009).
Oxidative stress and ROS may also lead to epigenetic changes including changes in the methylation of DNA and post-translational modifications of histones (methylation, tyrosine nitration, phosphorylation, ubiquitination, and SUMOylation), which may then affect gene expression and thus protein expression. For example, in the case of corticosteroid resistance, peroxynitrite nitrates tyrosine residues on histone deacetylase-2 leading to its inactivation and subsequent decrease in its ability to deacetylate histones. This in turn leads to the increased expression of the inflammatory genes associated with the histones (Barnes 2009).
Inactivation of antioxidant enzymes by electrophilic air pollutants chemicals could also be an important mechanism of air pollutant-induced oxidative stress. In a repeated measures analysis of 60 elderly subjects with a history of coronary artery disease, we found inverse associations of air pollutants (especially ultrafine particles and markers of primary combustion) with the activities of key erythrocyte antioxidant enzymes (GPx-1 and Cu,Zn-SOD; Delfino et al. 2008, 2009, 2010a). However, sensitivity analyses showed a small group of seven subjects with strongly positive associations (Delfino et al. 2009). Antioxidant enzymes in erythrocytes such as GPx-1 and Cu,Zn-SOD are important in protection against oxidative stress (Cimen 2008; Minetti et al. 2007; Tsantes et al. 2006). Whereas increased antioxidant proteins through the induction of genes is expected in nucleated cells (e.g., leukocytes) in response to oxidative stress, erythrocytes are unable to do so. It is possible that certain particle components can inactivate enzymes due to their electrophilic properties. We hypothesized that erythrocyte antioxidant enzyme inactivation by electrophilic components in air pollution is a possible explanation for our findings. Oxidative stress would be more likely as a result. Specifically, erythrocyte antioxidant inactivation could lead to pollutant-related increase in biomarkers of inflammation such as interleukin-6, which we also found was positively associated with air pollutants (Delfino et al. 2009). Interestingly, GPx-1 was also inversely associated with interleukin-6.
In vitro data has shown enzyme inactivation by pollutant components or ultrafine particles (Hatzis et al. 2006; Pigeolet et al. 1990). We recently conducted preliminary in vitro tests that showed pollutant-induced inactivation of the GPx-1 enzyme (Staimer et al. 2010). Compared with control conditions, GPx-1 activity significantly decreased when treated with an electrophilic chemical (p-Benzoquinone) and with extracts of PM2.5 particles collected on quartz filters at a downtown Los Angeles monitoring location. Other in vitro studies have shown irreversible inactivation of the thiol enzyme GAPDH by extracts of concentrated ambient ultrafine particles and diesel exhaust particles (Shinyashiki et al. 2008, 2009) as well as vapor phase ambient chemicals (Eiguren-Fernandez et al. 2010; Iwamoto et al. 2010). Some electrophilic chemicals in traffic exhaust are anticipated to lead to the inactivation of enzyme’s catalytic activity by covalent bond formation with nucleophilic (electron donor) functions of proteins. Enzyme inactivation may also occur through oxidation of protein cysteine residues by oxidant chemicals in vehicular emissions leading to irreversible oxidation products (Kumagai and Shimojo 2002).
Importance of particle size and composition to oxidative stress responses
In vitro data suggests that organic constituents and transition metals (e.g., Fe, Cu, Ni, and Zn) in PM are capable of generating ROS directly (Ayres et al. 2008; Cho et al. 2005; Ntziachristos et al. 2007) or as a result of their capacity to activate neutrophils or other leukocytes, alveolar macrophages, respiratory epithelial cells, and endothelial cells. Transition metals have known potential to induce oxidative stress through Fenton reactions. Cellular generation of ROS from exposure to PM mixtures has been demonstrated using an in vitro system of rat alveolar macrophages (Verma et al. 2009a, b). Important reactive chemicals may include PM2.5 organic components such as quinones that are themselves oxidizing species or PAH that undergo bio-transformation by cytochrome P-450 1A1 to quinones, which can induce oxidative stress (Bonvallot et al. 2001). Diesel exhaust particles are enriched in these redox cycling components (Shinyashiki et al. 2009). Automobile exhaust produces nearly the same set of components (Phuleria et al. 2007) and is responsible for a majority of mobile-source PM emissions in the USA (Gertler 2005).
Experimental data show that PAH and oxidized PAH (e.g., quinones) from diesel exhaust particles promote the generation of ROS leading to oxidative stress and NFκB activation with resultant inflammatory responses (Riedl and Diaz-Sanchez 2005). These exposures also lead to changes in antioxidant enzyme expression as shown in studies of airway responses in human subjects (Behndig et al. 2006) and with in vitro data (Li et al. 2002).
Such effects may underlie epidemiologic findings (including our own) of increased markers of vascular hypercoagulability and circulating inflammatory markers in relation to exposures to markers of traffic-related air pollutants (e.g., elemental carbon) in cohort panel studies (Delfino et al. 2008, 2009, 2010a, d; Ruckerl et al. 2006; Yue et al. 2007), in cross-sectional studies (Madrigano et al. 2009; O’Neill et al. 2007). This view is supported by results from a panel study of 60 elderly subjects described above showing that associations of plasma interleukin-6 (IL-6) and TNF-RII with PM0.25 mass were completely confounded by the PAH fraction of PM0.25 (Delfino et al. 2010d). Both indoor and outdoor home concentrations of low, medium, and high molecular weight PAHs were significantly associated with these biomarkers of systemic inflammation. Chemical mass balance model results using source tracers suggested that a majority of the PAH came from vehicular sources (Arhami et al. 2010; Delfino et al. 2010d). Another interesting study found associations between the traffic-related air pollutant, black carbon (BC), with plasma homocysteine levels, which is a risk factor for cardiovascular disease (Park et al. 2008). Authors suggested mechanisms to explain their findings, including an involvement of pollutant-generated ROS or inactivation of enzymes involved in homocysteine remethylation. These findings are consistent with a recent extensive review of the literature that concluded “vehicular emissions are a major environmental cause of cardiovascular mortality and morbidity in the United States” and that a PM2.5 BC standard should be considered under the US National Ambient Air Quality Standards (Grahame and Schlesinger 2010).
Particle size fraction is also an important determinant of the dose of redox active chemicals delivered to target organs. The ultrafine (<0.1 μm in diameter) and accumulation modes (0.1–2.5 μm) make up total PM2.5 mass (fine particles), which is regulated by the US Environmental Protection Agency (EPA). However, ultrafine particles are expected to induce greater responses per unit mass than larger particles that dominate PM2.5 mass. This has been attributed to higher deposition and retention in the lungs, ability to evade phagocytosis by surface macrophages, and magnitudes higher particle number and surface area than larger particles, thus delivering to the lungs greater concentrations of toxic components (Ayres et al. 2008; Delfino et al. 2005; Mills et al. 2009; Oberdörster et al. 2005; Schmid et al. 2009; Geiser and Kreyling 2010). Because of this surface area, ultrafine particles carry redox active organic chemicals (e.g., PAH) and transition metals in much higher concentrations than larger particles (Sioutas et al. 2005). Even for soot particles with low organic content, ultrafine particle–cell interactions can be an important pro-oxidant mechanism inducing inflammatory responses, especially in the lungs, possibly due to the large reactive surface area of carbon (Schmid et al. 2009). Therefore, toxic particle components and effective surface area are not well-represented by EPA-regulated PM2.5 and PM10.
Nevertheless, a few cohort panel studies still showed associations of biomarkers of oxidative stress with PM2.5 or PM10 mass (Chuang et al. 2007; Liu et al. 2007; Sørensen et al. 2003). However, there is limited epidemiologic evidence in humans relating well-characterized PM exposure to increases biomarkers of oxidative stress in blood. Two small panel cohort studies showed oxidative DNA damage (mainly 8-oxodG) in lymphocytes from exposure to increased ultrafine particle number concentrations (Vinzents et al. 2005) and PM metals (Sørensen et al. 2005). However, sample sizes were small in part due to the difficulty in performing the 8-oxodG assay on freshly isolated lymphocytes.
Translocation of ultrafine particles into the circulation has been shown in several studies, and despite the very low overall rate of perhaps ≤1%, the effect on target organs may be important given the high surface area and long retention times (Schmid et al. 2009; Geiser and Kreyling 2010). The high retention of ultrafine particles in the lungs shown in a recent study (Möller et al. 2008) suggests that a much greater impact is via the translocation of particle components to the circulation. This may lead to sustained effects through the gradual transfer of potentially redox active components to the circulation over many days. This could be particularly important for chemicals like PAH that require biotransformation by phase I enzymes (Fig. 1). Larger particles, including coarse particles, may have a greater impact in the lungs given that pro-inflammatory components in fine or coarse particles are efficiently delivered to the target airways. This is supported by an experimental study showing that aspiration of coarse particles collected both near and far from a busy highway led to inflammatory responses in the lungs of mice whereas only ultrafine particles from both locations led to cardiac reperfusion injury (Cho and Kleeberger 2010). Results in the cohort panel study described above for size-fractionated particles showed that only quasi-ultrafine particles <0.25 μm (PM0.25) were significantly associated with systemic biomarkers of inflammation [IL-6 and tumor necrosis factor-α receptor II] (Delfino et al. 2009) and with ischemic ST-segment depression measured with ambulatory electrocardiographs (Delfino et al. 2010c).
Furthermore, the potentially toxic components of carbonaceous aerosols are numerous and also vary considerably by location and by time of day and season, many being volatile during periods of low photochemical activity and near sources. In particular, the effects of combustion-related primary organic aerosols (POA) and their semivolatile organic compounds (SVOC) such as PAH may differ from effects of photochemically related secondary organic aerosol (SOA), which are in part derived from aged primary SVOC (Robinson et al. 2007). POA are formed during or shortly (within seconds) after the combustion of fossil fuels and are enriched in the ultrafine mode. SOA are formed from gas-to-particle conversion when volatile reactive organic gasses and SVOC are oxidized to form low-volatility products that condense to produce SOA, which is enriched in the accumulation mode (0.1–2.5 μm in diameter). Although a large part of the regulatory effort to protect public health involves regulating some of the gasses involved in SOA formation, there is little data on the importance of this characteristic of PM2.5 to cardiovascular health. Given results from experimental studies in ApoE mice (Campen et al. 2010; Lund et al. 2007), concentrations of volatile and semi-volatile organic precursor components from the combustion of fossil fuel are also likely important determinants of vascular oxidative responses important in the development of cardiovascular disease. However, there is little data regarding this for ambient or traffic-related exposures in human populations (Rossner et al. 2008a, b).
We have presented data showing that biomarkers of systemic inflammation (Delfino et al. 2008, 2009, 2010a, d), ambulatory blood pressure (Delfino et al. 2010b), and ischemic ST segment depression in ambulatory ECG (Delfino et al. 2010c) are more strongly associated with POA than SOA exposure markers and chemical components, whereas a biomarker of airway inflammation (fractional exhaled NO) was more strongly associated with SOA and ozone (Delfino et al. 2010d). We hypothesized that many SOA components, which are hydrophilic and highly oxidized, have immediate pro-oxidant effects on the airways, whereas many POA components (e.g., PAH), are hydrophobic and require biotransformation to pro-oxidant species (this can occur after systemic distribution of these chemicals). These findings do not support the belief that activation of leukocytes in the lungs, followed by the release of pro-inflammatory mediators is a major (indirect) pathway to the systemic inflammatory effects of particulate air pollution (Fig. 1; Mills et al. 2009).
Assessing the oxidative potential of PM components
This area of research has been extensively reviewed elsewhere (Ayres et al. 2008) and will only briefly be addressed here. The dithiothreitol (DTT) assay has been used to assess the redox potential from organic components in PM. The assay shows the capacity of PM to transfer electrons from DTT to oxygen resulting in the generation of superoxide. This redox activity is highest in ultrafine PM and is strongly correlated with elemental carbon and PAH fractions suggesting that traffic sources are important (Cho et al. 2005). Several in vitro and in vivo animal studies have shown that DTT activity of particle samples (along with strongly correlated concentrations of PAH, oxy-PAH, and polar organic chemicals) are related to ability of ultrafine PM and diesel exhaust particles to induce oxidative stress and inflammatory responses (Li et al. 2003, 2009; Shinyashiki et al. 2009). However, another potentially important mechanism of oxidative stress response to PM chemicals is the production of ROS by cells. One validated method is to assess this by incubation of rat alveolar macrophages in vitro with particle extracts and the ROS probe DCFH-DA under experimental conditions where cell injury is negligible, and then measuring fluorescence intensity to represent the oxidative generating capacity of particle extracts (Landerman et al. 2008). Macrophages are the first line of defense against pulmonary injury, including injury from pollutant particles. After phagocytizing particles, macrophages generate ROS in an oxidative burst. This is followed by cytokine cascades that result in inflammation and additional ROS production. The cell-based ROS activity induced by aqueous PM extracts in this assay has been correlated with transition metal and OC concentration in several studies in Los Angeles (Hu et al. 2008; Verma et al. 2009a) and elsewhere (Shafer 2010; Zhang et al. 2008). One of the LA studies also showed greater ROS activity for PM0.25 than larger particle size fractions (Hu et al. 2008).
We recently published among the first supportive evidence for the use of PM oxidative potential as a biomarker of exposure in epidemiologic research (Delfino et al. 2010d). We evaluated the relation of airway and systemic inflammatory responses to the potential of collected particles to induce cellular production of ROS. Sixty elderly subjects were monitored with 12 weekly measurements of both plasma IL-6 (a biomarker of systemic inflammation) and offline fractional exhaled NO (a biomarker of airway inflammation). To assess PM oxidative potential, ROS production was measured as described above (Landerman et al. 2008) by exposing rat alveolar macrophages (NR8383) in vitro to aqueous extracts of PM0.25 filters collected outdoors at the subjects’ retirement community. Both exhaled NO and IL-6 was positively associated with levels of macrophage ROS generation. Co-regression models of PM0.25 mass with ROS production suggested that oxidative potential explained most of the association of IL-6 with PM0.25 mass. The association of exhaled NO with ROS activity was not reflected by PM0.25 mass concentration. We concluded that the ability of particles to induce ROS generation by macrophages is associated with both systemic and airway biomarkers of inflammation, and this association is nominally reflected by particle mass concentration.
Potentially useful biomarkers of oxidative stress status
There is no universal biomarker of oxidative stress or oxidative status. Only a carefully selected panel of markers tailored for specific study goals may reflect different types of oxidative stress (Dotan et al. 2004). Furthermore, no biomarker meets all the technical requirements (cheap, non-invasive, easy to detect, chemically and biologically stable) as outlined by Dalle-Donne et al. (2006). More specifically, rapid screening methods are needed in large-scale epidemiological studies. Ideally, these methods should be standardized and cost effective with respect to biospecimen collection, processing, storage, and analysis. Thus, some biomarkers are better suited for epidemiologic studies than others. The following summarizes some of the more widely used and feasible indices of oxidative stress in large-scale human studies using blood samples (based, in part, on Dotan et al. 2004; Dalle-Donne et al. 2006, and Romieu et al. 2008) and highlights immunochemical methods for biomarker detection when applicable. Immunoassays are based on selective antibodies directed towards specific target analytes and they continue to be the method of choice for field screening methods.
Glutathione
GSH is the most commonly used index of biological stress status (Dalle-Donne et al. 2006). It is an important intracellular scavenger of free radicals and neutralizes peroxides in conjunction with GPx-1 and glutathione reductase, thus maintaining the redox balance of cells. Furthermore, GSH detoxifies electrophiles either directly or via GST-catalyzed conjugation (Keen and Jakoby 1978). Electrophilic compounds can enter erythrocytes in vitro and deplete erythrocyte GSH, which accounts for >98% of the whole blood GSH store (Palmen and Evelo 1996). Because erythrocytes are the main source of GSH in blood, GSH level will be significantly lower in anemic patients with or without oxidative stress, therefore the whole blood GSH concentration should be normalized against hemoglobin level. Glutathione deficiency in whole blood is associated with aging and the pathogenesis of cardiovascular disease (Kharb 2003; Muda et al. 2003). Conversely, high levels of total GSH in blood is found in healthy elderly subjects (Pastore et al. 2003). Oxidative stress caused by tobacco smoke has been reflected by lowered levels of whole blood GSH, which increases 3 weeks after smoking cessation (Lane et al. 1996). Likewise oxidative stress in chronic kidney disease is associated with reduced blood and tissue GSH levels (Vaziri 2004).
Measurement of both reduced and oxidized glutathione (GSH/GSSG ratio) by spectrophotometry, spectrofluorometry, and gas chromatography–mass spectrometry may be a useful tool to assess the oxidative stress status in human blood. However, the determination of GSSG in human blood could easily be overestimated if samples are not properly pre-treated with antioxidants during blood collection and blood sample preparation (Rossi et al. 2002).
Lipid peroxidation products
Isoprostanes
A comprehensive review on the analysis of isoprostanes by immunologic, gas chromatography/mass spectrometry (GC/MS), and high-performance liquid chromatography (HPLC)/MS assays has been provided by Lawson and FitzGerald (2002). 8-isoPGF2α is a major component of a family of prostaglandin-like compounds generated in vivo by nonenzymatic free radical-catalyzed peroxidation product of arachidonic acid (Taber et al. 1997). Most of the methods for F2-isoprostane quantification have focused on the measurement of 8-isoPGF2α in biological fluids (including human plasma) and are regarded as the gold standard for the assessment of free radical-mediated lipid peroxidation. 8-isoPGF2α determinations are carried out by ultra-sensitive analytical techniques such as GC-MS or HPLC-MS-MS (Milne et al. 2007). These methods have the advantage to accurately quantify isoprostane concentrations down to the picomolar range, but require sample preparation and expensive instrumentation. Immunoaffinity chromatography is an efficient sample preparation method linked to instrumental detection (HPLC-MS) (Sircar and Subbaiah 2007). Less expensive antibody-based assays are commercially available but lack specificity and matrix effects of plasma samples can drastically affect the results. However, as with any analysis, the results are dependent on how the samples were collected or handled, no matter how sophisticated and expensive the applied analytical instrumentation might be. In the case of isoprostane, the possibility of artefactual isoprostane generation due to arachidonic acid autooxidation requires precautionary measures to prevent ex vivo oxidation in blood samples prior to analysis.
Malondialdehyde
Malondialdehyde (MDA) can be excessively generated after ROS-mediated lipid peroxidation resulting in MDA-LDL adducts and damage to cell membranes. MDA-LDL, in addition to oxidized low density lipoprotein (oxLDL), is a key player in the pathogenesis of atherosclerosis (Berliner and Heinecke 1996). Elevated plasma MDA levels were found in patients with chronic heart failure (Diaz-Velez et al. 1996) and in patients with end-stage renal disease (Vaziri 2004). An age-dependent increase of erythrocyte MDA associated with a decline of GSH has been observed (Gil et al. 2006; Rizvi and Maurya 2007). In addition, several studies have reported an increase of peripheral blood MDA in smokers (Nielsen et al. 1997), in workers exposed to heavy metals (Jiun and Hsien 1994), in nonsmoking bus drivers (Autrup et al. 1999), and in female students after environmental exposure to air pollutants (Sørensen et al. 2003).
The most widely used method to assess lipid peroxidation is the thiobarbituric acid reactive substances (TBARS) test. TBARS reacts with MDA to generate the MDA-dithiobarbituric acid adduct. However, the TBARS assay does not specifically measure MDA or lipid peroxidation and therefore it has been criticized (Halliwell and Gutteridge 1999). Significant improvement can be made to the TBARS technique by using HPLC to separate the MDA-dithiobarbituric acid adduct (Halliwell and Whiteman 2004). Therefore, lipid peroxidation in plasma should be evaluated by HPLC with fluorescence detection, which quantifies only the actual MDA-dithiobarbituric acid adduct (Lykkesfeldt 2001; Mao et al. 2006).
Oxidized low density lipoprotein
Increased LDL oxidation is believed to play a critical role in the pathogenesis of atherosclerosis (Steinberg 1997; Heinecke 1998). LDL can be oxidized to oxLDL in vitro and in vivo by several mechanisms that affect apolipoprotein B function (Steinbrecher et al. 1987; Mertens and Holvoet 2001). The oxidative modification of LDL results in a distinctive pattern of oxidation-specific epitopes for scavenger receptors leading to macrophage foam cell formation (a key event in the arterial subendothelium leading to the development of atherosclerosis). Many studies indicate that circulating oxLDL is significantly increased in patients with coronary artery disease (Holvoet et al. 1998a, b; Toshima et al. 2000; Shimada et al. 2004; Huang et al. 2008; Kazunori and Hiroshi 2004) and those with end-stage renal disease (Vaziri et al. 2009). Oxidized LDL was a predictor of incident coronary artery disease in one cohort study (Meisinger et al. 2005); but in a large prospective cohort study, it was not an independent predictor of coronary artery disease after adjustment for LDL cholesterol, HDL cholesterol, and apolipoprotein B (Wu et al. 2006). OxLDL may be important in acute inflammatory events and as such could be a useful biomarker of acute air pollution effects. Circulating oxLDL has been suggested to be involved in the upregulation of inflammatory genes (Berliner et al. 1995) and expression of cytokines in human blood (Hulthe and Fagerberg 2002). Binding of ox-LDL to its receptor on endothelial cells and on macrophages leads to the expression of monocyte chemotactic protein-1 and adhesion molecules. Additionally, ox-LDL inhibits nitric oxide production and leads to endothelial dysfunction and apoptosis (Li and Mehta 2000a, b) resulting in oxidative stress and inflammation and thus promoting atherogenesis (Cominacini et al. 2000; Mehta et al. 2006).
Oxidized LDL in plasma is usually measured with a standardized competitive enzyme-linked immunosorbent assay (ELISA) based on monoclonal antibodies directed against a neo-epitope in the aldehyde-substituted apoB-100 moiety of LDL (mAB-4E6; oxLDL-4E6 assay, Mercodia AB, Uppsala, Sweden).
Protein oxidation products
There is no single universal marker for oxidative (and/or) nitrosative protein modification, because 20 different amino acids may be attacked by ROS/RNS and there are 35 or more ways in which proteins can be oxidized (Madian and Regnier 2010). Nevertheless, protein oxidation products are stable, have long half lives and sensitive assays are available for detection (Dalle-Donne et al. 2006). Unlike lipid peroxidation products, protein oxidation products (such as carbonylated proteins and 3-nitrotyrosine) are not subject to artificial oxidation during sample preparation and during prolonged storage in a freezer.
Carbonylated proteins
Many of the oxidative modifications of proteins involve some type of carbonlyation, which may result in the irreversible loss of protein function (Madian and Regnier 2010). Thus, irreversible ROS-mediated protein oxidation may be reflected by the accumulation of protein carbonyl levels. An increase of carbonyl proteins has been associated with aging and several human diseases such as Alzheimer’s disease, diabetes, sepsis (Dalle-Donne et al. 2003; Levine 2002), and chronic kidney disease (Vaziri 2004). Protein carbonyl levels were significantly higher in patients with chronic obstructive pulmonary disease exposed to tobacco and biomass smoke than in a control group (Ceylan et al. 2006). This is consistent with previous findings showing increased carbonyls in human plasma induced by smoking cigarettes (Reznick et al. 1992; Marangon et al. 1998, 1999). A more recent study reported significant small positive correlations (R = 0.30–0.31) of PM2.5 and PM10 exposure with plasma carbonyl protein concentrations in bus drivers, but adjusted regression models were not tested (Rossner et al. 2007).
The protein carbonyl content is considered as the most general and most commonly used index for protein oxidation (Dalle-Donne et al. 2006). Furthermore, a meta-analysis of published results revealed that in many cases levels of protein carbonyls correlate directly with concentrations of lipid peroxidation products (Dotan et al. 2004). Commercially available protein carbonyl ELISA kits can be used for the direct measurement of DNPH-derivatized carbonyl proteins. These assays use anti-DNP antibodies, are reproducible, and correlate well with typical spectrophotometric DNP assays (Rossner et al. 2007).
3-Nitrotyrosine
Redox modifications of tyrosine residues to 3-nitrotyrosine by peroxynitrite and other reactive nitrogen species, and by myeloperoxidase-mediated pathways, may lead to disruption of signal transduction pathways mediated by tyrosine phosphorylation, changes in enzyme activities in biological systems, and nitrosative stress (Keng et al. 2000; MacMillan-Crow et al. 1996). Thus, nitrotyrosination is likely to be involved in atherogenesis and thus in the pathology of cardiovascular diseases including coronary artery disease (Beckmann et al. 1994; Shishehbor et al. 2003) among others. Exposure to cigarette smoke and diesel exhaust particles induces protein nitration in vitro and in animal models (Sanbongi et al. 2003; Xiao et al. 2005; Piperi et al. 2003; Yamaguchi et al. 2000). Furthermore, higher exposure to PM2.5 and PM10 showed significant small positive correlations (R = 0.29–0.30) with increased levels of 3-nitrotyrosine in plasma samples of bus drivers, but adjusted regression models were not tested (Rossner et al. 2007).
The recommended method for plasma nitrotyrosine detection in plasma requires GC/MS or GC/MS/MS instrumentation (e.g., Tsikas and Caidahl 2005). However, less expensive ELISA methods for screening 3-nitrotyrosine plasma have been reported (Khan et al. 1998).
Plasma fluorescent oxidation products
Fluorescent oxidation products in human plasma may represent a more global marker of oxidative stress for epidemiologic studies reflecting stable protein oxidation, advanced glycation end products, and advanced lipoxidation end products (e.g., malondialdehyde; Wu et al. 2007a). They have been associated with risk of coronary artery disease and they were associated with several known promoters of oxidative stress such as smoking and hypertension (Wu et al. 2007b; Mezzetti et al. 2001). The chromophoric structures responsible for the fluorescence are conjugated Schiff bases. However, fluorescent oxidation products in plasma samples cannot be measured directly but require extraction with suitable organic solvents before spectrofluorometric analysis (Shimasaki 1994; Wu et al. 2004).
Biomarkers of antioxidant activity
Humans have evolved various enzymatic defenses against ROS generated endogenously (e.g., ROS generated by mitochondrial electron respiratory chain, by monooxygenase enzymes and by cell signaling processes) or originating from external sources (e.g., exposure to redox cycling agents present in air pollutants). More specifically, cellular SOD (MnSOD, Cu,ZnSOD) reduces O −2 to H2O2, then, in turn, catalase reduces H2O2 to water and molecular oxygen in order to minimize oxidative damage. Among other regulating functions, the selenium-dependent GPx-1 catalyzes the reduction of H2O2 and lipid peroxides to water and lipid alcohols by using the reducing power of GSH.
Commercial test kits are available for measuring the activities of SOD, GPX, and catalase in biological fluids by monitoring the generation of end-products (colorimetric end-point detection) or the decrease in the levels of substrate (colorimetric, kinetic assays).
Other antioxidant enzymes that can be measured and are potentially involved in effects of air pollutant exposures include NQ01, HO-1, and PON1 as discussed above.
Transcriptional oxidative-stress response
The peripheral blood transcriptome could serve as a useful biomarker of oxidative stress-related defense mechanisms in combination with protein expression levels. Systemic responses to air pollution can be measured in human experimental or epidemiologic studies using gene expression levels in leukocytes. In vitro data shows that inflammatory response genes and antioxidant enzyme genes (CuZn-SOD, HO-1, NQO1) are upregulated in human endothelial cells exposed to diesel exhaust particles (Gong et al. 2007). These effects were synergistically upregulated with co-exposure to an oxidized phospholipid. HO-1 was also upregulated in liver tissue of hypercholesterolemic mice exposed to urban ultrafine particles (Gong et al. 2007). Most gene expression and air pollution data has come from in vitro studies (Gong et al. 2007; Hirano et al. 2003; Li et al. 2003; Wan and Diaz-Sanchez 2006) or animal models (Gong et al. 2007; Kooter et al. 2005), but it is unclear whether these findings can be extrapolated to humans. There is little gene expression data from human subjects to support the relevance of experimental results (Wang et al. 2005; Peretz et al. 2007). An experimental study of five subjects showed upregulation of oxidative stress-related genes including HO-1 after diesel exhaust exposure (Peretz et al. 2007). An occupational study of welders linked air pollutant exposure (metal fumes) to within-subject changes in whole blood gene expression related to inflammation and oxidative stress (Wang et al. 2005). Despite a small sample size of 15 exposed welders, they found that changes in gene expression profiling could be measured in whole-blood total RNA using only one pair of pre- and post-work shift samples. Most of the observed expression variations were from only nine nonsmoking welders. They found more up- or down-regulated genes (p < 0.05) in welders (533 genes) than in seven non-exposed controls (86 genes).
An inherent limitation is that expression on a transcription level does not necessarily reflect changes on the (functional) protein level. Many transcripts are not being actively translated. Moreover, functional expression depends on protein-protein interactions and/or post-translational modifications.
Conclusions
Most epidemiologic research has focused on available exposure data including regulated PM2.5 mass and gasses such as ozone, or general markers of fossil fuel and other combustion sources such as black carbon. However, there is mounting experimental evidence that particle components that contribute little to overall particle mass have the potential for pro-oxidant effects in the body (such as PAH) leading to antioxidant responses, which when overwhelmed at higher pollutant concentrations results in oxidative stress and then inflammation. It is also possible that ultrafine particles are more likely to induce systemic oxidative stress and inflammation than larger particles and that this is attributable to greater surface area and higher concentrations of redox active components compared with the accumulation mode fraction of PM2.5. However, there is little data on this in human populations to conclude whether different particle size fractions have different magnitudes of association with oxidative stress outcomes. Similarly, it is unclear whether there are differences in oxidative stress effects for two general characteristics of organic PM and their precursor volatile and semivolatile components, namely, primary organic aerosols and associated chemicals from fossil fuel combustion such as PAH (that have oxidant potential after biotransformation) versus photochemical oxidants on secondary aerosols such as polar organics. Transition metals may have additional impacts on oxidative stress. There is also limited data on the effects of volatile and semi-volatile organic air pollutants in human populations, especially electrophiles. Finally, the overall oxidant potential of particulate and volatile pollutant mixtures may be more informative with regard to health effects that any specific set of components or source markers (although these are clearly important to identify in efforts to protect public health).
A variety of methods are available to measure oxidative stress in the circulation. The best choice of biomarkers for epidemiologic studies will depend on the research aims, but the decision may be difficult based on the current literature. In an epidemiological study where a large number of biospecimens are collected, more accurate measurement methods may be too costly. However, employing methods that are more feasible (e.g., antibody-based assays for 8-isoPGF2α) may result in semi-quantitative data and increased type II error since the measurement error is likely to be unrelated to exposure and the bias is thus towards the null hypothesis. A large sample size may or may not overcome this type of error. Furthermore, the ability to detect an exposure threshold where the balance between oxidant defense and oxidation of cell constituents is tipped toward oxidative stress may be adversely influenced by the limit of detection for the biomarker assay. Therefore, we believe there is a need for a comprehensive assessment of the reliability and validity of biomarkers of oxidative stress for large-scale field studies.
The use of a longitudinal design with repeated measures can enhance the representativeness of measurements and information content, and thus increase the utility of more feasible methods for the measurement of oxidative stress. The cohort panel study is a design that is particularly well-suited to the study of acute effects because with repeated measures, each subject can act as their own control, thus reducing variability from between-subject determinants of oxidative stress. Panel studies are particularly suited to assess the time course of exposure-response relations (from hourly to daily or weekly averages and lags) and to evaluate whether one biomarker of oxidative stress or antioxidant response is more closely associated with specific pollutant components than another is using within-subject data. Systemic inflammatory and thrombotic mediators in the blood as well as clinical outcomes should be measured simultaneously to assess the impact of air pollutant-related oxidative stress. Finally, transcriptomic and epigenetic approaches can be used to test and explore biological pathways that are relevant to these clinical and biological outcomes.
References
Adameova A, Xu YJ, Duhamel TA et al (2009) Anti-atherosclerotic molecules targeting oxidative stress and inflammation. Curr Pharm Des 15:3094–3107
Adkins S, Ganm KN, Mody M et al (1993) Molecular basis for the polymorphic forms of human serum paraoxonase/arylesterase: glutamine or arginine at position 191, for the respective A or B allozymes. Am J Hum Genet 52:598–608
Alexeeff SE, Litonjua AA, Wright RO et al (2008) Ozone exposure, antioxidant genes, and lung function in an elderly cohort: VA normative aging study. Occup Environ Med 65:736–742
Araujo JA, Nel AE (2009) Particulate matter and atherosclerosis: role of particle size, composition and oxidative stress. Part Fibre Toxicol 6:24
Araujo JA, Barajas B, Kleinman M et al (2008) Ambient particulate pollutants in the ultrafine range promote early atherosclerosis and systemic oxidative stress. Circ Res 102:589–596
Arhami M, Minguillón MC, Polidori A et al (2010) Organic compound characterization and source apportionment of indoor and outdoor quasi-ultrafine PM in retirement homes of the Los Angeles basin. Indoor Air 20:17–30
Autrup H, Daneshvar B, Dragsted LO et al (1999) Biomarkers for exposure to ambient air pollution-comparison of carcinogen-DNA adduct levels with other exposure markers and markers for oxidative stress. Environ Health Perspect 107:233–238
Ayres JG, Borm P, Cassee FR et al (2008) Evaluating the toxicity of airborne particulate matter and nanoparticles by measuring oxidative stress potential—a workshop report and consensus statement. Inhal Toxicol 20:75–99
Barnes PJ (2009) Targeting the epigenome in the treatment of asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc 6:693–696
Beckmann JS, Ye YZ, Anderson PG et al (1994) Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol Chem 375:81–88
Behndig AF, Mudway IS, Brown JL et al (2006) Airway antioxidant and inflammatory responses to diesel exhaust exposure in healthy humans. Eur Respir J 27:359–365
Berliner J, Heinecke JW (1996) The role of oxidized lipoproteins in atherogenesis. Free Radic Biol Med 20:707–727
Berliner JA, Navab M, Fogelman AM et al (1995) Atherosclerosis: basic mechanisms. Oxidation, inflammation, and genetics. Circulation 91:2488–2496
Bhaskaran K, Hajat S, Haines A et al (2009) Effects of air pollution on the incidence of myocardial infarction. Heart 95:1746–1759
Bogaards JJ, Venekamp JC, van Bladeren PJ (1997) Stereoselective conjugation of prostaglandin A2 and prostaglandin J2 with glutathione, catalysed by the human glutathione S-transferases A1-1, A2-2, M1a-1a, and P1-1. Chem Res Toxicol 10:310–317
Bonvallot V, Baeza-Squiban A, Baulig A et al (2001) Organic compounds from diesel exhaust particles elicit a proinflammatory response in human airway epithelial cells and induce cytochrome p450 1A1 expression. Am J Respir Cell Mol Biol 25:515–521
Brook RD, Rajagopalan S, Pope CA 3rd et al (2010) Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation 121:2331–2378
Campen MJ, Lund AK, Knuckles TL et al (2010) Inhaled diesel emissions alter atherosclerotic plaque composition in ApoE(−/−) mice. Toxicol Appl Pharmacol 242:310–317
Ceylan E, Kocyigit A, Gencer M et al (2006) Increased DNA damage in patients with chronic obstructive pulmonary disease who had once smoked or been exposed to biomass. Respir Med 100:1270–1276
Chahine T, Baccerelli A, Litonjua A et al (2007) Particulate air pollution, oxidative stress genes, and heart rate variability in an elderly cohort. Environ Health Perspect 115:1617–1622
Chen LC, Nadziejko C (2005) Effects of subchronic exposures to concentrated ambient particles (CAPs) in mice. V. CAPs exacerbate aortic plaque development in hyperlipidemic mice. Inhal Toxicol 17:217–224
Cho HY, Kleeberger SR (2010) Nrf2 protects against airway disorders. Toxicol Appl Pharmacol 244:43–56
Cho AK, Sioutas C, Miguel AH et al (2005) Redox activity of airborne particulate matter at different sites in the Los Angeles Basin. Environ Res 99:40–47
Chuang KJ, Chan CC, Su TC et al (2007) The effect of urban air pollution on inflammation, oxidative stress, coagulation, and autonomic dysfunction in young adults. Am J Respir Crit Care Med 176:370–376
Cimen MY (2008) Free radical metabolism in human erythrocytes. Clin Chim Acta 390:1–11
Cominacini L, Pasini AF, Garbin U et al (2000) Oxidized low density lipoprotein (ox-LDL) binding to ox-LDL receptor-1 in endothelial cells induces the activation of NF-kappaB through an increased production of intracellular reactive oxygen species. J Biol Chem 275:12633–12638
Cooke MS, Lunec J, Evans MD (2002) Progress in the analysis of urinary oxidative stress damage. Free Radic Biol Med 33:1601–1614
Dalle-Donne I, Giustarini D, Colombo R et al (2003) Protein carbonylation in human diseases. Trends Mol Med 9:169–176
Dalle-Donne I, Rossi R, Ceciliani F et al (2006) Proteins as sensitive biomarkers of human conditions associated with oxidative stress. In: Dalle-Donne I, Scaloni A, Butterfiled DA (eds) Redox Proteomics: from protein modifications to cellular dysfunction and diseases. Wiley, Hoboken
Decker M, Arand M, Cronin A (2009) Mammalian epoxide hydrolases in xenobiotic metabolism and signalling. Arch Toxicol 83:297–318
Delfino RJ, Sioutas C, Malik S (2005) Potential role of ultrafine particles in associations between airborne particle mass and cardiovascular health. Environ Health Perspect 113:934–946
Delfino RJ, Staimer N, Tjoa T et al (2008) Circulating biomarkers of inflammation, antioxidant activity, and platelet activation are associated with urban air pollution in elderly subjects with a history of coronary artery disease. Env Health Perspect 116:898–906
Delfino RJ, Staimer N, Tjoa T et al (2009) Air pollution exposures and circulating biomarkers of effect in a susceptible population: clues to potential causal component mixtures and mechanisms. Environ Health Perspect 117:1232–1238
Delfino RJ, Staimer N, Tjoa T et al (2010a) Association of biomarkers of systemic effects with organic components and source tracers in quasi-ultrafine particles. Environ Health Perspect 118:756–762
Delfino RJ, Tjoa T, Gillen D et al (2010b) Traffic-related air pollution and blood pressure in elderly subjects with coronary artery disease. Epidemiology 21:396–404
Delfino RJ, Gillen DL, Tjoa T et al (2010c) Electrocardiographic ST segment depression and exposure to traffic-related aerosols in elderly subjects with coronary artery disease. Environ Health Perspect (in press)
Delfino RJ, Staimer N, Tjoa T et al (2010d) Airway and systemic inflammation are differently associated with primary and secondary organic aerosols in an elderly panel cohort. Epidemiol (in press)
Dhalla NS, Temsah RM, Netticadan T (2000) Role of oxidative stress in cardiovascular diseases. J Hypertens 18:655–673
Diaz-Velez CR, Garcia-Castineiras S, Mendoza-Ramos E et al (1996) Increased malondialdehyde in peripheral blood of patients with congestive heart failure. Am Heart J 131:146–152
Dotan Y, Lichtenberg D, Pinchuk I (2004) Lipid peroxidation cannot be used as a universal criterion of oxidative stress. Prog Lipid Res 43(3):200–227
Dubowsky SD, Suh H, Schwartz J et al (2006) Diabetes, obesity, and hypertension may enhance associations between air pollution and markers of systemic inflammation. Environ Health Perspect 114:992–998
Eiguren-Fernandez A, Shinyashiki M, Schmitz DA et al (2010) Redox and electrophilic properties of vapor- and particle-phase components of ambient aerosols. Environ Res 110:207–212
Espinola-Klein C, Rupprecht HJ, Bickel C et al (2007) Glutathione peroxidase-1 activity, atherosclerotic burden, and cardiovascular prognosis. Am J Cardiol 99:808–812
Fattman CL, Schaefer LM, Oury TD (2003) Extracellular superoxide dismutase in biology and medicine. Free Radic Biol Med 35:236–256
Flores-Mateo G, Carrillo-Santisteve P, Elosua R et al (2009) Antioxidant enzyme activity and coronary heart disease: meta-analyses of observational studies. Am J Epidemiol 170:135–147
Förstermann U (2008) Oxidative stress in vascular disease: causes, defense mechanisms and potential therapies. Nat Clin Pract Cardiovasc Med 5:338–349
Frampton MW (2006) Inflammation and airborne particles. Clin Occup Environ Med 5:797–815
Fryer AA, Hume R, Strange RC (1986) The development of glutathione S-transferase and glutathione peroxidase activities in human lung. Biochim Biophys Acta 883:448–453
Fukai T, Folz RJ, Landmesser U et al (2002) Extracellular superoxide dismutase and cardiovascular disease. Cardiovasc Res 55:239–249
Geiser M, Kreyling WG (2010) Deposition and biokinetics of inhaled nanoparticles. Part Fibre Toxicol 20;7:2
Gertler AW (2005) Diesel vs. gasoline emissions: does PM from diesel or gasoline vehicles dominate in the US? Atmos Environ 39:2349–2355
Gianazza E, Crawford J, Miller I (2007) Detecting oxidative post-translational modifications in proteins. Amino Acids 33:51–56
Gil L, Siems W, Mazurek B et al (2006) Age-associated analysis of oxidative stress parameters in human plasma and erythrocytes. Free Radic Res 40:495–495
Gilliland FD, McConnell AR, Peters J et al (1999) A theoretical basis for investigating ambient air pollution and children’s respiratory health. Environ Health Perspect 107(suppl 3):403–407
Gong KW, Zhao W, Li N et al (2007) Air-pollutant chemicals and oxidized lipids exhibit genome-wide synergistic effects on endothelial cells. Genome Biol 8:R149
Grahame TJ, Schlesinger RB (2010) Cardiovascular health and particulate vehicular emissions: a critical evaluation of the evidence. Air Qual Atmos Health 3(1):3–27
Gurgueira SA, Lawrence J, Coull B et al (2002) Rapid increases in the steady-state concentration of reactive oxygen species in the lungs and heart after particulate air pollution inhalation. Environ Health Perspect 110:749–755
Halliwell B, Gutteridge JMC (1999) Free radicals in biology and medicine, 3rd edn. Oxford University Press, New York
Halliwell B, Whiteman M (2004) Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? Br J Pharmacol 142:231–255
Hatzis C, Godleski JJ, Gonzalez-Flecha B et al (2006) Ambient particulate matter exhibits direct inhibitory effects on oxidative stress enzymes. Environ Sci Technol 15:2805–2811
Hayes JD, McLellan LI (1999) Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic Res 31:273–300
Hayes JD, Flanagan JU, Jowsey IR (2005) Glutathione transferases. Annu Rev Pharmacol Toxicol 45:51–88
Heinecke JW (1998) Oxidants and antioxidants in the pathogenesis of atherosclerosis: implications for the oxidized low density lipoprotein hypothesis. Atherosclerosis 141:1–15
Hirano S, Furuyama A, Koike E et al (2003) Oxidative stress potency of organic extracts of diesel exhaust and urban fine particles in rat heart microvessel endothelial cells. Toxicology 187:161–170
Hoffmann B, Moebus S, Möhlenkamp S et al (2007) Residential exposure to traffic is associated with coronary atherosclerosis. Circulation 116:489–496
Holvoet P, Stassen JM, Van Cleemput J et al (1998a) Oxidized low density lipoproteins in patients with transplant-associated coronary artery disease. Arterioscler Thromb Vasc Biol 18:100–107
Holvoet P, Vanhaecke J, Janssens S et al (1998b) Oxidized LDL and malondialdehyde-modified LDL in patients with acute coronary syndromes and stable coronary artery disease. Circulation 98:1487–1494
Hu S, Polidori A, Arhami M et al (2008) Redox activity and chemical speciation of size fractioned PM in the communities of the Los Angeles–Long Beach harbor. Atmos Chem Physics 8:6439–6451
Huang H, Mai W, Liu D et al (2008) The oxidation ratio of LDL: a predictor for coronary artery disease. Dis Markers 24(6):341–349
Hulthe J, Fagerberg B (2002) Circulating oxidized LDL is associated with subclinical atherosclerosis development and inflammatory cytokines (AIR study). Arterioscler Thromb Vasc Biol 22:1162–1167
Islam T, McConnell R, Gauderman WJ et al (2008) Ozone, oxidant defense genes, and risk of asthma during adolescence. Am J Respir Crit Care Med 177:388–395
Islam T, Berhane K, McConnell R et al (2009) Glutathione-S-transferase (GST) P1, GSTM1, exercise, ozone and asthma incidence in school children. Thorax 64:197–202
Iwamoto N, Nishiyama A, Eiguren-Fernandez A et al (2010) Biochemical and cellular effects of electrophiles present in ambient air samples. Atmos Environ 44:1483–1489
Jiun YS, Hsien LT (1994) Lipid peroxidation in workers exposed to lead. Arch Environ Health 49:256–259
Kazunori S, Hiroshi M (2004) Circulating oxidized low-density lipoprotein is an independent predictor for cardiac event in patients with coronary artery disease. Atherosclerosis 174:343–347
Keen JH, Jakoby WB (1978) Glutathione transferases. Catalysis of nucleophilic reactions of glutathione. J Biol Chem 253:5654–57
Keng T, Privalle CT, Gilkeson GS et al (2000) Peroxynitrite formation and decreased catalase activity in autoimmune MRLlpr/lpr mice. Mol Med 6:779–792
Kensler TW, Wakabayashi N, Biswal S (2007) Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol 47:89–116
Khan J, Brennan DM, Bradley N et al (1998) 3-Nitrotyrosine in the proteins of human plasma determined by an ELISA method. Biochem J 330:795–801
Kharb S (2003) Low blood glutathione levels in acute myocardial infarction. Indian J Med Sci 57:335–337
Kim MK, Oh S, Lee JH et al (2004) Evaluation of biological monitoring markers using genomic and proteomic analysis for automobile emission inspectors and waste incinerating workers exposed to polycyclic aromatic hydrocarbons or 2, 3, 7, 8,-tetracholrodedibenzo-p-dioxins. Exp Mol Med 36:396–410
Kooter I, Pennings J, Opperhuizen A et al (2005) Gene expression pattern in spontaneously hypertensive rats exposed to urban particulate matter (EHC-93). Inhal Toxicol 17:53–65
Kumagai Y, Shimojo N (2002) Possible mechanisms for induction of oxidative stress and suppression of systemic nitric oxide production caused by exposure to environmental chemicals. Environ Health Prev Med 7:141–150
Künzli N, Jerrett M, Mack WJ et al (2005) Ambient air pollution and atherosclerosis in Los Angeles. Environ Health Perspect 113:201–206
Künzli N, Jerrett M, Garcia-Esteban R et al (2010) Ambient air pollution and the progression of atherosclerosis in adults. PLoS ONE 5(2):e9096. doi:10.1371/journal.pone.0009096
Lai C-H, Liou S-H, Lin H-C et al (2005) Exposure to traffic exhausts and oxidative DNA damage. Occup Environ Med 62:216–222
Laing S, Wang G, Briazova T et al (2010) Airborne particulate matter selectively activates endoplasmic reticulum stress response in the lung and liver tissues. Am J Physiol Cell Physiol (in press)
Landerman AP, Shafer MM, Hemming JC et al (2008) A macrophage-based method for the assessment of the oxidative stress activity of atmospheric particulate matter (PM) and application to routine (daily 24-hour) aerosol monitoring studies. Aerosol Sci Technol 42:946–957
Landmesser U, Merten R, Spiekerman S et al (2000) Vascular extracellular superoxide dismutase activity in patients with coronary artery disease: relation to endothelium-dependent vasodilation. Circulation 101:2264–2270
Lane JD, Opara EC, Rose JE et al (1996) Quitting smoking raises whole blood glutathione. Physiol Behavior 60:1379–1381
Lawson JA, FitzGerald GA (2002) Analysis of isoprostanes. Eur J Lipid Sci Technol 104:429–435
Levine RL (2002) Carbonyl modified proteins in cellular regulation, aging, and disease. Free Radical Biol Med 32:790–796
Li D, Mehta JL (2000a) Antisense to LOX-1 inhibits oxidized LDL-mediated upregulation of monocyte chemoattractant protein-1 and monocyte adhesion to human coronary artery endothelial cells. Circulation 101:2889–2895
Li D, Mehta JL (2000b) Upregulation of endothelial receptor for oxidized LDL (LOX-1) by oxidized LDL and implications in apoptosis of human coronary artery endothelial cells: evidence from use of antisense LOX-1 mRNA and chemical inhibitors. Arterioscler Thromb Vasc Biol 20:1116–1122
Li N, Kim S, Wang M et al (2002) Use of a stratified oxidative stress model to study the biological effects of ambient concentrated and diesel exhaust particulate matter. Inhal Toxicol 14:459–486
Li N, Sioutas C, Cho A et al (2003) Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ Health Perspect 111:455–460
Li N, Wang M, Bramble LA et al (2009) The adjuvant effect of ambient particulate matter is closely reflected by the particulate oxidant potential. Environ Health Perspect 117:1116–1123
Limón-Pacheco J, Gonsebatt ME (2009) The role of antioxidants and antioxidant-related enzymes in protective responses to environmentally induced oxidative stress. Mutat Res 674:137–147
Liu L, Ruddy TD, Dalipaj M et al (2007) Influence of personal exposure to particulate air pollution on cardiovascular physiology and biomarkers of inflammation and oxidative stress in subjects with diabetes. J Occup Environ Med 49:258–265
Lund AK, Knuckles TL, Obot Akata C et al (2007) Gasoline exhaust emissions induce vascular remodeling pathways involved in atherosclerosis. Toxicol Sci 95:485–94
Lykkesfeldt J (2001) Determination of malondialdehyde as dithiobarbituric acid adduct in biological samples by HPLC with fluorescence detection: comparison with ultraviolet-visible spectrophotometry. Clin Chem 47:1725–1727
Mackness B, Mackness MI, Arrol S et al (1998) Effect of the human serum paraoxonase 55 and 192 genetic poIymorphisms on the protection by high density lipoprotein against low density lipoprotein oxidative modification. FEBS Lett 423:57–60
MacMillan-Crow LA, Crow JP, Kerby JD et al (1996) Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc Natl Acad Sci 93:11853–11858
Madian AG, Regnier FE (2010) Proteomic identification of carbonylated proteins and their oxidation sites. J Proteome Res 9:3766–3780.
Madrigano J, Baccarelli A, Wright R et al (2009) Air pollution, obesity, genes, and cellular adhesion molecules. Occup Environ Med. doi:10.1136/oem.2009.046193
Mann GE, Niehueser-Saran J, Watson A et al (2007) Nrf2/ARE regulated antioxidant gene expression in endothelial and smooth muscle cells in oxidative stress: implications for atherosclerosis and preeclampsia. Acta Physiol Sin 59:117–127
Mao J, Zhang H, Luo J et al (2006) New method for HPLC separation and fluorescence detection of malonaldehyde in normal human plasma. J Chromatogr B 832:103–108
Marangon K, Herbeth B, Lecomte E et al (1998) Diet, antioxidant status, and smoking habits in French men. Am J Clin Nutr 67:231–239
Marangon K, Devaraj S, Jialal I (1999) Measurement of protein carbonyls in plasma of smokers and in oxidized LDL by an ELISA. Clin Chem 45:577–578
Mehta JL, Chen J, Hermonat PL et al (2006) Lectin-like, oxidized low-density lipoprotein receptor-1 (LOX-1): a critical player in the development of atherosclerosis and related disorders. Cardiovasc Res 69:36–45
Meisinger C, Baumert J, Khuseyinova N et al (2005) Plasma oxidized low-density lipoprotein, a strong predictor for acute coronary heart disease events in apparently healthy, middle-aged men from the general population. Circulation 112:651–657
Mertens A, Holvoet P (2001) Oxidized LDL and HDL: antagonists in atherothrombosis. FASEB J 15(12):2073–2084
Mezzetti A, Zuliani G, Romano F et al (2001) Vitamin E and lipid peroxide plasma levels predict the risk of cardiovascular events in a group of healthy very old people. J Am Geriatrics Soc 49:533–537
Mieyal JJ, Gallogly MM, Qanungo S et al (2008) Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid Redox Signal 10:1941–1988
Mills NL, Donaldson K, Hadoke PW et al (2009) Adverse cardiovascular effects of air pollution. Nat Clin Pract Cardiovasc Med 6:36–44
Milne GL, Sanchez SC, Musiek ES et al (2007) Quantification of F2-isoprostanes as a biomarker of oxidative stress. Nat Protoc 2:221–226
Minetti M, Agati L, Malorni W (2007) The microenvironment can shift erythrocytes from a friendly to a harmful behavior: pathogenetic implications for vascular diseases. Cardiovasc Res 75:21–28
Möller W, Felten K, Sommerer K et al (2008) Deposition, retention, and translocation of ultrafine particles from the central airways and lung periphery. Am J Respir Crit Care Med 177:426–432
Monks TJ, Hanzlik RP, Cohen GM et al (1992) Quinone chemistry and toxicity. Toxicol Appl Pharmacol 112:2–16
Muda P, Kampus P, Zilmer M et al (2003) Homocysteine and red blood cell glutathione as indices for middle-aged untreated essential hypertension patients. J Hypertension 21:2329–2333
Nielsen F, Mikkelsen BB, Nielsen JB et al (1997) Plasma-malondialdehyde as biomarker for oxidative stress: reference interval and effects of life-style factors. Clin Chem 43:1209–1214
Ntziachristos L, Froines JR, Cho AK et al (2007) Relationship between redox activity and chemical speciation of size-fractionated particulate matter. Part Fibre Toxicol 4:5
Oberdörster G, Oberdörster E, Oberdörster J (2005) Nanotoxicology: an emerging discipline evolving from studies of ultrafine particles. Environ Health Perspect 113:823–839
O’Donnell VB, Freeman BA (2001) Interactions between nitric oxide and lipid oxidation pathways: implications for vascular disease. Circ Res 88:12–21
O’Neill MS, Veves A, Sarnat JA et al (2007) Air pollution and inflammation in type 2 diabetes: a mechanism for susceptibility. Occup Environ Med 64:373–379
Pai JK, Pischon T, Ma J et al (2004) Inflammatory markers and the risk of coronary heart disease in men and women. N Engl J Med 351:2599–2610
Palmen NGM, Evelo CTA (1996) Glutathione depletion in human erythrocytes and rat liver: a study on the interplay between bioactivation and inactivation functions of liver and blood. Toxicol In Vitro 10:273–282
Panagiotakos DB, Pitsavos C, Chrysohoou C et al (2004) Effect of exposure to secondhand smoke on markers of inflammation: the ATTICA study. Am J Med 116:145–150
Park SK, O’Neill MS, Wright RO et al (2006) HFE genotype, particulate air pollution, and heart rate variability: a gene–environment interaction. Circulation 114:2798–2805
Park SK, O’Neill MS, Vokonas PS et al (2008) Traffic-related particles are associated with elevated homocysteine: the VA normative aging study. Am J Respir Crit Care Med 178:283–289
Pastore A, Federici G, Bertini E et al (2003) Analysis of glutathione: implication in redox and detoxification. Clin Chim Acta 333:19–39
Peretz A, Peck EC, Bammler TK et al (2007) Diesel exhaust inhalation and assessment of peripheral blood mononuclear cell gene transcription effects: an exploratory study of healthy human volunteers. Inhal Toxicol 19:1107–1119
Phuleria HC, Sheesley RJ, Schauer JJ et al (2007) Roadside measurements of size-segregated particulate organic compounds near gasoline and diesel-dominated freeways in Los Angeles, CA. Atmos Environ 41:4653–4671
Pigeolet E, Corbisier P, Houbion A et al (1990) Glutathione peroxidase, superoxide dismutase, and catalase inactivation by peroxides and oxygen derived free radicals. Mech Ageing Dev 51:283–297
Piperi C, Pouli AE, Katerelos NA et al (2003) Study of the mechanisms of cigarette smoke gas phase cytotoxicity. Anticancer Res 23:2185–2190
Pope CA 3rd, Dockery DW (2006) Health effects of fine particulate air pollution: lines that connect. J Air Waste Manag Assoc 56:709–742
Rahman I, Biswas SK, Kode A (2006) Oxidant and antioxidant balance in the airways and airway diseases. Eur J Pharmacol 533:222–239
Ren C, Park SK, Vokonas PS et al (2010) Air Pollution and homocysteine: more evidence that oxidative stress-related genes modify effects of particulate air pollution. Epidemiology. doi:10.1097/EDE.0b013e3181cc8bfc
Repine JE, Bast A, Lankhorst I (1997) Oxidative stress in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 156:341–357
Reznick AZ, Cross CE, Hu ML et al (1992) Modification of plasma proteins by cigarette smoke as measured by protein carbonyl formation. Biochem J 286(2):607–611
Riedl M, Diaz-Sanchez D (2005) Biology of diesel exhaust effects on respiratory function. J Allergy Clin Immunol 115:221–228
Rizvi SI, Maurya PK (2007) Markers of oxidative stress in erythrocytes during aging in humans. Ann NY Acad Sci 1100:373–382
Robinson AL, Donahue NM, Shrivastava MK et al (2007) Rethinking organic aerosols: semivolatile emissions and photochemical aging. Science 315:1259–1262
Romieu I, Castro-Giner F, Kunzli N et al (2008) Air pollution, oxidative stress and dietary supplementation: a review. Eur Resp J 31:179–196
Ross D, Kepa JK, Winski SL et al (2000) NAD(P)H:quinone oxidoreductase 1 (NQO1): chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem Biol Interact 129:77–97
Rossi R, Aldo M, Dalle-Donne I et al (2002) Blood glutathione disulfide: in vivo factor or in vitro artifact? Clin Chem 48:742–753
Rossner P, Svecova V, Milcova A et al (2007) Oxidative and nitrosative stress markers in bus drivers. Mutat Res 617:23–32
Rossner P Jr, Svecova V, Milcova A et al (2008a) Seasonal variability of oxidative stress markers in city bus drivers. Part I. Oxidative damage to DNA. Mutat Res 642:14–20
Rossner P Jr, Svecova V, Milcova A et al (2008b) Seasonal variability of oxidative stress markers in city bus drivers. Part II. Oxidative damage to lipids and proteins. Mutat Res 642:21–27
Ruckerl R, Ibald-Mulli A, Koenig W et al (2006) Air pollution and markers of inflammation and coagulation in patients with coronary heart disease. Am J Respir Crit Care Med 173:432–441
Ryter SW, Alam J, Choi AMK (2006) Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 86:583–650
Sanbongi C, Takano H, Osakabe N et al (2003) Rosmarinic acid inhibits lung injury induced by diesel exhaust particles. Free Radical Biol Med 34:1060–1069
Schindhelm RK, van der Zwan LP, Teerlink T et al (2009) Myeloperoxidase: a useful biomarker for cardiovascular disease risk stratification? Clin Chem 55:1462–1470
Schmid O, Möller W, Semmler-Behnke M et al (2009) Dosimetry and toxicology of inhaled ultrafine particles. Biomarkers 14(Suppl 1):67–73
Schwartz J, Park SK, O’Neill MS et al (2005) Glutathione-S-transferase M1, obesity, statins, and autonomic effects of particles: gene-by-drug-by-environment interaction. Am J Respir Crit Care Med 172:1529–1533
Shafer MM, Perkins DA, Antkeweitz DS et al (2010) Reactive oxygen species activity and chemical speciation of size-fractionated atmospheric particulate matter from Lahore Pakistan: an important role for transition metals. J Environ Monit 12:704–715
Sharman JE, Coombes JS, Geraghty DP et al (2002) Exposure to automotive pollution increases plasma susceptibility to oxidation. Arch Environ Health 57:536–540
Shimada K, Mokuno H, Matsunaga E et al (2004) Predictive value of circulating oxidized LDL for cardiac events in type 2 diabetic patients with coronary artery disease. Diab Care 27:843–844
Shimasaki H (1994) Assay of fluorescent lipid peroxidation products. Methods Enzymol 233:338–346
Shinyashiki M, Rodriguez CR, Di Stefano EM et al (2008) On the interaction between glyceraldehyde-3-phosphate dehydrogenase and airborne particles: Evidence for electrophilic species. Atmos Environ 42:517–529
Shinyashiki M, Eiguren-Fernandez A, Schmitz DA et al (2009) Electrophilic and redox properties of diesel exhaust particles. Environ Res 109:239–244
Shishehbor MH, Aviles RJ, Brennan M-L et al (2003) Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy. JAMA 289:1675–1680
Silkoff PE, Erzurum SC, Lundberg JO et al (2006) ATS workshop proceedings: exhaled nitric oxide and nitric oxide oxidative metabolism in exhaled breath condensate. Proc Am Thorac Soc 3:131–145
Singh R, Sram RJ, Binkova B et al (2007) The relationship between biomarkers of oxidative DNA damage, polycyclic aromatic hydrocarbon DNA adducts, antioxidant status and genetic susceptibility following exposure to environmental air pollution in humans. Mutat Res 620:83–92
Sioutas C, Delfino RJ, Singh M (2005) Exposure assessment for atmospheric ultrafine particles (UFP) and implications in epidemiological research. Environ Health Perspect 113:947–955
Sircar D, Subbaiah PV (2007) Isoprostane measurement in plasma and urine by liquid chromatography–mass spectrometry with one-step sample preparation. Clin Chem 53:251–258
Soares SR, Carvalho-Oliveira R, Ramos-Sanchez E et al (2009) Air pollution and antibodies against modified lipoproteins are associated with atherosclerosis and vascular remodeling in hyperlipemic mice. Atherosclerosis 207:368–373
Sørensen M, Daneshvar B, Hansen M et al (2003) Personal PM2.5 exposure and markers of oxidative stress in blood. Environ Health Perspect 111:161–166
Sørensen M, Schins RP, Hertel O et al (2005) Transition metals in personal samples of PM2.5 and oxidative stress in human volunteers. Cancer Epidemiol Biomark Prev 14:1340–1343
Staimer N, Cho A, Delfino RJ (2010) A new assay to screen for the inhibitory capacity of air pollutant components on antioxidant enzyme activities. Paper accepted for presentation at the American thoracic Society International Conference, New Orleans, LO
Steinberg D (1997) Oxidative modification of LDL and atherogenesis. Circulation 95:1062–1071
Steinbrecher UP, Witztum JL, Parthasarathy S et al (1987) Decrease in reactive amino groups during oxidation or endothelial cell modification of LDL. Correlation with changes in receptor-mediated catabolism. Arteriosclerosis 7:135–143
Sun Q, Wang A, Jin X et al (2005) Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. JAMA 294:3003–3010
Suwa T, Hogg JC, Quinlan KB et al (2002) Particulate air pollution induces progression of atherosclerosis. J Am Coll Cardiol 39:935–942
Taber DF, Morrow JD, Roberts LJ 2nd (1997) A nomenclature system for the isoprostanes. Prostaglandins 53:63–67
Toshima S, Hasegawa A, Kurabayashi M et al (2000) Circulating oxidized low density lipoprotein levels. A biochemical risk marker for coronary heart disease. Arterioscler Thromb Vasc Biol 20:2243–2247
Townsend DM (2007) S-glutathionylation: indicator of cell stress and regulator of the unfolded protein response. Mol Interv 7:313–324
Tsantes AE, Bonovas S, Travlou A et al (2006) Redox imbalance, macrocytosis, and RBC homeostasis. Antioxid Redox Signal 8:1205–1216
Tsikas D, Caidahl K (2005) Recent methodological advances in the mass spectrometric analysis of free and protein-associated 3-nitrotyrosine in human plasma. J Chromatogr B 814:1–9
Utell MJ, Frampton MW, Zareba W et al (2002) Cardiovascular effects associated with air pollution: potential mechanisms and methods of testing. Inhal Toxicol 14:1231–1247
Vaziri ND (2004) Oxidative stress in uremia. Nature, mechanisms and potential consequences. Semin Nephrol 24:469–473
Vaziri ND, Rodríguez-Iturbe B (2006) Mechanisms of disease: oxidative stress and inflammation in the pathogenesis of hypertension. Nat Clin Pract Nephrol 2:582–593
Vaziri ND, Moradi H, Pahl MV, Fogelman AM, Navab M (2009) In vitro stimulation of HDL anti-inflammatory activity and inhibition of LDL pro-inflammatory activity in the plasma of patients with end-stage renal disease by an apoA-1 mimetic peptide. Kidney Int 76:437–444
Verma V, Ning Z, Schauer JJ et al (2009a) Redox activity of urban quasi-ultrafine particles from primary and secondary sources. Atmos Environ 43:6360–6368
Verma V, Polidori A, Schauer JJ et al (2009b) Physicochemical and toxicological profiles of particulate matter in Los Angeles during the October 2007 southern California wildfires. Environ Sci Technol 43:954–960
Viña J, Gomez-Cabrera MC, Borras C (2007) Fostering antioxidant defences: up-regulation of antioxidant genes or antioxidant supplementation? Br J Nutr 98(Suppl 1):S36–S40
Vinzents PS, Møller P, Sørensen M et al (2005) Personal exposure to ultrafine particles and oxidative DNA damage. Environ Health Perspect 113:1485–1490
Wan J, Diaz-Sanchez D (2006) Phase II enzymes induction blocks the enhanced IgE production in B cells by diesel exhaust particles. J Immunol 177:3477–3483
Wang L, Groves MJ, Hepburn MD et al (2000) Glutathione S-transferase enzyme expression in hematopoietic cell lines implies a differential protective role for T1 and A1 isoenzymes in erythroid and for M1 in lymphoid lineages. Haematologica 85:573–579
Wang Z, Neuburg D, Li C et al (2005) Global gene expression profiling in whole-blood samples from individuals exposed to metal fumes. Environ Health Perspect 113:233–241
Watson AD, Berliner JA, Hama SY et al (1995) Protective effect of high density lipoprotein associated paraoxonase. Inhibition of the biological activity of minimally oxidized low density lipoprotein. J Clin Invest 96:2882–2891
Watterson TL, Hamilton B, Martin R et al (2009) Urban particulate matter causes ER stress and the unfolded protein response in human lung cells. Toxicol Sci 112:111–122
Wenger NK (2007) Do diet, folic acid, and vitamins matter? What did we learn from the Women’s Health Initiative, the Women’s Health Study, the Women’s Antioxidant and Folic Acid Cardiovascular Study, and other clinical trials? Cardiol Rev 15:288–290
Wu T, Rifai N, Roberts LJ II et al (2004) Stability of measurements of biomarkers of oxidative stress in blood over 36 hours. Cancer Epidemiol Biomark Prev 13:1399–1402
Wu T, Willett WC, Rifai N et al (2006) Is plasma oxidized low-density lipoprotein, measured with the widely used antibody 4E6, an independent predictor of coronary heart disease among US men and women? J Am Coll Cardiol 48:973–979
Wu T, Willett WC, Rifai N et al (2007a) Plasma fluorescent oxidation products as potential markers of oxidative stress for epidemiologic studies. Am J Epidemiol 166:552–560
Wu T, Rifai N, Willett WC et al (2007b) Plasma fluorescent oxidation products: independent predictors of coronary heart disease in men. Am J Epidemiol 166:544–551
Xia T, Kovochich M, Nel A (2006) The role of reactive oxygen species and oxidative stress in mediating particulate matter injury. Clin Occup Environ Med 5:817–836
Xiao GG, Nel AE, Loo JA (2005) Nitrotyrosine-modified proteins and oxidative stress induced by diesel exhaust particles. Electrophoresis 26:280–292
Yamaguchi KS, Haginaka J et al (2000) Peroxynitrite-generating species: good candidate oxidants in aqueous extracts of cigarette smoke. Jpn J Pharmacol 82:78–81
Yue W, Schneider A, Stolzel M et al (2007) Ambient source-specific particles are associated with prolonged repolarization and increased levels of inflammation in male coronary artery disease patients. Mutat Res 621:50–60
Zhang Y, Schauer JJ, Shafer MM et al (2008) Source apportionment of in vitro reactive oxygen species bioassay activity from atmospheric particulate matter. Environ Sci Technol 42:7502–7509
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Financial Support
Supported by grant number ES012243 from the National Institute of Environmental Health Sciences (RJD and NS), and grant number RD83241301 from the US Environmental Protection Agency (RJD, NS, and NV).
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Delfino, R.J., Staimer, N. & Vaziri, N.D. Air pollution and circulating biomarkers of oxidative stress. Air Qual Atmos Health 4, 37–52 (2011). https://doi.org/10.1007/s11869-010-0095-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11869-010-0095-2