
Abstract
Air Pollution has been associated with significant adverse health effects leading to increased morbidity and mortality. Cumulative epidemiological and experimental data have shown that exposure to air pollutants lead to increased cardiovascular ischemic events and enhanced atherosclerosis. It appears that these associations are much stronger with the air particulate matter (PM) component and that in urban areas, the smaller particles could be more pathogenic, as a result of their greater propensity to induce systemic prooxidant and proinflammatory effects. Much is still unknown about the toxicology of ambient particulates as well as the pathogenic mechanisms responsible for the induction of adverse cardiovascular health effects. It is expected that better understanding of these effects will have large implications and may lead to the formulation and implementation of new regulatory policies. Indeed, we have found that ultrafine particles (<0.18 μm) enhance early atherosclerosis, partly due to their high content in redox cycling chemicals and their ability to synergize with known proatherogenic mediators in the promotion of tissue oxidative stress. These changes take place in parallel with increased evidence of phase 2 enzymes expression, via the electrophile-sensitive transcription factor, p45-NFE2 related transcription factor 2 (Nrf2). Exposure to ultrafine particles also results in alterations of the plasma HDL anti-inflammatory function that could be indicative of systemic proatherogenic effects. This article reviews the epidemiological, clinical and experimental animal evidence that support the association of particulate matter with atherogenesis. It also discusses the possible pathogenic mechanisms involved, the physicochemical variables that may be of importance in the greater toxicity exhibited by a small particle size, interaction with genes and other proatherogenic factors as well as important elements to consider in the design of future mechanistic studies.
Extensive epidemiological evidence supports the association of air pollution with adverse health effects [1–3]. It is increasingly being recognized that such effects lead to enhanced morbidity and mortality, mostly due to exacerbation of cardiovascular diseases and predominantly those of ischemic character [4]. Indeed, in addition to the classical risk factors such as serum lipids, smoking, hypertension, aging, gender, family history, physical inactivity and diet, recent data have implicated air pollution as an important additional risk factor for atherosclerosis. This has been the subject of extensive reviews [5, 6] and a consensus statement from the American Heart Association [7]. This article reviews the supporting epidemiological and animal data, possible pathogenic mechanisms and future perspectives.
Similar content being viewed by others
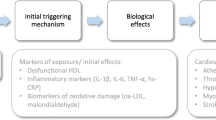


Exposure to air particulate matter leads to increased cardiovascular morbidity and mortality
While air pollution is a complex mixture of compounds in gaseous (ozone, CO and nitrogen oxides) and particle phases [8], the strongest evidence among several hundred epidemiological studies linking air pollution with human health effects, centers around the particulate components [7–13]. Particulate matter (PM) is comprised of heterogenous compounds varying in size, number, chemical composition, surface area, concentration and source [7, 8]. Some atmospheric particles are liquid, some are solid and others may contain a solid core surrounded by liquid. PM includes primary particles that are emitted directly from sources such as fossil-fuel combustion (e.g. diesel exhaust particles) and secondary particles that are generated from gases through chemical reactions involving atmospheric oxygen (O2), water vapor (H2O), reactive species such as ozone (O3), free radicals such as hydroxyl (.OH) and nitrate (.NO3) radicals, pollutants such as sulfur dioxide (SO2), nitrogen oxides (NOx), and organic gases from natural and anthropogenic sources [8].
Particles are also classified according to their aerodynamic diameter into size fractions such as PM10 ("thoracic" particles, < 10 μm), PM2.5-10 ("coarse" particles, 2.5 to 10 μm), PM2.5 (fine particles, < 2.5 μm and UFP (ultrafine particles, < 0.1 μm). These particles are derived from various sources and by various mechanisms as shown in Table 1, producing distinct lognormal modes in the particle size distributions by number and volume (nucleation, Aitken mode, accumulation and coarse modes) (Figure 1) [8]. UFP correspond to the nucleation and Aitken modes. They are typically derived from combustion of fossil-fuels and are the result of nucleation of gas phase species to form condensed phase species in newly formed particles which have had little chance to grow (nucleation mode) or in recently formed particles that are actively undergoing coagulation (Aitken mode). Some particle fractions are inclusive of others like PM2.5 which contain particles in the nucleation and Aitken mode (UFP) as well as particles in the accumulation mode (> 0.1 μm), where particles grow in size and "accumulate" by coagulation (two particles combining to form one) or by condensation (gas molecules condensing on a particle). The coarse particles correspond to the coarse mode, typically derived from soil, agricultural and road dust, tire wear emissions, construction debris, mining operations and are mainly formed by the mechanical breakdown (crushing, grinding, abrasion of surfaces) of crustal material, minerals and organic debris [8]. Major sources of PM2.5 include power plants, oil refinery and metal processing facilities, wildfires, tailpipe and brake emissions from mobile sources, residential fuel combusion. UFP are mostly generated through tailpipe emissions from mobile sources (motor vehicles, aircrafts, and marine vessels) [14].

Idealized particle size distribution that might be observed in traffic. Particles from different sizes are generated by four modes: nucleation, aitken, accumulation and coarse mode. Also shown are the major formation and growth mechanisms of the four modes of ambient particles. V = volume, Dp = particle diameter. Source: U.S. EPA [8].
Multiple studies show that short-term exposure to PM10 results in small increases in cardiovascular mortality. Thus, in approximately 50 million people from NMMAPS (National Morbidity, Mortality, and Air Pollution Study), it was found that daily cardiopulmonary mortality increased by 0.6% for every 20 μg/m3 increase in PM10 load [15]. This association was confirmed in Europe, where ~43 million adults in 29 European cities from the APHEA2 (Air Pollution and Health: A European Approach) study exhibited a 1.5% increase in daily cardiovascular mortality for every 20 μg/m3 increase in PM10 [16]. However, studies have also shown that the magnitude of these associations are bigger with the smaller PM2.5 fraction. Thus, 12,865 patients enrolled in IHCS (Intermountain Heart Collaborative Study) had a daily increase of 4.5% in acute ischemic coronary events per 10 μg/m3 increase in ambient PM2.5 [17]. Associations of air pollution with cardiovascular events have also been demonstrated in longer-term exposure studies. The latest report of the American Cancer Society II study demonstrated in ~500,000 adults, a 12% increase in cardiovacular deaths per 10 μg/m3 increase in long-term (annual average) PM2.5 exposure [4]. Importantly, this study showed that deaths from ischemic heart disease was the single largest cause of mortality (18% increase). This association was even stronger in the extended analysis of the Harvard Six Cities Study that showed a 28% increase in cardiovascular (CV) deaths per 10 μg/m3 increase in long-term PM2.5 exposure [18], and even stronger in the Women Health Initiative Study [19], which showed in 65,893 post-menopasual women, a 24% and 76% increase in the incidence of CV events and CV mortality, respectively. It appears that as the studies have gotten better tailored to assess CV morbility and mortality, the strength and magnitude of associations have become stronger. In addition, the extended analysis of the Harvard Six Cities Study showed in 8,096 participants from six US cities followed up over two time periods (1974 through 1989 and 1990-1998) that a decrease in CV mortality paralleled a decline in annual mean PM2.5 concentrations. CV mortality exhibited a 31% reduction per each 10-μg/m3 improvement in city-specific mean PM2.5 in between the two periods [18]. More importantly, the drop in the adjusted mortality rate was largest in the cities with the largest reductions in PM2.5, underscoring the importance of PM monitoring, regulation and acting upon this problem, given the potential and substantial benefits in Public Health outcomes. It has been suggested that reduction in the level of air PM may even account for some of the reduction observed in US mortality rates over the last 2 decades [20]. Although the epidemiological studies with PM10 and PM2.5 data support the notion that a smaller particle size correlates with bigger cardiovascular effects, there are few reports supporting the association of UFP with increased total or cardio-respiratory mortality [21, 22]. Lack of studies is partly due to the difficulty of quantifying UFP exposure due to the lack of routine air pollution monitoring equipment to assess UFP parameters.
Exposure to ambient PM leads to enhanced CV morbidity and mortality by a variety of proposed mechanisms, summarized in Figure 2. CV ischemic events may be due to a myriad of both acute and chronic effects. Thus, acute exposure to PM has been associated with the triggering of acute myocardial infarction [23–25], discharge of implanted automatic cardioverter defibrillators [26], hospitalizations for acutely decompensated congestive heart failure [27] and ischemic stroke [28]. Such acute effects may involve prothrombotic mechanisms such as elevation of fibrinogen [29], increased platelet aggregation and factors II, VII and X activity [30] or alterations of arterial vasoconstriction [13], leading to greater propensity for the development of ischemia in subjects with coronary artery disease [31, 32]. The accumulation of acute and sub-acute effects or the chronic exposure to PM may also result in the promotion of atherosclerosis which can be evidenced over months or years.

Possible mechanisms that link air pollution with increased cardiovascular morbidity and mortality. Air pollutants may trigger pulmonary reflexes that can activate the autonomic nervous system and result in alterations of heart rate variability and induction of dysrhythmias (in green). Air pollutants may directly affect cardiac tissue leading to the induction of dysrhythmias and/or cardiomyopathy (in yellow). Air pollutants can also enhance atherothrombotic processes that lead to the development of acute coronary syndromes (ACS) via the generation of pulmonary inflammation or direct translocation into the systemic circulation (in blue). Atherothrombotic processes include increased vascular oxidative stress, endothelial cell dysfunction, atherosclerosis and increased platelet aggregability and coagulation, among others.
The first study to find an association of PM exposure with atherosclerosis in human subjects was performed in the University of Southern California by Kunzli et al [33], a cross-sectional study that included 798 individuals. They correlated the degree of carotid intima-media thickness (CIMT) with levels of annual mean concentrations of ambient PM2.5, estimated by geocoding of the subjects' residential areas (Table 2). This study showed a 5.9% increase in carotid intima-medial thickness for every 10 μg/m3 rise in PM2.5 levels [33]. Likewise, a prospective German cohort study supported an association in between long-term residential exposure to high traffic, levels of PM2.5 and coronary atherosclerosis as assessed by coronary artery calcification scores [34]. Hoffmann et al found in 4,494 participants, that compared with subjects living > 200 meters away to a major road, subjects living within 50, 51 to 100 and 101 to 200 meters showed 63%, 34% and 8% increase in the probability of having a high coronary artery calcification (CAC) score, respectively. Nevertheless, the strength of associations between PM and atherosclerosis in humans is still being debated. While Hoffmann et al found positive associations with the distance to a major road, there were non-statistically significant associations with PM2.5 exposure. In addition, data from the Multi-Ethnic Study of Atherosclerosis [35] show some weaker associations than those shown by the previous studies (Table 2). Hence, long-term (20-year) exposure to PM was estimated on the basis of a residential history and the use of a spatio-temporal model to predict PM10 and PM2.5 exposures for each participant-month based on the geographic location of each participant's residence each month. Three measures of subclinical atherosclerosis, such as CIMT, coronary artery calcification and ankle-brachial index (ABI) were employed in 5,172 US participants. While no associations were present with either coronary calcium or ABI measures, PM10 exposures (20-year means and 2001 mean) and 20-year PM2.5 exposures did correlate with 1-3% increase in CIMT per 21 μg/m3 increase in PM10 or 12.5 μg/m3 increase in PM2.5, respectively.
Based on this evidence, it would appear that PM2.5 exposure may contribute to enhancement of human atherosclerosis, although the strength and relative importance of this mechanism in contributing to cardiovascular morbidity and mortality is still to be ascertained. Just as the strength of the associations of PM with cardiovascular outcomes increased when moving from PM10 to PM2.5 metrics, it has been suggested that these associations could be even stronger with the UFP fractions for reasons that are discussed below. Although there are no studies available yet that have evaluated the effects of UFP exposures on human atherosclerosis, recent findings from the Southern California Particle Center (SCPC) are consistent with the notion of UFP's greater proatherogenic potential. Delfino et al studied residents in independent living facilities of two retirement communities in Los Angeles [36]. 31 subjects with history of CAD were followed up over 7-month periods with a very detailed pollutant exposure characterization and blood collection for the determination of systemic inflammatory, antioxidant and coagulation markers. They found positive associations of particle number (dominated by UFPs) and outdoor quasi-ultrafine PM0.25 (< 0.25 μm) with biomarkers of systemic inflammation such as C-reactive protein (CRP), interleukin (IL-6) and soluble tumour necrosis factor receptor II (sTNF-RII). By contrast, 4-day average outdoor accumulation-mode PM0.25-2.5 showed an unexpected inverse association instead [36]. This study is in agreement with a previous report where exposure to UFP correlated with increased plasma levels of soluble CD40 ligand (sCD40L), a marker for platelet activation that reflects increased coagulation and inflammation [37]. Thus, there is need for human studies that evaluate the potential links between UFP exposures and clinical measures of atherosclerosis.
Studies in animal models support a causal association for PM and atherosclerosis
Work with experimental animals have been important to establish a causal association between exposure to PM and atherosclerosis. The first evidence was from work at the University of British Columbia in Ontario, Canada [38]. Suwa et al exposed female Watanabe heritable hyperlipidemic rabbits to biweekly intrapharingeal instillations of urban air PM10. The degree of aortic and coronary atherosclerosis was evaluated by estimating the atherosclerotic lesion volumes in sections from the various vessels. Qualitative analysis included the assessment of the types of plaques according to AHA guidelines [39, 40], plaque composition and cellularity. They determined that although there were no differences in the frequency of plaques in either the coronaries or aorta, PM10 administration resulted in a 71% significant increase in the relative volume of atherosclerotic lesions in the coronary arteries and a 62% increase in the relative volume of extracellular lipids in the aorta. Plaques were also more advanced and with a greater content of smooth muscle cells in the coronaries. Interestingly, the burden of atherosclerotic lesions correlated with the percentage of alveolar macrophages positive for particles.
The effects of inhaled PM2.5 on aortic atherosclerosis have been assessed on apoE null mice subject to long-term exposures to concentrated ambient particles (CAPs) performed at the A.J. Lanza Exposure Laboratory of New York University in Tuxedo, NY (Table 3). The first study reported in 2005 showed that 39-41 week-old apoE-/- mice fed a chow diet and exposed to 10× ambient concentrations of PM2.5 for 6 hours per day, 5 days per week for 5 months exhibited a 57% increase in the percentage of atherosclerotic plaque area in the aortic root [41] (Table 3, Figure 3). However, there were no differential effects in the context of extreme hyperlipidemia exhibited by apoE-/-, LDL-/- mice exposed to the same conditions [41]. A second study reported the same year showed that younger 6-week-old apoE-/- mice fed a chow diet and exposed to similar conditions for 6 months displayed a 45% increase in the percentage of aortic atherosclerotic plaque area that was not statistically significant [42]. In this study, however, marked hyperlipidemia achieved by the feeding of a high fat diet resulted in a greater and significant promotion of atherosclerosis as CAPs-exposed mice showed a 58% significant increase in aortic root plaque area (Figure 3) [42], together with impaired vasomotor response. More recently, a third study of apoE-/- mice fed a high fat diet and exposed to PM2.5 in the same facility confirmed the enhancement of aortic atherosclerosis as assessed by the percentage of the plaque area in the aortic arch by ultrasound bio-microscopy [43] (Table 3, Figure 3). In this study, PM2.5 exposures also led to atherosclerotic plaques that were richer in tissue factor [43], which could play a causative role or simply be an indicator of greater atherosclerotic plaque burden. One limitation of these studies is the relatively small number of sex-matched animals (n < 10/group) used to assess vascular pathology that has a large phenotypic variance. However, the consistent reproducibility of PM2.5 proatherogenic effects in all three studies decrease the potential for type I error.

PM effects on atherosclerotic lesions in animal studies. The relative increment in atherosclerotic lesions induced by PM exposures over filtered-air controls is displayed (%) Atherosclerotic lesions were assessed by histological analysis of aortic cross-sections, en-face assessment of the whole aorta or ultrasound biomicroscopy depending on the study. Mode of atherosclerotic lesion assessment and references for each study are shown in Table 3. HFD = High fat diet. * p < 0.05 in comparison with the corresponding filtered-air controls.
In another animal study in the Southern California Particle Center space (SCPC) and the University of California in Los Angeles (Table 3) [44], we tested the notion that the oxidant potential and size of ambient particles play an important role in determining atherosclerotic lesion development, with a hypothesis that UFP should trigger more plaque development than bigger particles thanks to their greater prooxidant potential, as it will be discussed below. Araujo et al exposed apoE-/- mice fed a chow diet to concentrated PM2.5, UFP or filtered air for 5 hours/day, 3 days per week for 5 weeks. UFP-exposed mice developed 25% and 55% more aortic atherosclerosis, assessed by the mean atherosclerotic lesional area in the aortic root, compared to PM2.5 or FA-exposed mice, respectively [44]. Although PM2.5 exposures tended to enhance lesion formation over FA controls, resulting in a 23% increment in lesional area, these effects were relatively smaller than in the previous studies (Figure 3). One possible explanation could be the shorter duration of exposures (5 weeks instead of 5-6 months). This is exemplified by the data in Figure 3 showing that the degree of PM2.5-induced atherogenesis increases with the length of the exposures. In addition, the magnitude of these effects is enhanced by UFP exposures to yield a comparable degree of atherosclerosis enhancement as long-term PM2.5 exposures (Figure 3).
One limitation of the SCPC study is that the concentrator technology employed (VACES: Versatile Aerosol Concentration Enrichment System) generates overlapping CAPs aerosols. The fact that the sub-2.5 μm aerosol included UFP made straight comparison with the UFP aerosol difficult since particle mass in the former aerosol was mostly determined by the accumulation mode particles (0.1-2.5 μm). The more abundant UFP contributed to less than a fifth of the PM2.5 aerosol's total mass. It is possible that the true relative proatherogenic effects of UFP, compared to the larger 0.1-2.5 μm particles are bigger than can be inferred from our study. When normalized by mass, atherosclerotic lesion scores of UFP-exposed mice were 460% greater than scores exhibited by PM2.5-exposed mice, an extrapolation that is not entirely adequate given that both aerosols overlapped in size. On the other hand, the UFP aerosol contained 44% more particles than the number of sub-0.18 μm particles present in the PM2.5 aerosol which could explain the observed difference, provided that the sub-0.18 μm components accounted for all or most of the PM2.5-induced proatherogenic effects. Any of these extrapolations could enhance the approximately 25% difference in the proatherogenic potentials of UFP versus the larger 0.1-2.5 μm particles. An accurate estimate of the relative potency of UFP vs. 0.1-2.5 μm particles would require a different concentrator technology with selective exposures to UFP and 0.1-2.5 μm particles as well as the use of four parallel exposure groups where both particle mass and particle number concentrations were matched to quantify the relative importance of UFP.
An additional limitation of this study is that it only evaluated a single dose at only one time point, which impedes the assessment of threshold, latency of effects or potency. Furthermore, the particles collected in Los Angeles have not been compared to other locations in the world where unique aerosol compositions in each size range could have different outcomes. It will be important to conduct similar studies in different places where the effects of different particle compositions on atherosclerosis, not only size, can be assessed.
Proposed oxidative stress paradigm for understanding of PM proatherogenic effects
A number of mechanisms have been proposed to explain the cardiovascular effects of ambient PM (Figure 2), which has been the subject of recent extensive reviews [5, 6, 45]. At the cellular level, these various mechanisms involve free radical production (by transition metals and organic compounds), oxidative stress, cytokine release, inflammation, endotoxin-mediated damage, stimulation of capsaicin receptors, autonomic nervous system activity, covalent modification of key cellular molecules and increased pro-coagulant activity [5, 6, 45, 46].
Diesel exhaust particles (DEP) have been used as a model pollutant and extensively studied in airway inflammation. We and others have shown that the pro-inflammatory effects of DEP and concentrated air particles (CAPs), that result in enhancement of allergic inflammation [47, 48], acute asthma exacerbation and bronchitis flares [3, 49] are linked to ROS production [46, 48, 50]. This includes O2.- production in macrophages, bronchial epithelial cells and lung microsomes incubated with the particles or their organic extracts [51, 52]. Moreover, we have demonstrated that thiol antioxidants, which are highly effective in suppressing reactive oxygen species (ROS) production by organic DEP chemicals in vitro, interfere in the adjuvant effects of DEP in a murine allergen sensitization model [53]. We have shown evidence that DEP induce oxidative stress and cellular effects in target cells that can be better understood according to a stratified oxidative stress model, aka as the hierarchical oxidative stress hypothesis [54] (Figure 4). When ROS production overwhelms the antioxidant defenses, a state of cellular oxidative stress develops [55], characterized by a depletion of intracellular GSH, leading to GSSG accumulation and a drop in the GSH/GSSG ratio [55]. This state of redox disequilibrium initiates additional redox-sensitive signaling pathways, including activation of several MAP kinases as well as the NF-κB cascade. These cascades act in synergistic fashion to activate cytokine and adhesion molecule expression through appropriate promoter response elements. Indeed, a number of laboratories have demonstrated that DEP and ambient PM induce JNK, p38MAPK and NF-κB activities, which are functionally linked to the cytokine and chemokine production in vivo and in vitro [56, 57]. DEP has also been shown to elicit ROS production in vascular cells such as pulmonary artery endothelial cells [58], rat heart microvessel endothelial cells [59] and human microvascular endothelial cells [60]. Furthermore, it appears that DEP could induce oxidative stress, inflammatory and propapoptotic signals in endothelial cells in a dose-dependent hierarchical fashion as incremental DEP concentrations result in a larger recruitment of genes and toxicity [60].

Stratified hierarchical response to air pollutants. Macrophages exposed to particulate matter components (e.g. DEP) respond in a dose-dependent fashion via the activation of pathways and mechanisms at various levels of action (tiers). For example, Nrf2 can induce the transcription of antioxidant genes via the antioxidant response element (ARE) in the earliest level of defense (tier 1). When the mechanisms of defense involved in one tier are not sufficient to contain the injurious stimuli, signaling pathways of the subsequent(s) tier of action may get activated, such as MAPK pathways that regulate proinflammatory genes via Activating Protein (AP) transcription factors (tier 2) or proapoptotic signals triggered by mitochondrial perturbation that can lead to activation of the mitochondrial permeability transition (PT) pore and cellular toxicity (tier 3). Modified from Li et al [123].
We have shown that the aromatic and polar DEP fractions, which are enriched in polycyclic aromatic hydrocarbons (PAHs) and quinones, respectively, are most active in the induction of heme oxygenase 1 (HO-1), glutathione S-transferase (GST), and other phase II enzymes that correspond to the first tier of defense in macrophages (Figure 4) and epithelial cells [61]. We have also demonstrated that the triggering of this antioxidant defense is regulated by the transcription factor p45-NFE2 related transcription factor 2 (Nrf2) by modulation of its proteasomal degradation in the cytosol and translocation into the nucleus [61]. Interestingly, ambient PM also triggers Nrf2-regulated genes in response to its content of prooxidative and electrophilic chemicals. Various particle sizes have marked differences in their chemical composition as a result of their different sources of origin and modes of generation previously described. These differences result in different redox activity and ability to cause mitochondrial damage as shown in Table 4. Thus, ambient UFP triggers the induction of Nrf2-regulated HO-1 in macrophages to a greater degree than ambient PM2.5 or coarse particles [61]. The same can also be shown in vivo using a transgenic mouse model in which the heme oxygenase promoter was linked to a luciferase reporter gene [62]. Exposure of these animals in a mobile animal trailer showed that the promoter reporter gene construct is more readily induced in animals exposed to concentrated UFP compared to animals simultaneously exposed to a concentrated atmosphere of PM2.5 or filtered air (Figure 5). This greater responsiveness was in relation with the greater prooxidant potential of ultrafine PM due to their higher content of redox cycling organic compounds.

Ambient UFP triggers prooxidant effects in-vivo. Representative dorsal photograph of HO-luc transgenic mice that were exposed to concentrated UFP, concentrated PM2.5 or filtered-air (FA) for 5 hours in a mobile exposure laboratory located in downtown Los Angeles, ~300 meters away from the I-110 freeway. Increased bioluminescence was due to a larger HO-1 upregulation response generated in tissues subject to greater oxidative stress. Mice were generated and obtained from Dr. Christopher Contag at Stanford University [62]. They contain a modified coding sequence of the luciferase gene under the control of the full HO-1 promoter. Whole-body images were acquired within 3 hours of the exposure using a cooled charged-couple device (CCD) camera at the Small Animal Imaging Center of the UCLA Crump Institute for Molecular Imaging [124] as previously described [125]. The bioluminescence signal was recorded as maximum photons/sec/cm2/steradian (p/sec/cm2/sr). Notice that the UFP-exposed mouse displays increased luciferase emissions both in the thorax and abdomen as compared with the PM2.5 or FA-exposed mice.
UFP exhibit greater relative content of elemental and organic carbon [44, 63] that vary depending on the sources. Urban UFP contain a higher content per unit mass of polycyclic aromatic hydrocarbons (PAH), which are relevant organic constituents since they can induce oxidative stress and electrophilic chemistry in tissues [46, 64] after conversion to quinones by the involvement of cytochrome P450 1A1, epoxide hydrolase and dihydrodiol dehydrogenase [65]. Some quinone species can act as redox cycling catalysts, leading to the formation of O2,- when undergoing one-electron reductions by NADPH cytochrome P450 reductase to form semiquinones [66] and recycling to the original quinones [50, 66]. There is an excellent correlation between the PAH content of UFP and their ability to engage in redox cycling reactions in-vitro. Indeed, UFP samples from the Los Angeles basin are more potent than PM2.5 or coarse PM at inducing oxidative stress as measured by the dithiothreitol (DTT) assay, extent of HO-1 induction, reduced glutathione to oxidized glutathione (GSH/GSSG) ratios and the ability to cause mitochondrial damage (Table 4). These biological endpoints are correlated to greater organic PM carbon and PAH contents, suggesting a role of these organic substances in generating redox activity [67–69]. Furthermore, UFP collected from various locations differ significantly in their ability to trigger Nrf2-regulated responses, also in strong relation with their chemical composition and electrophilic potential [67]. Thus, the greater prooxidant effects of the smaller particles appear to be partly derived from their higher content of prooxidant compounds.
PM ability to induce ROS generation appears to be key in promoting atherosclerosis, which could be the result of PM-mediated systemic prooxidant and proinflammatory effects. Atherosclerosis is a vascular inflammatory process where lipid deposition and oxidation in the artery wall constitutes a disease hallmark [39, 40, 70–73] (Figure 6). Infiltrating lipids come from low density lipoprotein (LDL) particles that travel into the arterial wall and get trapped in a three-dimensional cage-work of extracellular fibers and fibrils in the subendothelial space [74, 75], where they are subject to oxidative modifications [76–78] leading to the generation of "minimally modified" LDL (mm-LDL). Such oxidized LDL is capable of activating the overlying endothelial cells to produce pro-inflammatory molecules such as adhesion molecules, macrophage colony-stimulating factor (M-CSF) and monocyte chemotactic protein-1 (MCP-1) [79–81] that contribute to atherogenesis by recruiting additional monocytes and inducing macrophage differentiation [70, 71, 73]. Later stages of the disease involve the proliferation of smooth muscle cells, formation of fibrous caps, induction of apoptosis, hemorrhage and calcification [73]. Animal models have been useful not only to demonstrate the PM proatherosclerotic effects as described in the previous section, but also to get some insights into the possible mechanisms involved. It is possible that the different proatherogenic effects of particles of different sizes and compositions could at least be partly related to their different abilities to elicit prooxidant and proinflammatory reactions in vascular cells.

Pathogenesis of atherosclerosis. Lipid infiltration of the artery wall originating from circulating LDL followed by oxidative modification in the subendothelial space, monocyte chemotaxis and foam cell formation are among the earliest events in atherogenesis. Monocytes differentiate into macrophages, followed by release of inflammatory mediators and a vicious cycle of inflammation. More advance stages of the disease include smooth muscle cell proliferation, formation of fibrous caps, necrotic cores, calcification, rupture, hemorrhage and thrombosis. Possible mechanisms how PM enhances atherosclerosis include: 1) Systemically translocated UFP or their chemical constituents may synergize with ox-PAPC generated within ox-LDL in the activation of proatherogenic molecular pathways in endothelial cells, 2) Inflammatory mediators released from the lungs may promote monocyte chemotaxis into the vessels, 3) PM can induce HDL dysfunction with loss of its antiinflammatory properties. Modified from Araujo and Lusis [126].
How do the prooxidative effects of PM promote atherogenesis?
The promotion of atherogenesis by PM is likely the result of systemic prooxidant and proinflammatory effects at vascular sites. In support of this notion, Nurkiewicz et al have shown that intratracheal instillation of residual oil fly ash (ROFA) [82, 83] in rats led to greater vascular ROS generation, as assessed by the tetranitrobluetetrazolium (TNBT) reduction method [82], resulting in a dose-dependent impairment of systemic endothelium-dependent arterial dilation, increased leukocyte rolling and adhesion as well as deposition of myeloperoxidase in the spinotrapezius muscle microcirculation [82, 83]. Likewise, long-term PM2.5 inhaled exposures that promote atherosclerosis, also lead to enhanced ROS generation in the aortic plaques and increased formation of 3-nitrotyrosine residues [42].
How does PM inhalation in the lung lead to the systemic effects on the vasculature?. One possibility is that prooxidant PM deposited in the lungs lead to the release of proinflammatory cytokines (Figure 6) into the blood stream by a series of steps such as: a) Deposition of inhaled particles in the respiratory tract, b) Ability to trigger or enhance free-radical production, c) Development of pulmonary inflammation, d) Release of inflammatory mediators into the systemic circulation. Inhaled small particles deposit in the respiratory tract via diffusion due to displacement when they collide with air molecules [84]. According to the International Commission on Radiological Protection (IRCP) 1994 model for particle deposition in the respiratory tract [85], inhaled particles deposit in various segments of the human respiratory tract that can be grossly divided into three anatomical regions: the nasopharyngeal, tracheobronchial and alveolar regions [85]. Once particles get deposited in the lungs, they could trigger or enhance ROS formation when in contact with epithelial cells, alveolar macrophages, pneumocytes, etc. as discussed in the previous section. This could lead to development of interstitial and/or alveolar inflammation with the subsequent release of inflammatory mediators into the systemic circulation. While particle deposition (step a) has been amply studied in both animals and humans as well as their ability to induce free radical reactions in pulmonary cells (step b), the remaining steps c and d are still awaiting definite confirmation.
There is clear evidence that particle deposition can lead to systemic inflammatory events. Suwa et al reported that their PM10-exposed animals exhibited elevation of polymorphonuclear cells and circulating band cell counts 2 weeks after starting the exposures, with an increase in the size of the bone marrow mitotic pool as assessed by BrdU-labeling [38]. Although they claimed that their work support the notion of PM-induced systemic inflammation in response to local pulmonary inflammation, they did not provide evidence of increased systemic proinflammatory markers that could be linked to both effects. Likewise, none of the CAPs studies provided direct evidence for such mediators in apoE-/- mice. However, a recent study of C57BL6 mice exposed to CAPs for 6 months did result in increased plasma concentrations of inflammatory biomarkers such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), E-selectin and intracellular adhesion molecule-1 (ICAM-1), indicative of systemic inflammatory effects [86]. In that study, PM2.5 exposures also led to increased visceral adiposity and insulin resistance, suggesting a possible link between air pollutants and type 2 diabetes mellitus [86]. Despite the lack for direct evidence of systemic inflammatory mediators in apoE-/- mice, Araujo et al showed that CAPs exposures led to the development of dysfunctional HDL with a decrease or loss of its anti-inflammatory properties as exhibited by PM2.5 and UFP-exposed mice, respectively [44]. Furthermore, UFP exposures led to HDL that exhibited proinflammatory properties and could therefore contribute to enhanced disease pathogenesis (Figure 6).
PM-mediated generation of dysfunctional HDL could boost and propagate systemic inflammatory effects or at least function as a marker of systemic inflammation. It is well established that plasma HDL cholesterol and apoA1 levels are inversely correlated with the risk for coronary artery disease [87–89]. In addition to the well-characterized ability to promote reverse cholesterol transport [90], HDL has been reported to have antioxidant, antiinflammatory and antithrombotic activities [91–93]. However, high levels are not always protective in subjects, suggesting that not all HDLs prevent atherosclerosis [94]. Furthermore, dysfunctional proinflammatory HDL may serve as a useful marker for predicting susceptibility for atherosclerosis in humans [95] and in rabbits [96], where it has been found to be a better predictor han total or LDL cholesterol levels. Thus, it will be necessary to characterize the type of alterations that PM exposures induce in HDL particles and confirm whether those happen in human subjects also. This can help to elucidate pathogenesis and may even lead to the identification of a potential biomarker.
Interestingly, these proinflammatory effects occurred in the absence of significant pulmonary inflammation which raises the possibility that the lungs might not need to be an active player or mediator in the induction of systemic vascular inflammation. Alternatively, particles could be endocytosed by alveolar macrophages (Figure 7), which could trigger activation of molecular inflammatory pathways that could be transduced systemically even in the absence of obvious alveolitis or interstitial pneumonitis. Another potential mechanism is that once particles get deposited in the lungs, they could undergo transcytosis across epithelia of the respiratory tract into the interstitium and access the blood circulation directly or via lymphatics (Figure 6), or could be taken up by sensory nerve endings embedded in airway epithelia, followed by axonal translocation to ganglionic and CNS structures [84]. While there are multiple reports that support the notion of systemic translocation of synthetic nanoparticles in experimental models [52, 97, 98] and humans [99], no convincing demonstration has been provided about this route for inhaled ambient particles. In addition, in case nanoparticles do translocate to the systemic circulation, no studies have shown that the translocated particles actually reach or deposit in blood vessels, perhaps due to the inability to detect them at these sites because of small size. There could be alternative ports of entry such as ingestion and absorption via the gastrointestinal tract or direct contact through dermal exposure [84]. However, there is no direct evidence for the significance of these routes in the exposure to ambient particles and although their feasibility has been supported by studies with engineered nanoparticles, these are particles that fall outside the scope of this review.

UFP are endocytosed by alveolar macrophages. Electron microscopy photographs of a lipid-laden alveolar macrophage harvested from an apoE-/- mouse exposed to concentrated ambient ultrafine particles (<0.18 μm). Red arrows indicate electron-dense material corresponding to intracellular aggregates of nanoparticles. Notice the subcellular mitochondrial localization encircled by the red dotting. Source: Journal cover of Araujo et al [44].
An alternative possibility is that particles deposited in the lungs get internalized into the cells (e.g. endocytosis by alveolar macrophages) and their chemicals could get translocated into the systemic circulation allowing them to reach target tissues (Figure 6). Whether the whole particles or chemical constituents access the systemic targets, they can activate important proatherogenic molecular pathways once in contact with cells from the vascular endothelium [60]. Although we have not found direct evidence for increased aortic oxidative stress, we showed that both PM2.5 and UFP exposures led to increased hepatic oxidative stress [44]. In addition, UFP triggered the upregulation of Nrf2-regulated antioxidant genes and unfolded protein response (UPR) genes in the liver, suggesting that PM exposure leads to both systemic prooxidant and proinflammatory effects [44].
It is clear that inhaled particles do get deposited in the lungs (step a), with the potential to exert prooxidative effects and activate molecular pathways in response (step b), even in the absence of obvious clinical inflammation (step c). Somehow, systemic vascular inflammation and prooxidative effects follow via a mechanism(s) still to be elucidated. In addition, oxidative stress and inflammation are strongly connected and either one can lead to the other. It is likely that the prooxidant potential of ambient particles determine their ability to activate pathways that lead to prooxidant and proinflammatory effects in the vasculature and promotion of atherosclerosis. However, in spite of this logical inference, it still remains unclear whether PM-induced prooxidant effects trigger or are the consequence of vascular inflammatory effects. Further research is required to elucidate these mechanisms and to prove whether particle-mediated proinflammatory effects are due indeed to its prooxidant potential.
Why are UFP more proatherogenic than PM2.5?
A number of factors other than particle size need to be considered to explain why UFP may be more atherogenic than PM 2.5 (Table 4, Figure 8). This includes consideration of particle numbers, physicochemical composition, prooxidant potential, bioavailability and pulmonary retention. A brief discussion of how these factors could contribute to the increased atherogenic potential of UFP include:

Factors that may explain greater UFP's proatherogenic potential. UFP (in red) are the smallest, most numerous particles and with best pulmonary retention (top panel). UFP exhibit greater relative content of redox-active compounds (e.g. PAHs, in green) than bigger particles (mid panel). UFP's greater surface-to-mass ratio may allow reactive compounds (in green) to have increased localization towards the surface of the particle (in red) and be more bioavailable for free-radical reactions when in contact with cells (bottom panel). Estimates for the increased content of prooxidant PAHs and surface-to-mass ratio in comparison to PM2.5are from the SCPC study [44].
a) Larger particle number
Exposure to PM has been traditionally assessed by measures of mass per space (e.g. daily, annual, average or peak μg/m3). Adverse health effects have been correlated linearly with various measures of exposure, without any clear threshold below which PM levels have been found to be consistently safe [7, 8, 100]. However, particles < 0.1-0.2 μm, which contribute very little to overall PM2.5 mass, represent > 85-90% of the total PM2.5 particle number [101]. Therefore, it is conceivable that larger particle numbers in UFP atmospheres, despite a smaller mass, could result in larger biological effects. In our study, development of larger atherosclerotic lesions in the UFP exposures correlated with increased particle numbers rather than with PM mass. Thus, the 25% increase in atherosclerotic lesion area in UFP vs. PM2.5-exposed mice occurred in parallel with a 44% increase in the number of particles < 0.18 μm in spite of a 4-fold smaller PM mass [44].
b) Larger content of redox active compounds
PM is composed of hundreds of organic chemicals and transition metals that could trigger or exacerbate free radical reactions contributing to systemic proatherogenic effects. As discussed in section 3, UFP are enriched in organic carbon content as well as prooxidative PAHs (Table 4) that promote oxidative stress and inflammation. This could explain the increase in atherosclerotic lesion development. Whether or not these compounds are the actual toxicants contributing to atherogenesis, they may serve as a proxy for the substances ultimately involved. For instance, PAHs can be converted to quinones and other redox cycling substances. In addition, it appears that while transition metals are also important catalysts for free radical reactions in-vitro, they may be less critical for the in-vivo systemic effects, as UFP exhibited greater proatherogenic potential despite a lower metal content than PM2.5 [44].
c) Greater bioavailability
An important feature derived from the small size of UFP is that their large surface-to-mass ratio along with increased number could lead to a sizable increase in bioavailable surface [102]. The number of atoms or molecules that are displayed and packaged on the surface of particles increase exponentially as the size shrinks below 100 nm [84]. Thus, the presence of bioreactive chemicals (e.g. PAHs, transition metals) on the large surface area of UFP could make these chemicals more bioavailable at the contact sites of the particles with cells and tissues where prooxidant effects take place (Figure 8). The surface area or more accurately the bioreactive surface area of UFP constitutes an exposure parameter that may more accurately depict small particle dosimetry than the current calculation of PM exposure by mass [31, 103, 104]. Unfortunately there are no easy ways to calculate reactive surface area at present.
d) Greater lung retention
Small particle size allows better penetration and diffusion into the lungs according to the IRCP 1994 model for particle deposition in the respiratory tract [85]. The fractional deposition of particles during nose breathing into the three anatomical regions in this model (nasopharyngeal, tracheobronchial and alveolar regions) varies significantly depending on the aerodynamic diameter of the inhaled particles, respiratory frequency, pattern of breathing and respiratory co-morbid conditions. Indeed, UFP exhibit a greater fractional deposition in the tracheobronchial and alveolar regions than larger particles. For instance, 20-nm synthetic particles, which correspond to the particle peak size of ambient urban aerosols, exhibit the highest fractional deposition efficiency in the alveolar region (~50%) [84, 102]. In addition, the UFP fractional deposition increases markedly during exercise in healthy subjects [105] when the respiratory frequency is greater and the pattern of breathing is altered. Furthermore, pulmonary conditions such as chronic obstructive pulmonary disease (COPD) or asthma that are characterized by airway constriction with changes in air flow dynamics and increases in lung residual volume, have been shown to result in increased fractional deposition of UFP [106, 107], likely as a result of greater contact with the airway wall and enhanced diffusional deposition. Better airway deposition could translate into better retention, cellular uptake and greater propensity to induce systemic effects, a notion that remains to be proven. The greater retention of UFP could be partly due to increased Van der Waals forces or electrostatic interactions, all of which contributes to "adhesive interactions" [108, 109]. Once deposited in the alveoli, particles are wetted by the surfactant film and could be taken up by alveolar macrophages by physico-chemical forces rather than receptor-ligand interactions that play a role in phagocytosis of larger particles. Whether these particles or chemical constituents enter the systemic circulation is a matter of intense debate that requires further investigation. It will be important to determine whether all these factors confer greater toxicity to UFP in human subjects since they may imply the need for adjusting the metrics of exposure to take account of particle number, surface area and oxidant potential.
Relationship of PM to other proatherogenic factors: Gene-environment interactions
The development of PM-induced proatherogenic effects likely depends not only on exposure characteristics such as dose, duration and location of exposure as discussed above but also on the differential susceptibility of exposed individuals [110], according to a gene-environmental paradigm that recognizes that atherosclerosis is a complex disease that involves a multiplicity of genetic and environmental factors [73]. In fact, exposure to PM is now considered as a novel risk factor that could contribute to the composite clinical risk in synergy with well known cardiovascular risk factors such as hypertension, hypercholesterolemia, smoking, diabetes and/or obesity among others. However, this synergy has only been studied to a limited degree. While subjects with advanced age [111, 112], obesity [19] or congestive heart failure [113] have been reported to exhibit increased susceptibility to PM-mediated cardiovascular effects, only one study among those evaluating the impact of PM on atherosclerosis found predisposing conditions such as age and sex [33]. However, the number of studies and number of subjects are very small and it is still too early to draw any conclusions. Further research is required with larger number of individuals that undergo extensive characterization of clinical traits to maximize the possibility of unveiling gene-environment interactions.
Cellular and experimental animal work can be instrumental to identify conditions that help establish the molecular and genetic pathways that are involved in the interaction of PM with other risk factors. While the individual contribution of air pollutants to atherogenesis might be small, their effects could be exacerbated in synergy with other known proatherogenic factors. These interactions could be more evident on molecular pathways known to mediate disease pathogenesis. One possibility is that PM proatherogenic effects depend on generation of oxidative stress. PM could exacerbate the biological activity of well-known prooxidative and proatherogenic factors such as ox-LDL. We tested this hypothesis by evaluating the antioxidant response of microvascular endothelial cells to DEP in the absence and presence of oxidized PAPC (1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine), one of the key prooxidative components generated in LDL particles. While both DEP and ox-PAPC have been shown to exert prooxidative effects in vascular cells [58, 59], the combination of both stimuli synergize in increasing antioxidant gene expression, including HO-1 [60]. In addition, analysis of the genomic profiles of cells subjected to the various treatments unveiled a large number of genes where the same DEP and ox-PAPC synergy was present. These genes were grouped into clusters that were enriched in proinflammatory, apoptotic and UPR pathways, all important players in atherogenesis. These gene clusters are reminiscent of the different elements of the hierarchical oxidative stress paradigm discussed earlier on. Some representative examples include proinflammatory genes such as Interleukin 8 (IL-8) and chemokine (C-X-C motif) ligand 1 (CXCL1) production, immune response genes such as Interleukin 11 (IL-11) and UPR genes such as activating transcription factor 4 (ATF 4), heat shock 70 kDa protein 8 (HSPA8) and X-box binding protein 1 (XBP1). Importantly, some of these same genes were found to be preferentially upregulated in the livers of apoE-/-mice exposed to UFP, supporting the validity of in-vitro generated data to predict relevant in-vivo outcomes.
The identification of participant gene pathways would pave the way to study gene-environment interactions in the development of PM proatherogenic effects. For instance, it is conceivable that modulation in the expression levels of antioxidant genes as a result of gene polymorphisms could alter the magnitude of effects caused by PM. Indeed, polymorphic variants of the glutathione-S-transferase genes GSTP1 and GSTM1 [114] or HO-1 [115] have been reported to influence the risk of developing new onset asthma associated to air pollutants. It appears that gene-gene and gene-environment interactions could determine the fate of individuals to develop new disease under specific circumstances. Thus, in a population-based cohort study in Southern California, child carriers of the short GT-repeat polymorphism that results in higher HO-1 expression, exhibited decreased risk of new-onset asthma as compared with carriers of the non short GT alleles [115]. This association was only present in non-hispanic white children but not in hispanic subjects and especially among those residing in low ozone communities. These findings illustrate the high level of complexity present in the interaction between genetic and environmental factors that could be similar to the interactions between particulate pollutants and atherosclerosis-related genes. Interestingly, one study reported inverse associations of exposure to primary aerosols with the antioxidant erythrocyte enzyme Cu/Zn-SOD and GPx-1 in subjects with coronary artery disease [36]. Polymorphisms and variations in the expression of these phase 2 enzymes could potentially lead to increased susceptibility to adverse cardiovascular events. It is therefore necessary to perform large-scale studies where in addition to detailed characterization of pollutant exposures, individuals are genotyped and extensively phenotyped for clinical traits and gene expression levels. This will enhance our understanding of individual susceptibility to PM-mediated adverse cardiovascular effects and may also shed light on the adaptation mechanisms of people who live in a polluted environment.
Multi-pollutant effects contributing to atherosclerosis
Although most of the published data attributes the cardiovascular effects to the particulate matter components, these associations have been questioned on the basis of publication bias. While it is extremely unlikely that publication bias could explain the overall consistency, coherence and strength of those associations [6, 116], it is conceivable that other co-pollutants could play a role. Work from the Lovelace Respiratory Institute has shown that exposure of apoE-/- mice to both whole and particle-free gasoline exhausts results in the induction of aortic metalloproteinases (MMP)-2 and -9 after 7 weeks [117] and as early as after 7 days [118]. This suggests that gaseous components are responsible for those effects. Interestingly, gasoline exhaust emissions appear to trigger both ROS and endothelin 1 (ET-1) mediated pathways with a complex degree of interconnectivity. Increased expression and activity of MMP-9 was shown to be ET-1 mediated and also present in a small cohort of humans exposed to diesel exhausts [118]. While ET-1-mediated pathways triggered by either gasoline and/or diesel exhausts could play a role in atherogenesis, increased MMP-9 could be modulating plaque stability [119] and induce plaque rupture and subsequent development of acute coronary syndromes [120]. It is possible thus that gaseous and particulate pollutants could exert different but cooperative effects, which will need to be explored in more details in the future.
Future research perspectives
We have seen in recent years an enormous improvement in our understanding of the cardiovascular effects of air pollution. Human epidemiological studies together with experimental animal data discussed above support a causal association between air PM and atherosclerosis. Consequently, PM exposure is now considered a novel risk factor [5]. Much work is still needed however, in the identification of those factors that are clinically relevant. This work will require the collaboration of investigators from diverse disciplines that span from epidemiology, toxicology and engineering to pathology, genetics, vascular and molecular biology. Better understanding of the toxic air components, toxicology and pathogenesis will allow us to formulate better ways to monitor and control air pollution as well as to avoid or decrease their effects on cardiovascular health.
Although most of the epidemiological work has been focused on PM mass exposure parameters of the various size fractions, other physicochemical parameters such as chemical composition, particle number, surface area, surface-to-mass area need to be considered in future studies as discussed above. It may require us to develop new parameters or composite metrics that can combine all these various factors to best capture the overall PM-related cardiovascular toxicity. This may help to elucidate why exposure to PM, in strong association with CV events in the Northeast, Midwest and Southern U.S. regions, appears not be associated with excess risk in the Northwestern U.S. regions [121, 122].
Animal data that supports the notion of UFP's greater proatherogenic potential and development of HDL dysfunction are based on one single study [44] and require confirmation by further studies, including in human subjects. Dose-response studies are also needed to determine various toxicological parameters (e.g. threshold, latency of effects, etc.) and whether proatherogenic effects are in relation to the whole burden of PM exposure or the kinetics of those exposures. Since air pollutants include hundreds of different compounds, it is unlikely that one single or a few chemical constituents could be responsible for all pathogenic effects but it could be possible to identify groups of compounds that work in an additive or synergistic fashion (e.g. PAHs).
While exposure to PM leads to both prooxidant and proinflammatory effects, there is a great degree of interconnection in between oxidative stress and vascular inflammation that makes it very difficult to determine whether the induction of vascular oxidative stress precedes or follows inflammation as discussed above. The use of antioxidants as well as transgenics or knockouts for critical antioxidant genes or transcription factors (e.g. Nrf2, HO-1, superoxide dismutase) or prooxidant genes (e.g. NADPH oxidase) may help to clarify this. Since the portal of entry may determine the pathogenic mechanism and the mode for transduction into systemic effects, it will be important to fully characterized the role of the lungs as a mediator and to assess alternative ports of entry such as the gastrointestinal tract by ingestion of particles or skin by contact.
Exposure to PM likely leads to important gene-environment interactions that will need to be addressed in both human and animal studies to better define conditions of increased susceptibility. The use of gene profiling, analysis of gene expression by system biology approaches, genetically-engineered animals as well as mouse and human whole genome association approaches are needed to identify genes and/or pathways that either mediate the effects of air pollutants, are influenced by them or could be targeted for intervention in the future.
Conclusions
Cumulative epidemiological data support the association of exposure to air pollution with cardiovascular morbidity and mortality, mostly in relation to its particulate matter components. Experimental animal work using hypercholesterolemic rabbits and apoE null mice shows that ambient PM exposure promotes atherosclerosis and that the smaller the particles, the greater the proatherogenic effects. Enhancement of atherosclerosis correlates with the induction of systemic prooxidant and proinflammatory effects although the mechanism for transduction of these effects is not clear. UFP particles may be more toxic based on their greater number, larger content of redox active compounds such as PAHs, greater surface-to-mass ratio and bioavailability of chemically active constituents. Much work is needed to better characterize the main toxic compounds, mechanism(s) of pathogenesis, types of genetic susceptibility that exposed individuals may exhibit, relevant gene-environment interactions in the induction of those effects and development of a biomarker of exposure, degree of damage or susceptibility.
References
Pope CA 3rd, Thun MJ, Namboodiri MM, Dockery DW, Evans JS, Speizer FE, Heath CW Jr: Particulate air pollution as a predictor of mortality in a prospective study of U.S. adults. Am J Respir Crit Care Med 1995,151(3 Pt 1):669–674.
Samet JM, Dominici F, Curriero FC, Coursac I, Zeger SL: Fine particulate air pollution and mortality in 20 U.S. cities, 1987–1994. N Engl J Med 2000,343(24):1742–1749. 10.1056/NEJM200012143432401
Dockery DW, Pope CA 3rd, Xu X, Spengler JD, Ware JH, Fay ME, Ferris BG Jr, Speizer FE: An association between air pollution and mortality in six U.S. cities. N Engl J Med 1993,329(24):1753–1759. 10.1056/NEJM199312093292401
Pope CA 3rd, Burnett RT, Thurston GD, Thun MJ, Calle EE, Krewski D, Godleski JJ: Cardiovascular mortality and long-term exposure to particulate air pollution: epidemiological evidence of general pathophysiological pathways of disease. Circulation 2004,109(1):71–77. 10.1161/01.CIR.0000108927.80044.7F
Bhatnagar A: Environmental cardiology: studying mechanistic links between pollution and heart disease. Circ Res 2006,99(7):692–705. 10.1161/01.RES.0000243586.99701.cf
Brook RD: Cardiovascular effects of air pollution. Clin Sci (Lond) 2008,115(6):175–187. 10.1042/CS20070444
Brook RD, Franklin B, Cascio W, Hong Y, Howard G, Lipsett M, Luepker R, Mittleman M, Samet J, Smith SC Jr, et al.: Air pollution and cardiovascular disease: a statement for healthcare professionals from the Expert Panel on Population and Prevention Science of the American Heart Association. Circulation 2004,109(21):2655–2671. 10.1161/01.CIR.0000128587.30041.C8
USEPA: Air quality criteria for particulate matter (Final Report, Oct 2004). U.S. Environmental Protection Agency, Washington, DC, EPA 600/P-99/002aF-bF; 2004.
Pope CA 3rd, Verrier RL, Lovett EG, Larson AC, Raizenne ME, Kanner RE, Schwartz J, Villegas GM, Gold DR, Dockery DW: Heart rate variability associated with particulate air pollution. Am Heart J 1999,138(5 Pt 1):890–899. 10.1016/S0002-8703(99)70014-1
Gold DR, Litonjua A, Schwartz J, Lovett E, Larson A, Nearing B, Allen G, Verrier M, Cherry R, Verrier R: Ambient pollution and heart rate variability. Circulation 2000,101(11):1267–1273.
Peters A, Perz S, Doring A, Stieber J, Koenig W, Wichmann HE: Increases in heart rate during an air pollution episode. Am J Epidemiol 1999,150(10):1094–1098.
Ibald-Mulli A, Stieber J, Wichmann HE, Koenig W, Peters A: Effects of air pollution on blood pressure: a population-based approach. Am J Public Health 2001,91(4):571–577. 10.2105/AJPH.91.4.571
Brook RD, Brook JR, Urch B, Vincent R, Rajagopalan S, Silverman F: Inhalation of Fine Particulate Air Pollution and Ozone Causes Acute Arterial Vasoconstriction in Healthy Adults. Circulation 2002,105(13):1534–1536. 10.1161/01.CIR.0000013838.94747.64
Simkhovich BZ, Kleinman MT, Kloner RA: Air pollution and cardiovascular injury epidemiology, toxicology, and mechanisms. J Am Coll Cardiol 2008,52(9):719–726. 10.1016/j.jacc.2008.05.029
Dominici F, McDermott A, Daniels M, Zeger SL, Samet JM: Mortality among residents of 90 cities. In Revised Analyses of Time-Series Studies of Air Pollution and Health. Boston, MA: Health Effects Institute; 2003:9–24.
Analitis A, Katsouyanni K, Dimakopoulou K, Samoli E, Nikoloulopoulos AK, Petasakis Y, Touloumi G, Schwartz J, Anderson HR, Cambra K, et al.: Short-term effects of ambient particles on cardiovascular and respiratory mortality. Epidemiology 2006,17(2):230–233. 10.1097/01.ede.0000199439.57655.6b
Pope CA 3rd, Muhlestein JB, May HT, Renlund DG, Anderson JL, Horne BD: Ischemic heart disease events triggered by short-term exposure to fine particulate air pollution. Circulation 2006,114(23):2443–2448. 10.1161/CIRCULATIONAHA.106.636977
Laden F, Schwartz J, Speizer FE, Dockery DW: Reduction in fine particulate air pollution and mortality: Extended follow-up of the Harvard Six Cities study. Am J Respir Crit Care Med 2006,173(6):667–672. 10.1164/rccm.200503-443OC
Miller KA, Siscovick DS, Sheppard L, Shepherd K, Sullivan JH, Anderson GL, Kaufman JD: Long-term exposure to air pollution and incidence of cardiovascular events in women. N Engl J Med 2007,356(5):447–458. 10.1056/NEJMoa054409
Kabir Z: Decrease in U.S. deaths from coronary disease. N Engl J Med 2007,357(9):941. author reply 941. 10.1056/NEJMc071905
Stolzel M, Breitner S, Cyrys J, Pitz M, Wolke G, Kreyling W, Heinrich J, Wichmann HE, Peters A: Daily mortality and particulate matter in different size classes in Erfurt, Germany. J Expo Sci Environ Epidemiol 2007,17(5):458–467. 10.1038/sj.jes.7500538
Wichmann HE, Spix C, Tuch T, Wolke G, Peters A, Heinrich J, Kreyling WG, Heyder J: Daily mortality and fine and ultrafine particles in Erfurt, Germany part I: role of particle number and particle mass. Res Rep Health Eff Inst 2000, (98):5–86. discussion 87–94.
Peters A, Dockery DW, Muller JE, Mittleman MA: Increased particulate air pollution and the triggering of myocardial infarction. Circulation 2001,103(23):2810–2815.
Peters A, von Klot S, Heier M, Trentinaglia I, Hormann A, Wichmann HE, Lowel H: Exposure to traffic and the onset of myocardial infarction. N Engl J Med 2004,351(17):1721–1730. 10.1056/NEJMoa040203
D'Ippoliti D, Forastiere F, Ancona C, Agabiti N, Fusco D, Michelozzi P, Perucci CA: Air pollution and myocardial infarction in Rome: a case-crossover analysis. Epidemiology 2003,14(5):528–535. 10.1097/01.ede.0000082046.22919.72
Rich DQ, Schwartz J, Mittleman MA, Link M, Luttmann-Gibson H, Catalano PJ, Speizer FE, Dockery DW: Association of short-term ambient air pollution concentrations and ventricular arrhythmias. Am J Epidemiol 2005,161(12):1123–1132. 10.1093/aje/kwi143
Wellenius GA, Bateson TF, Mittleman MA, Schwartz J: Particulate air pollution and the rate of hospitalization for congestive heart failure among medicare beneficiaries in Pittsburgh, Pennsylvania. Am J Epidemiol 2005,161(11):1030–1036. 10.1093/aje/kwi135
Wellenius GA, Schwartz J, Mittleman MA: Air pollution and hospital admissions for ischemic and hemorrhagic stroke among medicare beneficiaries. Stroke 2005,36(12):2549–2553. 10.1161/01.STR.0000189687.78760.47
Ghio AJ, Hall A, Bassett MA, Cascio WE, Devlin RB: Exposure to concentrated ambient air particles alters hematologic indices in humans. Inhal Toxicol 2003,15(14):1465–1478. 10.1080/08958370390249111
Mutlu GM, Green D, Bellmeyer A, Baker CM, Burgess Z, Rajamannan N, Christman JW, Foiles N, Kamp DW, Ghio AJ, et al.: Ambient particulate matter accelerates coagulation via an IL-6-dependent pathway. J Clin Invest 2007,117(10):2952–2961. 10.1172/JCI30639
Mills NL, Tornqvist H, Gonzalez MC, Vink E, Robinson SD, Soderberg S, Boon NA, Donaldson K, Sandstrom T, Blomberg A, et al.: Ischemic and thrombotic effects of dilute diesel-exhaust inhalation in men with coronary heart disease. N Engl J Med 2007,357(11):1075–1082. 10.1056/NEJMoa066314
Chuang KJ, Coull BA, Zanobetti A, Suh H, Schwartz J, Stone PH, Litonjua A, Speizer FE, Gold DR: Particulate Air Pollution as a Risk Factor for ST-Segment Depression in Patients With Coronary Artery Disease. Circulation 2008,118(13):1314–1320. 10.1161/CIRCULATIONAHA.108.765669
Kunzli N, Jerrett M, Mack WJ, Beckerman B, LaBree L, Gilliland F, Thomas D, Peters J, Hodis HN: Ambient air pollution and atherosclerosis in Los Angeles. Environ Health Perspect 2005,113(2):201–206.
Hoffmann B, Moebus S, Mohlenkamp S, Stang A, Lehmann N, Dragano N, Schmermund A, Memmesheimer M, Mann K, Erbel R, et al.: Residential exposure to traffic is associated with coronary atherosclerosis. Circulation 2007,116(5):489–496. 10.1161/CIRCULATIONAHA.107.693622
Diez Roux AV, Auchincloss AH, Franklin TG, Raghunathan T, Barr RG, Kaufman J, Astor B, Keeler J: Long-term exposure to ambient particulate matter and prevalence of subclinical atherosclerosis in the Multi-Ethnic Study of Atherosclerosis. Am J Epidemiol 2008,167(6):667–675. 10.1093/aje/kwm359
Delfino RJ, Staimer N, Tjoa T, Polidori A, Arhami M, Gillen DL, Kleinman MT, Vaziri ND, Longhurst J, Zaldivar F, et al.: Circulating biomarkers of inflammation, antioxidant activity, and platelet activation are associated with primary combustion aerosols in subjects with coronary artery disease. Environ Health Perspect 2008,116(7):898–906.
Ruckerl R, Phipps RP, Schneider A, Frampton M, Cyrys J, Oberdorster G, Wichmann HE, Peters A: Ultrafine particles and platelet activation in patients with coronary heart disease--results from a prospective panel study. Part Fibre Toxicol 2007, 4: 1. 10.1186/1743-8977-4-1
Suwa T, Hogg JC, Quinlan KB, Ohgami A, Vincent R, van Eeden SF: Particulate air pollution induces progression of atherosclerosis. J Am Coll Cardiol 2002,39(6):935–942. 10.1016/S0735-1097(02)01715-1
Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W Jr, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW: A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1995,92(5):1355–1374.
Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W Jr, Rosenfeld ME, Schaffer SA, Schwartz CJ, Wagner WD, Wissler RW: A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1994,89(5):2462–2478.
Chen LC, Nadziejko C: Effects of subchronic exposures to concentrated ambient particles (CAPs) in mice. V. CAPs exacerbate aortic plaque development in hyperlipidemic mice. Inhal Toxicol 2005,17(4–5):217–224. 10.1080/08958370590912815
Sun Q, Wang A, Jin X, Natanzon A, Duquaine D, Brook RD, Aguinaldo JG, Fayad ZA, Fuster V, Lippmann M, et al.: Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. Jama 2005,294(23):3003–3010. 10.1001/jama.294.23.3003
Sun Q, Yue P, Kirk RI, Wang A, Moatti D, Jin X, Lu B, Schecter AD, Lippmann M, Gordon T, et al.: Ambient air particulate matter exposure and tissue factor expression in atherosclerosis. Inhal Toxicol 2008,20(2):127–137. 10.1080/08958370701821482
Araujo JA, Barajas B, Kleinman M, Wang X, Bennett BJ, Gong KW, Navab M, Harkema J, Sioutas C, Lusis AJ, et al.: Ambient particulate pollutants in the ultrafine range promote early atherosclerosis and systemic oxidative stress. Circ Res 2008,102(5):589–596. 10.1161/CIRCRESAHA.107.164970
Mills NL, Donaldson K, Hadoke PW, Boon NA, MacNee W, Cassee FR, Sandstrom T, Blomberg A, Newby DE: Adverse cardiovascular effects of air pollution. Nat Clin Pract Cardiovasc Med 2009,6(1):36–44. 10.1038/ncpcardio1399
Nel AE, Diaz-Sanchez D, Ng D, Hiura T, Saxon A: Enhancement of allergic inflammation by the interaction between diesel exhaust particles and the immune system. J Allergy Clin Immunol 1998,102(4 Pt 1):539–554. 10.1016/S0091-6749(98)70269-6
Frampton MW: Systemic and cardiovascular effects of airway injury and inflammation: ultrafine particle exposure in humans. Environ Health Perspect 2001,109(Suppl 4):529–532. 10.2307/3454664
Nel AE, Diaz-Sanchez D, Li N: The role of particulate pollutants in pulmonary inflammation and asthma: evidence for the involvement of organic chemicals and oxidative stress. Curr Opin Pulm Med 2001,7(1):20–26. 10.1097/00063198-200101000-00004
Utell MJ, Frampton MW: Acute health effects of ambient air pollution: the ultrafine particle hypothesis. J Aerosol Med 2000,13(4):355–359. 10.1089/jam.2000.13.355
Lippmann M, Frampton M, Schwartz J, Dockery D, Schlesinger R, Koutrakis P, Froines J, Nel A, Finkelstein J, Godleski J, et al.: The US Environmental Protection Agency particulate matter health effects research centers program: A midcourse report of status, progress, and plans. Environ Health Persp 2003, 111: 1074–1092.
Hiura TS, Kaszubowski MP, Li N, Nel AE: Chemicals in diesel exhaust particles generate reactive oxygen radicals and induce apoptosis in macrophages. J Immunol 1999,163(10):5582–5591.
Li N, Oberley TD, Sempf JM, Nel AE: Comparison of the pro-oxidative and proinflammatory effects of organic diesel exhaust particle chemicals in bronchial epithelial cells and macrophages. J Immunol 2002,169(8):4531–4541. 10.1080/00984100290071658
Whitekus MJ, Li N, Zhang M, Wang M, Horwitz MA, Nelson SK, Horwitz LD, Brechun N, Diaz-Sanchez D, Nel AE: Thiol antioxidants inhibit the adjuvant effects of aerosolized diesel exhaust particles in a murine model for ovalbumin sensitization. J Immunol 2002,168(5):2560–2567.
Xiao GG, Wang M, Li N, Loo JA, Nel AE: Use of proteomics to demonstrate a hierarchical oxidative stress response to diesel exhaust particle chemicals in a macrophage cell line. J Biol Chem 2003,278(50):50781–50790. 10.1074/jbc.M306423200
Bowler RP, Crapo JD: Oxidative stress in allergic respiratory diseases. J Allergy Clin Immunol 2002,110(3):349–356. 10.1067/mai.2002.126780
Li N, Kim S, Wang M, Froines J, Sioutas C, Nel A: Use of a stratified oxidative stress model to study the biological effects of ambient concentrated and diesel exhaust particulate matter. Inhal Toxicol 2002,14(5):459–486. 10.1080/089583701753678571
Takizawa H, Ohtoshi T, Kawasaki S, Kohyama T, Desaki M, Kasama T, Kobayashi K, Nakahara K, Yamamoto K, Matsushima K, et al.: Diesel exhaust particles induce NF-kappa B activation in human bronchial epithelial cells in vitro: importance in cytokine transcription. J Immunol 1999,162(8):4705–4711.
Bai Y, Suzuki AK, Sagai M: The cytotoxic effects of diesel exhaust particles on human pulmonary artery endothelial cells in vitro: role of active oxygen species. Free Radic Biol Med 2001,30(5):555–562. 10.1016/S0891-5849(00)00499-8
Hirano S, Furuyama A, Koike E, Kobayashi T: Oxidative-stress potency of organic extracts of diesel exhaust and urban fine particles in rat heart microvessel endothelial cells. Toxicology 2003,187(2–3):161–170. 10.1016/S0300-483X(03)00053-2
Gong KW, Zhao W, Li N, Barajas B, Kleinman M, Sioutas C, Horvath S, Lusis AJ, Nel A, Araujo JA: Air-pollutant chemicals and oxidized lipids exhibit genome-wide synergistic effects on endothelial cells. Genome Biol 2007,8(7):R149. 10.1186/gb-2007-8-7-r149
Li N, Alam J, Venkatesan MI, Eiguren-Fernandez A, Schmitz D, Di Stefano E, Slaughter N, Killeen E, Wang X, Huang A, et al.: Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J Immunol 2004,173(5):3467–3481.
Zhang W, Contag PR, Hardy J, Zhao H, Vreman HJ, Hajdena-Dawson M, Wong RJ, Stevenson DK, Contag CH: Selection of potential therapeutics based on in vivo spatiotemporal transcription patterns of heme oxygenase-1. J Mol Med 2002,80(10):655–664. 10.1007/s00109-002-0375-x
Tolocka MP, Lake DA, Johnston MV, Wexler AS: Size-resolved fine and ultrafine particle composition in Baltimore, Maryland. Journal of Geophysical Research-Atmospheres 2005.,110(D7):
Kumagai Y, Arimoto T, Shinyashiki M, Shimojo N, Nakai Y, Yoshikawa T, Sagai M: Generation of reactive oxygen species during interaction of diesel exhaust particle components with NADPH-cytochrome P450 reductase and involvement of the bioactivation in the DNA damage. Free Radic Biol Med 1997,22(3):479–487. 10.1016/S0891-5849(96)00341-3
Penning TM, Burczynski ME, Hung CF, McCoull KD, Palackal NT, Tsuruda LS: Dihydrodiol dehydrogenases and polycyclic aromatic hydrocarbon activation: generation of reactive and redox active o-quinones. Chem Res Toxicol 1999,12(1):1–18. 10.1021/tx980143n
Monks TJ, Hanzlik RP, Cohen GM, Ross D, Graham DG: Quinone chemistry and toxicity. Toxicol Appl Pharmacol 1992,112(1):2–16. 10.1016/0041-008X(92)90273-U
Li N, Sioutas C, Cho A, Schmitz D, Misra C, Sempf J, Wang M, Oberley T, Froines J, Nel A: Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ Health Perspect 2003,111(4):455–460.
Ntziachristos L, Froines J, Cho A, Sioutas C: Relationship between redox activity and chemical speciation of size-fractionated particulate matter. Particle and Fibre Toxicology 2007,4(1):5. 10.1186/1743-8977-4-5
Ayres JG, Borm P, Cassee FR, Castranova V, Donaldson K, Ghio A, Harrison RM, Hider R, Kelly F, Kooter IM, et al.: Evaluating the Toxicity of Airborne Particulate Matter and Nanoparticles by Measuring Oxidative Stress Potential - A Workshop Report and Consensus Statement. Inhalation Toxicology 2008,20(1):75–99. 10.1080/08958370701665517
Ross R: Atherosclerosis--an inflammatory disease. N Engl J Med 1999,340(2):115–126. 10.1056/NEJM199901143400207
Glass CK, Witztum JL: Atherosclerosis. the road ahead. Cell 2001,104(4):503–516. 10.1016/S0092-8674(01)00238-0
Watson AD, Berliner JA, Hama SY, La Du BN, Faull KF, Fogelman AM, Navab M: Protective effect of high density lipoprotein associated paraoxonase. Inhibition of the biological activity of minimally oxidized low density lipoprotein. J Clin Invest 1995,96(6):2882–2891. 10.1172/JCI118359
Lusis AJ: Atherosclerosis. Nature 2000,407(6801):233–241. 10.1038/35025203
Camejo G, Olofsson SO, Lopez F, Carlsson P, Bondjers G: Identification of Apo B-100 segments mediating the interaction of low density lipoproteins with arterial proteoglycans. Arteriosclerosis 1988,8(4):368–377.
Frank JS, Fogelman AM: Ultrastructure of the intima in WHHL and cholesterol-fed rabbit aortas prepared by ultra-rapid freezing and freeze-etching. J Lipid Res 1989,30(7):967–978.
Navab M, Berliner JA, Watson AD, Hama SY, Territo MC, Lusis AJ, Shih DM, Van Lenten BJ, Frank JS, Demer LL, et al.: The Yin and Yang of oxidation in the development of the fatty streak. A review based on the 1994 George Lyman Duff Memorial Lecture. Arterioscler Thromb Vasc Biol 1996,16(7):831–842.
Steinberg D: Low density lipoprotein oxidation and its pathobiological significance. J Biol Chem 1997,272(34):20963–20966. 10.1074/jbc.272.34.20963
Khoo JC, Miller E, McLoughlin P, Steinberg D: Enhanced macrophage uptake of low density lipoprotein after self-aggregation. Arteriosclerosis 1988,8(4):348–358.
Quinn MT, Parthasarathy S, Fong LG, Steinberg D: Oxidatively modified low density lipoproteins: a potential role in recruitment and retention of monocyte/macrophages during atherogenesis. Proc Natl Acad Sci USA 1987,84(9):2995–2998. 10.1073/pnas.84.9.2995
Rajavashisth TB, Andalibi A, Territo MC, Berliner JA, Navab M, Fogelman AM, Lusis AJ: Induction of endothelial cell expression of granulocyte and macrophage colony-stimulating factors by modified low-density lipoproteins. Nature 1990,344(6263):254–257. 10.1038/344254a0
Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, Rollins BJ: Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell 1998,2(2):275–281. 10.1016/S1097-2765(00)80139-2
Nurkiewicz TR, Porter DW, Barger M, Millecchia L, Rao KM, Marvar PJ, Hubbs AF, Castranova V, Boegehold MA: Systemic microvascular dysfunction and inflammation after pulmonary particulate matter exposure. Environ Health Perspect 2006,114(3):412–419.
Nurkiewicz TR, Porter DW, Barger M, Castranova V, Boegehold MA: Particulate matter exposure impairs systemic microvascular endothelium-dependent dilation. Environ Health Perspect 2004,112(13):1299–1306.
Oberdorster G, Oberdorster E, Oberdorster J: Nanotoxicology: an emerging discipline evolving from studies of ultrafine particles. Environ Health Perspect 2005,113(7):823–839.
International Commission on Radiological Protection: Human respiratory model for radiological protection. Ann IRCP 1994, 24: 1–300.
Sun Q, Yue P, Deiuliis JA, Lumeng CN, Kampfrath T, Mikolaj MB, Cai Y, Ostrowski MC, Lu B, Parthasarathy S, et al.: Ambient air pollution exaggerates adipose inflammation and insulin resistance in a mouse model of diet-induced obesity. Circulation 2009,119(4):538–546. 10.1161/CIRCULATIONAHA.108.799015
Assmann G, Gotto AM Jr: HDL cholesterol and protective factors in atherosclerosis. Circulation 2004,109(23 Suppl 1):III8–14.
Barter PJ, Rye KA: Relationship between the concentration and antiatherogenic activity of high-density lipoproteins. Curr Opin Lipidol 2006,17(4):399–403. 10.1097/01.mol.0000236365.40969.af
Rader DJ: Mechanisms of disease: HDL metabolism as a target for novel therapies. Nat Clin Pract Cardiovasc Med 2007,4(2):102–109. 10.1038/ncpcardio0768
Fielding CJ, Fielding PE: Molecular physiology of reverse cholesterol transport. J Lipid Res 1995,36(2):211–228.
Nofer JR, Kehrel B, Fobker M, Levkau B, Assmann G, von Eckardstein A: HDL and arteriosclerosis: beyond reverse cholesterol transport. Atherosclerosis 2002,161(1):1–16. 10.1016/S0021-9150(01)00651-7
Barter PJ, Nicholls S, Rye KA, Anantharamaiah GM, Navab M, Fogelman AM: Antiinflammatory properties of HDL. Circ Res 2004,95(8):764–772. 10.1161/01.RES.0000146094.59640.13
Nicholls SJ, Zheng L, Hazen SL: Formation of dysfunctional high-density lipoprotein by myeloperoxidase. Trends Cardiovasc Med 2005,15(6):212–219. 10.1016/j.tcm.2005.06.004
Ansell BJ, Navab M, Hama S, Kamranpour N, Fonarow G, Hough G, Rahmani S, Mottahedeh R, Dave R, Reddy ST: Inflammatory/antiinflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation 2003,108(22):2751–2756. 10.1161/01.CIR.0000103624.14436.4B
Van Lenten BJ, Wagner AC, Navab M, Antharamaiah GM, Hama ST, Fogelman AM, Reddy ST, et al.: Lipoprotein inflammatory properties and serum amyloid A levels but not cholesterol levels predict lesion area in cholesterol-fed rabbits. J Lipid Res 2007 2003,48(11):2344–2353. 10.1194/jlr.M700138-JLR200
Kreyling WG, Semmler M, Erbe F, Takenaka S, Mayer P, Schulz H, Oberdorster G, Ziesenis A: Translocation of ultrafine insoluble iridium particles from lung epithelium to extrapulmonary organs is size dependent but very low. Journal of Toxicology and Environmental Health-Part A 2002,65(20):1513–1530. 10.1194/jlr.M700138-JLR200
Nemmar A, Vanbilloen H, Hoylaerts MF, Hoet PH, Takenaka S, Verbruggen A, Nemery B: Translocation of ultrafine insoluble iridium particles from lung epithelium to extrapulmonary organs is size dependent but very low. Am J Respir Crit Care Med 2001,164(9):1665–1668.
Oberdorster G, Sharp Z, Atudorei V, Elder A, Gelein R, Lunts A, Kreyling W, Cox C: Extrapulmonary translocation of ultrafine carbon particles following whole-body inhalation exposure of rats. Journal of Toxicology and Environmental Health-Part A 2002,65(20):1531–1543. 10.1080/00984100290071658
Nemmar A, Hoet PH, Vanquickenborne B, Dinsdale D, Thomeer M, Hoylaerts MF, Vanbilloen H, Mortelmans L, Nemery B: Passage of inhaled particles into the blood circulation in humans. Circulation 2002,105(4):411–414. 10.1161/hc0402.104118
Pope CA 3rd, Dockery DW: Health effects of fine particulate air pollution: lines that connect. J Air Waste Manag Assoc 2006,56(6):709–742.
Sioutas C, Delfino RJ, Singh M: Exposure assessment for atmospheric ultrafine particles (UFPs) and implications in epidemiologic research. Environ Health Perspect 2005,113(8):947–955.
Oberdorster G: Pulmonary effects of inhaled ultrafine particles. Int Arch Occup Environ Health 2001,74(1):1–8. 10.1007/s004200000185
Brown DM, Wilson MR, MacNee W, Stone V, Donaldson K: Size-dependent proinflammatory effects of ultrafine polystyrene particles: a role for surface area and oxidative stress in the enhanced activity of ultrafines. Toxicol Appl Pharmacol 2001,175(3):191–199. 10.1006/taap.2001.9240
Tran CL, Buchanan D, Cullen RT, Searl A, Jones AD, Donaldson K: Inhalation of poorly soluble particles. II. Influence of particle surface area on inflammation and clearance. Inhal Toxicol 2000,12(12):1113–1126. 10.1080/08958370050166796
Daigle CC, Chalupa DC, Gibb FR, Morrow PE, Oberdorster G, Utell MJ, Frampton MW: Ultrafine particle deposition in humans during rest and exercise. Inhal Toxicol 2003,15(6):539–552. 10.1080/08958370304468
Donaldson K, Brown D, Clouter A, Duffin R, MacNee W, Renwick L, Tran L, Stone V: The pulmonary toxicology of ultrafine particles. J Aerosol Med 2002,15(2):213–220. 10.1089/089426802320282338
Chalupa DC, Morrow PE, Oberdorster G, Utell MJ, Frampton MW: Ultrafine particle deposition in subjects with asthma. Environ Health Perspect 2004,112(8):879–882.
Peters A, Veronesi B, Calderon-Garciduenas L, Gehr P, Chen L, Geiser M, Reed W, Rothen-Rutishauser B, Schurch S, Schulz H: Translocation and potential neurological effects of fine and ultrafine particles a critical update. Particle and Fibre Toxicology 2006,3(1):13. 10.1186/1743-8977-3-13
Geiser M, Rothen-Rutishauser B, Kapp N, Schurch S, Kreyling W, Schulz H, Semmler M, Im Hof V, Heyder J, Gehr P: Ultrafine particles cross cellular membranes by nonphagocytic mechanisms in lungs and in cultured cells. Environ Health Perspect 2005,113(11):1555–1560.
Briggs D: Environmental pollution and the global burden of disease. Br Med Bull 2003, 68: 1–24. 10.1093/bmb/ldg019
Koken PJ, Piver WT, Ye F, Elixhauser A, Olsen LM, Portier CJ: Temperature, air pollution, and hospitalization for cardiovascular diseases among elderly people in Denver. Environ Health Perspect 2003,111(10):1312–1317.
Anderson HR, Atkinson RW, Bremner SA, Marston L: Particulate air pollution and hospital admissions for cardiorespiratory diseases: are the elderly at greater risk? Eur Respir J Suppl 2003, 40: 39s-46s. 10.1183/09031936.03.00402203
Goldberg MS, Burnett RT, Valois MF, Flegel K, Bailar JC 3rd, Brook J, Vincent R, Radon K: Associations between ambient air pollution and daily mortality among persons with congestive heart failure. Environ Res 2003,91(1):8–20. 10.1016/S0013-9351(02)00022-1
Islam T, Berhane K, McConnell R, Gauderman WJ, Avol E, Peters JM, Gilliland FD: Glutathione-S-transferase (GST) P1, GSTM1, exercise, ozone and asthma incidence in school children. Thorax 2009,64(3):197–202. 10.1136/thx.2008.099366
Islam T, McConnell R, Gauderman WJ, Avol E, Peters JM, Gilliland FD: Ozone, oxidant defense genes, and risk of asthma during adolescence. Am J Respir Crit Care Med 2008,177(4):388–395. 10.1164/rccm.200706-863OC
Anderson HR, Atkinson RW, Peacock JL, Sweeting MJ, Marston L: Ambient particulate matter and health effects: publication bias in studies of short-term associations. Epidemiology 2005,16(2):155–163. 10.1097/01.ede.0000152528.22746.0f
Lund AK, Knuckles TL, Obot Akata C, Shohet R, McDonald JD, Gigliotti A, Seagrave JC, Campen MJ: Gasoline exhaust emissions induce vascular remodeling pathways involved in atherosclerosis. Toxicol Sci 2007,95(2):485–494. 10.1093/toxsci/kfl145
Lund AK, Lucero J, Lucas S, Madden MC, McDonald JD, Seagrave JC, Knuckles TL, Campen MJ: Vehicular Emissions Induce Vascular MMP-9 Expression and Activity Associated With Endothelin-1-Mediated Pathways. Arterioscler Thromb Vasc Biol 2009.
de Nooijer R, Verkleij CJ, von der Thusen JH, Jukema JW, Wall EE, van Berkel TJ, Baker AH, Biessen EA: Lesional overexpression of matrix metalloproteinase-9 promotes intraplaque hemorrhage in advanced lesions but not at earlier stages of atherogenesis. Arterioscler Thromb Vasc Biol 2006,26(2):340–346. 10.1161/01.ATV.0000197795.56960.64
Newby AC: Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol Rev 2005,85(1):1–31. 10.1152/physrev.00048.2003
Dominici F, Peng RD, Bell ML, Pham L, McDermott A, Zeger SL, Samet JM: Fine particulate air pollution and hospital admission for cardiovascular and respiratory diseases. Jama 2006,295(10):1127–1134. 10.1001/jama.295.10.1127
Sullivan J, Ishikawa N, Sheppard L, Siscovick D, Checkoway H, Kaufman J: Exposure to ambient fine particulate matter and primary cardiac arrest among persons with and without clinically recognized heart disease. Am J Epidemiol 2003,157(6):501–509. 10.1093/aje/kwg015
Li N, Hao M, Phalen RF, Hinds WC, Nel AE: Particulate air pollutants and asthma. A paradigm for the role of oxidative stress in PM-induced adverse health effects. Clin Immunol 2003,109(3):250–265. 10.1016/j.clim.2003.08.006
Stout DB, Chatziioannou AF, Lawson TP, Silverman RW, Gambhir SS, Phelps ME: Small animal imaging center design: the facility at the UCLA Crump Institute for Molecular Imaging. Mol Imaging Biol 2005,7(6):393–402. 10.1007/s11307-005-0015-2
Bhaumik S, Gambhir SS: Optical imaging of Renilla luciferase reporter gene expression in living mice. Proc Natl Acad Sci USA 2002,99(1):377–382. 10.1073/pnas.012611099
Araujo JA, Lusis AJ: Atherosclerosis. In Encyclopedia of Endocrine Diseases. Volume 1. Edited by: Martini L. Elsevier Inc; 2004:299–294.
Acknowledgements
This study was supported by US Public Health Service Grants to the UCLA Asthma and Allergic Disease Clinical Research Center (U19 AI070453), RO1 ES13432, RO1 ES015498, RO1 ES 016746, RO1 ES016959 as well as the U.S. EPA STAR award (RD-83241301) to the Southern California Particle Center. This work has not been subjected to the EPA for peer and policy review. JAA was also supported by a Robert Wood Johnson Foundation AMFDP Award. We thank Dr. Aldons J. Lusis for his valuable support and intellectual contribution to our studies.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Both JAA and AEN participated in the conception, design, drafting and critical revision of the manuscript. Both authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Araujo, J.A., Nel, A.E. Particulate matter and atherosclerosis: role of particle size, composition and oxidative stress. Part Fibre Toxicol 6, 24 (2009). https://doi.org/10.1186/1743-8977-6-24
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-8977-6-24