
Abstract
Purpose
Pheochromocytoma (PCC) and paraganglioma (PG) are evaluated and treated similarly. This study evaluates the hypothesis that tumor characteristics and outcome of patients with PCC and PG are equivalent.
Methods
Records of patients from a single institution undergoing resection of PCC or PG from 1999 to 2010 were reviewed. Data were collected for demographics, operative records, laboratory and pathologic results, adjuvant and palliative therapy given, recurrence, and length of survival. Descriptive statistics were used to describe differences between patients with benign and malignant PCC and PG. Analysis was performed using the Wilcoxon–Mann–Whitney test with p = 0.05 considered as significant.
Results
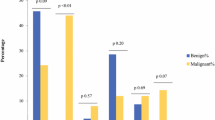
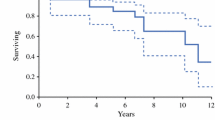
One hundred fifteen patients were identified (106 PCC and nine PG). Of the tumors, 5.2% were bilateral and 10.4% were malignant. Forty-three of the 115 patients underwent genetic testing; 21out of 37 (56.8%) PCC and five out of six (83.3%) PG had a genetic mutation. Twelve patients (seven PCC and five PG) had malignant tumors. Malignant PG (mPG) exhibited more invasive pathologic characteristics. The median sizes of benign and malignant PCC (mPCC) were 4.0 (0.7–14 cm) and 5.5 cm (3.7–11.2 cm), respectively, p = 0.03. The median sizes of benign and mPG were 4.1 (2.7–5.4 cm) and 5.8 cm (4–6.2 cm), respectively, p = 0.11. Sites of recurrence were similar between the groups. Patients with mPG received chemotherapy more often than those with mPCC. With a median follow-up of 54.7 months (2.0–185.3), two out of five mPG and zero out of seven mPCC had died of the disease.
Conclusion
Tumor size does not appear to correlate with malignancy in a clinically significant manner. Malignant paraganglioma may be more aggressive than malignant pheochromocytoma and is frequently offered more adjuvant therapy. PCC and PG should be evaluated separately in future analyses of these diseases.


Similar content being viewed by others

References
Golden SH, Robinson KA, Saldanha I, Anton B, Ladenson PW (2009) Clinical review: prevalence and incidence of endocrine and metabolic disorders in the United States: a comprehensive review. J Clin Endocrinol Metab 94(6):1853–1878
Reisch N, Peczkowska M, Januszewicz A, Neumann HP (2006) Pheochromocytoma: presentation, diagnosis and treatment. J Hypertens 24(12):2331–2339
Anderson GH Jr, Blakeman N, Streeten DH (1994) The effect of age on prevalence of secondary forms of hypertension in 4429 consecutively referred patients. J Hypertens 12(5):609–615
Omura M, Saito J, Yamaguchi K, Kakuta Y, Nishikawa T (2004) Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res 27(3):193–202
Mantero F, Terzolo M, Arnaldi G, Osella G, Masini AM, Ali A, Giovagnetti M, Opocher G, Angeli A (2000) A survey on adrenal incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. J Clin Endocrinol Metab 85(2):637–644
Mansmann G, Lau J, Balk E, Rothberg M, Miyachi Y, Bornstein SR (2004) The clinically inapparent adrenal mass: update in diagnosis and management. Endocr Rev 25(2):309–340
Lenders JW, Eisenhofer G, Mannelli M, Pacak K (2005) Phaeochromocytoma. Lancet 366(9486):665–675
Jimenez C, Cote G, Arnold A, Gagel RF (2006) Review: should patients with apparently sporadic pheochromocytomas or paragangliomas be screened for hereditary syndromes? J Clin Endocrinol Metab 91(8):2851–2858
Welbourn RB (1987) Early surgical history of phaeochromocytoma. Br J Surg 74(7):594–596
Thompson LD (2002) Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol 26(5):551–566
Sutton MG, Sheps SG, Lie JT (1981) Prevalence of clinically unsuspected pheochromocytoma. Review of a 50-year autopsy series. Mayo Clin Proc 56(6):354–360
Hartley L, Perry-Keene D (1985) Phaeochromocytoma in Queensland—1970–83. Aust N Z J Surg 55(5):471–475
Linnoila RI, Keiser HR, Steinberg SM, Lack EE (1990) Histopathology of benign versus malignant sympathoadrenal paragangliomas: clinicopathologic study of 120 cases including unusual histologic features. Hum Pathol 21(11):1168–1180
van Heerden JA, Roland CF, Carney JA, Sheps SG, Grant CS (1990) Long-term evaluation following resection of apparently benign pheochromocytoma(s)/paraganglioma(s). World J Surg 14(3):325–329
Goldstein RE, O’Neill JA Jr, Holcomb GW 3rd, Morgan WM 3rd, Neblett WW, Oates JA, Brown N, Nadeau J, Smith B, Page DL et al (1999) Clinical experience over 48 years with pheochromocytoma. Ann Surg 229(6):755–764, Discussion 764–756
Ayala-Ramirez M, Feng L, Johnson MM, Ejaz S, Habra MA, Rich T, Busaidy N, Cote GJ, Perrier N, Phan A et al (2011) Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab 96(3):717–725
Maher ER, Eng C (2002) The pressure rises: update on the genetics of phaeochromocytoma. Hum Mol Genet 11(20):2347–2354
Strong VE, Kennedy T, Al-Ahmadie H, Tang L, Coleman J, Fong Y, Brennan M, Ghossein RA (2008) Prognostic indicators of malignancy in adrenal pheochromocytomas: clinical, histopathologic, and cell cycle/apoptosis gene expression analysis. Surgery 143(6):759–768
Agarwal A, Mehrotra PK, Jain M, Gupta SK, Mishra A, Chand G, Agarwal G, Verma AK, Mishra SK, Singh U (2010) Size of the tumor and pheochromocytoma of the adrenal gland scaled score (PASS): can they predict malignancy? World J Surg 34(12):3022–3028
Blake MA, Kalra MK, Maher MM, Sahani DV, Sweeney AT, Mueller PR, Hahn PF, Boland GW (2004) Pheochromocytoma: an imaging chameleon. Radiographics 24(Suppl 1):S87–S99
Shen WT, Sturgeon C, Clark OH, Duh QY, Kebebew E (2004) Should pheochromocytoma size influence surgical approach? A comparison of 90 malignant and 60 benign pheochromocytomas. Surgery 136(6):1129–1137
Miller BS, Ammori JB, Gauger PG, Broome JT, Hammer GD, Doherty GM (2010) Laparoscopic resection is inappropriate in patients with known or suspected adrenocortical carcinoma. World J Surg 34(6):1380–1385
Schteingart DE, Doherty GM, Gauger PG, Giordano TJ, Hammer GD, Korobkin M, Worden FP (2005) Management of patients with adrenal cancer: recommendations of an international consensus conference. Endocr Relat Cancer 12(3):667–680
Shen WT, Grogan R, Vriens M, Clark OH, Duh QY (2010) One hundred two patients with pheochromocytoma treated at a single institution since the introduction of laparoscopic adrenalectomy. Arch Surg 145(9):893–897
Chrisoulidou A, Kaltsas G, Ilias I, Grossman AB (2007) The diagnosis and management of malignant phaeochromocytoma and paraganglioma. Endocr Relat Cancer 14(3):569–585
Erickson D, Kudva YC, Ebersold MJ, Thompson GB, Grant CS, van Heerden JA, Young WF Jr (2001) Benign paragangliomas: clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab 86(11):5210–5216
John H, Ziegler WH, Hauri D, Jaeger P (1999) Pheochromocytomas: can malignant potential be predicted? Urology 53(4):679–683
Nomura K, Kimura H, Shimizu S, Kodama H, Okamoto T, Obara T, Takano K (2009) Survival of patients with metastatic malignant pheochromocytoma and efficacy of combined cyclophosphamide, vincristine, and dacarbazine chemotherapy. J Clin Endocrinol Metab 94(8):2850–2856
Gimenez-Roqueplo AP, Lehnert H, Mannelli M, Neumann H, Opocher G, Maher ER, Plouin PF (2006) Phaeochromocytoma, new genes and screening strategies. Clin Endocrinol (Oxf) 65(6):699–705
Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K et al (2002) Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 346(19):1459–1466
Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de Paillerets B, Chabre O, Chamontin B, Delemer B, Giraud S, Murat A et al (2005) Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol 23(34):8812–8818
Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley TA et al (2004) Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 292(8):943–951
Brouwers FM, Eisenhofer G, Tao JJ, Kant JA, Adams KT, Linehan WM, Pacak K (2006) High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: implications for genetic testing. J Clin Endocrinol Metab 91(11):4505–4509
Burnichon N, Rohmer V, Amar L, Herman P, Leboulleux S, Darrouzet V, Niccoli P, Gaillard D, Chabrier G, Chabolle F et al (2009) The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J Clin Endocrinol Metab 94(8):2817–2827
Amar L, Baudin E, Burnichon N, Peyrard S, Silvera S, Bertherat J, Bertagna X, Schlumberger M, Jeunemaitre X, Gimenez-Roqueplo AP et al (2007) Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab 92(10):3822–3828
Scott HW Jr, Halter SA (1984) Oncologic aspects of pheochromocytoma: the importance of follow-up. Surgery 96(6):1061–1066
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was presented at the ESES workshop in Lyon, France, May 2011.
Rights and permissions
About this article
Cite this article
Laird, A.M., Gauger, P.G., Doherty, G.M. et al. Paraganglioma: not just an extra-adrenal pheochromocytoma. Langenbecks Arch Surg 397, 247–253 (2012). https://doi.org/10.1007/s00423-011-0871-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00423-011-0871-y