
Abstract
Background
Pheochromocytoma and paraganglioma are rare neuroendocrine tumors of the chromaffin tissue, which may produce catecholamines. The aim of our study was to analyze the clinical and para-clinical aspects as well as the therapeutic and evolutionary aspects of pheocromocytomas and paragangliomas based on a series of 40 cases.
Methods
Our retrospective population-based research study includes 40 patients. Then, a statistical analysis was carried out using the SPSS software (version21).
Results
Our study involves 40 patients, including 23 women (57, 5%) and 17 men (42,5%). The mean age at the time of the diagnosis was 43.8 ± 16.8 years. The circumstances of the discovery were mainly characterized by adrenal incidentaloma and hypertension. The biological diagnosis was based on the dosage of urinary metanephrines and plasma-free metanephrines in, respectively, 61.5% and 18% of cases. A computerized tomography scan and/or a magnetic resonance imaging scan could help to locate the tumor in 100% of cases. Our series includes 3 cases of bilateral pheochromocytoma, 3 cases of paragangliomas and 1 case of malignant pheochromocytoma, while a hereditary form was retained in 3 patients. In fact, thirty-two patients were operated; cure was clinically labeled in 100% and biologically in 87.5% of patients.
Conclusions
The main points for improvement that our study has revealed are; a patient follow-up after surgery, which was not always regular, and an insufficient screening for genetic diseases associated with pheochromocytomas and paragangliomas.
Similar content being viewed by others


1 Background
Pheochromocytomas and paragangliomas (PPGL) are rare neuroendocrine tumors of chromaffin cells that produce catecholamines [1]. More precisely, Pheocromocytomas (PHEO) are tumors that arise from chromaffin cells within the adrenal medulla, whereas paragangliomas (PGL) arise from extra-adrenal chromaffin cells of the sympathetic or parasympathetic paravertebral ganglia in the chest, abdomen, and pelvis. PGL may also arise from the chromaffin cells of the parasympathetic ganglia of the head and neck. Approximately, 80 to 85% of chromaffin cell tumors are PHEO (intra-adrenal), and 15 to 20% are PGL (extra-adrenal) [2, 3].
According to the current literature, the estimated prevalence of PPGL ranges from 0.1 to 0.6% of patients with hypertension in a general outpatient clinic [4, 5]. The clinical presentation is highly heterogeneous, ranging from normotensive (having normal blood pressure) cases to patients with sustained hypertension and others with dramatic swings in blood pressure (BP) and even involving hypertensive crises [6, 7]. Over time, non-physiological and excessive release of catecholamines can lead to cardiovascular complications and death. In fact, an adrenalectomy (surgical removal of one or both adrenal glands) is the appropriate treatment [8, 9]. Moreover, postoperative monitoring is clinical and biological in order to detect recurrence and possible metastases. Therefore, through a retrospective study, we propose to analyze the clinical and para-clinical aspects as well as the therapeutic and evolutionary aspects of pheocromocytomas and paragangliomas based on a series of 40 cases.
2 Methods
Our retrospective study includes all patients diagnosed with PPGLs during the period extending from January 2000 to December 2019.
Inclusion criteria: Any patient with PPGL confirmed by the results of biological or anatomopathological assessments. We excluded cases whose investigations were incomplete.
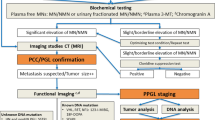
The collected data were epidemiological, clinical, biological, radiological and anatomopathological. They were identified from selected files and transferred to a patient’s standardized record card. As for the biochemical testing for the diagnosis of PPGL, it includes measurements of plasma free metanephrines (PFM) or urinary fractionated metanephrines (UFM). In order to make the interpretation of the results easier, they were reported to the upper cutoffs (UCF) given by the laboratory. In fact, a level four times higher than the UCF enables the diagnosis of the PPGL to be retained [10]. Furthermore, imaging studies are mostly conducted once there is biochemical evidence. The 123I-metaiodobenzylguanidine (MIBG) scintigraphy scan is indicated in our series to detect extra-adrenal forms or metastatic secondary locations.
The statistical analysis was carried out using the SPSS software (version21). Then, the description of the qualitative variables was achieved by determining the absolute and relative frequencies while the quantitative variables were driven by the averages and standard deviations.
3 Results
In fact, our study involves 40 patients, including 23 women (57,5%) and 17 men (42,5%). On the other hand, the average age at the time of diagnosis was 43.8 ± 16.8 years, with extremes that ranged from 10 to 74 years for the 40 patients.
Fourteen patients (35%) of our sample were found to be hypertensive for an average of 6 years before the diagnosis of PPGL (extremes: 6 months to 15 years). Moreover, different antihypertensive drugs were administered to patients, among whom six (42.8%) were on monotherapy, 3 on a combination therapy, 4 on a triple therapy and 1 on a quadritherapy.
On the other hand, the circumstances of discovery (COD) were mainly represented by adrenal incidentalomas (AI) in 23 cases (57,7%). Then, PHEO was discovered on the occasion of an investigation of hypertension (HT) among 12 patients (5 cases of HT among young patients, 5 cases of HT associated with paroxysmal PHEO signs, and 2 cases of drug-resistant HT). In addition, hypertension complications constituted a COD in 3 cases. Furthermore, an ectopic PPGL was discovered fortuitously during an anatomopathological examination of a biopsy of a right renal mass in a 63-year-old patient followed for a large cell B lymphoma. Finally, PPGL was diagnosed as a part of an extension review in a 46-year-old woman with syndromic features, wowing a multiple endocrine neoplasia type 2A (Medullary thyroid cancer, primary hyperparathyroidism, and cutaneous lichen amyloidosis).
In fact, different functional signs were reported by our patients. Palpitation was the most described sign (67.5%) followed by sweat (62.5%) then, headache (57.7%) while the Menard triad was present in 15 patients (37.5%). The UFM dosage was performed in 29 cases (72.5%). An elevation of this latter greater than 4 times the UCF was observed in 23 patients, while five patients showed an elevation between 1.4 and 3 times the UCF. Then, the amount of urine metanephrines was 18.2 times as high as that of the UCF (extreme: 1.4 − 80), while the average level of normetanephrine in urine was 23.2 times as high as that of the UCF (extremes: 1.5 − 165). On the other hand, PFM were dosed in 10 patients (25%). It was elevated in 90% of cases. Moreover, the levels of metanephrine (MNp) and normetanephrine (NMNp) in the blood were 5.7 times as high as those of the UCF for both parameters, with extremes ranging from 2 to 11 and 1.4 to 15 times, respectively. The elevation of PFM was 4 times as high as the UCF in 70% of cases and discrete between 1.4 and 3 times the UCF in 2 cases. On the other hand, the level of Chromogranine A (CGA) in the blood was dosed in seven cases, where it was normal in 2 patients and high in 5 patients with an average of 437.4 µg/l (extremes: 112–1650 µg/l). The Vanillylmandelic acid (VMA) was dosed in only one case in our series. This dosage allowed the diagnosis to be retained with an elevation of 3.8 times the upper reference limit. In general, the biological diagnosis in our series was based on the dosage of the UFM, PFM, and VMA, respectively, in 61.5%, 18% and 2.5% of cases. In fact, the associated dosages help retain the diagnosis at 18%.
The PPGL are a part of a genetic disease in 7,5% in our series (1 case of multiple endocrine neoplasia, type 2A, 1 case of multiple endocrine neoplasia, type 2B and 1 patient diagnosed with neurofibromatosis type 1).
An abdominal pan scan (CT) with contrast was performed in 39 patients. It was prescribed as a first-line imaging modality in 13 patients (32.5%) and preceded by an abdominal ultrasound in 26 patients. In fact, it helps locate the tumor in 100% of cases. The average tumor size was 55.2 mm (extreme: 16-157 mm). The mass was hypodense in 33.3% of cases and heterogeneous in 18% of cases. On the other hand, moderate to intense heterogeneous enhancement was objectified in 97.4% of cases. Abdominal magnetic resonance imaging (MRI) was performed on 15 patients (37.5%). MRI could help locate the tumor in 100% of cases. The tumor was found to have a low signal, T1 and a high signal T2 for all patients. Then, an enhancement was noted in 100% of cases. In total, CT and/or MRI were used to locate the tumor in all patients. The location was intra-adrenal in 92.5%: right adenoma in 23 patients, left adenoma in 11 patients and bilateral in 3 patients. Extra-adrenal location was objectified in 3 patients (pararenal, para-aortic and prostatic PGL). The MIBG scan was performed in 16 patients and increased radiotracer hyperfixation was noted in 93.7% of cases consistent with CT and/or MRI data.
Moreover, surgery was indicated in all patients: Thirty-two of our patients (80%) were operated, 6 patients refused the surgery, adrenalectomy was scheduled for 1 patient, and 1 patient died of a complicated pulmonary embolism with acute lung edema. All of our patients received a preoperative medical preparation for at least 7 days. Adrenergic blocking agents, also known as alpha-adrenergic antagonists, were prescribed as a first-line treatment in 8 cases then, associated with β-adrenergic receptor blockers or calcium channel blockers in 15 cases. The time between the biological diagnosis and the surgery was 6 months on average (extremes: 15 days–2 years). The gesture consisted of the following: a one-sided total adrenalectomy in 90% of cases (29 patients), a two-stage bilateral adrenalectomy was suggested for one patient and tumor resection was performed on 2 patients (2 PGL).
In fact, twenty-nine patients had at least one consultation for postoperative evaluation. The short-term evolution was marked by the disappearance of paroxysmal signs in almost all cases with the exception of one patient, who kept palpitations and for whom a β-blocker treatment was prescribed. An average weight gain of 4 kg (extremes: 1–10 kg) was objectified in 16 patients. Thirteen patients had normal blood pressure with the discontinuation of antihypertensive treatments after the surgery. Then, an improvement in blood pressure with therapeutic digression was observed in 6 patients, and persistent hypertension only in one patient (Fig. 1).

Differences in tumor size and urinary metanephrines according to the evolution of hypertension. HT: Hypertension, TMNu: Total Urinary Metanephrines, N: Number of times, UCF: upper cutoffs
Moreover, metanephrines (urinary or plasma) were controlled in 24 patients during the first postoperative year. The results were as follows: Normalization in 21 patients (87.5%) and persistence of moderate elevation (< 4 times the upper limit) in three patients (12.5%). The CGA was dosed in 2 cases after the surgery but came back high in both cases. Therefore, an adrenal CT scan, performed on 5 patients (3 with high metanephrines and 2 with high levels of CGA), gave normal results in all cases. Moreover, fourteen patients were regularly monitored for long-term follow-up. The mean duration of postoperative follow-up was 3 years. Hence, a biological recurrence was observed in 3 cases (1 case with malignant PHEO, 1 patient with a normal radiological investigation and one patient whose investigation is ongoing).
4 Discussion
In fact, PPGLs are rare tumors [1]. For example, in our series, they were observed in 2.4 cases/year, up from 1.4 case/year in a study conducted in the same department between 1997 and 2012 [11]. Moreover, PPGLs can be observed at any age. However, exceptional observations were reported in newborns [12] and beyond the age of 80 [13]. The average age at the time of the diagnosis in our series was 43.8 years with extremes ranging from 10 to 74 years, which is consistent with the results of the largest series of the literature. Furthermore, PPGLs affect both sexes, while several authors have reported a certain female predominance in their series.
HT is the master symptom of PPGL [6]. In our series, it represented the COD in 30% of cases. These results are consistent with data from the Charles Nicolle Hospital series in Tunis [14]. As regards to AI, it accounts for 25–58% of COD. In fact, its prevalence in the general population is between 1 and 8.7% [15]. However, it has increased over the past two decades with the expansion of the prescription of radiological exams to reach 10% of the elderly [16, 17]. It is estimated that PHEO represents between 1.5 and 23% of AI [18]. As a result, any patient suffering from AI should undergo a thorough investigation [1]. In our series, 57% of PHEO were discovered incidentally. In addition, HT complications constituted a COD of PPGL in 3 cases.
PPGLs are catecholamine-producing (adrenaline, norepinephrine or dopamine) neuroendocrine tumors. The symptomatology depends on the secretory profile, hence the great variability in clinical expression gave PPGL the appellation "the great mimic" [3]. The low specificity of these different symptoms explains the delay between the onset of symptomatology and the diagnosis [19].
The biological diagnosis of PPGLs is based on the evidence of an excess of secretion of catecholamines or their metabolites in the blood or urine. The existence of various metabolic pathways has led to different approaches [10]. According to the recommendations of the Endocrine Society [1], it is the dosage of PFM or UFM that must be performed in the first place. Indeed, the PFM have the highest diagnostic sensitivity [20, 21], especially in hereditary contexts [22, 23]. However, their measurement may fail to identify tumors that secrete small amounts of catecholamines or those that produce only dopamine [24]. In our series, this dosage, which was performed on 10 patients (25%), was found to be high in 90% of cases. Therefore, UFM have a better specificity than PFM [25]. However, the main pitfall of this test is the difficulty of completing the 24-h urine collection.
As for CGA, it is a biomarker widely used for the evaluation of neuroendocrine tumors, mainly of gastro-entero-pancreatic origin [26]. The sensitivity of CGA for the diagnosis of the PPGL is 90%, which is still lower than the sensitivity of the PFM. However, its specificity is 99%, which gives it an excellent negative predictive value. Hence, the interest of its dosage in cases of moderate elevation of PFM [27]. CGA was dosed in seven patients (17.5%) in our series. It was high in five patients, with an average elevation of 437,4 µg/l.
Regarding the topographical diagnosis, the Endocrine Society [1] recommends CT scan as a first-line exam, which is characterized by a sensitivity of 90 to 100% and a specificity of 70 to 80% [19, 28]. In fact, it is preferred to the MRI because of its excellent spatial resolution for the thorax, the abdomen and the pelvis. As for the skull base and the neck PGL, the sensitivity of the MRI is between 90 and 95% [29]. There is a characteristic T2 hyper-signal (adrenal/liver ratio > 3) and a rapid and intense enhancement with gadolinium injection [30]. As for the 123I-MIBG scintigraphy scan, it is mainly indicated in recurrent, multiple or metastatic forms [19, 31]. In our series, CT scan and/or MRI could locate the tumor in 100% of cases. The MIBG scan was performed on 16 patients.
On the other hand, the management of patients with PPGLs requires a multidisciplinary medical and surgical team while the only curative treatment remains surgical excision [1]. Moreover, the medical preparation before the surgery is a very essential measure. The risk of recurrence requires strict and prolonged monitoring of operated patients. In our series, cured HT was observed in 66,6% of cases, and a normalization of metanephrines during the first postoperative year was observed in 87,5%. However, only one case of malignant PHEO was objectified, which was a contralateral recurrence with lymph node metastasis 12 years after the adrenalectomy, and the patient refused the treatment.
5 Conclusions
In conclusion, PPGLs are rare neuroendocrine tumors of the chromaffin tissue of the sympathetic and parasympathetic nervous system. However, their early diagnosis is crucial given their curable nature and their lethal potential. In fact, the standard treatment is surgical and requires medical preparation. Moreover, regular subsequent follow-up is essential because of the risk of recurrence and metastases. The essential points for improvement that our study has revealed are: patient follow-up after surgery, which was not always regular, and insufficient screening for genetic diseases associated with PPGLs.
Availability of data and material
The datasets generated during and/or analyzed during the current study are available on demand.
Abbreviations
- PPGL:
-
Pheochromocytomas and paragangliomas
- PHEO:
-
Pheochromocytomas
- PGL:
-
Paragangliomas
- BP:
-
Blood pressure
- UHC:
-
University Hospital Center
- PFM:
-
Plasma-free metanephrines
- UFM:
-
Urinary fractionated metanephrines
- UCF:
-
Upper cutoffs
- MIBG:
-
123I-metaiodobenzylguanidine
- COD:
-
Circumstances of discovery
- AI:
-
Adrenal incidentalomas
- HT:
-
Hypertension
- MNp:
-
Plasma metanephrines
- NMNp:
-
Plasma normetanephrines
- CGA:
-
Chromogranine A
- VMA:
-
Vanillylmandelic acid
References
Lenders JWM, Duh Q-Y, Eisenhofer G, Gimenez-Roqueplo A-P, Grebe SKG, Murad MH et al (2014) Pheochromocytoma and Paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 99(6):1915–1942
Pacak K, Linehan WM, Eisenhofer G, Walther MM, Goldstein DS (2001) Recent advances in genetics, diagnosis, localization, and treatment of pheochromocytoma. Ann Intern Med 134(4):315
Lenders JW, Eisenhofer G, Mannelli M, Pacak K (2005) Phaeochromocytoma. Lancet août 366(9486):665–675
Kantorovich V, Pacak K (2010) Pheochromocytoma and Paraganglioma. In: Progress in Brain Research [Internet]. Elsevier; [cité 4 févr 2021]. p. 343‑73. Disponible sur: https://linkinghub.elsevier.com/retrieve/pii/S0079612310820151
Omura M, Saito J, Yamaguchi K, Kakuta Y, Nishikawa T (2004) Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res 27(3):193–202
Zuber SM, Kantorovich V, Pacak K (2011) Hypertension in pheochromocytoma: characteristics and treatment. Endocrinol Metab Clin North Am 40(2):295–311
Kopetschke R, Slisko M, Kilisli A, Tuschy U, Wallaschofski H, Fassnacht M et al (2009) Frequent incidental discovery of phaeochromocytoma: data from a German cohort of 201 phaeochromocytoma. Eur J Endocrinol 161(2):355–361
Hodin R, Lubitz C, Phitayakorn R, Stephen A (2014) Diagnosis and management of pheochromocytoma. Current Probl Surg 51(4):151–187
Chen H, Sippel RS, O’Dorisio MS, Vinik AI, Lloyd RV, Pacak K (2010) The North American neuroendocrine tumor society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas 39(6):775–783
Rouaix-Emery N, Tierny-Fontalirand C, Cardot-Bauters C, Carnaille B, Wemeau J-L, d’Herbomez M (2014) Biological diagnosis of pheochromocytoma in 2014. Ann Biol Clin 72(1):7–13
Ben Amar B (2013) Les phéochromocytomes : à propos de 21 cas. Thèse de médecine : faculté de médecine de Sfax
Kaufman BH, Telander RL, van Heerden JA, Zimmerman D, Sheps SG, Dawson B (1983) Pheochromocytoma in the pediatric age group: Current status. J Pediatr Surg 18(6):879–884
Buffet A, Morin A, Castro-Vega L-J, Habarou F, Lussey-Lepoutre C, Letouzé E et al (2018) Germline Mutations in the Mitochondrial 2-Oxoglutarate/Malate Carrier SLC25A11 Gene Confer a Predisposition to Metastatic Paragangliomas. Cancer Res 78(8):1914–1922
El Amri D (2017) Les aspects thérapeutiques et évolutifs des phéochromocytomes : à propos de 30 cas. Thèse de médecine : faculté de médecine de Tunis
Mansmann G, Lau J, Balk E, Rothberg M, Miyachi Y, Bornstein SR (2004) The clinically inapparent adrenal mass: update in diagnosis and management. Endocr Rev 25(2):309–340
Mantero F, Terzolo M, Arnaldi G, Osella G, Masini AM, ALı A, et al. (2000) A survey on adrenal incidentaloma in Italy. 85(2):8
Bovio S, Cataldi A, Reimondo G, Sperone P, Novello S, Berruti A et al (2006) Prevalence of adrenal incidentaloma in a contemporary computerized tomography series. J Endocrinol Invest 29(4):298–302
Terzolo M, Stigliano A, Chiodini I, Loli P, Furlani L, Arnaldi G et al (2011) AME position statement on adrenal incidentaloma. Euro J Endocrinol 164(6):851–870
Amar L, Servais A, Gimenez-Roqueplo A-P, Zinzindohoue F, Chatellier G, Plouin P-F (2005) Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. J Clin Endocrinol Metab 90(4):2110–2116
Grouzmann E, Drouard-Troalen L, Baudin E, Plouin P-F, Muller B, Grand D et al (2010) Diagnostic accuracy of free and total metanephrines in plasma and fractionated metanephrines in urine of patients with pheochromocytoma. Eur J Endocrinol 162(5):951–960
Shah U, Giubellino A, Pacak K (2012) Pheochromocytoma: implications in tumorigenesis and the actual management. Minerva Endocrinol 37(2):141–156
Lenders JWM, Pacak K, Walther MM, Linehan WM, Mannelli M, Friberg P, et al. (2002) Biochemical Diagnosis of Pheochromocytoma: Which Test Is Best? JAMA [Internet]. 20 mars 2002 [cité 17 janv 2021];287(11). Disponible sur: https://doi.org/10.1001/jama.287.11.1427
Eisenhofer G (2012) Screening for pheochromocytomas and paragangliomas. Curr Hypertens Rep avr 14(2):130–137
Chen Y, Xiao H, Zhou X, Huang X, Li Y, Xiao H et al (2017) Accuracy of plasma free metanephrines in the diagnosis of pheochromocytoma and paraganglioma: a systematic review and meta-analysis. Endocr Pract 23(10):1169–1177
Brain KL, Kay J, Shine B (2006) Measurement of urinary metanephrines to screen for pheochromocytoma in an unselected hospital referral population. Clin Chem 52(11):2060–2064
Di Giacinto P, Rota F, Rizza L, Campana D, Isidori A, Lania A, et al. Chromogranin A: From laboratory to clinical aspects of patients with neuroendocrine tumors. Int J Endocrinol [Internet]. 2 juill 2018 [cité 17 janv 2021];2018. Disponible sur: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6051263/
Algeciras-Schimnich A, Preissner CM, Young WF, Singh RJ, Grebe SKG (2008) Plasma Chromogranin A or Urine fractionated metanephrines follow-up testing improves the diagnostic accuracy of plasma fractionated metanephrines for pheochromocytoma. J Clin Endocrinol Metab 93(1):91–95
Noblet-Dick M, Grunenberger F, Brunot B, Jaeck D, Schlienger J-L (2003) Le phéochromocytome en médecine interne : particularités et place de la scintigraphie à la MIBG 123. La Revue de Médecine Interne juin 24(6):358–365
Gimenez-Roqueplo A-P, Caumont-Prim A, Houzard C, Hignette C, Hernigou A, Halimi P, et al. (2013) Imaging work-up for screening of paraganglioma and pheochromocytoma in SDHx mutation carriers: a multicenter prospective study from the PGL.EVA investigators. J Clin Endocrinol Metab. 98(1):E162–E173
Menegaux F, Chéreau N, Peix J-L, Christou N, Lifante J-C, Paladino N-C et al (2014) Conduite à tenir devant un incidentalome surrénalien. Journal de Chirurgie Viscérale 151(5):366–376
Jacobson AF, Deng H, Lombard J, Lessig HJ, Black RR (2010) Iodobenzylguanidine Scintigraphy for the detection of neuroblastoma and pheochromocytoma: results of a meta-analysis. J Clin Endocrinol Metab 95(6):2596–2606
Acknowledgements
None.
Funding
The authors declare that they have not received any funding.
Author information
Authors and Affiliations
Contributions
AS conceived the paper and wrote most of the drafts, including statistics, illustrations and references. BF performed data collection jointly with KB FH, NC and MA reviewed the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Ethical approval was obtained from the ethics committee of our institution, named “Le comité de protection des personnes Sud” on November 23, 2021, for the realization and publication of this study. The reference number of the approval is CPP SUD N 0368/2021. Consent to participate is not applicable as it is a retrospective study.
Consent to participate
Not applicable.
Consent for publication
Not applicable. Our manuscript does not contain any individual person's data. All authors have approved the manuscript for submission and publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
kacem, F.H., Salah, A., Fathallah, B. et al. Presentation and management of pheochromocytomas and paragangliomas: about 40 cases. Afr J Urol 27, 160 (2021). https://doi.org/10.1186/s12301-021-00265-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12301-021-00265-4