
Abstract
Plastic pollution is becoming a major health issue due to the recent discovery of microplastics and nanoplastics in living organisms and the environment, calling for advanced technologies to remove plastic waste. Here we review enzymes that degrade plastics with focus on plastic properties, protein engineering and polymers such as poly(ethylene terephthalate), poly(butylene adipate-co-terephthalate), poly(lactic acid), polyamide and polyurethane. The mechanism of action of natural and engineered enzymes has been probed by experimental and computation approaches. The performance of polyester-degrading enzymes has been improved via directed evolution, structure-guided rational design and machine learning-aided strategies. The improved enzymes display higher stability at elevated temperatures, and tailored substrate-binding sites.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Plastic materials are utilized almost everywhere in our daily lives, which have also brought tremendous amounts of waste that approaches 400 million tons each year (Lampitt et al. 2023). Currently, less than 20% of plastic waste is recycled, and the rest of it is disposed of in landfills or incinerated. Plastics are extremely recalcitrant to the natural processes; thus, the disposed postconsumer plastic wastes could exist in the environment for centuries without being decomposed significantly. The massive production of plastics also consumes non-renewable petroleum resources given that most plastics are produced using fossil-derived feedstocks. Although biobased plastics from renewable feedstocks and biodegradable plastics have been produced, they comprise only 1% of all plastics produced annually. Some types of biobased plastics are not biodegradable and may still show negative effects on the environment (Ali et al. 2023). Therefore, the need for incineration or recycling will remain for many years.
Recycling postconsumer plastics to recover the raw materials for resynthesizing new products is the ideal route in terms of sustainable utilization of plastics (Luo et al. 2023). Mechanical recycling has been implemented, but physical processes could devastate the properties of polymers and lead to downcycling. For instance, recycling poly(ethylene terephthalate) (PET) through mechanical grinding and melting reduces ductility from 310 to 218% after one cycle and to 2.9% after three cycles (Damayanti 2021; Dhaka et al. 2022). Chemical processes enable the bond cleavage to release the constitutive feedstocks for subsequent applications without compromising the material properties. However, chemical processes usually involve extreme reaction conditions and hazardous reagents (Barnard et al. 2021; Christopher et al. 2022; Thiounn and Smith 2020). Enzymes are renewable biocatalysts that catalyze specific reactions under mild conditions such as temperature, pH and pressure. Therefore, enzyme-mediated decomposition brings a lower environmental impact than chemical approaches, offering an eco-friendly approach to managing plastic waste.
The plastic decomposition efficacy of naturally derived enzymes, if any, is very low because plastics were designed to withstand biodegradation processes. Therefore, the plastic decomposition rate of candidate enzymes needs to be improved via protein engineering. In recent years, inspiring results have been produced in searching, characterizing and engineering enzymes that can decompose PET with state-of-the-art technologies. It has also been demonstrated that the terephthalic acid recovered from enzyme-mediated PET decomposition can produce virgin PET that exhibits features similar to those synthesized using petrochemical terephthalic acid (Lu et al. 2022; Tournier et al. 2020). Random mutagenesis as well as rational design with specific purposes such as optimizing substrate-binding and increasing enzyme thermostability have been exploited, and intensive structural and mechanistic investigations have been conducted to understand how these amino acid residues contribute to the enhanced performance.
This review provides a comprehensive summary of recent developments in the structural and mechanistic analysis of naturally occurring or artificially engineered enzymes that have been shown to possess the potential for decomposing plastics. The focus is primarily on the enzymatic degradation and depolymerization of the most commonly used commodity polymers, specifically polyesters, while also including descriptions of enzymes that exhibit significant activity toward other major polymers such as polyamides, polyurethanes and polyolefins. As the unique properties of the substrate can significantly affect the enzyme reaction, this review will also briefly introduce the physiochemical properties of the polymers. Additionally, the technologies used to identify and engineer plastic-degrading enzymes will be described, along with a discussion of the obstacles and limitations involved in the technical and economic viability of current strategies.
Physiochemical properties of plastics
The chemical composition of plastics is the primary determinant of their biodegradability. Specifically, polyolefins comprising carbon–carbon bond-linked polymers, including the most produced polyethylene (PE), polystyrene (PS) and polypropylene (PP), are recalcitrant to biodegradation. In contrast, plastics composed of heteroatom-based backbones, mainly C–O and C–N, are susceptible to enzyme action (Fig. 1). The polymers’ chemical composition also contributes to the physical properties of the plastics. For instance, the amide moieties distributed along polymers in polyamides form inter-polymer hydrogen bond interactions to assist the regular alignment of this polymer and thus increasing its strength.

Chemical composition of major synthetic polymers, including carbon–carbon-based and heteroatom polymers
Polymers can align in randomly organized amorphous or well-organized crystalline configurations (Fig. 2). Crystalline regions that are stiff and rigid can augment the strength of the materials (Nilsson et al. 2012). Based on the crystallinity, plastics can be divided into amorphous and semicrystalline. The former, including polystyrene (PS) and poly(vinyl) chloride (PVC), comprises polymers that only contain amorphous regions. The latter, exemplified by PET and polyethylene (PE), contains polymers arranged in amorphous and crystalline configurations. The degree of crystallinity (Xc) of semicrystalline plastics that corresponds to the relative content of the crystalline part is closely related to their resistance to enzyme reaction (Fig. 2).

Physical properties of selected plastics. The temperature-dependent polymer motility of semicrystalline plastics is displayed in the upper part. The random curves and aligned columns represent polymers in crystalline and amorphous regions, respectively. The blue and red curves represent amorphous polymer chains in low and high motility. Plastics are resting (glassy state) under Tg, partially mobile near Tg and completely resolved near Tm. The Tg and Tm of six major types of plastics are listed in the table. PE, polyethylene; PS, polystyrene; PP, polypropylene; PVC, poly(vinyl chloride); PET, poly(ethylene terephthalate); PU, polyurethane
Some physical properties of plastics limit the polymer accessibility and mobility and pose challenges for enzyme reactions. First, plastics are insoluble and densely packed materials, such that the enzyme action could be limited to the plastic surface without infiltrating into the interior of the polymers. Therefore, fragmentation or grinding of plastics to increase the enzyme-plastic contacts might be needed to accelerate the enzymatic degradation efficiency. Second, the polymers’ high molecular weight (Mw) lowers the relative surface area and slows enzyme-mediated reaction (Singh and Sharma 2008). Different from ordinary substances, the Mw of a given polymer is a range (e.g., 10,000–500,000 Da) as the polymer chain elongation is ruled by random events that may lead to various chain lengths. Therefore, the molecular weight distribution and number-averaged molecular mass (Mn) are also needed to be measured. Polydispersity, which is defined as the weighted average divided by the number of average molecular weight (Mw/Mn), provides an idea of the width of the molecular weight distribution. These parameters are important in describing the status of polymer integrity, especially when incomplete decomposition occurs. Third, polymers in the crystalline region are more stable than those in the amorphous part. Therefore, plastics that have higher Xc are more resistant to enzymatic degradation compared to those with lower Xc (Strobl 1997) (Fig. 2). Fourth, the polymer softening is represented by its glass transition temperature (Tg), where the amorphous regions in the materials are transformed from glassy to rubbery state (Fig. 2). Polymer chains are kinetically arrested and exhibit higher strengths at temperatures below Tg. Therefore, a degradation reaction conducted at a temperature near or higher than Tg of the polymers is considered advantageous. For instance, Tg of PET is around 70 °C (i.e., 75–80 °C in air and 65–70 °C in water); thus, thermostable or thermophilic enzymes should exhibit a higher PET decomposition rate (Alves et al. 2002; Müller et al. 2005). Notably, polymers in amorphous regions can transition to rigid fractions and even recrystallize at elevated temperatures, a process called “physical aging” (Zhao et al. 2002). Taking PET as an example, material Xc increased from 14.6 to 24.7% and 37.5% in 9 h at 72 °C and at 75 °C in 6 h, respectively (Tournier et al. 2020). Such an increase in Xc would complicate the enzyme-catalyzed depolymerization. Therefore, enzyme reaction temperature should be chosen cautiously, and monitoring the Xc alteration during reaction is recommended.
Polymer materials could differ considerably in terms of crystallinity, thickness, molecular mass and many other physical parameters, which make a direct comparison of data generated in different studies difficult. Therefore, benchmark substrates should be used wherever possible. Otherwise, explicit descriptions of substrate properties should be provided. Another concern is various additives (e.g., stabilizers, antioxidants and flame retardants), fillers (e.g., organic, mineral and glass fiber) and pigments that can be blended into the polymers to strengthen the plastic or optimize the manufacturing processes. It has been shown that the pigments of colored PET do not influence the enzyme reaction and can be separated from the PET hydrolytic products (Lu et al. 2022), while the effects of other additives on the enzyme activity remain unexplored.
Heteroatomic polymers
Poly(ethylene terephthalate)
PET represents the most abundant polyester, constituting nearly 18% of the global polymer production. As of 2021, its annual production volume reached 400 million tons (Markit 2021). PET has been extensively studied for its potential to be decomposed by enzyme-mediated reaction, largely owing to its backbone composition, which consists of terephthalic acid and ethylene glycol linked by ester bonds (Koshti et al. 2018; Zimmermann 2020). PET is theoretically amenable to the action of ester bond hydrolytic enzymes named esterases. As PET has Tg of around 70 °C, thermostable enzymes that can operate near this temperature have been considered to exhibit higher activity. Thus, several early research studies focused on searching for thermophilic enzymes for PET decomposition.
Esterases, including lipases (EC 3.1.1.3), carboxylesterases (EC 3.1.1.1) and cutinases (EC 3.1.1.74), have been reported to exhibit PET hydrolytic activity (Wei and Zimmermann 2017a, b). As the same catalytic mechanism is employed (Fig. 3), the PET hydrolytic efficacy of these esterases is determined by the conformation of their substrate-binding pocket. Lipases that catalyze the hydrolysis of long-chain triglycerides exhibit limited PET hydrolytic activity (Marten et al. 2005; Müller et al. 2005). This is mainly attributed to the lid structure that covers the active site and prevents enzymes from directly accessing the macromolecular substrates (Brzozowski et al. 2000). The active center of the other two types of esterases is exposed to the bulk solvent owing to the lack of a lid structure. Carboxylesterases form a narrow substrate-binding pocket and show higher activity on short-chain acyl esters (Biundo et al. 2016; Ribitsch et al. 2011), whereas cutinases from bacteria and fungi responsible for the degradation of a plant polyester known as cutin display activity toward long-chain substrates (Herrero Acero et al. 2011; Müller et al. 2005; Ronkvist et al. 2009). Based on existing knowledge, the most effective PET-degrading enzymes are cutinases- or cutinase-like enzymes (Chen et al. 2020; Wei and Zimmermann 2017b). The degree of PET decomposition can be evaluated by measuring the weight loss of PET substrate, observing the polymer surface via scanning electron microscope and measuring the soluble hydrolytic products that mainly include terephthalic acid, mono(2-hydroxyethyl) terephthalate (MHET) and bis(2-hydroxyethyl terephthalate).

Catalytic mechanism of serine hydrolases. The catalytic reaction is catalyzed by serine hydrolases that harbor a catalytic triad comprising Ser-His-Asp. The substrate is colored in green and the oxyanion hole comprising the main chain amino group of two amino acids is noted by red dashed curves
Cutinases are a type of serine hydrolases that employ a catalytic triad composed of a nucleophile (Ser), a base (His) and a deprotonated acid (Asp) to exert the hydrolytic reaction (Fig. 3). Cutinases adopt the canonical α/β-hydrolase fold, which contains a central β-sheet that comprises nine β-strands and flanked by three helices on either side (Fig. 4a, b). The catalytic triad and oxyanion hole are situated on the opposite wall of the substrate-binding cleft formed on the protein surface. The substrate binds with its carbonyl carbon (C) pointing toward the Ser –OH and the carbonyl oxygen (O) stabilized by the oxyanion hole comprising two backbone –NH groups. The Ser residue that is deprotonated by His serves as a nucleophile to attack the carbonyl carbon (C) of the substrate to afford an acyl-enzyme intermediate. Then, the R2 group is removed from the polymer in an alcohol form due to an elimination reaction. The acyl-enzyme intermediate is then attacked by a water molecule (H2O) to afford the acid form of R1, which regenerates the active center to the resting state.

The overall structure- and substrate-binding site of PET decomposing cutinases. The structures of representative a type I and b type II cutinases are illustrated. a. The overall structure of LCC (left, PDB ID, 4EB0) and the substrate-enzyme interaction network in the complex structure of LCC-ICCG and MHET (right, PDB ID, 7VVE). The β-strands are labeled numerically. Disulfide bonds (DS) and residues that constitute the catalytic triad and oxyanion hole are indicated. Dashed lines linking MHET and residues indicate a distance less than 3.5 Å. b. The overall structure of IsPETase (left, PDB ID, 5XG0) and its interaction network of MHET (PDB ID, 7XTW). Dashed lines measure distance as described in (a). The catalytic Ser in both complex structures was mutated to Ala. The locations of + 1 and + 2 subsites are displayed on the right. c. Three various conformations of W185 observed in IsPETase (PDB ID, 5XG0) are shown in the left panel. Right panel: The MHET complexes of IsPETase (cyan; PDB ID, 7XTW) and LCC-ICCG (orange; PDB ID, 7VVE) are superimposed. The residues that comprise catalytic triad, oxyanion hole and substrate-binding pocket are indicated by line models and MHETs are shown in sticks. PET, poly(ethylene terephthalate); MHET, mono(2-hydroxyethyl) terephthalate. LCC, leaf compost cutinase; LCC-ICCG, a thermostable variant of LCC that harbors F233I, D238C, S238C and Y127G alterations
While the PET hydrolytic activity of some fungal cutinases has been documented (Ribitsch et al. 2012a; Ronkvist et al. 2009), current research mainly focuses on bacterial cutinases. In 2016, a cutinase-like enzyme termed IsPETase was identified in Ideonella sakaiensis, a PET-assimilating bacterium isolated from a natural microbial community collected in a PET recycling factory that can grow on and use PET (1.9% Xc) as a carbon source (Yoshida et al. 2016). The optimal operation temperature of IsPETase is 30 °C, exhibiting the highest PET hydrolytic activity than other reported enzymes (Yoshida et al. 2016). IsPETase is classified as a subclass of cutinase (EC 3.1.1.101), which shares up to 47.6% of the sequence identity with conventional cutinases but harbors some different structural characteristics. The most explicit feature distinguishing canonical cutinases from IsPETase-like cutinases is the number of intramolecular disulfide (DS) bonds (Fig. 4a, b). Here, the conventional cutinases that contain one DS bond (DS1) are classified as type I and IsPETase-like cutinases that contain two DS bonds (DS1 and DS2) are type II. All currently known cutinases possess the conserved DS1 that links a α-helix to a loop on the C-terminus of the protein (Fig. 4a). On the other hand, IsPETase-like cutinases harbor an additional DS termed DS2 that has been proposed to stabilize an active site extended loop to constitute a putative substrate-binding cavity (Chen et al. 2018; Joo et al. 2018) (Fig. 4b).
Type I cutinases
The first enzymatic decomposition of PET film was reported by TfH or BTA1 from a soil cellulolytic actinomycete Thermobifida fusca (Lykidis et al. 2007), whose reaction caused weight loss of two kinds of PET films by around 40–50% at 55 °C in 3 weeks (Müller et al. 2005). Afterward, a series of cutinases from various Thermobifida species that exhibit PET hydrolytic activity were reported (Herrero Acero et al. 2011; Ribitsch et al. 2012a, b; Roth et al. 2014; Thumarat et al. 2015). Enzymes from other genera were also reported. Cut190 from Saccharomonospora viridis shares around 65% sequence identity with Thermobifida cutinases (Kawai et al. 2014), and its variant that contains mutations S226P and R228S exhibits up to 27% weight loss of PET film at 63 °C in three days. DmPETase from Deinococcus maricopensis was found to be less sensitive to the crystallinity degree and exhibits activity on semicrystalline sections of postconsumer PET bottles at 50 °C without the need for amorphization of the materials (Makryniotis et al. 2023). Other than the thermophilic enzymes, two cold-active cutinases derived from Antarctic bacterium Aequorivita sp. (PET27) and Kaistella jeonii (PET30) that showed depolymerizing activity on amorphous PET foil were also reported (Zhang et al. 2021). The PET hydrolytic activity of PET27 is 38-fold higher than PET30 at 30 °C, though both are very low relative to IsPETase.
In addition to microbial strains, metagenomes are important sources for searching candidate enzymes (Danso et al. 2018; Eiamthong et al. 2022; Erickson et al. 2022; Pfaff et al. 2022; Sonnendecker et al. 2022; Sulaiman et al. 2012). Leaf compost cutinase (LCC) identified from a leaf compost metagenome attracted much attention due to its superior activity over other reported PET hydrolytic enzymes (Tournier et al. 2020; Wei et al. 2019). A type I cutinase termed BhrPETase sourced from hot spring water metagenome is highly identical to LCC, such that only 16 various amino acids are identified in the mature peptides of these two enzymes. BhrPETase exhibits 11 °C higher Tm than LCC when expressed in Bacillus subtilis (Xi et al. 2021). PHL-7, named as PES-H1 in an independent study, which was identified from plant compost metagenome exhibits twofold higher activity compared with LCC at 75–80 °C (Pfaff et al. 2022; Sonnendecker et al. 2022). Notably, as high as 1 M phosphate buffer is demanded for PHL-7 to reach its optimal performance, which should be considered in further investigations and applications (Pfaff et al. 2022; Sonnendecker et al. 2022).
Type II cutinases
The identification, structural and biochemical analysis of IsPETase triggered the characterization of type II cutinases, which share a highly identical three-dimensional structure to type I cutinases while containing an additional DS2 (Fig. 4b) (Austin et al. 2018; Danso et al. 2018; Fecker et al. 2018; Han et al. 2017; Joo et al. 2018; Liu et al. 2018). Partial β6-corresponding fragment in IsPETase was judged as a loop in some deposited structures, yet the strand numbering used here follows those of type I cutinases to facilitate the parallel structural comparison. Type II cutinases appear to exist widely in the environment, and several have been identified in various bacteria strains and metagenomes (Blázquez-Sánchez et al. 2022; Chen et al. 2021a, b; Danso et al. 2018; Eiamthong et al. 2022; Sagong et al. 2021). A few type II cutinases that harbor a highly identical substrate-binding site composition to that of IsPETase were identified from GenBank (Joo et al. 2018), including those from Rhizobacter gummiphilus (RgPETase), Burkholderiales bacterium RIFCSPLOWO_02_FULL_57_36 (BurPL or BbPETase) and Polyangium brachysporum (PbPL or PET12). RgPETase shows the same thermal profile, comparable hydrolytic activity toward microcrystalline PET (milled commercial PET bottle, 17.1% Xc) and lower activity toward amorphous PET film (6% Xc) relative to IsPETase (Sagong et al. 2021). BurPL shows similar activity and higher thermal stability than IsPETase (Chen et al. 2021a, b; Sagong et al. 2022). BurPL has an N-terminal domain whose presence renders the enzyme higher thermal stability yet lower activity, probably owing to the steric hindrance that interferes with the enzyme attachment on PET (Sagong et al. 2022). Wild-type PbPL exhibits very low activity, but its performance can be elevated to a comparable level relative to IsPETase by substituting two active site-nearby residues to mimic that in IsPETase (see below) (Chen et al. 2021a, b). PE-H from a marine bacterium Pseudomonas aestusnigri exhibits PET hydrolytic activity at 30 °C (Bollinger et al. 2020). Thermophilic enzymes including two derived from metagenome (PET2 and PET6) and CtPL from Caldimonas taiwanensis show optimal activity at 55–70 °C (Chen et al. 2021a, b; Danso et al. 2018). MG8 identified from human saliva metagenome, exhibits optimal activity at 55 °C in the presence of sodium chloride (NaCl) at a concentration as high as 4 M (Eiamthong et al. 2022). Two psychrophilic enzymes from Antarctic bacteria show activity at 25 °C, with Mors1 showing comparable activity to IsPETase (Blázquez-Sánchez et al. 2022; Danso et al. 2018).
Substrate-binding modes of poly(ethylene terephthalate)-degrading cutinases
The substrate-binding pose of PET-degrading enzymes should be one of the most important subjects to address. For type I cutinases, molecular simulation and docking have been implemented to predict the substrate-binding pose of PET-degrading enzymes, mainly prompted by the attempt to identify the candidate residues for engineering (Chen et al. 2022; Kitadokoro et al. 2012; Tournier et al. 2020). However, only a few complex structures that contain PET degradation products have been reported: LCC-ICCG, a thermostable variant of LCC that harbors F233I, D238C, S238C and Y127G alterations, in complex with MHET, wild-type and variant TfCut in complex with MHET and PHL-7 in complex with terephthalic acid (Richter et al. 2023; Yang et al. 2023; Zeng et al. 2022) (Fig. 4a). LCC-ICCG can decompose more than 90% PET at 72 °C in 10 h (Tournier et al. 2020). In the crystal structure of the complex of LCC-ICCG and MHET, the MHET binds to a surface cleft termed + 1 subsite here, which is lined by Y95, T96, A97, H164, S165, M166, W190, V212, H242, I243 and N246. Y95, M166, W196 and V212 clamp the aromatic moiety of MHET, while others comprise a tunnel to accommodate the hydroxyethyl part. Notably, the F243I mutation widens the putative secondary subsite of the PET-binding tunnel, which was proposed to facilitate the binding of the bulkier PET and leads to higher PET hydrolytic activity (Zeng et al. 2022). The highly identical interaction network is observed in the complex of PHL-7 and terephthalic acid (Richter et al. 2023). Interestingly, the I243-corresponding residue in PHL-7 is L210. This residue has been verified to account for the high degradation efficiency of PHL-7, as substituting L210 to Phe, an equivalent in a highly homologous PHL-3 and most type I cutinases, reduced PET hydrolytic activity (Sonnendecker et al. 2022).
The overall fold and substrate-binding site composition of type II cutinases are highly identical to that of type I cutinases (Fig. 4b), but IsPETase has been found to exploit a unique substrate-binding behavior. The + 1 subsite composition of IsPETase has been revealed by the complex structures of MHET or other substrate analogues (Han et al. 2017; Yang et al. 2023) (Fig. 4b). Notably, W185 which provides a T-stacking force to the aromatic moiety of MHET displays a unique conformation that has not been seen in all known canonical cutinases (Chen et al. 2021a, b; Han et al. 2017). This pose, termed “B-type,” is one of the three conformations that were reported in our previous study (Han et al. 2017) (Fig. 4c). The W185 corresponding Trp in all cutinases is constrained by two nearby residues, His and Phe, such that only “C-type” conformation can be observed (Fig. 4c). In IsPETase, the His and Phe are replaced by Ser and Ile, respectively, whose side groups are smaller to allow W185 to wobble freely (Fig. 4c). Structural comparisons suggest that MHET adopts a more relaxed angle in IsPETase relative to that in LCC-ICCG (Fig. 4c). This suggests that the MHET-binding Trp in the C-type conformation should lead to a more rigid substrate-binding groove, such that polymers need to bend a bit to fit in. On the other hand, Trp in B-type renders the substrate-binding pocket higher flexibility. Notably, His-Phe is strictly conserved in all cutinases and Ser-Ile is an IsPETase-unique feature. Mutating His-Phe to Ser-Ile, a strategy termed DM, in several canonical cutinases renders higher PET hydrolytic activity though the thermostability is reduced, possibly owing to a higher degree of protein flexibility (Chen et al. 2021a, b).
Additional substrate-binding sites other than + 1 subsite in these PET-degrading cutinase have been proposed in a study, which conducted computational docking of an oligomeric substrate 2-hydroxyethyl-(monohydroxyethyl terephthalate)4-(2-HE(MHET)4) and claimed four MHET subsites (Joo et al. 2018). This hypothesis has been argued by a solid-state nuclear magnetic resonance (NMR) study, which suggests that PET adopts a highly rigid conformation at the reaction temperature of IsPETase (30 °C) and hardly forms the suggested conformation (Wei et al. 2019). A recent study revealed a similar scenario in the type I cutianse TfCut2, which traces the PET-binding mode of the enzyme in a real-time fashion with an advanced solid-state NMR method (Falkenstein et al. 2023). It suggests that only one PET monomer unit is bound to the enzyme during the degradation process while the rest of the PET chain is loosely confined to the active site.
Accordingly, the + 1 subsite shall be the most well-defined substrate-binding site of PET-degrading cutinases based on the current knowledge. Nonetheless, the presence of multiple binding sites can still provide non-catalytic benefits, considering that more enzyme-plastic contacts should facilitate enzyme action. These results might be supported by kinetic investigations to some extent. The inverse Michaelis–Menten framework, which has been applied to evaluate interfacial catalytic reactions such as enzyme-mediated degradation of cellulose or chitin, was applied to study the kinetics of IsPETase, which suggests the high efficiency of the enzyme at ambient conditions owing to a high kcat rather than a low KM (Bååth et al. 2021). In view of this, the Sabatier principle states that optimal catalysis occurs when interactions between catalyst and substrate are of intermediary strength, as the case in the heterogeneous catalyst of non-biochemical reactions might be appropriate to be applied to interfacial enzyme catalysis (Bååth et al. 2022; Kari et al. 2018).
Engineering of poly(ethylene terephthalate)-degrading enzymes
The PET hydrolytic activity of enzymes from various sources is still low and needs to be improved. Concerning the influence of polymer mobility on enzyme action and the durable reaction required for better decomposition, enhancing the enzyme thermostability is one major goal to pursue. Modifying the substrate-binding site is a logical rationale, given that PET is not the natural substrate for these enzymes. Structural analyses have illustrated the substrate-binding pattern in the + 1 subsite; thus, the + 1 subsite comprising residues are the most appealing targets of engineering to improve the enzyme activity. Increasing the protein thermostability is another main direction because PET polymers that gain mobility at elevated temperatures should benefit the enzyme reaction. In addition, modifying enzyme surface to increase enzyme-PET contact has also been implemented. This is based on the hypothesis that the negative charge of the PET surface should gradually increase during catalytic reaction and the higher amounts of anionic carboxyl on PET would attract PET hydrolases with positive surface charge.
Type I enzyme engineering
LCC had been considered the most potent PET hydrolytic enzyme and thus was subjected to engineering to enhance its performance (Tournier et al. 2020). The PET-binding groove was predicted through modeling a PET analog, and saturation mutagenesis was conducted to modify eleven tunnel-forming residues including Y95, F125, Y127, W190, A213, T96, H164, V212, S101, F243 and N246. Among 209 variants generated, only variants F243I (127.5 ± 6.4%) and F243W (118.4 ± 6.8%) exhibit higher activity than the wild-type enzyme (Fig. 5a, i). Then, a disulfide bond was introduced to a divalent ion-binding site, a feature shared by several type I cutinases (Fig. 5a, ii), to free the enzyme from the metal ion-dependent thermostability (Miyakawa et al. 2015; Ribitsch et al. 2017; Then et al. 2016). This disulfide bridge, termed DS3 herein, has been introduced in the equivalent location in several cutinases to enhance their thermostability, including LCC (D238C-S283C, ΔTm equals to 9.8 °C (Tournier et al. 2020)), TfCut2 (D204C-E253C, ΔTm equals to 24.9 °C (Chen et al. 2022; Then et al. 2016)), Cut190 (D250C-E296C, ΔTm equals to 23.1 °C (Emori et al. 2021)) and PHL-7 (R204C-S250C, ΔTm equals to 6.4 °C (Pfaff et al. 2022)). Introducing D238C-S283C mutations to the variant F243I lowered the enzyme Tm (ΔTm equals to 6.2 °C); thus, Y127G mutation that was identified in the first-round screening was introduced to rescue the protein thermostability (Fig. 5a, iii). Finally, the quadruple LCC-ICCG exhibits equal activity and 9.3 °C higher Tm relative to LCC and can depolymerize more than 90% PET (Xc lower than 15%) in 10 h at 72 °C with 3 mgenzyme gPET−1 enzyme. Furthermore, this study illustrates that terephthalic acid recovered from the enzyme-mediated hydrolytic reaction can be used to resynthesize virgin PET, proving the viability of implementing enzyme-mediated PET recycling.

Strategies applied to modify poly(ethylene terephthalate) (PET)-degrading cutinases. Selected protein engineering strategies that show beneficial effects in improving the performance of a type I (represented by LCC; PDB ID, 4EB0) and b type II (represented by IsPETase; PDB ID, 5GX0) PET hydrolytic cutinases. Some involved residues are displayed by sticks
In light of the beneficial effects of enhancing enzyme thermostability, LCC-ICCG was further subjected to structure-based rational design and machine learning-based modification (Ding et al. 2023; Zeng et al. 2022). Zeng et al. (2022) conducted rational design by adding non-covalent interactions to strengthen the interactions between the central sheet and the nearby helices (Fig. 5a, iv). Among 27 variants generated, six variants that exhibited improved thermostability were further combined to yield two triple mutants (A59K-V68I-N248P, A59R-V63I-N248P and A59K-V75R-N248P), which exhibit Tm that approaches 99 °C. Ding et al. (2023) applied two strategies based on a deep learning-based prediction tool, Preoptem and the differential evolutional analytical methods to identify 36 potential beneficial variants (Ding et al. 2023). Through experimental validation, six variants including D53T, S67L, S133R, T192P, E208Q and N248P exhibited higher activity than LCC-ICCG at 75 °C. (The amino acid numberings shown here are adjusted based on the full-length protein sequence.) A variant termed LCC-ICCG_I6M carrying six mutations was generated, which produces 3.64 times more hydrolytic products from digesting ground PET water bottles (31.3% Xc) when compared with LCC-ICCG (Ding et al. 2023). Residue 192 is located opposing to G127, thus is proposed to widen the putative − 1 subsite (Fig. 5a, iii), while the mechanism underlying the other three mutations that are located on the protein surface remains elusive. Notably, both studies modified N248 to Pro (Fig. 5a, v), an amino acid found in the corresponding location in LCC-homologous BhrPETase that shows higher thermostability than LCC. The N248P-mediated stabilization effects on β8-α6 loop have also been structurally validated (Zeng et al. 2022).
Many of the abovementioned strategies also show promise in modifying other type I cutinases. The + 1 subsite of TfCut2 was modified by mutating G62 to Ala in accordance with A97 in LCC to yield a variant that exhibits 2.7 times higher activity (Wei et al. 2016). Chen et al. (2022) exploited a directional-path modification strategy aiming to optimize the direct and indirect enzyme–substrate contacts of TfCut2 to generate a quadruple mutant Q92G-I213K-H184S-F209I (Chen et al. 2022). Residue H184, which is associated with W155 wobbling, was mutated to Ser, and residue F209, the equivalent of F243 in LCC, was mutated to Ile. The beneficial effect of Q92G remains elusive as the equivalent mutation in LCC (Y127G) was found to cause about 35% reduction in activity (Tournier et al. 2020). These mutations were introduced in a thermostable variant that harbors DS3 (generated by introducing D204C-E253C mutation) to afford TfCut2 4Mz. This variant is 30-fold more efficient than the wild type and can degrade 90% PET with 50 mgenzyme gPET−1 at 60 °C in 96 h (Chen et al. 2022). These four mutations also improve the performance of LCC, Est119 and BhrPETase, indicating the universal benefit of these substitutions in improving the properties of type I cutinases. TfCut2 was subjected to a machine learning method to yield a variant that carries S121P, D174S and D204P, which exhibits 9.3 °C higher Tm and 46.42-fold higher activity at 70 °C (Li et al. 2022). Notably, residue 204 is part of the DS3-forming residue of TfCut2, mutating D204 to Pro might provide local stabilization effects similar to that by introducing a disulfide bridge.
Type II enzyme engineering
Numerous efforts have been made to enhance the thermostability of IsPETase, whose PET decomposition efficacy is limited by low protein stability (Kawai et al. 2019). Through structure-guided rational design, variants P181A, S121D, S121E, D186H and R280A were generated to strengthen the local interactions, and a variant termed ThermoPETase that carries S121E, D186H and R280A exhibits 8.81 °C higher Tm and 14-fold higher activity at 40 °C was eventually obtained (Son et al. 2019). S121 and D186 are located in the putative −1 subsite (Fig. 5b, i) while R280 is located on the β6-β7 connecting loop that can be stabilized by DS3 in other cases (Fig. 5b, ii). A variant termed DuraPETase derived from a computational redesign program named GRAPE carries ten mutations and exhibits 31 °C higher in Tm (Cui et al. 2021). Its performance can be further increased by introducing DS3, restoring W185 wobbling (H214S) (Fig. 5b, iii) and increasing surface positive charge by introducing mutation S245R (Liu et al. 2022a) (Fig. 5b, iv). The resulting variant DuraPETase-4 M degrades up to 70% PET film (10.2% Xc) at 60 °C in 96 h, higher than DuraPETase which causes 12.9% decomposition under the same condition.
Compared with rational design, directed evolution offers a more comprehensive engineering approach. It is powerful but labor-intensive and demands an efficient high-throughput screening platform to evaluate the performance of many variants. This could be an obstacle for PET-degrading enzymes, as measuring PET hydrolytic products by high-performance liquid chromatography (HPLC) analysis is the standard method to probe the efficacy of these enzymes. Shi et al. (2023) developed a novel PET analogous substrate, bis (2-hydroxyethyl) 2-hydroxyterephthalate (Shi et al. 2023). The enzyme activity can be quickly determined by measuring the fluorescence emitted by the hydrolyzed product. With this method, the authors screened around 10,000 clones in three rounds of directed evolution and selected a variant termed DepoPETase that contains seven mutations (D186H, N233K, T88I, D220N, N246D, R260Y and S290P). This variant exhibits the same thermal profile and comparable activity of FAST-PETase (see below). A more straightforward screening method has been reported. Bell et al. incubated the lysates of protein expression hosts with the amorphous PET disk in 96-well plates and analyzed the released MHET and terephthalic acid with an integrated, automated system containing Ultra Performance Liquid Chromatograph (Bell et al. 2022). The authors claimed that over 2,000 reactions can be analyzed in two days. Using ThermoPETase as a starting template, six rounds of saturation mutagenesis were conducted to target residues involved in substrate binding or thermostability. After screening more than 13,000 variants using higher activity at elevated temperatures and extended reaction time as an evolutionary pressure, the authors eventually obtained a variant termed HotPETase that carries 18 mutations. HotPETase has an optimal temperature at 60–70 °C and exhibits higher activity than LCC-ICCG at 70 °C. Notably, most of the mutations in HotPETase are located outside the active center and are considered to play a role in stabilizing the protein structure. Notably, HotPETase also harbors the DS3 rendered by N233C-S282C mutations, further indicating the transferability of this feature.
A machine learning-aided engineering with a three-dimensional self-supervised, convolutional neural network termed MutCompute has been applied to identify suboptimal amino acids that could be modified to fit better than the parental ones in IsPETase (Lu et al. 2022). Eight out of ten predicated mutations showed improved thermostability and activity. After experimentally characterizing 159 single or combined mutations, four mutations including S121E, T140D, R224Q and N233K were the most beneficial alterations. Eventually, a variant termed FAST-PETase that was generated by introducing R224Q and N233K to ThermoPETase showed the highest performance. N233 is located on the DS3-equivalent site (Fig. 5b, ii), while the location of R224 does not belong to any category classified here. This variant shows superior PET hydrolytic activity relative to IsPETase and LCC variants, such that complete depolymerization of postconsumer-PET from 51 different PET products (Xc lower than 6.24%) can be achieved at 50 °C in one week.
The activity of CtPL derived from C. taiwanensis was increased by up to 7.5-fold by introducing the Ser-His mutation (CtPL-DM) (Fig. 5b, iii) (Chen et al. 2021a, b). F235L alteration further elevates the activity by 60% at 60 °C, and introducing DS3 by R230C-S284C mutation enhances the enzyme activity at 70 °C by more than 2.5-fold (Li et al. 2023). Notably, CtPL-DM showed no activity when expressed in Pichia pastoris. This has been suspected due to steric hindrance mediated by the N-glycosylation on N181 that constrains the flexibility of the + 1 subsite-forming Trp. N181A mutation that reduced glycosylation degree completely restored the enzyme activity (Li et al. 2023). These results have important application implications as P. pastoris is a workhorse in enzyme production industries and could be a candidate strain for large-scale production of PET decomposing enzymes. In this context, the influences of posttranslational modifications of enzymes on their decomposition efficacies deserve more investigation.
PET2 identified from marine metagenome was engineered by increasing positive charge on the protein surface. Two surface residues were mutated to basic amino acids in accordance with those in IsPETase (F105R and E110K) (Fig. 5b, iv). In addition, two amino acids located on loop regions were mutated to Pro (S156P and T297P), and one of the signature four-Gly residues that form an α-helix near the catalytic Ser was mutated to Ala for helix stabilization. In addition, introducing an additional disulfide bond termed DS4 via R47C and G89C mutation links the N-terminal loop and the β2-β3 loop slightly increased enzyme activity, indicating that stabilization of this region also provides merits (DS4, Fig. 5b, v) (Nakamura et al. 2021). The resulting variant termed PET2 7 M showed 6.7 °C higher Tm, 2.7-fold higher PET-binding affinity and 6.8-fold higher activity. Notably, S156P and T297P are located in α2-β5 loop and the DS3-corresponding loop, respectively (Fig. 5b, vi and ii).
Strategies other than amino acid alteration have been applied to improve the performance of PET-degrading enzymes. The Antarctic type II cutinase Mors1 was subjected to a chimeric design, which replaced the extended loop β8-ɑ6 of the enzyme with a shorter one from the thermophilic LCC (Blazquez-Sanchez et al. 2023). The optimal operation temperature of the chimeric Mors1 is elevated from 25 °C to 45 °C. Notably, the type II-unique DS2 was restored by introducing A266C mutation because depleting DS2 caused the loss of activity, a phenomenon that has also been reported in IsPETase (Chen et al. 2018). More recently, constructing IsPETase or FAST-PETase in cyclic monomer, cyclic dimer or catenane topology using SpyCatcher-SpyTag, p53 dimerization domain or enhanced GFP-nanobody scaffolds to increase enzyme resilience to heat and mechanical stresses have been reported (Hayes and Luk 2023; Sana et al. 2023). Sana et al. (2023) demonstrated that IsPETase in cyclic monomer shows higher thermostability but lower catalytic activity, while Hayes et al. (2023) indicated no enhancement in protein thermostability with all cyclization strategies.
Nonetheless, the latter report demonstrates that enzyme cyclization shows improvements toward agitation, which was proposed as a beneficial factor to the interfacial reactions. In comparison, using GFP-nanobody as a scaffold should be a promising strategy, such that the Tm of the dimeric enzymes of IsPETase and FAST-PETase are increased to 80 °C and 85 °C without compromising the catalytic activity. Many biomass-degrading enzymes possess auxiliary domains to promote enzyme–substrate contact efficiency, while most PET hydrolytic enzymes lack such domains (Atthoff and Hilborn 2007). Interestingly, carbohydrate-binding modules, the auxiliary domain of carbohydrate-degrading enzymes, have been reported to display PET-binding affinity (Weber et al. 2019). Accordingly, a series of wild-type and engineered carbohydrate-binding modules and chitin-binding domains from various sources were fused to PET hydrolytic enzymes to enhance their PET degradation efficacy (Dai et al. 2021; Xue et al. 2021; Zhang et al. 2013). Hydrophobins that promote the attachment to the hydrophobic polymer surface have also been used in conjugation with PET hydrolytic enzymes to enhance PET decomposition activity (Puspitasari et al. 2020, 2021; Ribitsch et al. 2015). In addition, amphiphilic anchor peptides have been fused to PET hydrolytic enzymes to promote their performance (Chen et al. 2021a, b; Liu et al. 2022a, b).
Other polyesters
Poly(butylene adipate-co-terephthalate)
PET exhibits excellent material properties but is highly resistant to biological processes, while aliphatic polyesters such as polycaprolactone, which are more susceptible to biodegradation, might lack important properties for applications (Bartnikowski et al. 2019). To harness robust material properties and better biodegradability, poly(butylene adipate-co-terephthalate) (PBAT) that contains flexible parts (butylene adipate) and rigid parts (terephthalic acid) was produced (Fig. 1), whose properties can be tuned by adjusting the length of the aliphatic parts and the ratio of aliphatic to aromatic dicarboxylic acids (Mueller 2006; Müller et al. 2001; Witt et al. 1997). One notable utility of PBAT is in plastic film mulching technology. Compared with conventional PE mulch materials, the ester bond-based structure should render PBAT with a faster biodegradation rate (Kader et al. 2017).
The PBAT decomposition is mainly evaluated by measuring the weight loss and the amounts of hydrolytic products. PBAT decomposition mainly involves the action of esterases, but the cleavage of ester bonds in PBAT polymers yields more complicated hydrolytic products than PET. With respect to the completeness of decomposition and monomer recycling, potent PBAT-degrading enzymes should be able to produce terephthalic acid as a main product. The first reported PBAT-degrading microorganism was Thermomonospora fusca isolated from a compost material (Kleeberg et al. 1998). Later, many other strains with various degrees of decomposition capacity have been identified to do the same (Jia et al. 2023; Meyer-Cifuentes et al. 2020; Muroi et al. 2017; Trinh Tan et al. 2008; Witt et al. 2001). Albeit lipase and esterase have also been demonstrated to exhibit PBAT degradation activity (Biundo et al. 2016; Perz et al. 2016a, 2016b; Wallace et al. 2017), cutinase should be the most effective PBAT-degrading enzyme (Chen et al. 2008; Kleeberg et al. 2005; Perz et al. 2016b; Suzuki et al. 2014; Yang et al. 2023).
It has been suggested that the hydrolysability of PBAT is related to polymer flexibility, as the enzyme-mediated decomposition rate increases with the decrease in the terephthalic acid-to-adipate ratio (Zumstein et al. 2017). This indicates that PET-degrading enzymes might be good candidates for PBAT decomposing enzymes. A recent study demonstrates that several PET hydrolytic enzymes including IsPETase, TfCut, TcCut and BurPL can decompose PBAT more efficiently relative to other esterases (Yang et al. 2023). Intriguingly, introducing IsPETase-unique Ser-Ile mutation to TfCut, TcCut and BurPL also elevates the PBAT decomposition rate of these enzymes. Moreover, the Ser-Ile variant of Thermobifida cutinases produces terephthalic acid as a major end product. Crystal structures of wild-type and Ser-Ile variant of TfCut in complex with MHET show the same substrate-binding pattern as in LCC-ICCG and IsPETase (Fig. 4c) (Yang et al. 2023). This supports that the wobbling Trp should afford a flexible substrate-binding site for terephthalic acid moiety to bind in an extended conformation, which might render the enzyme a preference to cleave the terephthalic acid-adjacent ester bond and yield terephthalic acid.
Poly(lactic acid)
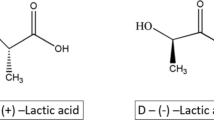
Poly(lactic acid) is an aliphatic polyester composed of lactic acid that can be derived from microbial fermentation using renewable resources (Ahmad et al. 2021; Nandhini et al. 2023). Despite its limited commercial applications due to brittleness, poor elasticity and low thermal stability, poly(lactic acid) is attractive owing to its biodegradability. Nonetheless, the natural degradation rate of poly(lactic acid) is still very slow and takes decades to achieve under environmental conditions (i.e., lower than 37 °C) and around 180 days in industrial composting facilities (i.e., higher than 60 °C) (Samantaray et al. 2021). Similar to the case for other plastics, the enzyme-mediated poly(lactic acid) decomposition efficacy is highly sensitive to the Xc of the materials (MacDonald et al. 1996; Reeve et al. 1994). It has been reported that the degradation rate of poly(lactic acid) drastically dropped with the Xc of around 26% (Li and McCarthy 1999).
Poly(lactic acid) depolymerases are mainly serine hydrolases that are classified as lipase-type (i.e., esterases, lipases, cutinases) (Akutsu-Shigeno et al. 2003; Hu et al. 2010; Masaki et al. 2005) and protease-type (Lim et al. 2005; Oda et al. 2000; Pranamuda et al. 2001; Williams 1981). These enzymes employ the same catalytic triad that comprises Ser-His-Asp to conduct bond cleavage (Fig. 2) but have different active site topologies and display different degradation activity toward two optical isomers of poly(lactic acid). Protease-type enzymes exclusively hydrolyze poly(L-lactic acid) and lipase-type enzymes are more flexible but prefer to hydrolyze poly(D-lactic acid) (Fig. 1) (Kawai et al. 2011; Reeve et al. 1994). Many crystal structures of these poly(lactic acid) depolymerases have been reported, but the information on substrate-bound complexes is scarce. The complex of protease-type poly(lactic acid)-depolymerase is not available, such that the substrate-binding behavior toward poly(lactic acid) can only be predicted through simulation (Kawai et al. 2011). The crystal structure of a cutinase termed Est119 that contains a D-type ethyl lactate and a lactate has been reported, consistent with the finding that the enzyme hydrolyzes poly(D-lactic acid) more easily than poly(L-lactic acid) (Hu et al. 2010; Kitadokoro et al. 2019). A variant of cutinase Thc_Cut2 (R29N-A30V) was found to release 4.5 times more lactic acid from poly(L-lactic acid) than the wild-type enzyme (Ribitsch et al. 2017). Small-angle X-ray scattering (SAXS) data collected from the enzymes in solution indicated that the mutations might change the electrostatic and hydrophobic properties in regions that involve polymer-binding surfaces.
Two microbial carboxylesterases that exhibit high poly(L-lactic acid) depolymerization activity were identified from screening microbial hydrolases. The crystal structure of the one termed RPA1511 has been resolved, and the model of the other was predicted (Hajighasemi et al. 2016). Later, the same group solved the crystal structure of MGS0156, an esterase with potent poly(L-lactic acid) decomposition activity identified from a metagenome from paper mill waste-degrading microbial community (Hajighasemi et al. 2018). All these enzymes cover the hydrophobic active site with a U-shaped lid domain. Although no complex structure is available, the role of the active site residues of these enzymes has been probed by mutagenesis experiments. It has been reported that variant V202A of RPA1511 and L169A of MGS0156 showed higher poly(L-lactic acid) decomposition activity (Hajighasemi et al. 2018). However, the underlying mechanism of the enhanced activity remains elusive.
Polyamide
Polyamide, known as nylon, is widely used in automotive, electrical, construction, packaging and fabric applications. As the world demand for polyamides increases yearly, the issue of recycling polyamide waste has attracted increasing attention. The majority of polyamide is composed of short aliphatic chains, including polycaprolactam (i.e., PA-6 or nylon-6) and polyhexamethylene adipamide (i.e., PA66 or nylon-66) (Fig. 1). Although the amide bond of polyamide is common to that links amino acids in proteins, the biological degradation rate toward PA-6 and PA66 is very low, such that complete depolymerization of polyamide fibers by enzymes has not been documented. This could be attributed to the high crystallinity of polyamide resulting from the abundant hydrogen bonds between polyamide chains.
Two types of enzymatic reactions are known to cause nylon degradation. One is hydrolases including proteases, cutinases and amidases. The action of these enzymes is limited to the polyamide surface, such that these enzymes can be applied to increase polyamide hydrophilicity to improve textile comfort by allowing better evaporation of perspiration or to increase dyeability (Kanelli et al. 2017; Silva et al. 2005). The other type is ligninolytic enzymes including laccases (EC 1.10.3.2) and manganese peroxidases (EC 1.11.1.13) (Deguchi et al. 1998; Fujisawa et al. 2001; Linko 1992; Negoro et al. 2021). Laccases are multicopper oxidases that oxidize phenolic substrates using O2 as the electron acceptor (Janusz et al. 2020). The active site of these enzymes consists of a type 1 (T1) copper site where the substrate is oxidized and a trinuclear copper cluster (T2 and T3) where the oxygen is activated and reduced. Laccases have broad substrate specificity but cannot oxidize substrates with high redox potential (e.g., non-phenolic substrates). This can be overcome by incorporating small aromatic molecules called “mediators.” In such a laccase-mediator system, laccases first oxidize the mediator and then the oxidized small molecules act as chemical oxidants to oxidize substrates (Hilgers et al. 2018). A laccase-mediator system coupled with hydroxybenzotriazole markedly reduced the elongation and tensile strength of PA66, such that the molecular weight and polydispersity of polymers were altered (Fujisawa et al. 2001). Manganese peroxidases are heme-containing peroxidases that utilize H2O2 as a cosubstrate to generate reactive manganese (Mn) that can oxidize the substrate (Hofrichter et al. 2010). Based on current knowledge, these ligninolytic enzymes should produce oxidative species to cause polymer chain breakage indirectly, and no evidence of direct enzyme–substrate interactions is reported (Chen et al. 2020).
Polyurethane
Polyurethanes (PU) are the sixth most used polymers with an annual consumption of more than 20 million metric tons, which are mainly used to produce flexible foams (e.g., mattresses and furniture) or rigid foams (e.g., insulation and construction materials) (Eling et al. 2020; Kemona and Piotrowska 2020). Polyurethane is composed of the carbamate-linked isocyanate and a polyester–(polyester PU) or polyether-containing diol (polyether PU), which comprise the hard and soft regions of the polymer, respectively (Fig. 1). Despite containing a heteroatom backbone, the biodegradation of polyurethane remains challenging and the current understanding of biological routes and enzymes from the degradative microbial communities is limiting (Liu et al. 2021). In most studies, polyurethane decomposition efficacy is probed using model substrates Impranil® DLN for aliphatic polyester PU and PolyLack for polyether PU. They can be dissolved as colloidal dispersions and become translucent when hydrolyzed, enabling a fast and easy observation of depolymerization events. Polyurethanes are highly recalcitrant to biodegradation, such that the best Impranil degrader ever reported, Cladosporium pseudocladosporioides strain T1.PL.1, demands two weeks to achieve 87% decomposition (Álvarez-Barragán et al. 2016).
Various enzyme activities have been proposed to account for the polyurethane decomposition in polyurethane-degrading microorganisms including esterase, lipase, protease, amidase and urease (Magnin et al. 2020). Nonetheless, no direct evidence is available for urease-mediated polyurethane degradation. Enzymes with esterase activity can hydrolyze the ester bond in polyester PU and reduce the turbidity of Impranil® DLN (Akutsu et al. 1998; Álvarez-Barragán et al. 2016; Schmidt et al. 2017). Similar effects were observed using amidase, also belonging to serine hydrolase, which might cleave both ester and urethane bonds (Gamerith et al. 2016; Magnin et al. 2019). Cutinases can hydrolyze the ester bonds in the soft segments, releasing carboxylic acid and alcohol end groups. However, the overall decomposition efficacy is very low relative to PET, such that LCC leads to up to 4.9% weight loss in 200 h at 70 °C (Schmidt et al. 2017). Some enzymes able to degrade polyurethane have been called polyurethaneases, but a detail characterization of their abilities to hydrolyze urethane bonds is not available. A true urethanase that can directly degrade the urethane bonds in polyurethane polymers to release aromatic moieties has not been undisputedly identified (Wei et al. 2020). Notably, thermophilic enzymes might be advantageous over those operating at lower temperatures, offering a possible direction to improving polyurethane decomposing enzymes (Schmidt et al. 2017). Relative to polyester PU, polyether PU is less susceptible to enzymatic attack and their degradation by reactive oxygen species produced by laccases and peroxidases has been proposed, although the degradation effects are still very low (Mahajan and Gupta 2015).
Other polymers
Other major plastic polymers include polyethylene (PE), polystyrene (PS), polypropylene (PP) and poly(vinyl chloride) (PVC), with polyethylene and polystyrene accounting for about 40% of the total plastic production. Effective biodegradation of these polymers is still highly challenging because the C–C bond is inert to the enzyme reaction (Zhang et al. 2022a, b). The natural decomposition of polyolefins proceeds at an extremely low rate and should demand a concerted action of multiple microorganisms and enzyme systems (Ali et al. 2021; Chow et al. 2022; Ru et al. 2020). Most polyolefin decomposition studies focus on polyethylene, with few reports describing polystyrene degradation. Polypropylene and poly(vinyl chloride) are extremely stable and their biodegradation, if it occurs at all, takes months to decades to become apparent (Chen et al. 2020). Even for polyethylene, abiotic factors such as mechanical forces, UV irradiation and oxidizing agents are usually employed in conjunction with biological agents to achieve meaningful results (Restrepo-Flórez et al. 2014). These pretreatments mediate oxidation to introduce carbonyl or olefin functional groups into the polymer backbone, making them more susceptible to enzyme-mediated events. Therefore, measuring the carbonyl index of the polymer, which is usually analyzed by Fourier transform infrared spectroscopy, is utilized to evaluate the polymer decomposition. In addition, surface erosion, weight loss and reduction of average molecular mass of PE also serve as indicators.
Polyethylene comprises a linear chain of carbons and can be produced with different chain arrangements. Linear polyethylene has high symmetry, and it is commonly known as high-density polyethylene, which is harder, offers higher strength and has better heat resistance. Low-density polyethylene contains long and short irregular branches and is widely used in plastic bags owing to its light and flexible properties. Current research on polyethylene decomposition is mainly toward low-density polyethylene. Many polyethylene-degrading microorganisms have been reported (Gao et al. 2022; Jayan et al. 2023; Kumar Sen and Raut 2015). Some show capability in metabolizing linear alkanes with a membranous enzyme alkane hydroxylase, a member of the alkB family (EC 1.14.15.3) that plays a key role in the aerobic degradation of alkanes and β-oxidation of fatty acids in bacteria. A complete alkane hydroxylase system contains three components: an electron-generating reductase (rubredoxin reductase, EC 1.18.1.1), an electron-transporting small Fe-binding protein termed rubredoxin and a membrane-bound non-heme diiron monooxygenase (alkB) (Beilen and Funhoff 2007). The rubredoxin reductase reduces the Fe(III)-Cys4 site of rubredoxin to its Fe(II)-Cys4 form, which, in turn, relays the electron to alkB. The reduced alkB then reacts with O2 to perform terminal or subterminal oxidation of hydrocarbon oligomers. The products can be further catalyzed to produce aldehydes and acids, eventually entering the intracellular β-oxidation process. This machinery operates terminal oxidation and thus should proceed at a slow rate. It has been reported that the reconstitution of alkB in E. coli BL21 resulted in converting 30% of low-molecular-weight polyethylene to CO2 in 80 days (Jeon and Kim 2015).
Microbial enzymes that can act to decompose plant lignin, including laccase, manganese peroxidase and lignin peroxidase (EC 1.11.1.14), are the most reported enzymes related to polyethylene decomposition (Chen et al. 2020; Wei and Zimmermann 2017b). These enzymes act to produce oxidative species to oxidize the polymer, but the degradation efficacy is still very low (Fujisawa et al. 2001; Santo et al. 2013). Compared to peroxidases, more studies on laccase-mediated PE decomposition are reported (Santo et al. 2013; Yao et al. 2022; Zhang et al. 2023). Laccase-mediator system constructed by laccases from the fungus Botrytis aclada and a bacterium B. subtilis in combination with three synthetic mediators shows oxidation, molecular weight reduction and surface erosion on low-density polyethylene films that were pretreated with high-temperature UV irradiation (Yao et al. 2022). A psychrophilic laccase from Antarctic bacterium Psychrobacter sp. NJ228 can achieve up to 13.2% weight loss of PE particles in 24 h (Zhang et al. 2022a, b). Two laccase-like multicopper oxidases induced in Rhodococcus opacus that can utilize polyethylene as the only carbon and energy source to grow can afford polyethylene oxidation, various alkyl compounds and oxygenated products in 48 h (Zampolli et al. 2023). A thermophilic laccase from polyethylene-degrading bacterium Lysinibaccillus fusiformis (LfLAC3) that was expected to possess a relatively open catalytic pocket was selected by using the computer-aided discovery and activity-based screening (Zhang et al. 2023). The formation of various functional groups was detected and the surface damage on the polyethylene films was observed, though only 3.75% weight loss of polyethylene films was achieved at 28 °C in eight weeks. Although the substrate-binding modes of LfLAC3 have been proposed by structure analysis, these enzymes possess small substrate-binding pockets that should only allow the accommodation of small molecules (Matera et al. 2008; Santacruz-Juarez et al. 2021; Sundaramoorthy et al. 2010). Therefore, the decomposition effects are more likely to occur indirectly.
Insects have been reported with the ability to degrade untreated polyolefins including polyethylene and polystyrene (Bombelli et al. 2017; Yang et al. 2020). Whether these capacities are mediated by the microorganisms in the worm gut, the invertebrate itself, or a concerted action of both has remained elusive. Recently, two enzymes belonging to the hexamerin/prophenoloxidase family have been reported as the main components in the saliva of Galleria mellonella larvae (wax worm) to cause polyethylene deterioration (Sanluis-Verdes et al. 2022). These enzymes are attractive for further applications as they cause oxidation and depolymerization of untreated polyethylene film within a few hours at room temperature. Although the mechanism of action of these enzymes in polyethylene modification remains unknown, targeting the aromatic additives blended in the plastics to form free radicals and initiate oxidative chain reactions is a possible route.
Conclusion
Enzyme-based decomposition and recycling of synthetic polymers, especially poly(ethylene terephthalate), have advanced at an unprecedented rate in recent years. These successes largely depend on developing technologies used in enzyme identification, molecular manipulation, protein engineering and high-throughput screening methods. The real-world application of the enzyme-mediated decomposition and recycling of poly(ethylene terephthalate) could be expected to occur within the next few years, whereas a longer time is required for other types of plastics. The cost of enzyme production should be lowered to trigger economic motivations, given that producing new plastic materials is cheaper than recycling the waste at the current stage. Therefore, evaluating the effectiveness of enzyme production on an industrial scale should be put on the schedule. Furthermore, pretreatments are required to unify the physical properties of postconsumer commodities, e.g., plastic size and crystallinity, to facilitate enzymatic degradation. It should be noted that enzyme-mediated degradation on nano- or microplastics that show a higher penetration rate to the living cells deserves more investigations (Anand et al. 2023; Sharma et al. 2023), as more accessible sites on such kinds of plastics could increase the enzyme reaction rate. In order to realize the long-term objectives, it is imperative to continue research endeavors to uncover enzyme properties, polymer-enzyme interactions and polymer behavior during catalytic reactions while simultaneously striving to augment enzyme performance. Although enzyme-mediated depolymerization toward other plastics is still in its infancy, efforts to identify and improve enzyme performance should also be encouraged. In this context, the enzyme properties revealed in poly(ethylene terephthalate)-related research might also benefit those for other types of plastics.
Funding
This work was supported by the National Key Research and Development Program of China (2022YFE0135300), Hubei Hongshan Laboratory (2022hszd030), the National Natural Science Foundation of China (32271318).
Abbreviations
- Da:
-
Dalton
- DS bond:
-
Disulfide bond
- EC number:
-
Enzyme commission number
- GFP:
-
Green fluorescence protein
- LCC:
-
Leaf compost cutinase
- M w :
-
Molecular weight
- M n :
-
Number-averaged molecular mass
- MHET:
-
Mono(2-hydroxyethyl) terephthalate
- PBAT:
-
Poly(butylene adipate terephthalate)
- PE:
-
Polyethylene
- PET:
-
Polyethylene terephthalate
- PP:
-
Polypropylene
- PS:
-
Polystyrene
- PVC:
-
Poly(vinyl) chloride
- PU:
-
Polyurethane
- T g :
-
Glass transition temperature
- T m :
-
Melting temperature
- UV:
-
Ultraviolet
- X c :
-
Degree of crystallinity
References
Ahmad A, Othman I, Rambabu K, Bharath G, Taher H, Hasan SW, Banat F (2021) Polymerization of lactic acid produced from food waste by metal oxide-assisted dark fermentation. Environ Technol Innovation 24:101862. https://doi.org/10.1016/j.eti.2021.101862
Akutsu Y, Nakajima-Kambe T, Nomura N, Nakahara T (1998) Purification and properties of a polyester polyurethane-degrading enzyme from Comamonas acidovorans TB-35. Appl Environ Microbiol 64(1):62–67. https://doi.org/10.1128/AEM.64.1.62-67.1998
Akutsu-Shigeno Y, Teeraphatpornchai T, Teamtisong K, Nomura N, Uchiyama H, Nakahara T, Nakajima-Kambe T (2003) Cloning and sequencing of a poly(DL-lactic acid) depolymerase gene from Paenibacillus amylolyticus strain TB-13 and its functional expression in Escherichia coli. Appl Environ Microbiol 69(5):2498–2504. https://doi.org/10.1128/AEM.69.5.2498-2504.2003
Ali SS, Elsamahy T, Al-Tohamy R, Zhu D, Mahmoud YA, Koutra E, Metwally MA, Kornaros M, Sun J (2021) Plastic wastes biodegradation: mechanisms, challenges and future prospects. Sci Total Environ 780:146590. https://doi.org/10.1016/j.scitotenv.2021.146590
Ali W, Ali H, Souissi S, Zinck P (2023) Are bioplastics an ecofriendly alternative to fossil fuel plastics? Environ Chem Lett 21(4):1991–2002. https://doi.org/10.1007/s10311-023-01601-6
Álvarez-Barragán J, Domínguez-Malfavón L, Vargas-Suárez M, González-Hernández R, Aguilar-Osorio G, Loza-Tavera H (2016) Biodegradative activities of selected environmental fungi on a polyester polyurethane varnish and polyether polyurethane foams. Appl Environ Microbiol 82(17):5225–5235. https://doi.org/10.1128/AEM.01344-16
Alves NM, Mano JF, Balaguer E, Meseguer Dueñas JM, Gómez Ribelles JL (2002) Glass transition and structural relaxation in semi-crystalline poly(ethylene terephthalate): a DSC study. Polymer 43(15):4111–4122. https://doi.org/10.1016/S0032-3861(02)00236-7
Anand U, Dey S, Bontempi E, Ducoli S, Vethaak AD, Dey A, Federici S (2023) Biotechnological methods to remove microplastics: a review. Environ Chem Lett 21(3):1787–1810. https://doi.org/10.1007/s10311-022-01552-4
Atthoff B, Hilborn J (2007) Protein adsorption onto polyester surfaces: is there a need for surface activation? J Biomed Mater Res Part B 80(1):121–130. https://doi.org/10.1002/jbm.b.30576
Austin HP, Allen MD, Donohoe BS, Rorrer NA, Kearns FL, Silveira RL, Pollard BC, Dominick G, Duman R, El Omari K, Mykhaylyk V, Wagner A, Michener WE, Amore A, Skaf MS, Crowley MF, Thorne AW, Johnson CW, Woodcock HL, McGeehan JE, Beckham GT (2018) Characterization and engineering of a plastic-degrading aromatic polyesterase. Proc Natl Acad Sci U S A 115(19):e4350–e4357. https://doi.org/10.1073/pnas.1718804115
Bååth JA, Borch K, Jensen K, Brask J, Westh P (2021) Comparative biochemistry of four polyester (PET) hydrolases. ChemBioChem 22(9):1627–1637. https://doi.org/10.1002/cbic.202000793
Bååth JA, Jensen K, Borch K, Westh P, Kari J (2022) Sabatier principle for rationalizing enzymatic hydrolysis of a synthetic polyester. JACS Au 2(5):1223–1231. https://doi.org/10.1021/jacsau.2c00204
Barnard E, Rubio Arias JJ, Thielemans W (2021) Chemolytic depolymerisation of PET: a review. Green Chem 23(11):3765–3789. https://doi.org/10.1039/D1GC00887K
Bartnikowski M, Dargaville TR, Ivanovski S, Hutmacher DW (2019) Degradation mechanisms of polycaprolactone in the context of chemistry, geometry and environment. Prog Polym Sci 96:1–20. https://doi.org/10.1016/j.progpolymsci.2019.05.004
Bell EL, Smithson R, Kilbride S, Foster J, Hardy FJ, Ramachandran S, Tedstone AA, Haigh SJ, Garforth AA, Day PJR, Levy C, Shaver MP, Green AP (2022) Directed evolution of an efficient and thermostable PET depolymerase. Nat Catal 5(8):673–681. https://doi.org/10.1038/s41929-022-00821-3
Biundo A, Hromic A, Pavkov-Keller T, Gruber K, Quartinello F, Haernvall K, Perz V, Arrell MS, Zinn M, Ribitsch D, Guebitz GM (2016) Characterization of a poly(butylene adipate-co-terephthalate)-hydrolyzing lipase from Pelosinus fermentans. Appl Environ Microbiol 100(4):1753–1764. https://doi.org/10.1007/s00253-015-7031-1
Blázquez-Sánchez P, Engelberger F, Cifuentes-Anticevic J, Sonnendecker C, Griñén A, Reyes J, Díez B, Guixé V, Richter PK, Zimmermann W, Ramírez-Sarmiento CA (2022) Antarctic polyester hydrolases degrade aliphatic and aromatic polyesters at moderate temperatures. Appl Environ Microbiol 88(1):e0184221. https://doi.org/10.1128/aem.01842-21
Blazquez-Sanchez P, Vargas JA, Furtado AA, Grinen A, Leonardo DA, Sculaccio SA, Pereira HD, Sonnendecker C, Zimmermann W, Diez B, Garratt RC, Ramirez-Sarmiento CA (2023) Engineering the catalytic activity of an Antarctic PET-degrading enzyme by loop exchange. Protein Sci 32(9):e4757. https://doi.org/10.1002/pro.4757
Bollinger A, Thies S, Knieps-Grunhagen E, Gertzen C, Kobus S, Hoppner A, Ferrer M, Gohlke H, Smits SHJ, Jaeger KE (2020) A novel polyester hydrolase from the marine bacterium Pseudomonas aestusnigri—structural and functional insights. Front Microbiol 11:114. https://doi.org/10.3389/fmicb.2020.00114
Bombelli P, Howe CJ, Bertocchini F (2017) Polyethylene bio-degradation by caterpillars of the wax moth Galleria mellonella. Curr Biol 27(8):R292-r293. https://doi.org/10.1016/j.cub.2017.02.060
Brzozowski AM, Savage H, Verma CS, Turkenburg JP, Lawson DM, Svendsen A, Patkar S (2000) Structural origins of the interfacial activation in Thermomyces (Humicola) lanuginosa lipase. Biochemistry 39(49):15071–15082. https://doi.org/10.1021/bi0013905
Chen S, Tong X, Woodard RW, Du G, Wu J, Chen J (2008) Identification and characterization of bacterial cutinase. J Biol Chem 283(38):25854–25862. https://doi.org/10.1074/jbc.M800848200
Chen CC, Han X, Ko TP, Liu W, Guo RT (2018) Structural studies reveal the molecular mechanism of PETase. FEBS J 285(20):3717–3723. https://doi.org/10.1111/febs.14612
Chen C-C, Dai L, Ma L, Guo R-T (2020) Enzymatic degradation of plant biomass and synthetic polymers. Nat Rev Chem 4(3):114–126. https://doi.org/10.1038/s41570-020-0163-6
Chen CC, Han X, Li X, Jiang P, Niu D, Ma L, Liu W, Li S, Qu Y, Hu H, Min J, Yang Y, Zhang L, Zeng W, Huang J-W, Dai L, Guo RT (2021a) General features to enhance enzymatic activity of poly(ethylene terephthalate) hydrolysis. Nat Catal 4(5):425–430. https://doi.org/10.1038/s41929-021-00616-y
Chen K, Hu Y, Dong X, Sun Y (2021b) Molecular insights into the enhanced performance of EKylated PETase toward PET degradation. ACS Catal 11(12):7358–7370. https://doi.org/10.1021/acscatal.1c01062
Chen X-Q, Guo Z-Y, Wang L, Yan Z-F, Jin C-X, Huang Q-S, Kong D-M, Rao D-M, Wu J (2022) Directional-path modification strategy enhances PET hydrolase catalysis of plastic degradation. J Hazard Mater 433:128816. https://doi.org/10.1016/j.jhazmat.2022.128816
Chow J, Perez-Garcia P, Dierkes R, Streit WR (2022) Microbial enzymes will offer limited solutions to the global plastic pollution crisis. Microb Biotechnol l16(2):195–217. https://doi.org/10.1111/1751-7915.14135
Christopher FJ, Kumar PS, Vo D-VN, Christopher FC, Jayaraman L (2022) Methods for chemical conversion of plastic wastes into fuels and chemicals. A Review. Environ Chem Lett 20(1):223–242. https://doi.org/10.1007/s10311-021-01329-1
Cui Y, Chen Y, Liu X, Dong S, Ye T, Qiao Y, Mitra R, Han J, Li C, Han X, Liu W, Chen Q, Wei W, Wang X, Du W, Tang S, Xiang H, Liu H, Liang Y, Houk KN, Wu B (2021) Computational redesign of a PETase for plastic biodegradation under ambient condition by the GRAPE strategy. ACS Catal 11(3):1340–1350. https://doi.org/10.1021/acscatal.0c05126
Dai L, Qu Y, Huang J-W, Hu Y, Hu H, Li S, Chen C-C, Guo R-T (2021) Enhancing PET hydrolytic enzyme activity by fusion of the cellulose–binding domain of cellobiohydrolase I from Trichoderma reesei. J Biotechnol 334:47–50. https://doi.org/10.1016/j.jbiotec.2021.05.006
Damayanti W-S (2021) Strategic possibility routes of recycled PET. Polymers 13(9):1475. https://doi.org/10.3390/polym13091475
Danso D, Schmeisser C, Chow J, Zimmermann W, Wei R, Leggewie C, Li X, Hazen T, Streit WR (2018) New insights into the function and global distribution of polyethylene terephthalate (PET)-degrading bacteria and enzymes in marine and terrestrial metagenomes. Appl Environ Microbiol 84(8):e02773-e12717. https://doi.org/10.1128/AEM.02773-17
Deguchi T, Kitaoka Y, Kakezawa M, Nishida T (1998) Purification and characterization of a nylon-degrading enzyme. Appl Environ Microbiol 64(4):1366–1371. https://doi.org/10.1128/AEM.64.4.1366-1371.1998
Dhaka V, Singh S, Anil AG, Sunil Kumar Naik TS, Garg S, Samuel J, Kumar M, Ramamurthy PC, Singh J (2022) Occurrence, toxicity and remediation of polyethylene terephthalate plastics. A Review. Environ Chem Lett 20(3):1777–1800. https://doi.org/10.1007/s10311-021-01384-8
Ding Z, Xu G, Miao R, Wu N, Zhang W, Yao B, Guan F, Huang H, Tian J (2023) Rational redesign of thermophilic PET hydrolase LCCICCG to enhance hydrolysis of high crystallinity polyethylene terephthalates. J Hazard Mater 453:131386. https://doi.org/10.1016/j.jhazmat.2023.131386
Eiamthong B, Meesawat P, Wongsatit T, Jitdee J, Sangsri R, Patchsung M, Aphicho K, Suraritdechachai S, Huguenin-Dezot N, Tang S, Suginta W, Paosawatyanyong B, Babu MM, Chin JW, Pakotiprapha D, Bhanthumnavin W, Uttamapinant C (2022) Discovery and genetic code expansion of a polyethylene terephthalate (PET) hydrolase from the human saliva metagenome for the degradation and bio-functionalization of PET. Angew Chem Int Ed 61(37):e202203061. https://doi.org/10.1002/anie.202203061
Eling B, Tomović Ž, Schädler V (2020) Current and future trends in polyurethanes: An industrial perspective. Macromol Chem Phys 221(14):2000114. https://doi.org/10.1002/macp.202000114
Emori M, Numoto N, Senga A, Bekker G-J, Kamiya N, Kobayashi Y, Ito N, Kawai F, Oda M (2021) Structural basis of mutants of PET-degrading enzyme from Saccharomonospora viridis AHK190 with high activity and thermal stability. Proteins Struct Funct Bioinf 89(5):502–511. https://doi.org/10.1002/prot.26034
Erickson E, Gado JE, Avilán L, Bratti F, Brizendine RK, Cox PA, Gill R, Graham R, Kim D-J, König G, Michener WE, Poudel S, Ramirez KJ, Shakespeare TJ, Zahn M, Boyd ES, Payne CM, DuBois JL, Pickford AR, Beckham GT, McGeehan JE (2022) Sourcing thermotolerant poly(ethylene terephthalate) hydrolase scaffolds from natural diversity. Nat Commun 13(1):7850. https://doi.org/10.1038/s41467-022-35237-x
Falkenstein P, Zhao Z, Di Pede-Mattatelli A, Künze G, Sommer M, Sonnendecker C, Zimmermann W, Colizzi F, Matysik J, Song C (2023) On the binding mode and molecular mechanism of enzymatic polyethylene terephthalate degradation. ACS Catal 13(10):6919–6933. https://doi.org/10.1021/acscatal.3c00259
Fecker T, Galaz-Davison P, Engelberger F, Narui Y, Sotomayor M, Parra LP, Ramirez-Sarmiento CA (2018) Active site flexibility as a hallmark for efficient PET degradation by I. sakaiensis PETase. Biophys J 114(6):1302–1312. https://doi.org/10.1016/j.bpj.2018.02.005
Fujisawa M, Hirai H, Nishida T (2001) Degradation of polyethylene and Nylon-66 by the laccase-mediator system. J Polym Environ 9(3):103–108. https://doi.org/10.1023/A:1020472426516
Gamerith C, Herrero Acero E, Pellis A, Ortner A, Vielnascher R, Luschnig D, Zartl B, Haernvall K, Zitzenbacher S, Strohmeier G, Hoff O, Steinkellner G, Gruber K, Ribitsch D, Guebitz GM (2016) Improving enzymatic polyurethane hydrolysis by tuning enzyme sorption. Polym Degrad Stab 132:69–77. https://doi.org/10.1016/j.polymdegradstab.2016.02.025
Gao R, Liu R, Sun C (2022) A marine fungus Alternaria alternata FB1 efficiently degrades polyethylene. J Hazard Mater 431:128617. https://doi.org/10.1016/j.jhazmat.2022.128617
Hajighasemi M, Nocek BP, Tchigvintsev A, Brown G, Flick R, Xu X, Cui H, Hai T, Joachimiak A, Golyshin PN, Savchenko A, Edwards EA, Yakunin AF (2016) Biochemical and structural insights into enzymatic depolymerization of polylactic acid and other polyesters by microbial carboxylesterases. Biomacromol 17(6):2027–2039. https://doi.org/10.1021/acs.biomac.6b00223
Hajighasemi M, Tchigvintsev A, Nocek B, Flick R, Popovic A, Hai T, Khusnutdinova AN, Brown G, Xu X, Cui H, Anstett J, Chernikova TN, Brüls T, Le Paslier D, Yakimov MM, Joachimiak A, Golyshina OV, Savchenko A, Golyshin PN, Edwards EA, Yakunin AF (2018) Screening and characterization of novel polyesterases from environmental metagenomes with high hydrolytic activity against synthetic polyesters. Environ Sci Technol 52(21):12388–12401. https://doi.org/10.1021/acs.est.8b04252
Han X, Liu W, Huang JW, Ma J, Zheng Y, Ko TP, Xu L, Cheng YS, Chen CC, Guo RT (2017) Structural insight into catalytic mechanism of PET hydrolase. Nat Commun 8(1):2106. https://doi.org/10.1038/s41467-017-02255-z
Hayes HC, Luk LYP (2023) Investigating the effects of cyclic topology on the performance of a plastic degrading enzyme for polyethylene terephthalate degradation. Sci Rep 13(1):1267. https://doi.org/10.1038/s41598-023-27780-4
Herrero Acero E, Ribitsch D, Steinkellner G, Gruber K, Greimel K, Eiteljoerg I, Trotscha E, Wei R, Zimmermann W, Zinn M, Cavaco-Paulo A, Freddi G, Schwab H, Guebitz G (2011) Enzymatic surface hydrolysis of PET: effect of structural diversity on kinetic properties of cutinases from Thermobifida. Macromolecules 44(12):4632–4640. https://doi.org/10.1021/ma200949p
Hilgers R, Vincken JP, Gruppen H, Kabel MA (2018) Laccase/mediator systems: their reactivity toward phenolic lignin structures. ACS Sustain Chem Eng 6(2):2037–2046. https://doi.org/10.1021/acssuschemeng.7b03451
Hofrichter M, Ullrich R, Pecyna MJ, Liers C, Lundell T (2010) New and classic families of secreted fungal heme peroxidases. Appl Microbiol Biotechnol 87(3):871–897. https://doi.org/10.1007/s00253-010-2633-0
Hu X, Thumarat U, Zhang X, Tang M, Kawai F (2010) Diversity of polyester-degrading bacteria in compost and molecular analysis of a thermoactive esterase from Thermobifida alba AHK119. Appl Microbiol Biotechnol 87(2):771–779. https://doi.org/10.1007/s00253-010-2555-x
Janusz G, Pawlik A, Świderska-Burek U, Polak J, Sulej J, Jarosz-Wilkołazka A, Paszczyński A (2020) Laccase properties, physiological functions, and evolution. Int J Mol Sci 21(3):966. https://doi.org/10.3390/ijms21030966
Jayan N, Skariyachan S, Sebastian D (2023) The escalated potential of the novel isolate Bacillus cereus NJD1 for effective biodegradation of LDPE films without pre-treatment. J Hazard Mater 455:131623. https://doi.org/10.1016/j.jhazmat.2023.131623
Jeon HJ, Kim MN (2015) Functional analysis of alkane hydroxylase system derived from Pseudomonas aeruginosa E7 for low molecular weight polyethylene biodegradation. Int Biodeterior Biodegrad 103:141–146. https://doi.org/10.1016/j.ibiod.2015.04.024
Jia X, Zhao K, Zhao J, Lin C, Zhang H, Chen L, Chen J, Fang Y (2023) Degradation of poly(butylene adipate-co-terephthalate) films by Thermobifida fusca FXJ-1 isolated from compost. J Hazard Mater 441:129958. https://doi.org/10.1016/j.jhazmat.2022.129958
Joo S, Cho IJ, Seo H, Son HF, Sagong HY, Shin TJ, Choi SY, Lee SY, Kim KJ (2018) Structural insight into molecular mechanism of poly(ethylene terephthalate) degradation. Nat Commun 9(1):382. https://doi.org/10.1038/s41467-018-02881-1
Kader MA, Senge M, Mojid MA, Ito K (2017) Recent advances in mulching materials and methods for modifying soil environment. Soil Tillage Res 168:155–166. https://doi.org/10.1016/j.still.2017.01.001
Kanelli M, Vasilakos S, Ladas S, Symianakis E, Christakopoulos P, Topakas E (2017) Surface modification of polyamide 6.6 fibers by enzymatic hydrolysis. Process Biochem 59:97–103. https://doi.org/10.1016/j.procbio.2016.06.022
Kari J, Olsen JP, Jensen K, Badino SF, Krogh KBRM, Borch K, Westh P (2018) Sabatier principle for interfacial (heterogeneous) enzyme catalysis. ACS Catal 8(12):11966–11972. https://doi.org/10.1021/acscatal.8b03547
Kawai F, Nakadai K, Nishioka E, Nakajima H, Ohara H, Masaki K, Iefuji H (2011) Different enantioselectivity of two types of poly(lactic acid) depolymerases toward poly(L-lactic acid) and poly(D-lactic acid). Polym Degrad Stab 96(7):1342–1348. https://doi.org/10.1016/j.polymdegradstab.2011.03.022
Kawai F, Oda M, Tamashiro T, Waku T, Tanaka N, Yamamoto M, Mizushima H, Miyakawa T, Tanokura M (2014) A novel Ca2+-activated, thermostabilized polyesterase capable of hydrolyzing polyethylene terephthalate from Saccharomonospora viridis AHK190. Appl Microbiol Biotechnol 98(24):10053–10064. https://doi.org/10.1007/s00253-014-5860-y
Kawai F, Kawabata T, Oda M (2019) Current knowledge on enzymatic PET degradation and its possible application to waste stream management and other fields. Appl Microbiol Biotechnol 103(11):4253–4268. https://doi.org/10.1007/s00253-019-09717-y
Kemona A, Piotrowska M (2020) Polyurethane recycling and disposal: methods and prospects. Polymers 12(8):1752. https://doi.org/10.3390/polym12081752
Kitadokoro K, Thumarat U, Nakamura R, Nishimura K, Karatani H, Suzuki H, Kawai F (2012) Crystal structure of cutinase Est119 from Thermobifida alba AHK119 that can degrade modified polyethylene terephthalate at 1.76 Å resolution. Polym Degrad Stab 97:771–775. https://doi.org/10.1016/j.polymdegradstab.2012.02.003
Kitadokoro K, Kakara M, Matsui S, Osokoshi R, Thumarat U, Kawai F, Kamitani S (2019) Structural insights into the unique polylactate-degrading mechanism of Thermobifida alba cutinase. FEBS J 286(11):2087–2098. https://doi.org/10.1111/febs.14781
Kleeberg I, Hetz C, Kroppenstedt RM, Muller RJ, Deckwer WD (1998) Biodegradation of aliphatic-aromatic copolyesters by Thermomonospora fusca and other thermophilic compost isolates. Appl Environ Microbiol 64(5):1731–1735. https://doi.org/10.1128/AEM.64.5.1731-1735.1998
Kleeberg I, Welzel K, VandenHeuvel J, Müller RJ, Deckwer WD (2005) Characterization of a new extracellular hydrolase from Thermobifida fusca degrading aliphatic–aromatic copolyesters. Biomacromol 6(1):262–270. https://doi.org/10.1021/bm049582t
Koshti R, Mehta L, Samarth N (2018) Biological recycling of polyethylene terephthalate: a mini-review. J Polym Environ 26(8):3520–3529. https://doi.org/10.1007/s10924-018-1214-7
Kumar Sen S, Raut S (2015) Microbial degradation of low density polyethylene (LDPE): a review. J Environ Chem Eng 3(1):462–473. https://doi.org/10.1016/j.jece.2015.01.003
Lampitt RS, Fletcher S, Cole M, Kloker A, Krause S, O’Hara F, Ryde P, Saha M, Voronkova A, Whyle A (2023) Stakeholder alliances are essential to reduce the scourge of plastic pollution. Nat Commun 14(1):2849. https://doi.org/10.1038/s41467-023-38613-3
Li S, McCarthy S (1999) Influence of crystallinity and stereochemistry on the enzymatic degradation of poly(lactide)s. Macromolecules 32(13):4454–4456. https://doi.org/10.1021/ma990117b
Li Q, Zheng Y, Su T, Wang Q, Liang Q, Zhang Z, Qi Q, Tian J (2022) Computational design of a cutinase for plastic biodegradation by mining molecular dynamics simulations trajectories. Comput Struct Biotechnol J 20:459–470. https://doi.org/10.1016/j.csbj.2021.12.042
Li X, Shi B, Huang J-W, Zeng Z, Yang Y, Zhang L, Min J, Chen C-C, Guo R-T (2023) Functional tailoring of a PET hydrolytic enzyme expressed in Pichia pastoris. Bioresour Bioprocess 10(1):26. https://doi.org/10.1186/s40643-023-00648-1
Lim HA, Raku T, Tokiwa Y (2005) Hydrolysis of polyesters by serine proteases. Biotechnol Lett 27(7):459–464. https://doi.org/10.1007/s10529-005-2217-8
Linko S (1992) Production of Phanerochaete chrysosporium lignin peroxidase. Biotechnol Adv 10(2):191–236. https://doi.org/10.1016/0734-9750(92)90003-r
Liu B, He L, Wang L, Li T, Li C, Liu H, Luo Y, Bao R (2018) Protein crystallography and site-direct mutagenesis analysis of the poly(ethylene terephthalate) hydrolase PETase from Ideonella sakaiensis. ChemBioChem 19(14):1471–1475. https://doi.org/10.1002/cbic.201800097
Liu J, He J, Xue R, Xu B, Qian X, Xin F, Blank LM, Zhou J, Wei R, Dong W, Jiang M (2021) Biodegradation and up-cycling of polyurethanes: progress, challenges, and prospects. Biotechnol Adv 48:107730. https://doi.org/10.1016/j.biotechadv.2021.107730
Liu Y, Liu Z, Guo Z, Yan T, Jin C, Wu J (2022a) Enhancement of the degradation capacity of IsPETase for PET plastic degradation by protein engineering. Sci Total Environ 834:154947. https://doi.org/10.1016/j.scitotenv.2022.154947
Liu Z, Zhang Y, Wu J (2022b) Enhancement of PET biodegradation by anchor peptide-cutinase fusion protein. Enzyme Microb Technol 156:110004. https://doi.org/10.1016/j.enzmictec.2022.110004
Lu H, Diaz DJ, Czarnecki NJ, Zhu C, Kim W, Shroff R, Acosta DJ, Alexander BR, Cole HO, Zhang Y, Lynd NA, Ellington AD, Alper HS (2022) Machine learning-aided engineering of hydrolases for PET depolymerization. Nature 604(7907):662–667. https://doi.org/10.1038/s41586-022-04599-z
Luo Y, Lin X, Lichtfouse E, Jiang H, Wang C (2023) Conversion of waste plastics into value-added carbon materials. Environ Chem Lett 21(6):3127–3158. https://doi.org/10.1007/s10311-023-01638-7
Lykidis A, Mavromatis K, Ivanova N, Anderson I, Land M, DiBartolo G, Martinez M, Lapidus A, Lucas S, Copeland A, Richardson P, Wilson David B, Kyrpides N (2007) Genome sequence and analysis of the soil cellulolytic actinomycete Thermobifida fusca YX. J Bacteriol 189(6):2477–2486. https://doi.org/10.1128/jb.01899-06
MacDonald RT, McCarthy SP, Gross RA (1996) Enzymatic degradability of poly(lactide): effects of chain stereochemistry and material crystallinity. Macromolecules 29(23):7356–7361. https://doi.org/10.1021/ma960513j
Magnin A, Pollet E, Perrin R, Ullmann C, Persillon C, Phalip V, Avérous L (2019) Enzymatic recycling of thermoplastic polyurethanes: synergistic effect of an esterase and an amidase and recovery of building blocks. Waste Manag 85:141–150. https://doi.org/10.1016/j.wasman.2018.12.024
Magnin A, Pollet E, Phalip V, Avérous L (2020) Evaluation of biological degradation of polyurethanes. Biotechnol Adv 39:107457. https://doi.org/10.1016/j.biotechadv.2019.107457
Mahajan N, Gupta P (2015) New insights into the microbial degradation of polyurethanes. RSC Adv 5(52):41839–41854. https://doi.org/10.1039/C5RA04589D
Makryniotis K, Nikolaivits E, Gkountela C, Vouyiouka S, Topakas E (2023) Discovery of a polyesterase from Deinococcus maricopensis and comparison to the benchmark LCCICCG suggests high potential for semi-crystalline post-consumer PET degradation. J Hazard Mater 455:131574. https://doi.org/10.1016/j.jhazmat.2023.131574
Markit IHS (2021) PET polymer: Chemical economics handbook. can be found under. https: //ihsmarkitcom/products/pet-polymer-chemical-economics-handbookhtml
Marten E, Müller R-J, Deckwer W-D (2005) Studies on the enzymatic hydrolysis of polyesters. II. Aliphatic–aromatic copolyesters. Polym Degrad Stab 88(3):371–381. https://doi.org/10.1016/j.polymdegradstab.2004.12.001
Masaki K, Kamini Numbi R, Ikeda H, Iefuji H (2005) Cutinase-like enzyme from the yeast Cryptococcus sp. strain S-2 hydrolyzes polylactic acid and other biodegradable plastics. Appl Environ Microbiol 71(11):7548–7550. https://doi.org/10.1128/AEM.71.11.7548-7550.2005
Matera I, Gullotto A, Tilli S, Ferraroni M, Scozzafava A, Briganti F (2008) Crystal structure of the blue multicopper oxidase from the white-rot fungus Trametes trogii complexed with p-toluate. Inorg Chim Acta 361(14):4129–4137. https://doi.org/10.1016/j.ica.2008.03.091
Meyer-Cifuentes IE, Werner J, Jehmlich N, Will SE, Neumann-Schaal M, Ozturk B (2020) Synergistic biodegradation of aromatic-aliphatic copolyester plastic by a marine microbial consortium. Nat Commun 11(1):5790. https://doi.org/10.1038/s41467-020-19583-2
Miyakawa T, Mizushima H, Ohtsuka J, Oda M, Kawai F, Tanokura M (2015) Structural basis for the Ca(2+)-enhanced thermostability and activity of PET-degrading cutinase-like enzyme from Saccharomonospora viridis AHK190. Appl Microbiol Biotechnol 99(10):4297–4307. https://doi.org/10.1007/s00253-014-6272-8
Mueller RJ (2006) Biological degradation of synthetic polyesters—Enzymes as potential catalysts for polyester recycling. Process Biochem 41(10):2124–2128. https://doi.org/10.1016/j.procbio.2006.05.018
Müller RJ, Kleeberg I, Deckwer WD (2001) Biodegradation of polyesters containing aromatic constituents. J Biotechnol 86(2):87–95. https://doi.org/10.1016/S0168-1656(00)00407-7
Müller RJ, Schrader H, Profe J, Dresler K, Deckwer WD (2005) Enzymatic degradation of poly(ethylene terephthalate): rapid hydrolyse using a hydrolase from T. fusca. Macromol Rapid Commun 26(17):1400–1405. https://doi.org/10.1002/marc.200500410
Muroi F, Tachibana Y, Soulenthone P, Yamamoto K, Mizuno T, Sakurai T, Kobayashi Y, Kasuya K-i (2017) Characterization of a poly(butylene adipate-co-terephthalate) hydrolase from the aerobic mesophilic bacterium Bacillus pumilus. Polym Degrad Stab 137:11–22. https://doi.org/10.1016/j.polymdegradstab.2017.01.006
Nakamura A, Kobayashi N, Koga N, Iino R (2021) Positive charge introduction on the surface of thermostabilized PET hydrolase facilitates PET binding and degradation. ACS Catal 11(14):8550–8564. https://doi.org/10.1021/acscatal.1c01204
Nandhini R, Sivaprakash B, Rajamohan N, Vo D-VN (2023) Lignin and polylactic acid for the production of bioplastics and valuable chemicals. Environ Chem Lett 21(1):403–427. https://doi.org/10.1007/s10311-022-01505-x
Negoro S, Kato DI, Ohki T, Yasuhira K, Kawashima Y, Nagai K, Takeo M, Shibata N, Kamiya K, Shigeta Y (2021) Structural and functional characterization of nylon hydrolases. Methods Enzymol 648:357–389. https://doi.org/10.1016/bs.mie.2020.11.004
Nilsson F, Lan X, Gkourmpis T, Hedenqvist MS, Gedde UW (2012) Modelling tie chains and trapped entanglements in polyethylene. Polymer 53(16):3594–3601. https://doi.org/10.1016/j.polymer.2012.05.045
Oda Y, Yonetsu A, Urakami T, Tonomura K (2000) Degradation of polylactide by commercial proteases. J Polym Environ 8(1):29–32. https://doi.org/10.1023/A:1010120128048
Perz V, Baumschlager A, Bleymaier K, Zitzenbacher S, Hromic A, Steinkellner G, Pairitsch A, Łyskowski A, Gruber K, Sinkel C, Küper U, Ribitsch D, Guebitz GM (2016a) Hydrolysis of synthetic polyesters by Clostridium botulinum esterases. Biotechnol Bioeng 113(5):1024–1034. https://doi.org/10.1002/bit.25874
Perz V, Bleymaier K, Sinkel C, Kueper U, Bonnekessel M, Ribitsch D, Guebitz GM (2016b) Substrate specificities of cutinases on aliphatic–aromatic polyesters and on their model substrates. New Biotechnol 33(2):295–304. https://doi.org/10.1016/j.nbt.2015.11.004
Pfaff L, Gao J, Li Z, Jäckering A, Weber G, Mican J, Chen Y, Dong W, Han X, Feiler CG, Ao Y-F, Badenhorst CPS, Bednar D, Palm GJ, Lammers M, Damborsky J, Strodel B, Liu W, Bornscheuer UT, Wei R (2022) Multiple substrate binding mode-guided engineering of a thermophilic PET hydrolase. ACS Catal 12(15):9790–9800. https://doi.org/10.1021/acscatal.2c02275
Pranamuda H, Tsuchii A, Tokiwa Y (2001) Poly (L-lactide)-degrading enzyme produced by Amycolatopsis sp. Macromol Biosci 1(1):25–29. https://doi.org/10.1002/1616-5195(200101)1:1%3c25::AID-MABI25%3e3.0.CO;2-3
Puspitasari N, Tsai S-L, Lee C-K (2020) Fungal hydrophobin RolA enhanced PETase hydrolysis of polyethylene terephthalate. Appl Biochem Biotechnol 193(5):1284–1295. https://doi.org/10.1007/s12010-020-03358-y
Puspitasari N, Tsai SL, Lee CK (2021) Class I hydrophobins pretreatment stimulates PETase for monomers recycling of waste PETs. Int J Biol Macromol 176:157–164. https://doi.org/10.1016/j.ijbiomac.2021.02.026
Reeve MS, McCarthy SP, Downey MJ, Gross RA (1994) Polylactide stereochemistry: effect on enzymic degradability. Macromolecules 27(3):825–831. https://doi.org/10.1021/ma00081a030
Restrepo-Flórez J-M, Bassi A, Thompson MR (2014) Microbial degradation and deterioration of polyethylene—a review. Int Biodeterior Biodegrad 88:83–90. https://doi.org/10.1016/j.ibiod.2013.12.014
Ribitsch D, Heumann S, Trotscha E, Herrero Acero E, Greimel K, Leber R, Birner-Gruenberger R, Deller S, Eiteljoerg I, Remler P, Weber T, Siegert P, Maurer KH, Donelli I, Freddi G, Schwab H, Guebitz GM (2011) Hydrolysis of polyethyleneterephthalate by p-nitrobenzylesterase from Bacillus subtilis. Biotechnol Prog 27(4):951–960. https://doi.org/10.1002/btpr.610
Ribitsch D, Acero EH, Greimel K, Eiteljoerg I, Trotscha E, Freddi G, Schwab H, Guebitz GM (2012a) Characterization of a new cutinase from Thermobifida alba for PET-surface hydrolysis. Biocatal Biotransform 30(1):2–9. https://doi.org/10.3109/10242422.2012.644435
Ribitsch D, Herrero Acero E, Greimel K, Dellacher A, Zitzenbacher S, Marold A, Rodriguez RD, Steinkellner G, Gruber K, Schwab H, Guebitz GM (2012b) A new esterase from Thermobifida halotolerans hydrolyses polyethylene terephthalate (PET) and polylactic acid (PLA). Polymers 4(1):617–629. https://doi.org/10.3390/polym4010617
Ribitsch D, Herrero Acero E, Przylucka A, Zitzenbacher S, Marold A, Gamerith C, Tscheliessnig R, Jungbauer A, Rennhofer H, Lichtenegger H, Amenitsch H, Bonazza K, Kubicek CP, Druzhinina IS, Guebitz GM (2015) Enhanced cutinase-catalyzed hydrolysis of polyethylene terephthalate by covalent fusion to hydrophobins. Appl Environ Microbiol 81(11):3586–3592. https://doi.org/10.1128/AEM.04111-14
Ribitsch D, Hromic A, Zitzenbacher S, Zartl B, Gamerith C, Pellis A, Jungbauer A, Łyskowski A, Steinkellner G, Gruber K, Tscheliessnig R, Herrero Acero E, Guebitz GM (2017) Small cause, large effect: structural characterization of cutinases from Thermobifida cellulosilytica. Biotechnol Bioeng 114(11):2481–2488. https://doi.org/10.1002/bit.26372
Richter PK, Blazquez-Sanchez P, Zhao Z, Engelberger F, Wiebeler C, Kunze G, Frank R, Krinke D, Frezzotti E, Lihanova Y, Falkenstein P, Matysik J, Zimmermann W, Strater N, Sonnendecker C (2023) Structure and function of the metagenomic plastic-degrading polyester hydrolase PHL7 bound to its product. Nat Commun 14(1):1905. https://doi.org/10.1038/s41467-023-37415-x
Ronkvist ÅM, Xie W, Lu W, Gross RA (2009) Cutinase-catalyzed hydrolysis of poly (ethylene terephthalate). Macromolecules 42(14):5128–5138. https://doi.org/10.1021/ma9005318
Roth C, Wei R, Oeser T, Then J, Föllner C, Zimmermann W, Sträter N (2014) Structural and functional studies on a thermostable polyethylene terephthalate degrading hydrolase from Thermobifida fusca. Appl Microbiol Biotechnol 98(18):7815–7823. https://doi.org/10.1007/s00253-014-5672-0
Ru J, Huo Y, Yang Y (2020) Microbial degradation and valorization of plastic wastes. Front Microbiol 11:442. https://doi.org/10.3389/fmicb.2020.00442
Sagong HY, Son HF, Seo H, Hong H, Lee D, Kim KJ (2021) Implications for the PET decomposition mechanism through similarity and dissimilarity between PETases from Rhizobacter gummiphilus and Ideonella sakaiensis. J Hazard Mater 416:126075. https://doi.org/10.1016/j.jhazmat.2021.126075
Sagong HY, Kim S, Lee D, Hong H, Lee SH, Seo H, Kim KJ (2022) Structural and functional characterization of an auxiliary domain-containing PET hydrolase from Burkholderiales bacterium. J Hazard Mater 429:128267. https://doi.org/10.1016/j.jhazmat.2022.128267
Samantaray PK, Little A, Wemyss AM, Iacovidou E, Wan C (2021) Design and control of compostability in synthetic biopolyesters. ACS Sustainable Chem Eng 9(28):9151–9164. https://doi.org/10.1021/acssuschemeng.1c01424
Sana B, Ding K, Siau JW, Pasula RR, Chee S, Kharel S, Lena JH, Goh E, Rajamani L, Lam YM, Lim S, Ghadessy JF (2023) Thermostability enhancement of polyethylene terephthalate degrading PETase using self- and nonself-ligating protein scaffolding approaches. Biotechnol Bioeng 120(11):3200–3209. https://doi.org/10.1002/bit.28523
Sanluis-Verdes A, Colomer-Vidal P, Rodriguez-Ventura F, Bello-Villarino M, Spinola-Amilibia M, Ruiz-Lopez E, Illanes-Vicioso R, Castroviejo P, Aiese Cigliano R, Montoya M, Falabella P, Pesquera C, Gonzalez-Legarreta L, Arias-Palomo E, Solà M, Torroba T, Arias CF, Bertocchini F (2022) Wax worm saliva and the enzymes therein are the key to polyethylene degradation by Galleria mellonella. Nat Commun 13(1):5568. https://doi.org/10.1038/s41467-022-33127-w
Santacruz-Juarez E, Buendia-Corona RE, Ramirez RE, Sanchez C (2021) Fungal enzymes for the degradation of polyethylene: molecular docking simulation and biodegradation pathway proposal. J Hazard Mater 411:125118. https://doi.org/10.1016/j.jhazmat.2021.125118
Santo M, Weitsman R, Sivan A (2013) The role of the copper-binding enzyme–laccase–in the biodegradation of polyethylene by the actinomycete Rhodococcus ruber. Int Biodeterior Biodegrad 84:204–210. https://doi.org/10.1016/j.ibiod.2012.03.001
Schmidt J, Wei R, Oeser T, Dedavid e Silva LA, Breite D, Schulze A, Zimmermann W, (2017) Degradation of polyester polyurethane by bacterial polyester hydrolases. Polymers 9(2):65. https://doi.org/10.3390/polym9020065
Sharma VK, Ma X, Lichtfouse E, Robert D (2023) Nanoplastics are potentially more dangerous than microplastics. Environ Chem Lett 21(4):1933–1936. https://doi.org/10.1007/s10311-022-01539-1
Shi L, Liu P, Tan Z, Zhao W, Gao J, Gu Q, Ma H, Liu H, Zhu L (2023) Complete depolymerization of PET wastes by an evolved PET hydrolase from directed evolution. Angew Chem Int Ed 62(14):e202218390. https://doi.org/10.1002/anie.202218390
Silva CM, Carneiro F, O’Neill A, Fonseca LP, Cabral JSM, Guebitz G, Cavaco-Paulo A (2005) Cutinase—a new tool for biomodification of synthetic fibers. J Polym Sci Part A Polym Chem 43(11):2448–2450. https://doi.org/10.1002/pola.20684
Singh B, Sharma N (2008) Mechanistic implications of plastic degradation. Polym Degrad Stab 93(3):561–584. https://doi.org/10.1016/j.polymdegradstab.2007.11.008
Son HF, Cho IJ, Joo S, Seo H, Sagong H-Y, Choi SY, Lee SY, Kim K-J (2019) Rational protein engineering of thermo-stable PETase from Ideonella sakaiensis for highly efficient PET degradation. ACS Catal 9(4):3519–3526. https://doi.org/10.1021/acscatal.9b00568
Sonnendecker C, Oeser J, Richter PK, Hille P, Zhao Z, Fischer C, Lippold H, Blazquez-Sanchez P, Engelberger F, Ramirez-Sarmiento CA, Oeser T, Lihanova Y, Frank R, Jahnke HG, Billig S, Abel B, Strater N, Matysik J, Zimmermann W (2022) Low carbon footprint recycling of post-consumer PET plastic with a metagenomic polyester hydrolase. Chemsuschem 15(9):e202101062. https://doi.org/10.1002/cssc.202101062
Strobl GR (1997) The physics of poymers: Concepts for understanding their structures and behavior. Springer, New York. https://doi.org/10.1007/978-3-540-68411-4
Sulaiman S, Yamato S, Kanaya E, Kim J-J, Koga Y, Takano K, Kanaya S (2012) Isolation of a novel cutinase homolog with polyethylene terephthalate-degrading activity from leaf-branch compost by using a metagenomic approach. Appl Environ Microbiol 78(5):1556–1562. https://doi.org/10.1128/aem.06725-11
Sundaramoorthy M, Gold MH, Poulos TL (2010) Ultrahigh (0.93Å) resolution structure of manganese peroxidase from Phanerochaete chrysosporium: Implications for the catalytic mechanism. J Inorg Biochem 104(6):683–690. https://doi.org/10.1016/j.jinorgbio.2010.02.011
Suzuki K, Noguchi MT, Shinozaki Y, Koitabashi M, Sameshima-Yamashita Y, Yoshida S, Fujii T, Kitamoto HK (2014) Purification, characterization, and cloning of the gene for a biodegradable plastic-degrading enzyme from Paraphoma-related fungal strain B47–9. Appl Microbiol Biotechnol 98(10):4457–4465. https://doi.org/10.1007/s00253-013-5454-0
Then J, Wei R, Oeser T, Gerdts A, Schmidt J, Barth M, Zimmermann W (2016) A disulfide bridge in the calcium binding site of a polyester hydrolase increases its thermal stability and activity against polyethylene terephthalate. FEBS Open Bio 6(5):425–432. https://doi.org/10.1002/2211-5463.12053
Thiounn T, Smith RC (2020) Advances and approaches for chemical recycling of plastic waste. J Polym Sci 58(10):1347–1364. https://doi.org/10.1002/pol.20190261
Thumarat U, Kawabata T, Nakajima M, Nakajima H, Sugiyama A, Yazaki K, Tada T, Waku T, Tanaka N, Kawai F (2015) Comparison of genetic structures and biochemical properties of tandem cutinase-type polyesterases from Thermobifida alba AHK119. J Biosci Bioeng 120(5):491–497. https://doi.org/10.1016/j.jbiosc.2015.03.006
Tournier V, Topham CM, Gilles A, David B, Folgoas C, Moya-Leclair E, Kamionka E, Desrousseaux ML, Texier H, Gavalda S, Cot M, Guemard E, Dalibey M, Nomme J, Cioci G, Barbe S, Chateau M, Andre I, Duquesne S, Marty A (2020) An engineered PET depolymerase to break down and recycle plastic bottles. Nature 580(7802):216–219. https://doi.org/10.1038/s41586-020-2149-4
Trinh Tan F, Cooper DG, Marić M, Nicell JA (2008) Biodegradation of a synthetic co-polyester by aerobic mesophilic microorganisms. Polym Degrad Stab 93(8):1479–1485. https://doi.org/10.1016/j.polymdegradstab.2008.05.005
van Beilen JB, Funhoff EG (2007) Alkane hydroxylases involved in microbial alkane degradation. Appl Microbiol Biotechnol 74(1):13–21. https://doi.org/10.1007/s00253-006-0748-0
Wallace PW, Haernvall K, Ribitsch D, Zitzenbacher S, Schittmayer M, Steinkellner G, Gruber K, Guebitz GM, Birner-Gruenberger R (2017) PpEst is a novel PBAT degrading polyesterase identified by proteomic screening of Pseudomonas pseudoalcaligenes. Appl Microbiol Biotechnol 101(6):2291–2303. https://doi.org/10.1007/s00253-016-7992-8
Weber J, Petrović D, Strodel B, Smits SHJ, Kolkenbrock S, Leggewie C, Jaeger K-E (2019) Interaction of carbohydrate-binding modules with poly(ethylene terephthalate). Appl Microbiol Biotechnol 103(12):4801–4812. https://doi.org/10.1007/s00253-019-09760-9
Wei R, Zimmermann W (2017a) Biocatalysis as a green route for recycling the recalcitrant plastic polyethylene terephthalate. Microb Biotechnol 10(6):1302–1307. https://doi.org/10.1111/1751-7915.12714
Wei R, Zimmermann W (2017b) Microbial enzymes for the recycling of recalcitrant petroleum-based plastics: how far are we? Microb Biotechnol 10(6):1308–1322. https://doi.org/10.1111/1751-7915.12710
Wei R, Oeser T, Schmidt J, Meier R, Barth M, Then J, Zimmermann W (2016) Engineered bacterial polyester hydrolases efficiently degrade polyethylene terephthalate due to relieved product inhibition. Biotechnol Bioeng 113(8):1658–1665. https://doi.org/10.1002/bit.25941
Wei R, Song C, Gräsing D, Schneider T, Bielytskyi P, Böttcher D, Matysik J, Bornscheuer UT, Zimmermann W (2019) Conformational fitting of a flexible oligomeric substrate does not explain the enzymatic PET degradation. Nat Commun 10(1):5581. https://doi.org/10.1038/s41467-019-13492-9
Wei R, Tiso T, Bertling J, O’Connor K, Blank LM, Bornscheuer UT (2020) Possibilities and limitations of biotechnological plastic degradation and recycling. Nat Catal 3(11):867–871. https://doi.org/10.1038/s41929-020-00521-w
Williams DF (1981) Enzymic hydrolysis of polylactic acid. Eng Med 10(1):5–7. https://doi.org/10.1243/EMED_JOUR_1981_010_004_02
Witt U, Müller R-J, Deckwer W-D (1997) Biodegradation behavior and material properties of aliphatic/aromatic polyesters of commercial importance. J Environ Polym Degrad 5(2):81–89. https://doi.org/10.1007/BF02763591
Witt U, Einig T, Yamamoto M, Kleeberg I, Deckwer WD, Müller RJ (2001) Biodegradation of aliphatic–aromatic copolyesters: evaluation of the final biodegradability and ecotoxicological impact of degradation intermediates. Chemosphere 44(2):289–299. https://doi.org/10.1016/S0045-6535(00)00162-4
Xi X, Ni K, Hao H, Shang Y, Zhao B, Qian Z (2021) Secretory expression in Bacillus subtilis and biochemical characterization of a highly thermostable polyethylene terephthalate hydrolase from bacterium HR29. Enzyme Microb Technol 143:109715. https://doi.org/10.1016/j.enzmictec.2020.109715
Xue R, Chen Y, Rong H, Wei R, Cui Z, Zhou J, Dong W, Jiang M (2021) Fusion of chitin-binding domain from Chitinolyticbacter meiyuanensis SYBC-H1 to the leaf-branch compost cutinase for enhanced PET hydrolysis. Front Bioeng Biotechnol 9:762854. https://doi.org/10.3389/fbioe.2021.762854
Yang Y, Wang J, Xia M (2020) Biodegradation and mineralization of polystyrene by plastic-eating superworms Zophobas atratus. Sci Total Environ 708:135233. https://doi.org/10.1016/j.scitotenv.2019.135233
Yang Y, Min J, Xue T, Jiang P, Liu X, Peng R, Huang J-W, Qu Y, Li X, Ma N, Tsai F-C, Dai L, Zhang Q, Liu Y, Chen C-C, Guo R-T (2023) Complete bio-degradation of poly(butylene adipate-co-terephthalate) via engineered cutinases. Nat Commun 14(1):1645. https://doi.org/10.1038/s41467-023-37374-3
Yao C, Xia W, Dou M, Du Y, Wu J (2022) Oxidative degradation of UV-irradiated polyethylene by laccase-mediator system. J Hazard Mater 440:129709. https://doi.org/10.1016/j.jhazmat.2022.129709
Yoshida S, Hiraga K, Takehana T, Taniguchi I, Yamaji H, Maeda Y, Toyohara K, Miyamoto K, Kimura Y, Oda K (2016) A bacterium that degrades and assimilates poly(ethylene terephthalate). Science 351(6278):1196–1199. https://doi.org/10.1126/science.aad6359
Zampolli J, Mangiagalli M, Vezzini D, Lasagni M, Ami D, Natalello A, Arrigoni F, Bertini L, Lotti M, Di Gennaro P (2023) Oxidative degradation of polyethylene by two novel laccase-like multicopper oxidases from Rhodococcus opacus R7. Environ Technol Innovation 32:103273. https://doi.org/10.1016/j.eti.2023.103273
Zeng W, Li X, Yang Y, Min J, Huang J-W, Liu W, Niu D, Yang X, Han X, Zhang L, Dai L, Chen C-C, Guo R-T (2022) Substrate-binding mode of a thermophilic PET hydrolase and engineering the enzyme to enhance the hydrolytic efficacy. ACS Catal 12(5):3033–3040. https://doi.org/10.1021/acscatal.1c05800
Zhang Y, Wang L, Chen J, Wu J (2013) Enhanced activity toward PET by site-directed mutagenesis of Thermobifida fusca cutinase–CBM fusion protein. Carbohydr Polym 97(1):124–129. https://doi.org/10.1016/j.carbpol.2013.04.042
Zhang H, Perez-Garcia P, Dierkes RF, Applegate V, Schumacher J, Chibani CM, Sternagel S, Preuss L, Weigert S, Schmeisser C, Danso D, Pleiss J, Almeida A, Hocker B, Hallam SJ, Schmitz RA, Smits SHJ, Chow J, Streit WR (2021) The bacteroidetes Aequorivita sp. and Kaistella jeonii produce promiscuous esterases with PET-hydrolyzing activity. Front Microbiol 12:803896. https://doi.org/10.3389/fmicb.2021.803896
Zhang A, Hou Y, Wang Q, Wang Y (2022a) Characteristics and polyethylene biodegradation function of a novel cold-adapted bacterial laccase from Antarctic sea ice psychrophile Psychrobacter sp. NJ228. J Hazard Mater 439:129656. https://doi.org/10.1016/j.jhazmat.2022.129656
Zhang Y, Pedersen JN, Eser BE, Guo Z (2022b) Biodegradation of polyethylene and polystyrene: from microbial deterioration to enzyme discovery. Biotechnol Adv 60:107991. https://doi.org/10.1016/j.biotechadv.2022.107991
Zhang Y, Plesner TJ, Ouyang Y, Zheng Y-C, Bouhier E, Berentzen EI, Zhang M, Zhou P, Zimmermann W, Andersen GR, Eser BE, Guo Z (2023) Computer-aided discovery of a novel thermophilic laccase for low-density polyethylene degradation. J Hazard Mater 458:131986. https://doi.org/10.1016/j.jhazmat.2023.131986
Zhao J, Wang J, Li C, Fan Q (2002) Study of the amorphous phase in semicrystalline poly(ethylene terephthalate) via physical aging. Macromolecules 35(8):3097–3103. https://doi.org/10.1021/ma011333z
Zimmermann W (2020) Biocatalytic recycling of polyethylene terephthalate plastic. Philos Trans R Soc A 378(2176):20190273. https://doi.org/10.1098/rsta.2019.0273
Zumstein MT, Rechsteiner D, Roduner N, Perz V, Ribitsch D, Guebitz GM, Kohler H-PE, McNeill K, Sander M (2017) Enzymatic hydrolysis of polyester thin films at the nanoscale: effects of polyester structure and enzyme active-site accessibility. Environ Sci Technol 51(13):7476–7485. https://doi.org/10.1021/acs.est.7b01330
Author information
Authors and Affiliations
Contributions
Rey-Ting Guo and Chun-Chi Chen were involved in conceptualization and writing—original draft preparation. Xian Li, Yu Yang, Jian-Wen Huang and Panpan Shen helped with writing—original draft preparation. Rock Keey Liew took part in reviewing and editing.
Corresponding authors
Ethics declarations
Declarations
RK declares that he is the Editor of Environmental Chemistry Letters.
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, RT., Li, X., Yang, Y. et al. Natural and engineered enzymes for polyester degradation: a review. Environ Chem Lett 22, 1275–1296 (2024). https://doi.org/10.1007/s10311-024-01714-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10311-024-01714-6