
Abstract
We have measured the dependence of the Fe–Mg interdiffusion coefficient, DFe-Mg, on the ferrosilite component in orthopyroxene, which so far has not been experimentally calibrated. Diffusion couples, consisting of approximately 1 µm thin-films were deposited by pulsed laser ablation on orthopyroxene crystals of En91Fs9 composition. Diffusion experiments were carried out at atmospheric pressure in vertical gas mixing furnaces (CO-CO2) at temperatures between 950 and 1100 °C at constant fO2 = 10–7 Pa. Using a focused ion beam-scanning electron microscope (FIB-SEM), FIB-foils were cut from diffusion couples before and after annealing. Diffusion profiles were extracted by using combined backscattered electron (BSE) imaging and energy dispersive X-ray (EDXS) mapping on FIB-foils which allowed to resolve concentration gradients within 20 nm. The microstructure of the diffusion experiments was investigated using transmission electron microscopy (TEM). Using this method, we obtained the first experimentally determined data on the dependence of DFe-Mg on the ferrosilite content in orthopyroxene at different temperatures, appearing to increase with increasing a temperature. For the temperature range (950 – 1100 °C), log fO2 = -7 Pa, along [001] in the composition Fs9, the log DFe-Mg yields the following Arrhenius equation:
The effect of XFe on DFe-Mg, given by \(D(X_{Fe} ) = D(X_{Fe} = 0.09) \cdot {10}^{m (X_{Fe} - 0.09)},\) can be calculated by the following parameterization where m follows a linear regression of m on temperature:
By considering the DFe-Mg dependence on XFe, the timescales of natural processes obtained from modelling the compositional zoning of natural crystal may considerably differ from previously estimated timescales.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diffusion modelling is a growing field in geosciences, owing to the broad spectrum of interest in petrological studies. Among other applications, it allows for the determination of time information in magmatic systems, such as magma mixing events, cooling rates or crystal residence times in magma chambers (e.g., see the reviews of Turner and Costa 2007; Chakraborty 2008; Costa et al. 2008; Costa and Morgan 2010; Dohmen et al. 2017). To do so, it is desirable to have a comprehensive data set of experimentally determined diffusion coefficients, measured as a function of the relevant thermodynamic conditions. Both the experimental determination of diffusion coefficients as well as the determination of timescales from diffusion modeling (“diffusion chronometry”) relies on the measurement of concentration profiles or maps in minerals with concentration gradients. Together with an appropriate diffusion model, it is then possible to determine timescales (or diffusion coefficients) by fitting modelled diffusion profiles to measured compositional gradients. One of the ongoing limitations of diffusion modelling is the spatial resolution and precision of the available analytical techniques. As an example, when measuring sharp concentration gradients, the spatial resolution of the method may not fully resolve the gradient, resulting in a convolved concentration profile. A mathematical treatment of this problem was presented in Ganguly et al. (1988) for electron microprobe measurements or in Hofmann (1994) for SIMS depth profiling. Evidently, if the “diffusion profile length” (loosely defined here as the distance over which the concentration was changed by diffusion within a detectable range) is even shorter than the spatial resolution, it is impossible to extract diffusion coefficients or timescales, leaving only the option to estimate a maximum diffusion coefficient or a maximum timescale. Therefore, the experimental determination of diffusion coefficients slower than approximately 10−18 m2/s presents a significant challenge due to the impractically long durations required for diffusion anneals to obtain µm-scale diffusion profiles, as highlighted in earlier works, e.g., Cherniak and Watson (1994), Dohmen et al. (2002). In these studies, Rutherford Backscattering (RBS) or Secondary Ion Mass Spectrometry (SIMS) depth profiling was used to measure diffusion profiles on the nm scale on powder source diffusion couples or thin film diffusion couples (see also Watson and Dohmen 2010). An additional complexity is that in some cases the diffusion coefficient is dependent on the concentration of the diffusing component, e.g., due to effects on the point defect chemistry or coupling with other diffusing components (e.g., Nakamura and Schmalzried 1984; Chakraborty and Ganguly 1991). A noteworthy example is the experimental determination of Fe–Mg interdiffusion coefficients in orthopyroxene. Because Fe–Mg diffusion profiles in orthopyroxene are generally much shorter than olivine, owing to slower diffusion rates (Ganguly & Tazzoli 1994; Schwandt et al. 1998; Stimpfl et al. 2005; Dohmen et al. 2016), the experimental calibration of DFe-Mg in orthopyroxene was met with some challenges in the past. One of the parameters that is currently not well constrained is the effect of the orthopyroxene composition (e.g., the effect of XMg or Al content) on DFe-Mg (Dohmen et al. 2016). However, kinetic experiments on order–disorder rates of Fe–Mg on the M-site in orthopyroxene indicate a much stronger dependence of DFe-Mg on XMg (Ganguly and Tazzoli 1994; Stimpfl et al. 2005) than those inferred in Dohmen et al. (2016) but similar to what has been found for olivine (Nakamura and Schmalzried 1984; Chakraborty 1997; Dohmen et al. 2007). These diffusion coefficients were extracted from compositional depth profiles using Rutherford Backscattering Spectroscopy (RBS) spectra on thin film diffusion couples (see also, Watson and Dohmen 2010; Dohmen et al. 2007, 2016). In this case, the spatial resolution of the method does not allow to resolve Fe profiles of more than 500 nm (including thin-film and source). Because it is essential to have relatively long diffusion profiles and a high density of data points within the diffusion zone to determine the compositional dependence, the method did not allow to calibrate the compositional dependence of DFe-Mg. Consequently, there is a pressing need for the development of analytical techniques and experimental setups capable of resolving diffusion profiles on the nanometric scale.
In the recent past there were many applications of DFe-Mg in orthopyroxene to determine timescales from Fe–Mg zoning in which different calibrations were used for DFe-Mg and its dependence on XMg (e.g., Saunders et al. 2012; Allan et al. 2013; Chamberlain et al. 2014; Kilgour et al. 2014; Cooper et al. 2017; Fabbro et al. 2017; Davydova et al. 2018; Flaherty et al. 2018; Pichavant et al. 2018; Conway et al. 2020; Solaro et al. 2020; Utami et al. 2021; Phillips and Till 2022; Ostorero et al. 2022; Moshrefzadeh et al. 2023). In most of these studies the XMg of orthopyroxene was significantly lower (XMg = 0.3–0.7) than those of the crystals used in Dohmen et al. (2016) and they used the calibration based on the kinetics of order–disorder data to consider the effect of XMg. Therefore, a direct and more accurate determination of the dependence of DFe-Mg on XMg is required.
Thin film diffusion couples produced by pulsed laser deposition in combination with high resolution analytical methods like TEM have been used in the past to measure interdiffusion in solids (e.g., Fe–Mg in olivine: Meissner et al. 1998; Y-Yb in YAG: Marquardt et al. 2010). In this work we further develop this general approach to measure the effect of XMg on DFe-Mg in orthopyroxene and address some of the remaining issues discussed above. A combination of techniques available on a DualBeam Focused Ion Beam – Secondary Electron Microscope (FIB-SEM) instrument were used to extract DFe-Mg interdiffusion coefficients as a function of XMg in orthopyroxene. We used various analytical tools of a SEM on a Dual Beam FIB system to characterize a specimen in detail, allowing the simultaneous acquisition of compositional information, crystallographic orientation and structural information using one single specimen. This includes secondary electron imaging (SEM), backscattered electron imaging (BSE), X-ray analysis (EDXS), electron backscattering diffraction (EBSD), and a scanning transmission electron microscope (STEM) detector. The zoning profiles were mainly extracted from BSE images and fitted by various numerical approaches to determine DFe-Mg. The aim of this work was to adapt known FIB-SEM preparation methods and explore the instrument range of capabilities on orthopyroxene Fe–Mg interdiffusion experiments. It will be shown that with this approach we were able to calibrate the effect of XMg on DFe-Mg and that this effect is broadly consistent with those inferred from the kinetic order–disorder rates of Fe–Mg in Opx, which we in addition re-evaluated.
Experimental setup and methodology
Diffusion couples
Gem-quality natural single crystals from Tanzania with an average composition of Mg1.824Fe0.183Ca0.005Al0.005Si1.985O6 were used for the diffusion experiments. The crystals are homogeneous in terms of major elements and close to Fe–Mg solid solutions with different mole fractions of the ferrosilite component. The chemical composition of the orthopyroxene crystals was measured by electron microprobe analysis (EPMA) using a Cameca microprobe (SX Five FE) operating at 15 keV and 15 nA hosted at Ruhr-Universität Bochum. The orthopyroxene crystals were crystallographically oriented using a Laue camera (Ruhr-Universität Bochum). The orientation was subsequently verified using electron backscatter diffraction (EBSD) on a Scios 2 DualBeam FIB-SEM (Ruhr-Universität Bochum). The crystal surfaces perpendicular to the [001] crystallographic axis according to the space group Pbca were mechanically polished and finished with a chemical–mechanical polishing step using colloidal silica compounds and cut into cuboids with an approximate dimension of 2 × 2 × 2 mm using a high-precision diamond wire saw. This orientation was chosen as diffusion along the c axis in pyroxene is known to be faster than along other crystallographic directions (Ganguly and Tazzoli 1994; Müller et al. 2013; Dohmen et al. 2016). The samples were cleaned in several steps with distilled water, acetone and ethanol. Crack-free samples with a pristine polished surface were selected using a reflected light microscope for the preparation of the diffusion couples. Diffusion couples consisting of ferrosilite-rich thin films with thicknesses that vary between ≈ 300 nm and 1 µm on the polished single crystals were produced by pulsed laser deposition (PLD). We used a synthetic polycrystalline pellet of orthopyroxene with 50 mol% ferrosilite and 50 mol% enstatite composition as target material. The desired thickness of the film was achieved by calibrating the ablation rate of the synthetic pellet on a Si-wafer for a determined amount of time. Prior to the deposition, the samples were heated for approximately 20 to 30 min at approximately 400 °C under vacuum (≈ 10–3 Pa) to remove any volatile adsorbents. A laser pulse with a wavelength of 193 nm at a frequency of 10 Hz was used to ablate the synthetic orthopyroxene pellet. When ablated, a plasma cloud is created, deposing an amorphous thin-film on the surface of the crystal (perpendicular to the [001] crystallographic axis), serving as an Fe-rich reservoir for the Fe–Mg exchange with the crystal during the diffusion experiments. One sample out of every deposition run was kept as a reference (Table 1). A more detailed description of the setup and the method can be found in Dohmen et al. (2002).
Diffusion experiments
Diffusion experiments were carried out at atmospheric pressure in vertical gas mixing furnaces (Ruhr-Universität Bochum) at temperatures between 950 and 1100 °C and at varying oxygen fugacity (fO2) conditions (same equipment as used for example in Dohmen et al. 2007, 2016). The diffusion couples were placed inside a small corundum crucible and suspended inside a larger corundum crucible, which was hanging on an alumina rod using Pt wire. The fO2 was controlled by a flowing gas mix of CO and CO2 and monitored throughout the experimental runs using a fO2 sensor (ZrO2). Temperature was controlled using a B-type thermocouple (Pt–Rh). The list of samples and corresponding experimental conditions are given in Table 1. Samples were quenched by simply removing them from the furnaces and cool them in air. The duration of the experiments was planned based on the data for DFe-Mg in Dohmen et al. (2016) in order to obtain profile lengths greater than 1 µm.
Annealed samples were characterized in respect to the surface topography, as well as structure and geometry. On a first approach, the surface topography of the diffusion couples was characterized using reflected light microscopy and phase shift interference light microscope (Ruhr-Universität Bochum). For this set of samples, a surface topography of ± 100–500 nm was determined. The surface roughness and structure of the thin-films of annealed samples was later closely inspected and confirmed using BSE imaging and STEM imaging on the FIB foils. The measured average roughness of the diffusion experiments excludes the possibility of using depth-profiling methods, such as RBS or SIMS to obtain well resolved concentration profiles on the resolution of a few nm. The only pre-requisite for FIB preparation (with in-situ and ex-situ lift-out of the foil) is a surface roughness of less than a micron (Wirth, 2009), making the samples suitable for preparation and extraction. Even if a surface roughness greater than 1 µm was locally measured, it was still possible to prepare a foil from the areas where a smooth surface was maintained.
Preparation using FIB-SEM
We prepared FIB-foils cross sections from reference samples (thin-film diffusion couples before diffusion anneal) and annealed samples using the Scios 2 DualBeam FIB-SEM at Ruhr-Universität Bochum (Fig. 1). The samples were first embedded in Indium inside a 1-inch holder in a way that only the surface remained exposed, which proved to be a reliable way to keep the samples in place and minimize charging effects. In addition, this setup keeps the sample surfaces at the same elevation (position in z-axis), which helps to prevent hitting of SEM-equipment (e.g., the electron column) during movement of the sample stage. The samples were subsequently coated by a 6 nm carbon layer. The fundamentals of the FIB lift-out technique (FIB LO) have been described in detail by several authors (e.g., Nastasi et al. 1996; Wirth 2009). In our setup we used Ga + ions that are accelerated at 30 kV. The ion beam current was changed during the different preparation steps according to a recipe given by the AUTO-TEM 4 software (1.5 pA to 65 nA). A fair amount of user interaction was necessary as well as several adjustments, which did not allow to use the AutoTEM 4 automated procedure throughout the whole preparation. Generally, the Pt-strip deposition and trench milling was done automatically whereas the lift-out, thinning, and final polishing were either guided or done manually. A 1.5 µm thick Pt layer was deposited using the ion beam, serving both as a protective layer and as an indicator of the roughness of the region of interest. The importance of the thickness of the Pt layer has been demonstrated in previous studies (e.g., Lee et al. 2007). Before depositing a 1.5 µm Pt layer using the ion beam, we deposited a thinner Pt layer using the electron beam to protect the top layer of the sample to avoid ion-induced damage on the first top nanometers of the sample. More details of the preparation procedure are given in the electronic material. Using this procedure, we prepared foils that have an average dimension of ≈ 10 × 5 µm with a typical thickness between 300–400 nm (Fig. 1). In addition, for sample Opx7_cOp_47, we prepared into the FIB foil a window with a thickness of 40 to 100 nm to obtain STEM images and investigate the structure of the thin films as well as interface features.

Schematics of the FIB foil extraction. a Position of the electron and ion beam in relation to the sample surface and area of extraction. The surface of the sample is perpendicular to the [001] crystallographic axis. b Illustration of the lift-out of the FIB-foil and dimensions. c and d SE images of the lift-out area (c) and d example of a FIB-foil prepared for STEM imaging
Scanning Electron Microscopy (SEM)
Grey-scale intensity profiles across the FIB foils were obtained by BSE imaging using the segmented lower in-lens Trinity Detector T1 (composite mode A + B) on the Scios 2 DualBeam FIB-SEM at Ruhr-Universität Bochum. This detector provides strong BSE contrast at accelerating voltage of 5 kV or higher. Because BSE contrast images are a function of the mean atomic number, they will not be strongly dependent on the Al, Mg and Si concentrations given that these elements have similar atomic numbers. Therefore, in the case of the chosen orthopyroxene compositions, BSE intensity profiles will be a direct response to the XMg (XMg = MgO/(MgO + FeOtotal)) (e.g., Morgan et al. 2004; Costa and Morgan 2010; Saunders et al. 2012). If correctly calibrated the spatial resolution of this method is significantly higher than EPMA analysis. Images were obtained at acceleration voltages of 5, 10 and 20 kV and at currents ranging from 0.1 nA to 1.6 nA. In addition, different imaging parameters (resolution and dwell time) were explored.
Transmission Electron Microscopy (TEM)
For detailed structural observations, the prepared foils of annealed samples were analysed using a scanning transmission electron microscopy detector (STEM3 +) on the Scios 2 DualBeam FIB-SEM system (Ruhr-Universität Bochum) with < 30 kV acceleration voltage. Sample Opx7_cOp_47 was analysed using transmission electron microscopy (TEM) and STEM on a JEOL JEM-ARM200F transmission electron microscope (TEM) operated at 200 kV (Ruhr-Universität Bochum). Scanning transmission electron microscopy bright field (BF), dark field (DF), angular dark field (ADF), high angle annular dark field (HAADF) techniques were used to characterize the structure of the thin-film upon annealing. The foil used for structural analysis had a window with a thickness of less than 100 nm and were subject to a final cleaning at procedure for optimal imaging at low voltages (5 and 10 kV) using an overtilt angle of 6°.
Energy-dispersive X-ray Spectroscopy (EDXS)
Energy-dispersive X-ray spectroscopy (EDXS) measurements and mapping were used to calibrate grey-scale intensity profiles of the diffusion experiments obtained through BSE imaging. The analysis were conducted using an Ultim Oxford spectrometer equipped on the Scios 2 DualBeam FIB-SEM system (Ruhr-Universität Bochum), acquired at 15 kV. The quantification followed X-Ray emission lines and included: O (K series), Fe (K series), Mg (K series), Al (K series), Si (K series), Mn (K series) and Ti (K series) using the Oxford internal quantification standardizations. Elements that were excluded were C (K series), Cu (L series), Ga (L series), and Pt (M series), as they are present on the sample holder, coating of the sample, protective strip and/or introduced during FIB preparation. For all samples, a Pt mapping acquired to provide a reference point for the sample surface. The quantification of the analysis was carried out using the internal Quant Oxford library. Intensity ratios were converted into concentration ratios following the Cliff-Lorimer (1975) equation:
where concentrations were obtained by EMPA analysis on a Opx7 single crystal and intensities by EDXS analysis on a reference (pre-annealed) Opx7 sample. By calculating the Cliff-Lorimer factor, kab, the unknown concentrations at the thin-film could be extracted for the samples where EDXS spectra was acquired. The XFe \(\left( {X_{Fe} = \frac{Fe}{{Fe + Mg}}} \right)\) can therefore be given by:
The calculated concentrations were used for comparison purposes with the concentrations obtained through the grey-scale integration. The comparison between EDXS-SEM concentration profiles and concentration profiles obtained from grey-scale intensities can be found in the supplementary material. Additionally, EDXS-TEM line-scan analysis and maps were acquired on a representative sample using a JEOL JEM-ARM200F transmission electron microscope (TEM) equipped with a EDAX EDXS system and operating at 200 kV (Ruhr-Universität Bochum). Line-scans were acquired at 60 × magnification in steps of 0.13 µm over a 2.5 µm long profile. Quantitative microanalyses were carried out using the EDAX library.
Conversion of grey scale from BSE images to X Fe
Grey-scale intensity profiles were extracted from the BSE images of the FIB-foils using the open source software ImageJ (Abràmoff et al. 2004). The grey-scale intensity is linearly correlated with the Fe content in Opx provided that the concentration of elements that can interfere with the brightness is low (e.g., Saunders et al. 2012). For the reference samples, we calibrated the grey scale from the known crystal composition (as measured using the microprobe) and the thin film composition by EDXS analysis. In the case of the annealed samples, we calibrated the grey-scale profiles by plotting them against the measured Fe content from EDXS analysis and/or by integrating the grey-scale profile. Given that the grey-scale intensity varies linearly in response to the Fe content, we obtain the following relationship between the integration of grey-scale intensity and the Fe-content along a profile:
where L corresponds to the length of the profile, Y to the grey-scale intensity value, and a and b are constants that define the linear relation between Y and XFe. To obtain a and b from Eq. 3 we apply the mass balance and use the known initial composition of the single crystal, XFe0, the initial film composition, XFefilm, and film thickness, h:
In addition, for the grey-scale in the crystal, Y0, not affected by diffusion we have the constraint:
From the combination of Eqs. 4, 5 we can derive the slope (a) of this linear relationship:
The constant b follows then simply from Eq. 5.
Diffusion model for fitting of the concentration profiles
The concentration profiles of the diffusion experiments were fitted by the one-dimensional diffusion equation following Fick’s second law (Crank 1975) with boundary conditions that account for mass balance and local equilibrium between film and substrate, as well as assuming a zero flux at the surface (e.g., see Dohmen et al. 2016; Crank 1975). As initial condition we used the step profile of the respective reference sample. Additionally, film and substrate were considered as two different diffusion media (i.e., the two media yield different Fe–Mg interdiffusion coefficients, Dsub for the crystal and Dfilm for the thin-film, with Dfilm > > Dsub) with the thin-film being considered as a layer of finite thickness at the surface of a semi-infinite media that remains unaffected at depth. According to earlier experimental data (e.g., Ganguly and Tazzoli 1994; Dohmen et al. 2016), it needs to be considered that Fe–Mg interdiffusion in orthopyroxene is dependent on the mole fraction of the Fe end-member. Similar to Dohmen et al. (2016) and to Fe–Mg diffusion in olivine (e.g., Chakraborty 1997), we used the following exponential form, where the compositional dependence is characterized by the factor m in the exponent,
with Do and m as fitting parameters. The evaluation of the compositional dependence will be defined by the detailed shape of the profiles, i.e., changes in the slope along the concentration profiles. Given that Do and m are related, for high compositional dependencies it is important to have diffusion profiles that are long enough to fully resolve such slope changes within the diffusion zone.
Since the diffusion coefficient is dependent on the concentration of the diffusing component it is necessary to use a numerical method for solving the diffusion equation and perform the fitting. We applied the finite difference method in its explicit form (Crank 1975). The DFe-Mg was calculated at each grid point along the profile to determine the concentration variation after each time step according to the expression:
On a first approach, the best fits were determined visually. The best fit of the selected experiments was subsequently determined using a regression procedure with m and Do as free parameters. The simulations were performed using a C ++ program compiled and linked with the Visual Studio 2022 C ++ programing environment. More details on the fitting procedure and a discussion on the uncertainties for the derived parameters are given in the electronic annex.
Consideration of the convolution effect
The grey-scale intensity images (and consequently the extracted grey-scale profiles) or concentration mappings using EDXS, represent a weighted spatial average of the composition around the point of incidence. This convolution effect will depend greatly on multiple factors intrinsic to the instrument or the analytical conditions, such as the excitation volume of the X-rays of the sample upon EDXS analysis. It follows that by analyzing the compositional profiles the convolution effect will result on an artificial broadening of the profiles, for example at the interface of the thin film and single crystal. This can lead to an overestimation of the diffusion coefficients (and underestimation of the compositional dependence), which is critical for the accuracy of the derived timescales (Ganguly et al. 1988; Costa and Morgan 2010; Morgan et al. 2004; Bradshaw and Kent 2017). The convolution effect cannot really be avoided but it can be accounted for using analytical and numerical approaches. In the case of a simple Gaussian beam, this has been derived (Ganguly et al. 1988; Arnould and Hild 2002) and is given by the convolution function, as follows:
with σ standing for the standard deviation of the distribution function for a given coordinate. For diffusion profiles the convolution effect can be also considered in a simplified way by using for the simulation of the concentration profile an effective diffusion coefficient D*, as determined by the following relationship (Ganguly et al. 1988),
with D being the real diffusion coefficient, σ corresponds to the standard deviation of the Gaussian error function density of the excited sample volume (as calibrated from the step profiles of the reference samples) and t is the time (here the duration of the experiment). For the diffusion coefficient depending on XFe (or XMg) this implies that the numerical solution of the diffusion equation is performed using
A simple consequence of this relation is that with increasing t, equivalent to an increasing diffusion profile length, the contribution of the convolution effect to the measured diffusion profile becomes smaller and for significantly large t, can be ignored.
Results
Starting material
With the reference samples we can identify analytical and preparation artifacts and determine the effective spatial resolution of the applied imaging method by simulating the convolution effect, as described in the previous section. It is known that the real interface between the two phases (thin film and crystal) is sharp, as defined by the surface roughness of the single crystal before deposition of the thin film (< 1 nm, see Dohmen et al. 2002) and hence the measured profile will be a result of the spatial averaging of the compositions at both sides of the interface (assuming that the FIB foil is perpendicular to the thin film-crystal interface). Different currents, voltages and image acquisition parameters were tested to determine the optimal conditions for imaging. For each set of conditions, the standard deviation of the Gaussian function was determined (σ). Generally, at accelerating voltages of 5 and 10 kV, beam currents of 0.1–0.8 nA, high resolution (6144 × 4096) and longer dwell times (45 µs) we produced the best results for the BSE images. However, if the preparation gives origin to surface imperfections or creates topography on the foil (e.g., gradient in foil thickness, particularly at the thin film/crystal interface due to material contrast of amorphous Fe-rich material vs. crystalline Mg-rich material) we can obtain drastic interferences on the grey-scale profile that can even dominate over the chemical contrast. This effect is simply due to the increased excitation volume for BSE, which therefore increases the brightness compared to thinner areas even if the chemical composition is the same. Such effects and beam aberrations were minimized by decreasing the accelerating voltage and probe current to 5 kV as illustrated in Fig. 2.

BSE images and extracted grey scale profiles using 10 kV (a and c) or 5 kV (b) and d acceleration voltage of the same reference sample (Opx7_cOp_24) in which the bottom half of the prepared foil is thicker than the upper part, i.e., variation in thickness perpendicular to the substrate boundary (400 nm vs. 300 nm, approximately). This reference sample was chosen to illustrate possible preparation artifacts and how to best avoid their interference on grey-scale profiles. The difference in thickness along the foil was due to inadequate over-tilt angles during the thinning and polishing steps. The yellow square represents the area over which the grey-scale intensity profiles were acquired. The variable thickness across the foil contributes in addition to the chemical step gradient significantly to the visible gradient on the grey-scale image. This effect is much more evident at the higher accelerating voltage of 10 kV c, in which the chemical gradient is basically invisible. Imaging at 5 kV significantly reduces the introduction of such artifacts on the extracted profiles (d). In addition, overall thicker foils (> 200 nm) and minimizing variations in foil thickness help to reduce these effects for acceleration voltages in the range of 5 – 10 kV. Simulation of the interaction volume for BSE using the software WinXRay 1.4.2 shows that the penetration depth for 5 kV is down to x nm (see electronic appendix). Therefore, the optimum foil thickness is > 200 nm for BSE images measured at 5 kV acceleration voltage
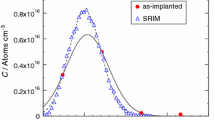
The chemical analysis by EDXS maps or EDXS linescans are significantly less sensitive to a variable foil thickness than BSE imaging at higher voltages. However, two additional points needs to be considered using EDXS analysis of FIB-foils: (i) the common software used does not consider the analysis of a thin specimen such that the correction algorithms (see for example, Lorimer 1987) for the matrix effects related to the different excitation volumes of the elements are not fully appropriate; (ii) to obtain the Kα-lines in the spectrum for elements like Fe an acceleration voltage of 15 kV is required, which considerably reduces the spatial resolution due to the larger excitation volume (e.g., Doig and Flewitt 1983; Boona 2021). Based on the comparison between EDXS point analysis of the foil in the substrate area and the microprobe analysis of the crystal before the thin film deposition, the error introduced by EDXS analysis of the thin specimen is relatively small (± 0.05 a.p.f.u. Fe and ± 0.03 a.p.f.u. Si) (Table 2). According to EDXS analysis, the thin film is slightly depleted in SiO2 compared to the nominal target composition (Fe0.5Mg0.5SiO3) and the Fe/Mg-ratio is slightly higher than the target (Table 2). Finally, for an acceleration voltage of 15 kV we obtain a spatial resolution (σ = 170 nm), which is considerably lower than BSE grey-scale intensity profiles at voltages in the range of 5–10 kV (Fig. 3). It was not possible to perfectly fit the profile from EDXS analysis with the convolution equation, as the concentration profile from the reference sample is not perfectly symmetrical, showing a longer tail in the region of the crystal (Fig. 3). We used the acquisition of EDXS spectra as a complement to grey-scale intensity profiles to calibrate the grey scale and identify artifacts from the thinning procedure.

Comparison between the different profiles and respective simulated σ values extracted from the grey-scale intensity profiles acquired though BSE imaging at 5 and 10 kV b and d and from EDXS Fe-mapping on the same non-annealed reference sample Opx7_cOp_24 (c)
Annealed diffusion couples
Textural observations
A combination of BSE and STEM imaging on FIB foils of the experiments revealed that the thin film crystallizes and forms a polycrystalline matrix upon annealing. The film is composed by a mixture of two chemically distinct types of crystals and pores (Figs. 4, 5). The majority of the film is composed of about 10 – 100 nm large crystals (allowing for EDXS analysis) that have Opx stoichiometry and locally some larger crystals (200 − 1 µm). The dimensions of these crystals do not allow for accurate EDXS point analysis as they are surrounded by Opx. EDXS mapping (Fig. 4b-d) shows that these crystals are depleted in Si and enriched in Fe and Mg relative to the Opx crystals, which indicates that these are composed of olivine. Since the initial film was depleted in SiO2, we can expect that upon crystallization the film layer forms a mixture of olivine and orthopyroxene (Fig. 4). In this case the SiO2 activity is fixed by the coexistence of olivine and orthopyroxene during the diffusion experiments as the thin-film is expected to rapidly crystallize for T > 900 °C (e.g., Milke et al. 2007; Dohmen et al. 2016). STEM images over the selected areas were acquired to confirm that the interface maintains its original geometry and to closely inspect the structure of the film upon annealing. The texture of the film varied depending on the temperature, the length of the annealing and the original film thickness. The influence of each parameter is unclear; however, in longer experiments the surface topography seems to get progressively more irregular due to grain growth (Ostwald ripening likely accommodated by the reaction Mg2SiO4 ↔ MgSiO3 + MgO). Regardless, no significant difference in the crystallinity of the film was observed for the annealed samples, as evidenced in the timeseries experiment we performed in sample Opx7_cOp_47 (see in electronic supplementary material Fig. 10). Furthermore, in most areas the film seemingly maintained a straight interface with the crystal surface (Fig. 5). To closely investigate the microstructure of the film and identify detailed interface features, TEM and high-resolution TEM (HR-TEM) images were acquired on sample Opx7_cOp_47. TEM BF images show that the film crystallizes into columnar Opx crystals with up to a few hundred nanometers, which is consistent with earlier observations for orthopyroxene thin films (Milke et al. 2007). HR-TEM images acquired close to the interface show that the crystallized thin-film shares the same orientation as the Opx crystal at the interface and that the crystal is free of linear or planar defects (Fig. 5). Additionally, no strain contrast features are visible which allows to exclude a mechanical contribution on the diffusion mechanism that could potentially affect the diffusion data close to the interface. Nanometric crystals were identified close to the surface of the thin-film but this feature had no significant influence on the diffusion front.

a SE image of the surface of sample Opx7_cOp_16 annealed at 1100 °C, log fO2 = −7 Pa for 96 h, where the surface roughness is the result of crystallization of the film during anneal. The brighter crystals in this image that are mostly responsible for the roughness are likely olivine, also visible in cross-section in b) BSE image of a foil cut out from the same sample. The interface between thin-film and Opx substrate is well defined (represented by the dashed line); however, some pores and larger crystals are visible on the crystallized film matrix. The same features are visible on EDXS maps in c, d, and e, notably affecting the diffusion front as seen in the EDXS Fe map c. The larger crystals are enriched in Fe and Mg but Si-poor relative to the film matrix (e). This suggests that upon annealing, the film crystallized into an olivine-orthopyroxene polycrystalline matrix. The polycrystalline nature of the film is clearly visible on the STEM BF image, but which also shows curtaining effects (f)

Several features of sample Opx7_cOp_47 annealed at 1050 °C, log fO2 = −7 Pa for 72 h. a TEM bright field image of thin-film and substrate. The surface roughness is not greater than 100 nm. Upon annealing, the film becomes polycrystalline with Opx crystals of up to a few hundred nanometers in diameter. Close to the interface there are no observable strain contrast features and thin-film and substrate share the same crystallographic orientation, as seen in the high resolution-TEM micrograph (b). c STEM3 + dark field image showing the straight and geometrically well-defined interface, as well as nanometric crystal aggregates that develop on the upper most part of the film (white square). This feature does not significantly increase the sample surface roughness and can be seen in detail in the TEM ADF micrograph displayed in d
The most important preparation artifact derived from the different material properties within the film was the so-called “curtaining effect”. This effect is particularly clear in samples in which the thin-film suffered significant re-crystallization and pores were formed (Fig. 4f). This leads to inhomogeneities of the specimen material properties, changing the angle of incidence and thus changing the sputtering yield. The direct consequence of this effect is the development of a sample surface with different thicknesses perpendicular to the interface between film and substrate, resembling a curtain after the successive thinning steps. This artifact can therefore influence the interpretation of the grey-scale image and possibly lead to the under or over-estimation of the diffusion profile length. In such cases, EDXS maps can be acquired to help identify this artifact by the comparison with the BSE image. Areas with visible porosities were avoided to ensure that there were no potential barriers for element transport within the selected area. If such features were not easily recognizable, any possible preparation artifacts, compositional inhomogeneities within the film, or a locally rough interface were accounted for and avoided by inspecting the sample with STEM imaging or EDXS elemental mapping over the area. Irrespective of local features, the samples are chemically and geometrically well-defined and therefore suited for diffusion studies.
Diffusion profiles as obtained from different analytical settings
To evaluate the reliability of the extracted profiles and exclude potential image artifacts, samples were compared with EDXS Fe line scans and profiles extracted from EDXS Fe elemental maps, acquired using EDXS-SEM over the same selected area to provide a direct comparison. A TEM approach is generally considered more reliable for samples with thicknesses below 500 nm (e.g., Boona 2021). The XFe values obtained through the integration of the grey-scale intensity profiles were compared to the composition given by EDXS-TEM spectra over the same area to provide an extra constraint over the profile calibration. There is very good agreement between XFe values obtained through the grey-scale integration of BSE images and the Fe content obtained through EDXS-TEM spectra, both in the film and substrate (Fig. 6). Additionally, the Fe concentration profiles obtained from EDXS Fe maps, EDXS-TEM line-scans and from BSE images acquired at 5 kV are recognizable similar in shape and length (Fig. 6). Given that our results were reproducible across these different methods, and because the acquisition of EDXS-TEM spectra are very time consuming, the calculation of the Fe concentration by grey-scale integration can be considered a more robust and expedite method. The BSE images that were ultimately chosen to extract grey-scale intensity profiles were obtained at 5 kV. These conditions were chosen based on the calculated spatial resolution, the minimization of interferences on the grey-scale profiles (discussed in previous sections) and Monte Carlo simulations of the backscattered electron trajectories using the software “Win X-Ray” (Version 1.4.2.1) for our compositions and considering the thickness of the produced foils (see supplementary material).

Annealed Opx7_cOp_47 sample at 1050°C, log fO2 = −7 Pa for 72 h; a EDXS TEM maps and line-scans were acquired over the same area where grey-scale profiles were extracted for direct comparison. The white dots correspond to the points where spectra for the line-scan were acquired. b Overlay of a compositional profile obtained through the calibration of a grey-scale intensity images acquired at 5 kV with a compositional profile extracted from EDXS TEM Fe mapping and line-scan
Profile fits and extracted diffusion coefficients
Overall the profile shapes in the crystal can be very well reproduced using the exponential dependence of DFe-Mg on XFe. The profile shapes are clearly sensitive to the factor m in the exponent but with the regression of the data we can constrain it to only within an interval of approximately ± 0.5 (see electronic supplementary material), depending also on the counting statistics of the BSE image. The values for D0 and m obtained from the regression procedure are reported in Table 1 and the corresponding fit profiles are shown in Fig. 7. The logDFe-Mg values could be reproduced within 0.16 log units and m within 0.1 on a time series experiment performed on sample Opx7_cOp_47 (Fig. 7). Even considering a relatively low accuracy for m (Fig. 8), we can observe a clear trend of a decreasing m with T (Fig. 9a). The other fitting parameter, D0, follow approximately an Arrhenius relationship (Fig. 9b). From D0 and m we calculated DFe-Mg at XFe = 0.1, 0.3, and 0.5, which are also shown in Fig. 9b. The implication of the increasing m with T is that the activation energy is apparently decreasing for lower Fe contents (Fig. 9b).

Profile fits for the four experiments (Opx7_cOp_16, Opx7_cOp_47, Opx7_cOp_9 and Opx7_cOp_22). Results for the time series experiment in Opx7_cOp_47 (log[Do(m2/s)] = −19.11 and m = 2.9) given in the corresponding panel. Note the trend of decreasing compositional dependence (m) with temperature. Interferences from the Pt protective strip were removed from the compositional profiles and not considered for the fitting procedure

Different fits for sample Opx7_cOp_47 with different input values for Dfilm, Do and m. The correlation between m and Do is here illustrated

a Values for m given by the regression procedure plotted against temperature and b DFe-Mg calculated for XFe = 0.1, XFe = 0.3 and XFe = 0.5, along with DFe-Mg for pure enstatite (in blue) in an Arrhenius plot. Grey squares in b correspond to the refitted data of Dohmen et al. (2016) using our m values and calculated for Fs10
Discussion
Methodological approach
To determine a compositional dependence of diffusion coefficients a long enough diffusion profile is required where the concentration of the diffusing component varies over the relevant compositional range and changes in the concentration gradient are well resolved (e.g., Chakraborty 1997). Alternatively, a considerable number of experiments could be performed with different crystals that cover the relevant compositional range (e.g., Cherniak and Watson 1994). The latter method is not always possible due to the limited availability of appropriate crystals for diffusion experiments and the experimental effort would be much higher than determining the compositional dependence from changes in the concentration gradient of one diffusion profile.
By using a combination of high-resolution analytical techniques on thin film diffusion couples, it was possible to obtain DFe-Mg in orthopyroxene and confidently determine its dependence on XFe in the binary solid solution. We have demonstrated that with our setup (thin-film diffusion couples, FIB-foil preparation, and BSE imaging) we can resolve Fe–Mg diffusion profiles on a scale shorter than 100 nm and quantify the effects of XFe on the profile.
Using depth profiling methods like RBS or SIMS we could, in the ideal case, obtain a slightly better spatial resolution (represented by σ = 5 nm, e.g., Dohmen et al. 2002, vs. 20 nm, present study) but this resolution can be only reached if the samples would have a perfect geometry, meaning that they have a surface and interface roughness smaller than a few nm. For our samples, this is clearly not the case (e.g., Figs. 4, 5) and it is often not the case for samples after diffusion experiments using thin-film diffusion couples, powder source or any other setup. In addition, with the present approach we have the advantage of being able to resolve short diffusion profiles, while simultaneously controlling and correcting for structural features that may arise from the crystallization of the films during the diffusion anneals. The method proved to be particularly advantageous for small sample volumes as it allowed us to acquire information on micro to nm-sized volumes without considerable preparation. Once a sample is analysed it can be used for a next diffusion anneal and a different location on the samples can be investigated to obtain the temporal evolution of the diffusion profile in the same sample, as we did here for sample Opx7_cOp_47 (see supplementary material).
The importance of a good control over the microstructure of the diffusion couples has been previously discussed (e.g., Watson and Dohmen 2010) but the general idea is that it is necessary to ensure that the profiles are extracted from areas where a time invariant geometry at the interface between film and substrate was maintained during thermal annealing to exclude any potential mechanical contribution (e.g., development of local stresses in the interface film-substrate contributing to the chemical potentials of the diffusing species). Temporal change of the texture in the film (e.g., pore evolution, grain size) has only a minor influence on the diffusion profile within the crystal. As long as the diffusive flux is not limited by the film, a temporal change in the effective diffusion properties of the film does not affect the extracted diffusion coefficients in the crystal (e.g., see Marquardt et al. 2010).
This was directly investigated by BSE imaging on cross-sections or by using the STEM detector, allowing to readily identify such features. In depth profiling methods, like RBS or SIMS, we cannot easily select the best areas for profile analysis, not only because of the difficulties selecting the profile location by optical inspection of the surface (e.g., with reflected microscope) but also due to the low lateral resolution of the methods (about a hundred µm for SIMS, 0.5 mm for RBS), which would integrate over texturally very different regions for the present samples. Nevertheless, in case of an ideal geometry, RBS and SIMS can be very useful to resolve shorter diffusion profiles (< 100 nm). A lower lateral resolution can be obtained by ToF–SIMS or NanoSIMS where the latter is usually not designed and applied for depth profiling. Newer generation of ToF–SIMS do have the advantage of 3D imaging during sputtering of the sample allowing to resolve chemical heterogeneities in the sample (e.g., in a thin film) on a scale of a few hundred nm (e.g., Beyer et al. 2019). Alternatively, NanoSIMS, which provides a high lateral resolution of around 50 nm and has been applied in diffusion studies (e.g., Saunders et al. 2012; Zhukova et al. 2014; Till et al. 2015; Jollands et al. 2018), could be used to analyse cross sections prepared by FIB-SEM.
Diffusion coefficients determined on thin-film diffusion couples with ideal geometry have been determined in Marquardt et al. (2010) for DY-Yb in YAG at 1450 °C using both RBS and on cross sections using ATEM. The DY-Yb measured with RBS was 0.5 log units smaller than those using ATEM but this difference is mostly attributed to the misfit of the ATEM profile (see Fig. 4a vs. 4b in Marquardt et al. 2010), which was based on an automatic regression procedure but does not reproduce the critical part of the diffusion profile around the inflection point.
When using electron induced analysis (EDXS, BSE), a correct characterization of the samples and interpretation of the results is only possible if the analysis is conducted on high-quality foils, preferably homogenous and free of other preparation artifacts. A good knowledge of the material properties and control over the operating conditions are therefore necessary to produce high-quality specimens and extract accurate diffusion profiles. The given estimate of the preparation steps is only a rough estimate based on a limited number of samples with similar compositions. The appropriate preparation and polishing conditions will depend on the methods and the sample quality they demand. For our set of samples, accelerating voltages of 5 kV, high resolution (6144 × 4096) and longer dwell times (45 µs) on acquisition yielded the best results with a spatial resolution of 20 nm. Because every sample exhibits different features, caution should be exercised when interpreting the grey-scale intensity images and when selecting the best area to extract the profiles. The obtained XFe (XFe = Fe/(Fe + Mg)) contents of the thin-film using this method are very similar to the concentrations obtained by EDXS point analysis (difference within 0.02 to 0.05 XFe) (Table 2). The chemical information obtained through these methods is less accurate than electron microprobe analysis, thus creating an additional source for analytical uncertainty. However, it has the added benefit of requiring a significantly smaller sample volume.
If the preparation method and imaging settings are adequate for the sample, the extraction of diffusion profiles using grey-scale intensity profiles from cut foils becomes very fast and can be used routinely to determine diffusion coefficients and acquire geometrical information of the diffusion couple.
Comparison with literature data for D Fe-Mg
Currently, the only available dataset of measured Fe–Mg interdiffusion coefficients in orthopyroxene that serve as an independent constraint is the one found in Dohmen et al. (2016) determined in the T range 870–1100 °C at fO2 varying between 10–7 and 10–11 Pa. However, the authors were not able to fully quantify the compositional dependence of DFe-Mg in orthopyroxene on the mole fraction of the Fe end-member. In Dohmen et al. (2016) the dependence of DFe-Mg on XMg could not be unambiguously determined from the profile shape (see Fig. 2b and related discussion in Dohmen et al. 2016). The main reason for this ambiguity is that the films were thinner and hence the diffusion reservoir in the experiment significantly smaller compared to this study. As a consequence, after the diffusion anneal the ferrosilite content was relatively low at the film-crystal interface and in addition, the profiles were much shorter (< 300 nm). In most cases, the best fit was obtained with values for m between 1 and 2. For the final fitting procedure and the reported values for DFe-Mg given for the crystal compositions (XFe = 0.09 and XFe = 0.01) the authors fixed the value for m = 1 to avoid artificial variations of the Do value due to the correlation of the m value and Do by the fitting procedure. For example, larger values for Do are obtained if the compositional dependence is underestimated (Fig. 8). Nonetheless, in Dohmen et al. (2016) when their derived DFe-Mg was directly compared for the two types of single crystals (with XFe = 0.09 vs. XFe = 0.01) used in their experiments at various T and fO2 = 10–7 Pa, the DFe-Mg of the more Fe-rich crystal is larger than average by 0.2 log units. Assuming that this behavior is purely related to the different ferrosilite contents, this would in fact imply a compositional dependence of m ≈ 2.5–3, similar to what has been found in this study at temperatures > 1000 °C. In comparing DFe-Mg obtained in this study to the ones in Dohmen et al. (2016), we observe that for the same XFe, T and fO2 conditions, our Fe–Mg diffusion rates are similar but becoming systematically slower with T up to 0.5 log units at T = 1100 ºC (Fig. 10a). This can be explained by the aforementioned correlation between the Do and m values on the fitting procedure, where a higher m value results in slower absolute diffusivities (Fig. 8). We have therefore corrected the DFe-Mg determined by Dohmen et al. (2016) for Opx7 (XFe = 0.09) by refitting their measured diffusion profiles measured at various T and fO2 = 10–7 Pa using now the newly determined values for m as a function of T. The DFe-Mg of Dohmen et al. (2016) now plot much closer to our results for XFe = 0.1 (Fig. 10a).

a The best-fit values of log DFe-Mg (m2/s) along the c axis and at logfO2 = −7 Pa are plotted in an Arrhenius plot with the appropriate compositional correction for different XFe compositions at different temperatures. For comparison we also plotted the results of Dohmen et al. (2016) measured at logfO2 = −7 Pa where a compositional dependence of m = 1 was suggested (original data) and the correction if m was fixed to the values we obtained for the corresponding temperatures (squares in grey filling). The new refitted data of Dohmen et al. (2016) now plots much closer to our results. We can observe that the larger compositional dependence obtained at higher temperatures results in significant shifts on DFe-Mg at T > 1000°C. b) DFe-Mg obtained through the kinetic data for Opx using the Kroll et al. (1997) recalculated values for XFe < 0.55. Here we can see that the data as recalculated by Kroll et al. (1997) for T = 500–800°C shows a slight decrease in the compositional dependence with increasing temperature, as opposed to the significant increase we observed for T > 950 °C
We performed a combined regression of DFe-Mg for XFe = 0.1 from this study and Dohmen et al. (2016), both determined at fO2 = 10–7 Pa and obtained the following Arrhenius relationship:
The activation energy is now 47 kJ/mol smaller when compared to Dohmen et al. (2016), which is partly a consequence of the different fitting procedures with m being a fixed parameter in Dohmen et al. (2016).
Based on our experiments the effect of XFe on DFe-Mg, as given by \(D\left({X}_{Fe}\right)= D({X}_{Fe}=0.1)\cdot {10}^{m({X}_{Fe}-0.1)}\) (Eq. 7), can be calculated for the temperature range 950–1100 °C by the following parameterization where it is possible to determine m following a linear regression of m on temperature:
It is worth mentioning here that the effect of XFe on DFe-Mg found here for Opx is similar to those previously found for DFe-Mg in olivine, characterized by m = 2.8 at T = 1000 °C in Chakraborty (1997) and m = 3 in Dohmen et al. (2007) for T = 700–1200 °C for log fO2 = −7 to −12 Pa. Most notably, the studies of Nakamura and Schmalzried (1984) and Chakraborty (1997) show an analogous increase of the effect of XFe on DFe-Mg with temperature.
Comparison with earlier estimates for effect of X Fe on D Fe-Mg
Another way of predicting DFe-Mg in Opx was based on the experimental determination of order–disorder rates in orthopyroxene, generally obtained from the isothermal annealing of crystals and characterized by a reaction constant (K+). According to Ganguly and Tazzoli (1994) it is possible to relate DFe-Mg with K+ through the following relationship:
where λ corresponds to the average jump distance between successive M1 and M2 sites and KD is the distribution coefficient between the M1 and M2 sites. Based on the experimental order–disorder data of Besancon (1981), Saxena et al. (1987), Anovitz et al. (1988) and Sykes-Nord and Molin (1993), an analytical expression was formulated by Ganguly and Tazzoli (1994) that predicts DFe-Mg in orthopyroxene as a function of temperature and XFe:
In fact, the compositional dependence is given here by an exponent of 2.6, which is close to those found here at T > 1050 °C. However, these data were determined in the T range 500–800 °C and mostly at an fO2 close to the IW buffer. The Eq. 15 of Ganguly and Tazzoli (1994) has been discussed in some detail in Dohmen et al. (2016). The central idea was that the exponent of 2.6 was mainly defined by the kinetic constants of Anovitz et al. (1988), which were criticized and corrected by Kroll et al. (1997) using a consistent modeling approach for all published order–disorder experiments with Opx. A new regression of the kinetic data for Opx with XFe < 0.55 by Kroll et al. (1997) reveals a new parameterization of the effect of T and XFe on K+:
The more recent kinetic data of Wang et al. (2005) shows that KD is independent of XFe. It follows that the compositional dependence of DFe-Mg on XFe directly relates to the dependence of the reaction constant on XFe. Thus, combining Eqs. 14 and 15 with the new calibration of KD from Wang et al (2005), we can recalculate DFe-Mg as a function of T and XFe. The main consequence of the new calibration of Kroll et al. (1997) for K+ is that the activation energy is a function of XFe and that the dependence of log DFe-Mg on XFe at constant T is not linear and becomes slightly smaller with increasing T. In Fig. 10b we plotted the predicted DFe-Mg as a function of XFe at four different T, characteristic for the domain of the experimental kinetic data. We made a linear regression of these data at 500 °C and 800 °C and obtained an equivalent exponent m = 2 and m = 1.5, respectively. It is difficult to evaluate if the experimental data for K+ strongly indicate a quadratic dependence on XFe but overall the effective dependence is smaller than those in Eq. 15 from Ganguly and Tazzoli (1994). The more recent kinetic data of Stimpfl et al. (2005) stands out in this regard, as the compositional dependence of DFe-Mg on XFe was found to be much larger than the one obtained by the optimized expression of Ganguly and Tazzoli (1994) (Eq. 15). However, the authors were not able to explain this discrepancy as the experiments were conducted at similar T and fO2 conditions, particularly for Opx with XFe = 0.1 where the data shows values 3 log units lower compared to the other literature data.
An additional complication arises when considering the effect of fO2 on the compositional dependence. Most of the kinetic experiments were performed without appropriate control over the fO2 conditions, which can result in variations in the defect concentration and consequently, the order–disorder kinetics. The disorder experiments of Anovitz et al. (1988) were conducted at fO2 of approximately 0.5 log units above the iron-wüstite (IW) buffer and the general expression derived by Ganguly and Tazzoli (1994) is valid for fO2 = IW to IW + 0.8. These are more reducing conditions and lower temperatures than the ones explored in this study, being a potential source for discrepancy. Here and in Dohmen et al. (2016), DFe-Mg was measured at constant fO2 = 10–7 and 10–11 Pa and Dohmen et al. (2016) found a week effect of fO2 for the more Fe-rich crystal (Opx7). Therefore, we cannot exclude the possibility that m is a function of fO2 and the results from the disordering experiments cannot be directly compared to our data. Furthermore, it should be mentioned that the kinetic experiments were not buffered for silica. This should also be considered, as silica activity (aSiO2) has potentially an effect on the point defect chemistry of Opx. For example, defect models for olivine have shown that from a theoretical perspective aSiO2 is a critical parameter to fully define the thermodynamic state of the crystal and hence its point defect chemistry and diffusion properties (e.g., Stocker and Smyth 1978). In fact, aSiO2 was shown to have an effect on Mg diffusion in pure forsterite (Muir et al. 2020, Zhukova et al. 2014) but its role for Fe–Mg diffusion in olivine with some fayalite seems to be less significant (see Nakamura and Schmalzried 1984, Dohmen and Chakraborty, 2007). Similar conclusions might be applicable to Opx.
The activation energies inferred from the kinetic experiments are comparable (251 kJ/mol for Fs9) to those measured here (261 ± 24 kJ/mol for Fs9) but in a different T ( 500–800 °C vs. 950-1100°C, respectively) and fO2 domain. This discrepancy between activation energies becomes obviously larger for higher Fe compositions (Fig. 10b) but are also comparable when we extrapolate to pure enstatite: 252 kJ/mol (as calculated from Kroll et al. 1997) and 210 kJ/mol (present study).
It should also be noted that the natural crystals used for the kinetic experiments were more Al-rich than the ones used for our experiments (octahedral Al: Saxena et al. (1987): ≈ 0.02–0.135 a.p.f.u.; Anovitz et al. (1988):0.002–0.02 a.p.f.u.; Sykes-Nord and Molin (1993): 0.00–0.04 a.p.f.u). Because the presence of Ca (larger concentrations) or aliovalent trace elements (e.g., Al or Cr) could potentially influence the point defect chemistry and hence the diffusion properties of Opx, further calibration would be necessary to apply the data in compositions where these elements are present in larger amounts, as well as for Opx compositions with Fs-contents > 50 mol%.
Irrespective of any conclusions one can draw from the comparison with previous published data, it is difficult to recognize or quantify potential sources of error or uncertainties associated with the determination of reaction constants obtained in kinetic studies. Some of these potential sources of uncertainty were discussed before but it is clear that further calibration of the compositional dependence as a function of fO2 or for compositions with Al, Cr or other aliovalent impurities in the structure would be useful.
Implications for diffusion chronometry using orthopyroxene
The direct implication of our data in terms of modelling natural processes is that by considering a stronger compositional dependence for the appropriate temperature range (for e.g., m = 3 for T = 1000–1050 °C) than previously suggested by experimental data (Dohmen et al. 2016), the variation of DFe-Mg for different Fs-contents will be larger (Fig. 10a). Following Eq. 7, and considering m = 3, the difference in DFe-Mg for Fs9 compositions can be of up to 0.3 log units faster compared to pure enstatite. Therefore, if the DFe-Mg dependence on XFe is corrected for, the timescales of natural processes obtained from modelling the compositional zoning of natural crystal would be shorter than the ones obtained considering the compositional dependence previously available in experimental data. The effect of temperature on the compositional dependence should also be considered but it is generally not recommended to extrapolate the values to temperatures below the experimental range, due to potential changes in the diffusion mechanism. In fact, the DFe-Mg calculated from order/disorder rates according to Kroll et al. (1997) indicates a more constant compositional dependence in the T range 500–800 °C and a smaller than those estimated from Ganguly and Tazoli (1994). Nonetheless, if we attempt to extrapolate our results, the compositional dependence would be seemingly less important at temperatures below 950 °C (for log fO2 = −7 Pa), possibly even ceasing to affect DFe-Mg at lower temperatures. For most volcanic systems the estimated temperatures often fall below 900 °C, which means that the modelling of timescales could be somewhat simplified as the compositional dependence on XFe is likely less relevant at such temperatures. As an illustration, taking the data and the fO2 parameterization of Allan et al. (2013) based on the derivation of Ganguly and Tazzoli (1994), in which the authors calculated a constant compositional dependence of m = 2.4, we recalculated the value of DFe-Mg in the orthopyroxenes of Fs45 composition for the final magmatic temperature conditions of the high-SiO2 rhyolites of the Oruanui mush zone at fO2 = NNO. The authors calculated a temperature of 770 °C which falls below our T experimental range. As previously mentioned, it is not advisable to extrapolate our results to different T conditions; however, and merely serving as a conceptual exercise, we assumed a value of m = 1 for XFe = 0.45 as suggested by Kroll et al. (1997), see also Fig. 10b. By making the compositional correction using Eq. 7, we obtained a log DFe-Mg (m2/s) value that is slower by 0.64 log units. Considering the peak probability age of 230 years for the extraction of melt and crystals of the Oruanui mush zone calculated by the authors, and using our recalculated DFe-Mg values, we obtain an approximate age of 981 years. In addition, the authors considered a larger effect of fO2 on DFe-Mg than what was experimentally found by Dohmen et al. (2016). Similarly, Saunders et al. (2012) obtained timescales of 4.6 to 5.7 ± 3.5 weeks prior to the 1982 May 14 Mount St. Helens eruption from modelling zoned orthopyroxene crystals using the parameterization of Ganguly and Tazzoli (1994). As shown above, this parameterization seemingly overestimates the compositional dependence of DFe-Mg on the Fe mole fraction for temperatures below 1000 ºC. If we correct the DFe-Mg values now using a compositional dependence of m = 1 for the temperature of 880 ºC, as calculated by the authors, and for the given Opx compositions (XFe ≈ 0.34), we obtain a log DFe-Mg (m2/s) that is slower by approximately 0.36 log units. This results in a timescale of ca. 10.6 ± 3.5 weeks of crystal residency in the magma chamber prior to the eruption, which is now closer to the last known episode of deep seismicity before the May 14 eruption, documented to have happened 9 weeks before (Weaver et al., 1983).
While using the parameterization of Ganguly and Tazzoli (1994) to model timescales will result in shorter timescales for temperatures < 1000 °C (temperature at which we determined a similar compositional dependence on the Fe mole fraction), using other DFe-Mg databases that do not account for the compositional dependence of DFe-Mg on XFe will have the opposite result. Timescales of magmatic processes in the Ruapehu volcano were calculated by Kilgour et al. (2014) using the estimated Fe–Mg interdiffusion coefficients for orthopyroxene from the Mg-tracer diffusion experiments of Schwandt et al. (1998) where no effect XFe on DFe-Mg is considered. We recalculated DFe-Mg for XFe ≈ 0.40, T = 958 °C and using a m value of 1.1. For most of the orthopyroxene crystals the authors obtained diffusion timescales of ca. 7 months while our recalculations give out timescales of ca. 3.3 months which is consistent with registered seismic swarms prior to eruption (Kilgour et al. 2014).
These results serve as an example, but they showcase the errors when incorrectly accounting for the effect of XFe on DFe-Mg can introduce in the derived timescales, even considering the conservative compositional dependence of m = 1. This reinforces the importance of experimentally constrain the DFe-Mg dependence on composition and to correct for it when modelling timescales of natural processes from the compositional zoning of orthopyroxenes. Evidently, if the chemical zoning of natural crystals showcases large enough variations of XFe and the profiles are long enough to be resolvable using the method here described, it is also possible to obtain constraints on the effect of XFe on DFe-Mg from modelling natural profiles.
Conclusions
1) We developed a new approach using thin film diffusion couples and BSE imaging of FIB-foils to measure Fe–Mg diffusion profiles in Opx and resolve concentration gradients within 20 nm while simultaneously account for the microstructure of the samples. This allowed to accurately determine diffusion coefficients and resolve the compositional dependence of DFe-Mg on XFe in Opx through the analysis of the detailed profile shape. Our study showcases the importance of analysis at high spatial resolution and with good constraints over the overall profile shape. This general approach could be also applied to other mafic minerals like olivine, clinopyroxene or potentially amphibole.
2) In the investigated T range 950–1100 °C the dependence of DFe-Mg on XFe was found to be a strong function of temperature. The influence of other parameters, such as fO2 or the presence of trivalent impurities remain to be considered in the calibration of DFe-Mg in orthopyroxene. For this reason, these effects cannot be combined to derive a general expression that describes the compositional dependence and that is generally applicable to natural systems.
3) If the compositional dependence of DFe-Mg on XFe is not accounted correctly for when modelling natural systems, it can introduce significant errors on the derived timescales. With our new calibration and considering in addition constraints for DFe-Mg from re-evaluated order–disorder rates it appears that at the most relevant temperature range for most applications in volcanology (< 900 ºC), the compositional dependence is smaller than those used in many earlier studies relying on the calibration of Ganguly and Tazzoli(1994). As a consequence, the correct diffusion coefficients are smaller than the used ones and the respective time scales longer than those obtained in these studies.
Data availability
The data of this study are availabe in the Electronic Supplementary Materials.
References
Abràmoff MD, Paulo MJ, Sunanda J (2004) Image processing with imageJ. Biophoton Intern 11(7):36–42
Allan ASR, Morgan DJ, Wilson CJN, Millet MA (2013) From mush to eruption in centuries: assembly of the super-sized Oruanui magma body. Contrib Miner Petrol 166:143–164. https://doi.org/10.1007/s00410-013-0869-2
Anovitz LM, Essene EJ, Dunham WR (1988) Order-disorder experiments on orthopyroxenes: implications for the orthopyroxene geospeedometer. Am Miner 73:1060–1073
Arnould O, Hild FË (2002) EPMA measurements of diffusion profiles at the submicrometre scale. Mikrochim Acta 139:3–10
Besencon JR (1981) Rate of cation disordering in orthopyroxenes. Am Miner 66:98
Beyer C, Dohmen R, Rogalla D, Becker HW, Marquardt K, Vollmer C, Chakraborty S (2019) Lead diffusion in CaTiO3: A combined study using Rutherford backscattering and TOF-SIMS for depth profiling to reveal the role of lattice strain in diffusion processes. Am Mineral 104(4):557–568
Boona SR (2021) Quantitative SEM-EDS analysis of semi-transparent samples. Micros Today 29:42–48. https://doi.org/10.1017/s1551929520001790
Bradshaw RW, Kent AJR (2017) The analytical limits of modeling short diffusion timescales. Chem Geol 466:667–677. https://doi.org/10.1016/j.chemgeo.2017.07.018
Buening DK, Buseck PR (1973) Fe-Mg lattice diffusion in olivine. J Geophys Res 78:6852–6862
Chakraborty S (2008) Diffusion in solid silicates: a tool to track timescales of processes comes of age. Annu Rev Earth Planet Sci 36:153–190. https://doi.org/10.1146/annurev.earth.36.031207.124125
Chakraborty S (1997) Rates and mechanisms of Fe-Mg interdiffusion in olivine at 980–1300°C. J Geophys Res b: Solid Earth 102:12317–12331. https://doi.org/10.1029/97jb00208
Chakraborty S, Ganguly J (1991) Compositional zoning and cation diffusion in garnets. In: Diffusion, atomic ordering, and mass transport: selected topics in geochemistry. Springer US, New York, NY, pp. 120–175
Chamberlain KJ, Morgan DJ, Wilson CJN (2014) Timescales of mixing and mobilisation in the Bishop Tuff magma body: perspectives from diffusion chronometry. Contrib Miner Petrol 168:1–24. https://doi.org/10.1007/s00410-014-1034-2
Cherniak DJ, Watson EB (1994) A study of strontium diffusion in plagioclase using Rutherford backscattering spectroscopy. Geochim Cosmochim Acta 58(23):5179–5190
Cliff G, Lorimer GW (1975) The quantitative analysis of thin specimens. J Microsc 103(2):203–207
Cooper GF, Morgan DJ, Wilson CJN (2017) Rapid assembly and rejuvenation of a large silicic magmatic system: Insights from mineral diffusive profiles in the Kidnappers and Rocky Hill deposits, New Zealand. Earth Planet Sci Lett 473:1–13
Conway CE, Chamberlain KJ, Harigane Y, Morgan DJ, Wilson CJ (2020) Rapid assembly of high-Mg andesites and dacites by magma mixing at a continental arc stratovolcano. Geology 48(10):1033–1037
Costa F, Dohmen R, Chakraborty S (2008) Time scales of magmatic processes from modeling the zoning patterns of crystals. Rev Mineral Geochem 69:545–594. https://doi.org/10.2138/rmg.2008.69.14
Costa F, Morgan D (2010) Time constraints from chemical equilibration in magmatic crystals. In: Timescales of Magmatic Processes: From Core to Atmosphere. pp 125–159
Crank J (1975) The mathematics of diffusion, 2nd Edition. Clarendon Press, Oxford, 144
Davydova VO, Shcherbakov VD, Plechov PY (2018) The timescales of magma mixing in the plumbing System of Bezymianny Volcano (Kamchatka): insights from diffusion chronometry. Mosc Univ Geol Bull 73:444–450. https://doi.org/10.3103/S0145875218050058
Dohmen R, Becker HW, Chakraborty S (2007) Fe-Mg diffusion in olivine I: experimental determination between 700 and 1200°C as a function of composition, crystal orientation and oxygen fugacity. Phys Chem Miner 34:389–407. https://doi.org/10.1007/s00269-007-0157-7
Dohmen R, Chakraborty S (2007) Fe–Mg diffusion in olivine II: point defect chemistry, change of diffusion mechanisms and a model for calculation of diffusion coefficients in natural olivine. Phys Chem Miner 34(6):409–430
Dohmen R, Becker H-W, Meißner E et al (2002) Production of silicate thin films using pulsed laser deposition (PLD) and applications to studies in mineral kinetics. Eur J Mineral 14:1155–1168. https://doi.org/10.1127/0935-1221/2002/0014-1155
Dohmen R, Faak K, Blundy JD (2017) Chronometry and speedometry of magmatic processes using chemical diffusion in olivine, plagioclase and pyroxenes. Rev Mineral Geochem 83:535–575. https://doi.org/10.2138/rmg.2017.83.16
Dohmen R, Ter Heege JH, Becker HW, Chakraborty S (2016) Fe-Mg interdiffusion in orthopyroxene. Am Miner 101:2210–2221. https://doi.org/10.2138/am-2016-5815
Doig P, Flewitt PEJ (1983) The role of specimen and instrumental parameters in STEM-EDS X-ray microanalysis of thin foils. J Microsc 130:377–388. https://doi.org/10.1111/j.1365-2818.1983.tb04557.x
Fabbro GN, Druitt TH, Costa F (2017) Storage and eruption of silicic magma across the transition from dominantly effusive to caldera-forming states at an arc volcano (Santorini, Greece). J Petrol 58:2429–2464. https://doi.org/10.1093/petrology/egy013
Flaherty T, Druitt TH, Tuffen H et al (2018) Multiple timescale constraints for high-flux magma chamber assembly prior to the Late Bronze Age eruption of Santorini (Greece). Contrib Miner Petrol 173:1–21. https://doi.org/10.1007/s00410-018-1490-1
Ganguly J, Bhattacharya RN, Chakraborty S (1988) Convolution effect in the determination of compositional profiles and diffusion coefficients by microprobe step scans. Am Miner 73:901-909
Ganguly J, Tazzoli V (1994) Fe2+-Mg interdiffusion in orthopyroxene: retrieval from the data on intracrystalline exchange reaction. Am Miner 79:930–937
Hofmann S (1994) Atomic mixing, surface roughness and information depth in high-resolution AES depth profiling of a GaAs/AlAs superlattice structure. Surf Interface Anal 21:673–678
Jollands MC, Hanger BJ, Yaxley GM et al (2018) Timescales between mantle metasomatism and kimberlite ascent indicated by diffusion profiles in garnet crystals from peridotite xenoliths. Earth Planet Sci Lett 481:143–153. https://doi.org/10.1016/j.epsl.2017.10.021
Kilgour GN, Saunders KE, Blundy JD et al (2014) Timescales of magmatic processes at Ruapehu volcano from diffusion chronometry and their comparison to monitoring data. J Volcanol Geoth Res 288:62–75. https://doi.org/10.1016/j.jvolgeores.2014.09.010
Kroll H, Lueder T, Schlenz H, Kirfel A, Vad T (1997) The Fe2+, Mg distribution in orthopyroxene: a critical assessment of its potential as a geospeedometer. Euro J Mineral 9:705–733
Lee MR, Brown DJ, Smith CL et al (2007) Characterization of mineral surfaces using FIB and TEM: a case study of naturally weathered alkali feldspars. Am Miner 92:1383–1394. https://doi.org/10.2138/am.2007.2453
Lorimer GW (1987) Quantitative X-ray microanalysis of thin specimens in the transmission electron microscope; a review. Mineral Mag 51:49–60. https://doi.org/10.1180/minmag.1987.051.359.05
Marquardt K, Petrishcheva E, Abart R et al (2010) Volume diffusion of Ytterbium in YAG: thin-film experiments and combined TEM-RBS analysis. Phys Chem Miner 37:751–760. https://doi.org/10.1007/s00269-010-0373-4
Meissner E, Sharp TG, Chakraborty S (1998) Quantitative measurement of short compositional profiles using analytical transmission electron microscopy. Am Miner 83:546-552
Milke R, Dohmen R, Becker HW, Wirth R (2007) Growth kinetics of enstatite reaction rims studied on nano-scale, Part I: methodology, microscopic observations and the role of water. Contrib Miner Petrol 154:519–533. https://doi.org/10.1007/s00410-007-0207-7
Morgan DJ, Blake S, Rogers NW et al (2004) Time scales of crystal residence and magma chamber volume from modelling of diffusion profiles in phenocrysts: vesuvius 1944. Earth Planet Sci Lett 222:933–946. https://doi.org/10.1016/j.epsl.2004.03.030
Moshrefzadeh J, Izbekov P, Loewen M, Larsen J, Regan S (2023) Dating individual zones in phenocrysts from the 2016–2017 eruption of Bogoslof volcano provides constraints on timescales of magmatic processes. J Volcanol Geoth Res 435:107741
Muir JMR, Jollands M, Zhang F, Walker AM (2020) Explaining the dependence of M-site diffusion in forsterite on silica activity: a density functional theory approach. Phys Chem Miner 47:1–16. https://doi.org/10.1007/s00269-020-01123-5
Müller T, Dohmen R, Becker HW et al (2013) Fe-Mg interdiffusion rates in clinopyroxene: experimental data and implications for Fe-Mg exchange geothermometers. Contrib Miner Petrol 166:1563–1576. https://doi.org/10.1007/s00410-013-0941-y
Nakamura A, Schmalzried H (1984) On the Fe2+ –Mg2+-Interdiffusion in Olivine (II). Ber Bunsenges Phys Chem 88:140–145
Nastasi M, Mayer JW, Hirvonen JK (1996) Ion-solid interactions: fundamentals and applications. Cambridge University Press
Ostorero L, Balcone-Boissard H, Boudon G, Shapiro NM, Belousov A, Belousova M, Auer A, Senyukov SL, Droznina SY (2022) Correlated petrology and seismicity indicate rapid magma accumulation prior to eruption of Kizimen volcano, Kamchatka. Commun Earth Environ 3(1):290
Petrone CM, Bugatti G, Braschi E, Tommasini S (2016) Pre-eruptive magmatic processes re-timed using a non-isothermal approach to magma chamber dynamics. Nat Commun 7:1–11. https://doi.org/10.1038/ncomms12946
Phillips M, Till CB (2022) Crustal storage and ascent history of the Mt. Shasta primitive magnesian andesite: implications for arc magma crustal flux rates. Contrib Mineral Petrol 177(1):9
Pichavant M, Poussineau S, Lesne P et al (2018) Experimental parametrization of magma mixing: Application to the AD 1530 eruption of La Soufrière, Guadeloupe (Lesser Antilles). J Petrol 59:257–282. https://doi.org/10.1093/petrology/egy030
Saunders K, Rinnen S, Blundy J et al (2012) TOF-SIMS and electron microprobe investigations of zoned magmatic orthopyroxenes: first results of trace and minor element analysis with implications for diffusion modeling. Am Miner 97:532–542. https://doi.org/10.2138/am.2012.3893
Saxena SK, Tazzoli V, Domeneghetti MC (1987) Fe2+ - Mg distribution in aluminous orthopyroxenes. Phys Chem Miner 15:140–147
Schwandt CS, Cygan RT, Westrich HR (1998) Magnesium self-diffusion in orthoenstatite. Contrib Miner Petrol 130:390–396. https://doi.org/10.1007/s004100050373
Solaro C, Balcone-Boissard H, Morgan DJ, Boudon G, Martel C, Ostorero L (2020) A system dynamics approach to understanding the deep magma plumbing system beneath Dominica (Lesser Antilles). Front Earth Sci 8:574032
Stimpfl M, Ganguly J, Molin G (2005) Kinetics of Fe2+-Mg order-disorder in orthopyroxene: experimental studies and applications to cooling rates of rocks. Contrib Miner Petrol 150:319–334. https://doi.org/10.1007/s00410-005-0016-9
Stocker RL, Smyth DM (1978) Effect of enstatite activity and oxygen partial pressure on the point-defect chemistry of olivine. Phys Earth Planet Inter 16:145
Sykes-Nord J, Molin M (1993) Mg-Fe order-disorder reaction in Fe-rich orthopyroxene: structural variations and kinetics. Am Miner 78:921–993
Till CB, Vazquez JA, Boyce JW (2015) Months between rejuvenation and volcanic eruption at Yellowstone caldera, Wyoming. Geology 43:695–698. https://doi.org/10.1130/G36862.1
Turner S, Costa F (2007) Measuring timescales of magmatic evolution. Elements 3:267–272
Utami SB, Costa F, Lesage P, Allard P, Humaida H (2021) Fluid fluxing and accumulation drive decadal and short-lived explosive basaltic andesite eruptions preceded by limited volcanic unrest. J Petrol 62(11):egab086
Wang L, Moon N, Zhang Y et al (2005) Fe-Mg order-disorder in orthopyroxenes. Geochim Cosmochim Acta 69:5777–5788. https://doi.org/10.1016/j.gca.2005.08.011
Watson EB, Dohmen R (2010) Non-traditional and emerging methods for characterizing diffusion in minerals and mineral aggregates. Rev Mineral Geochem 72:61–105. https://doi.org/10.2138/rmg.2010.72.3
Weaver CS, Zollweg JE, Malone SD (1983) Deep earthquakes beneath Mount St. Helens: evidence for magmatic gas transport? Science 221(4618):1391–1394
Wirth R (2009) Focused Ion Beam (FIB) combined with SEM and TEM: Advanced analytical tools for studies of chemical composition, microstructure and crystal structure in geomaterials on a nanometre scale. Chem Geol 261(3–4):217–229
Zhukova I, StC O’Neill H, Cambell IH, Kilburn MR (2014) The effect of silica activity on the diffusion of Ni and Co in olivine. Contrib Miner Petrol 168:1–15. https://doi.org/10.1007/s00410-014-1029-z
Acknowledgements
Niels Jöns, René Hoffmann and Sabine Weisel are thanked for their analytical support and initial assistance with FIB-SEM work. The authors would like to thank Aleksander Kostka for the help provided on the TEM imaging and analysis. M.A. Dias would like to thank C. Hirschle for Laue analysis for crystal orientation. We thank the editor, Othmar Müntener for handling the manuscript and the reviewers, Christy Till and Jim Van Orman for their thorough reviews that helped to improve the manuscript. This project was funded by the DFG project DO777/8-1 (part of the 748 Forschungsgruppe FOR 2881 "Diffusion chronometry of magmatic systems").
Funding
Open Access funding enabled and organized by Projekt DEAL. Deutsche Forschungsgemeinschaft, DO777/8-1 Research unit: FOR2881, Ralf Dohmen.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Othmar Müntener.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dias, M.A., Dohmen, R. Experimental determination of Fe–Mg interdiffusion in orthopyroxene as a function of Fe content. Contrib Mineral Petrol 179, 36 (2024). https://doi.org/10.1007/s00410-024-02110-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-024-02110-7