
Abstract
Emerging and re-emerging illnesses will probably present a new hazard of infectious diseases and have fostered the urge to research new antiviral agents. Most of the antiviral agents are analogs of nucleosides and only a few are non-nucleoside antiviral agents. There is quite a less percentage of marketed/clinically approved non-nucleoside antiviral medications. Schiff bases are organic compounds that possess a well-demonstrated profile against cancer, viruses, fungus, and bacteria, as well as in the management of diabetes, chemotherapy-resistant cases, and malarial infections. Schiff bases resemble aldehydes or ketones with an imine/azomethine group instead of a carbonyl ring. Schiff bases have a broad application profile not only in therapeutics/medicine but also in industrial applications. Researchers have synthesized and screened various Schiff base analogs for their antiviral potential. Some of the important heterocyclic compounds like istatin, thiosemicarbazide, quinazoline, quinoyl acetohydrazide, etc. have been used to derive novel Schiff base analogs. Keeping in view the outbreak of viral pandemics and epidemics, this manuscript compiles a review of Schiff base analogs concerning their antiviral properties and structural-activity relationship analysis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The worldwide emergence of viral pandemic and epidemic threats and the development of resistance to available therapeutics have turned into a severe menace. A sizable fraction of pandemics and/or epidemics that have dogged humanity throughout history are caused by infectious illnesses that are either from existing or newly emerging pathogenic viruses [1, 2]. At the end of 2019, atypical pneumonia caused by the novel coronavirus-2 or severe acute respiratory syndrome (SARS) within a population in Wuhan (China) was reported and is now known as COVID-19. The infection has been observed with a range of illnesses, from minor symptoms to life-threatening diseases, or it can be asymptomatic [3,4,5]. The pathogenic nature and mortality rate has warranted an urgent need to identify preventive and therapeutic interventions. In less than a year, the US Food and Drug Administration (USFDA) granted emergency use status to the vaccine developed by Pfizer/BioNTech based on a new platform consisting of mRNA encoding the virus spike protein, and therapeutics have now been approved for clinical use [6, 7].
Even though people are living longer and infectious outbreaks are curbed within a few years, the new hazards of infectious diseases will likely come from developing and resurfacing infections [8]. In the upcoming decades, the danger of disease outbreaks will grow due to climate change, increased urbanization, and shifting land use practices [9]. For instance, climate change may modify the range of global pathogens, allowing infections, especially vector-borne infections, to spread to new areas [10,11,12]. This has increased the desire to investigate novel antiviral medicines that target untapped pharmacological mechanisms with enhanced safety and limited toxicity [13,14,15]. Only a few non-nucleoside antiviral medicines are currently commercialized, and the majority of them are nucleoside analogs (example: Catherine, Fig. 1) [16].

Chemical structures of some antiviral agents
Zidovudine, a Human Immunodeficiency Virus (HIV) nucleoside reverse transcriptase inhibitor, received FDA approval in 1987, which sped up the efforts toward the development of nucleoside-based antiviral agents [17]. Several combination medicines are now being researched to stop the activity of the same or different viral macromolecules during various stages of their life cycle. They aim to obstruct the replication of the viral genome and thus control infection [18, 19]. Since the majority of antiviral medications only work against specific viral strains, it is crucial to continue looking for antiviral substances being active over a wide range of genotypes or subtypes of viruses [20, 21]. Efforts also aim to develop broad-spectrum antiviral agents for various genotypes or subtypes. As an illustration, many antiviral medications, such as Amprenavir, solely suppress HIV-1 and do not affect HIV-2 [22]. Numerous Hepatitis C Virus (HCV) inhibitors have been authorized exclusively for HCV genotype 1, and no other genotypes [23, 24]. However, several viral inhibitors (including Valacyclovir, PegIFN-2a, PegIFN-2b, Acyclovir, TDF, Foscarnet, Famciclovir, Lamivudine, and Ribavirin) have received approval to treat multiple viruses, which support the hypothesis that antiviral medications for a variety of infectious diseases may 1 day be developed [25]. In 1997, fixed-dose treatment combined with lamivudine/zidovudine was approved for use in HIV-infected patients [26, 27]. As a monotherapy against HCV, sofosbuvir was also authorized in 2013 [28, 29]. Imiquimod was authorized for the topical medication of solar keratoses, shallow basal cell carcinoma, and vaginal and perianal warts in 1997 [30,31,32]. In 2000, Docosanol was authorized for use as a supracutaneous cream to cure herpes labialis brought on by Herpes Simplex Virus (HSV) infections [33]. In 1998, it was thought that ribavirin worked through a variety of mechanisms, with modulation of the immune system and suppression of inosine-5’-monophosphate dehydrogenase (IMPDH) in humans [34, 35]. Topical sine catechins, an aqueous extract of leaves of green tea, were approved in 2006 for the cure of papilloma in genitalia brought on by HIV without having a known mechanism of action [36]. Polymerase inhibitors are known to be the most prevalent type of virus-targeting therapeutics and are classified as nucleoside and non-nucleoside analogs. Acyclovir, Zalcitabine, Didanosine, Lamivudine, Emtricitabine, Tenofovir, and Diisopropyl fumarate belong to the category of nucleoside analogs, whereas Nevirapine, Efavirenz, Rilpivirine hydrochloride, Delavirdine mesylate, and Etravirine are from the category of non-nucleoside analogs. For the treatment of HCV infection, only one non-nucleoside polymerase inhibitor is currently approved which is Dasabuvir [37, 38]. Saquinavir, a peptidomimetic-designed HIV-1 protease inhibitor, received approval in 1995 [39]. Raltegravir along with Elvitegravir was approved in 2007 and 2012, respectively as first-generation integrase inhibitors [40]. Second-generation integrase inhibitor Raltegravir has a greater genetic resistance barrier and maintains antiviral efficacy [41]. In 2014/2015, the FDA approved the first NS5A inhibitors, Ledipasvir, and Daclatasvir dihydrochloride followed by Ombitasvir, Velpatasvir, Pibrentasvir, and Elbasvir in 2016 and 2017 [42]. There are around 100 active medication candidates in research as of January 2018, with 29, 46, and 25 in Phase I, II, and III, respectively of clinical trials [43].
Imine or azomethine groups have been substituted for the group where a carbon atom is double bonded to an oxygen atom (C=O) to create the nitrogen cognate known as Schiff base (Fig. 2) [44, 45]. Frequently utilized carbon-based substances are Schiff bases. Their biological effects range from fungicidal to bactericidal, in treating malaria, suppressing cell growth, treating inflammation, treating viral diseases, and also in lowering the body temperature. They serve as catalysts, intermediates, pigments, and dyes in the production of chemicals [46,47,48]. Numerous naturally occurring, naturally produced, and artificially created substances contain imine or azomethine groups. For these molecules to have biological effects, an imine group must be present [49,50,51].

Basic structure of Schiff base
In the field of pharmaceutical sciences, Schiff base is used for a variety of purposes, including the following: as anticancer, antiviral, antifungal, and antibacterial chemicals; to treat HIV infections and diabetes; to combat cancer medication resistance; as antimalarial agents; and to immobilize enzymes [52,53,54,55].
In a study, a Schiff base ligand was produced by mixing the substances 5-nitropyridine-2-amine and 4-hydroxy-3-methoxy benzaldehyde in a 2:1 stoichiometric ratio (2HL: M). BHA was utilized as a positive control in the in vitro assay, which was carried out similarly to those for DPPH, ABTS, and superoxide. When compared to the ligand, it was discovered that the in vitro glucosidase inhibitory activities had a significant amount of inhibitory potential [56].
It is reported in a study that (E)-ethyl 2-(4-methoxybenzylidene) hydrazine carboxylate and 4-(nitro benzaldehyde) ethyl carbamate were synthesized and characterized as a new Schiff base ligand. Density functional theory and results from experiments limited to establishing the compound’s structure have been found to have a positive association. The effectiveness of the Schiff base as a bioactive molecule has been demonstrated by molecular docking and antibacterial tests [57, 58].
A study discovered a connection between the zinc-Schiff base and the use of the peptide Novicidin that penetrates the membrane as a transporter for the delivery of zinc to cells of the prostate. It was discovered that the potency was toward PC3 tumor cells and the toxicity was normal in PNT1A cells. Additionally, both cell lines confirm zinc uptake. The combination has demonstrated a significant possibility for treating cancer, the activation of zinc stress (e.g., ZnT-1) and apoptosis was verified through a molecular analysis (e.g., CASP-1) [59, 60].
To create Schiff’s bases, a series of (S)-flurbiprofen derivatives were synthesized using various aromatic or aliphatic aldehydes and ketones. Their structure was then validated using HR-ESI-MS, 1H, and 13C-NMR spectroscopy. It was shown that the newly synthesized compounds had considerable -gluta oxidase inhibiting actions. All of the active compounds’ modes of binding were identified using the molecular docking method, and the compounds displayed potential anti-diabetic properties by obstructing high binding energies (between 7.51 and 3.36 kcal/mol) in the active site, which hinders the enzyme’s capacity to operate [61]. (E)-2-((2-hydroxybenzylidene) amino)-4-methyl phenol (SL1) and (E)-2-((2-hydroxybenzylidene) amino) are two Schiff bases. The synthesis of -4-methyl phenol (SL2) and their solvent-free, non-toxic inclusion complexes with cyclodextrin (-CD) was carried out. Since these insertion complexes result in apoptosis, SL2—CD is more effective at combating cancer. For the first time, the bio-potency of CD was boosted by the addition of nanocrystalline Schiff bases [62,63,64].
The Schiff base has increased the curiosity of researchers immensely these days because of its versatile properties and applications in different areas. Some of the important applications of reported Schiff base derivatives are depicted in (Fig. 3).

Diverse applications of Schiff base analogs
Keeping in view the outbreak of COVID-19 and the requirement for potent antiviral drugs, the goal of the current manuscript is to give a thorough analysis of various significant chemical core’s Schiff base analogs which have demonstrated antiviral properties in pre-clinical evaluation. It has focused on some important chemical cores, i.e., istatin, thiosemicarbazide, hydroxyguanidine tosylate, quinazoline, and quinolinyl to hydrazide been derivatized to prepare Schiff base analogs. It has also included details of some potential metal complexes with inhibition properties.
Antiviral Schiff bases
Isatin and its Schiff base derivatives
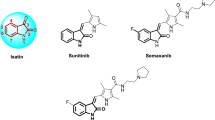
Isatin (1H-indole-2,3-dione) is a heterocyclic moiety having a well-demonstrated role in the control of numerous viral infections [65] Some of the well-reported antiviral agents with isatin nuclei are Sunitinib, Semaxanib, and Toceranib (Fig. 4).

Chemical structures of antiviral agents having isatin nuclei
Pandeya et al. synthesized norfloxacin-isatin mannich base for checking anti-HIV activity [66]. Here two compounds 1a and 1b (Fig. 5) showed maximum potency with EC50 values of 11.3 and 13.9 µg/ml and protection in the range of 70–95%. Substitution of trimethoprim at the 3rd position and electron-withdrawing group at the 5th position showed maximum potency as compared to the other substituent.

Details of chemical structures of isatin and its Schiff base analogs
In another series, 3(N)-sulphadoxine of Isatin derivatives (2a–c) were tested for their ability to inhibit HIV using the assay of MTT 3-(4,5-dimethyl thiazol-2-yl) (Fig. 5). It was found that a compound containing piperidinomethyl group (2a; YP-04) has shown activity up to 16% against HIV-2 (ROD) strain at an EC50 > 2 µg/ml and about 12% were effective against HIV-1 (IIIB) strain. In SAR, it was found that methyl-substituted isatin has shown less activity compared to the unsubstituted isatin derivatives, and 3-methyl morpholine substitution at position-1 of isatin showed better potency as compared to others. Two drug moieties of sulphadoxine and pyrimethamine (2b; YP-8) showed 13% protection against HIV-2 (ROD) at an EC50 of more than 2 µg/ml [67].
Isatinyl thiosemicarbazone Schiff base derivatives
The N-methyl-isatin ß-thiosemicarbazone i.e., Methisazone is an FDA-approved antiviral drug (Fig. 6) [68] Studies have demonstrated viral inhibitory properties of various thiosemicarbazone derivatives namely HSV-1, HSV-2, respiratory syncytial virus (RSV), dengue virus bovine viral diarrhea virus (BVDV), encephalitis virus, hepatitis C virus (HCV), and influenza virus [69,70,71,72,73,74,75]. Efforts have been envisaged to investigate the viral inhibitory potential of its Schiff base analogs and these are reported here.

Example of clinically approved antiviral drug with thiosemicarbazone nuclei
In an investigation conducted by Pandeya et al., 4-chloro acetophenone was used to manufacture N-[4-(4’-Chlorophenyl) thiazol-2-yl] thiosemicarbazides (3a–l; Fig. 7) [76]. In the human T cell line, the replication-inhibiting properties of HIV-1 were evaluated for the synthesized compounds. It was observed that none of them have substantial anti-HIV potential (Table 1). There was a belief that medications might not be able to stop HIV-1 reverse transcriptase (RT). It was assumed that a small hydrophobic molecule would be necessary for RT inhibitory activity, but the thiosemicarbazide end of these compounds has a large p-chlorophenyl thiazolyl group which may not favor the inhibition of HIV-1 reverse transcriptase.

Chemical structure of N-[4-(4’-Chlorophenyl) thiazol-2-yl] thiosemicarbazide (3a–l)
Also, Banerjee et al. synthesized a series of isatinyl thiosemicarbazide analogs of 60 compounds and checked their anti-HIV and anti-tubercular activity. The EC50 values were found to be in the range of 1.69–129.96 µM. Out of these, hydroxy-substituted thiosemicarbazone compound 4b showed EC50 4.18 µM, and methoxy thiosemicarbazone analogs compound 4a showed EC50 1.69 µM (Fig. 8). So, methoxy thiosemicarbazones showed a better inhibitory profile and higher activity as compared to hydroxy thiosemicarbazones. On studying SAR, it was found that the chlorine present at the 4th position of phenyl piperazinyl methyl shows lipophilic interaction at the active site. The Methyl group at the 5th position of indolyl moiety shows interaction with the aromatic residue of the enzyme [77].

Chemical structure of isatinyl thiosemicarbazide analogs
Quinoline and quinazoline Schiff base derivatives
Quinoline and quinazoline are heterocyclic moieties with well-reported antiviral properties. During investigation of a potential drug to control the pandemic caused by SARS-CoV2, it has been found that RNA-dependent RNA polymerase (RdRp) has a pivotal viral life cycle and quinoline and quinazoline derivatives not only inhibit RNA virus-like HIV-1, Ebola virus, Influenza virus but also shows activity against DNA viruses such as HSV (Herpes simplex virus) and Hepatitis B virus [78,79,80,81,82]. In continuation of exploring this moiety, Zhao et al. synthesized 101 quinoline and quinazoline derivatives and on screening using a cell-based assay, it was found that the inhibition activity shown by the compounds are 5e = 95.03%, 5h = 92.85%, and 5i = 74.94%. On further study, it was found that these compounds 5e, 5h, and 5i target the RNA replication which is driven by RdRp (Fig. 9). In respect of SAR, at the 2nd position of the quinoline ring, when the pyrrolidinyl group is replaced by another group like boc-piperazinyl (5e) and (s)-boc-3-amino-pyrrodinyl (5h) derivatives, a significant increase in activity was seen [83].

Chemical structure of quinoline and quinazoline derivatives
Through the development of 3-amino-2-phenyl quinazolines which is a Schiff base, a fresh sequence of 3-(benzylidene amino)-2-phenyl quinazolines (4(3H)-ones (6a–l; Fig. 10) was created [84]. Spectral analyses were used to interpret their chemical structures and viruses like HSV-1, Coxsackievirus, and coronavirus subtypes, etc. were tested for cytotoxicity and antiviral activity. Quinazoline derivatives were tested for the anti-influenza activity and cell-destroying capacity in the culture of the (MDCK) kidney cell line with the help of the MTS assay. It was found when compound 6a was substituted with the hydroxyl group at R’ position, the 50% cytotoxic concentration was found to be 2.6 µg/ml, and minimal cytotoxic concentration was found to be ≥0.8 µg/ml, while compounds 6b, 6d, 6e, 6h, and 6i were found to have CC50, >100.0 µg/ml when substituted with nitro, dimethyl amine, chlorine, and methyl group, respectively. Also, compounds 6c, 6f, 6g, 6j, and 6k have shown activity against influenza virus with CC50 in the range of 8.0 to 50.0 µg/ml when substituted with methoxy, hydrogen, and a hydroxyl group at R’ position. However, these compounds did not find to be active at a higher concentration for the anti-influenza activity of the H1N1 subtype, and H3N2 subtypes. When cytotoxicity and antiviral activity were calculated for the compounds 6a–l in cell cultures of HEL, Compounds 6a and 6c–6l were found to have a minimum cytotoxic concentration ±100.0 µg/ml when substituted with hydroxy, methoxy, dimethyl amine, chlorine or H at R’ while compound 6b showed ≥20 µg/ml when substituted with the nitro group at R’ position. While 50% effective concentration was calculated for compound 6a substituted with R’ = hydroxy group showing good activity with the quantity of 1.0 µg/ml against HSV-1 (KOS), HSV-1 TK-KOS ACVr, and HSV-2(G), 0.8 µg/ml for vaccinia virus and 15 µg/ml for vesicular stomatitis virus. Compound 6a among all others demonstrated superior antiviral efficacy against all tested viruses. Compounds 6a–l were also found to be effective at a culture of the Vero cell lineage extracted from the kidney’s epithelial cells. It was found that the minimum cytotoxic concentration of compounds 6a, 6c, 6d, 6e, 6g, 6h, 6k, and 6l was found to be 100.0 µg/ml when substituted with hydroxy, methoxy, dimethylamine, chlorine, and H at R’ position and H and methyl at R position. The 50% effective concentration to reduce virus-induced cytopathogenicity for compounds 6a, 6c, 6d, 6e, 6g, 6h, 6k, and 6l was found >20.0 µg/ml when substituted with hydroxy, methoxy, dimethylamine, chlorine, or H at R’ position for Para influenza-3 virus, while it is >4.0 µg/ml for compounds 6b, 6f, and 6j. For Reovirus-1, the EC50 was found to be 10.0 µg/ml in compound 6a, >4.0 µg/ml in compounds 6b, 6f, 6j and >20.0 µg/ml in compound 6c, 6d, 6e, 6g, 6h, 6k and 6l. In case of Sindbis virus, the EC50 was shown to be 5 µg/ml by compound 6a, >4 µg/ml for compound 6b, 6f, and 6j, >20.0 µg/ml for 6c, 6d, 6e, 6g, 6h, 6k and 6l, and >100.0 µg/ml for compound 6i. So, it was seen that the substitution of the nitro or chloro group was found to be more effective as compared to the substitution of the hydroxy or methoxy group.

Chemical structure and substitutions for compounds 6(a–l)
Compound 6a was discovered to work well against viruses in culture developed from the Hela cell line at a minimum cytotoxic dosage of 4.0 g/ml, with an EC50 (g/ml) value of >0.8 for Vesicular stomatitis virus, >0.8 for Coxsackievirus B4, and >0.8 for the respiratory syncytial virus when substituted with a hydroxy group i.e., 2-OH at R’ position.
In CRFK cell culture (FIPV), a minimum cytotoxic concentration of 11.2 µg/ml of compound 6a was found to be effective against the virus with EC50 (µg/ml) value of >4.0 for Feline Coronavirus (FIPV) and >4.0 for Feline Herpes virus when substituted with a hydroxy group i.e., 2-OH at R’ position.
In another study against viruses including feline herpes and feline coronavirus by synthetic 2-phenyl-3-substituted quinazoline-4(3H)-ones (7a–h, Fig. 11) by Krishnan et al. [85] structural characteristics were found to be correlated with the biological activity. It was observed that compound 7a when substituted with 2-methyl furan (CC50 = 14.1 ± 2.3 µg/ml, EC50 = 4.0 ± 0.5 µg/ml) 7b, when substituted with 1-methyl-4-nitrobenzene (CC50 13.8 ± 1.8 µg/ml, EC50 = 4.0 ± 0.3 µg/ml), 7d when substituted with 1,2-dimethoxy-4-methylbenzene (CC50 = 13.8 ± 0.5 µg/ml, EC50 = 4.0 ± 0.2 µg/ml) and 7g when substituted with 5-Bromo-3-methylidene-1,3-dihydro-2H-indol-2-one/5-bromoisatin (CC50 = 16.2 ± 0.8 µg/ml, EC50 > 4.0 µg/ml) were close to each other while compound 7e when substituted with 1-chloro-4-methylbenzene (CC50 = 42.2 ± 4.7 µg/ml, EC50 = 42.2 ± 4.7 µg/ml) which was higher as compared to 7a, 7b and 7d. It was found that the minimum cytotoxic concentration for compounds 7f (substituted with 3-methylidene-1,3-dihydro-2H-indol-2-one)/isatin and 7h (methylidene cyclohexane) was equal to that of HHA (CC50 > 100.0 µg/ml, EC50 2.6 ± 0.8 µg/ml), UDA (CC50 > 100 µg/ml, EC50 117.6 ± 1.3 µg/ml) and Ganciclovir (CC50 > 100 µM, EC50 > 100.0 µg/ml). The selectivity index of the compounds 7c and 7g was found to be 4.0 which is much less as compared to the compounds HHA (SI 38), and UDA (SI 9).

Structural details of 3-(benzylidene amino)-2-phenyl. quinazoline-4(3H)-ones (7a–h) structure sizes proportion needs to be consistent
On cultivated HeLa cells, the minimum concentration required to alter the morphology of the cells for compound 7a was 100 ± 5.4 µg/ml, and 7g was 100 ± 5.4 µg/ml when substituted with furan and indole derivatives at the R position. This concentration was reduced to CC50 ≥ 4.0 µg/ml when substituted with elongated alkyl chain [(1E)-prop-1-en-1-yl] benzene in compound 7c, the least cytotoxic concentration for the compounds 7d, 7f, and 7e was near to 20.0 µg/ml when substituted with 1,2-dimethoxy-4-methylbenzene, 3-methylidene-1,3-dihydro-2H-indol-2-one, and 1-chloro-4-methylbenzene, respectively. Compound 7c has shown the least value of less than 4.0 µg/ml for HeLa cells. The EC50 of the compounds for Vesicular stomatitis virus, Coxsackie virus, and RSV was found near 4 µg/ml for compounds 7a, 7b, 7c, 7d, 7e, and 7f when substituted with 2-methyl furan, 1-methyl-4-nitrobenzene, [(1E)-prop-1-en-1-yl]benzene, 1,2-dimethoxy-4-methylbenzene, 1-chloro-4-methylbenzene, and, 3-methylidene-1,3-dihydro-2H-indol-2-one, respectively, this value went to 20.0 µg/ml for compound 7g when substituted with 5-bromo-3-methylidene-1,3-dihydro-2H-indol-2-one, the maximum value for EC50 was more than 100 µg/ml for the compound 7h when substituted with methylidene cyclohexane. All the compounds from 7a–7h were found to have a good response when compared with (S)-DHPA and Ribavirin with a minimum cytotoxic concentration above 250 µg/ml and EC50 in the range of 20 to 150 µg/ml for different viruses in Cultivated HeLa cells. Compound 7a showed the highest selectivity index against 25 which is much higher as compared to reference compounds DS-5000, (S)-DHPA, and Ribavirin with selectivity index 0, 0, and 11, respectively.
Compounds 7a, 7d, 7g, and 7h in the cultivated cell lineage of Vero were having the minimum concentration to alter the normal cell morphology by 50% around 100 g/ml when substituted with (2-methyl furan), (1,2-dimethoxy-4-methylbenzene), (5-bromo-3-methylidene-1,3-dihydro-2H-indol-2-one), and methylidene cyclohexane) respectively. The effective concentration of compounds 7b, 7d, 7e, and 7f to reduce the virus-induced pathogenicity in cells was found to be around 4 µg/ml when substituted with (1-methyl-4-nitrobenzene), (1,2-dimethoxy-4-methylbenzene), (1-chloro-4-methylbenzene) and, (3-methylidene-1,3-dihydro-2H-indol-2-one), respectively and above 20 µg/ml for the compounds 7a, 7c, 7g, and 7h when substituted with (2-methyl furan), ([(1E)-prop-1-en-1-yl]benzene), (5-bromo-3-methylidene-1,3-dihydro-2H-indol-2-one), and (methylidene cyclohexane) respectively in Para influenza-3, Reovirus-1, Sindbis, Punta Toro virus, and Coxsackie virus. The selectivity index value of compound 7d was found to be 25 which is higher as compared to the reference.
Myricetin Schiff bases with triazole substitutions
A polyphenol flavonoid, called myricetin has a wide range of documented actions, including antiviral effects [86,87,88,89,90,91,92]. Similarly, heterocyclic compounds based on the derivatives of 1,2,4-triazole have high efficiency and a broad spectrum and are very popular. To test their potential for having antiviral effects against the tobacco mosaic virus (TMV), 1,2,4-Triazole Schiff base-based derivatives of myricetin were synthesized and evaluated [93]. Substitution at R with Phenyl; 3,4-dichloro phenyl; 3,4-dimethoxy phenyl; and 2-thiophene are some of the significant structural traits identified for effective therapeutic and protective effect. It was found when compounds 8a and 8t were substituted with a phenyl group, it showed curative percentage inhibition of around 52%, protection inhibition percentage of around 52% and 56%, respectively and inactivation inhibition percentage of around 67%, and 57% respectively, compound 8g and 8w, when substituted with 3,4-dimethoxy phenyl, showed curative percentage inhibition of around 53 for both, protection inhibition percent of around 55 and 58, respectively and inactivation inhibition percent of around 88 and 43 approximately. Compound 8j, when substituted with 2-thiophene, showed a curative inhibition percentage of around 53%, a protection inhibition percentage of around 55, and an inactivation percentage inhibition of around 68. The antiviral activities of the synthesized compounds were compared against TMV (in vivo, 500 µg/ml). The above-mentioned results were compared with Ribavirin and Ningnanmycin and Myricetin. For Ribavirin and Ningnanmycin, curative percentage inhibition was found to be around 32% and 53, protection percentage inhibition, around 52 and 66, and inactivation percentage inhibition, around 74 and 92, respectively. For Myricetin, the curative percentage inhibition percentage was around 32, the protection inhibition percentage around 52, and the inactivation inhibition percentage around 52. Derivatives 8g and 8j were found to have a better therapeutic effect, 8p, 8w, and 8f were having a good protective effect, and 8g had a good passivation effect (Fig. 12) and have shown higher inhibitory percentage than that for Myricetin and Ribavirin while it is close to Ningnanmycin.

Structural details for Compounds 8a–w
Copper complex [Cu(L)(phen)] with mixed ligands
According to a recent study, the primary protease (Mpro) of the COVID-19 virus is what causes the virus to become proteolytic and is crucial to the virus’s life cycle [94,95,96,97]. Schiff bases have been discovered to be very significant in the medical and pharmaceutical industries, and the effectiveness of their complexes against COVID has been investigated. Computer modeling was done to determine the possibility against the virus by the compound [Cu(L)(phen)] of mixed ligands for copper (II) that was created by Bharti et al. (Fig. 13) shows the coordination of the Cu (II) ion with the Schiff base ligand (H2L) in the ONS-tridentate mode [98]. The complex formed with the copper having 8 molecules in one cell was having a square pyramidal shape when observed through X-ray crystallographic examination. Additionally, the functional characteristics of [Cu(L)(phen)] were investigated using quantum chemical calculations. The [Cu(L)(phen)] was practically created and developed in computer and assessed with the help of Swiss ADME, allowing for simple parameter analysis. The ligand and its copper complex underwent a molecular docking analysis, which reveals an attraction for the primary protease (Mpro) which is a spike protein for COVID-19. The results were quite positive when the binding affinity and inhibition constants were compared with current antiviral medications. According to the reports, the ligand’s inhibition constant was 5.82 µM and the binding affinity was −7.14 kcal/mol for the H2L molecule (a Schiff base ligand) with Mpro, while the inhibition constant was 0.76 µM and the binding affinity was −6.18 kcal/mol for the complex [Cu(L)(phen)] with Mpro protein. The study is anticipated to provide scientific solutions and be found to be effective against COVID-19 viral suppression as a consequence of the recent issues it has posed throughout the world [99]. In comparison to previously developed antiviral medications, the binding affinity was fairly good. For instance, it was discovered that the ligand and complex binding affinity was higher than the docking results of the drugs chloroquine, hydroxychloroquine, and remdesivir with the Mpro protein (−6.293 kcal/mol, −5.573 kcal/mol, and −6.352 kcal/mol, respectively).

Chemical structure of [Cu(L)(phen)]
Metallized Schiff base tridentate
A Schiff base ligand has a tendency to bind metal ions with a nitrogen atom of the azomethine group [100]. The main motive of this work was to create a Schiff base ligand. Using the crystal-structure inhibitor named UAW247, a molecular docking investigation looked at the produced compounds’ affinity for the major SARS-CoV-2 protease (PDB-ID: 6XBH). The docking studies show the best conformation, free binding energy, and stability of the ligand along with other complexes and target proteins. The compounds in the formularies utilized to treat COVID-19 were predicted in silico to have antiviral activity [101,102,103]. Inhibitor UAW247 used molecular docking research to determine the ligand’s and its metal complexes’ mechanisms of attachment toward the complex’s major protease. The binding models were found to have best confirmations with binding energies of 2.88, 3.38, 3.77, 2.86, 3.31, 3.08, 3.06, and 3.34 Å distances, respectively. In 6XBH docking, many ligands were confirmed for binding like Fe (III), Nickel (III), Copper (II), Zinc (II), and Chromium (III). As per the interaction energies, it was found that Cr (III) complex was highly stable and has lesser binding energies. According to many studies, the ligand was successfully useful in combating coronavirus, and Chromium (III) complex was proven a strong antiviral because of its lower binding energy. This research opens a new door for the future development of coronavirus treatment [104].
Hydroxyguanidine tosylate Schiff base derivatives
1-Amino-3-hydroxyguanidine tosylate-salicylaldehyde Schiff bases are excellent starting points for the creation of novel antiviral medications [105]. When employed at levels as low as 3.2 µM, compound (9a) is one of the varieties of Schiff bases (9a–l, Fig. 14) synthesized from 1-amino-3-hydroxyguanidine tosylate, was found to be especially effective against mouse hepatitis virus, suppressing the half of the growth. Schiff bases (9i–l) obtained from abacavir were synthesized and tested for activity against the virus by Sriram et al. [106]. These substances belong to a brand-new class of abacavir prodrugs. Reverse transcriptase activity can be inhibited by the nucleoside analog abacavir. HIV-1 was strongly inhibited by compounds 9i–l. These Schiff bases generated from Abacavir required a lower effective concentration of 6.0 µM to protect human leukemia cells from the cytotoxic effects of HIV half of its value. Molecule 9d when substituted with H at R1 and 4-CH3C6H at R2 was discovered to be the most effective Schiff base, showing activity against CEM cells at levels greater than 100 µM and being effective at 50 nM, demonstrating its probability as a prime substance for the creation of new medications effective against HIV.

Chemical structures of Compounds 9a–l
Quinolonylacetohydrazides
Quinolines/quinolones and their structural analogs have exhibited inhibitory properties against HIV, human cytomegalovirus (HCMV), SARS coronavirus, Zika virus, Chikungunya virus, hepatitis C virus (HCV), and Ebola virus [107,108,109,110,111,112]. The mechanism of action of antiviral quinolone remains unclear. Specific studies aimed at understanding the nature of drug’s targets at the molecular level indicated that quinolones inhibit viral transcription [113]. Alshammari et al. created a novel of quinolonylacetohydrazides Schiff base derivatives (10a–k). Different spectroscopic techniques, such as NMR (1H, 13C, 15N, 2D), elemental analysis, and mass spectroscopy, were used to determine the chemical structures [114].
Using Autodock 4.2.6, calculations for docking were used to determine the attraction for binding of the synthesized molecules (10a–k; Fig. 15) with the Coronavirus-2 major protease (Mpro) and RNA polymerase (RdRp) with values between −7.0 and −9.7 kcal/mol, compounds (10a–k) have demonstrated strong binding tendencies for the Mpro of the Coronavirus-2. Compound (10a–k) had lower binding affinities for RdRP compared to Mpro, with docking values between −6 and −8 kcal/mol, whereas Remdesivir had affinities value of binding between −8.5 and −5.6 kcal/mol for Mpro and RdRp, respectively. Adenosine analog Remdesivir works well opposing various viruses that have RNA, including severe acute respiratory syndrome (SARS), Middle East Respiratory Syndrome (MERS), and Coronavirus [115,116,117]. It is under clinical development for the treatment of the infection caused by the Ebola virus [118]. Studies reported compound (10d) had the highest ability to bind with Mpro of Coronavirus-2 causing SARS and could create four crucial hydrogen bonds with the amino acids LEU141, SER144, HIS163, and GLU166, with lengths of 2.02, 2.22, 1.83, and 2.07 Å, respectively. To determine, how alternative quinolone derivatives can be utilized as a cure for infection brought on by coronavirus, more research on these chemicals is necessary.

N-substituted-2-quinolonylacetohydrazides (10a–k)
Toward termination and control of the epidemic, researchers from many medical specialties have worked together. Numerous research institutions and pharmaceutical businesses have recently presented 115 vaccine candidates and 249 potential therapies for COVID-19 disorders at various pre-clinical and clinical research stages. Around 330 clinical trials and 249 potential treatments for COVID-19 disorders were ongoing as of April 2020. This pandemic has posed a significant obstacle to the quick development of medications to combat this deadly illness [119]. This requirement has bent the interest of researchers toward metallodrugs, which could offer good opportunities to achieve the desired goals [120, 121]. Micronutrients like Zn, Se, and Vitamin D can intervene to stop the progression of COVID-19 [122, 123]. In addition to regulating the inflammatory response, zinc is known to control the effect of viruses, and bacteria. Along with all, the degree of COVID-19 severity can be altered, for which Schiff base shows itself to be a remarkably good candidate. Furthermore, co-ligands including NO, CO, and H2S may strengthen the coronavirus impact against coronavirus. The interaction with spike protein and stopping RNA replication of coronavirus has a huge impact on the growth of anti-COVID agents [124]. Therefore, the use of Schiff base complexed metals in metallodrug-based design for the treatment of viral infection may be advantageous [125, 126].
Schiff bases are adaptable ligands that interact with several different metal ions. All transition metals, along with lanthanoids, are capable of forming complexes with Schiff bases. Because of their diverse biological characteristics, When Schiff bases made from sulfonamides form a metal complex it shows a specific role in pharmaceutical chemistry. For instance, different biological parameters were revealed by metal complexes of 5-chloro-2-hydroxybenzylidene aminobenzene sulfonamides [127], 4-(2-aminoethyl) benzenesulfonamide, 4-(2-aminomethyl) benzenesulfonamide, and sulfisoxazole [128]. In addition to this, the metal Schiff base complexes formed when various sulfonamides were combined with 5-chlorosalicyladehyde or indole-3-carboxaldehyde have demonstrated antibacterial and antifungal activities [129]. Additionally, carbonic anhydrase, an enzyme containing zinc, is significantly inhibited by Schiff bases [130,131,132,133,134]. There have been numerous Schiff base compounds so far. At the University of Manitoba, Malcolm Xing patented Schiff-base hydrogel as a potential wound treatment [135, 136]. Imine salts which are made from new pyrimidine have been patented to find against the pests that harm animals [137]. Consequently, it has been discovered that Schiff base has a variety of therapeutic properties; ranging from inhibitory action against bacteria, fungus, viruses, malaria, cancer, pests, inflammation, oxidation, and many enzymes [138,139,140].
Conclusion
Schiff bases have thus far been extensively investigated in numerous industrial and pharmacological applications. However, additional research is still needed to fully understand this compound class and its biological function. The formation of metal complexes by Schiff bases was found to be an important area for investigation in this review. Evaluation based on theoretical chemistry is preferred these days before attempting to synthesize a chemical that may ultimately produce useful outcomes. Although there are already analyzed molecules available, the biological properties of these Schiff base-metal complexes that have been made to be further investigated, and new complexes must be made with more features. Analysis of the links between the Schiff bases and the compounds mechanisms of action will be necessary for this field to advance. Additionally, molecular docking studies enable identifying potential binding mechanisms of highly effective drugs easier. The computation of potential receptor and ligand docking modes of novel/compounds is achievable with many molecular docking tools and molecular graphic programs. Pre-clinical and clinical reports are opening the doors of research for exploring the biological activity of Schiff base via several aspects.
References
Spernovasilis N, Tsiodras S, Poulakou G. Emerging and re-emerging infectious diseases: humankind’s companions and competitors. Microorganisms. 2022;10:1–5.
Samudrala PK, Kumar P, Choudhary K. Virology, pathogenesis, diagnosis and in-line treatment of COVID-19. Eur J Pharmacol. 2020;883:173375.
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–33.
Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA. 2020;324:782–93.
Yasuhara J, Kuno T, Takagi H, Sumitomo N. Clinical characteristics of COVID-19 in children: a systematic review. Pediatr Pulmonol. 2020;55:2565–75.
Verma S, Patil VM, Gupta MK. Mutation informatics: SARS-CoV-2 receptor-binding domain of the spike protein. Drug Discov Today. 2022;27:103312.
Fortner A, Schumacher D. First COVID-19 vaccines receiving the US FDA and EMA emergency use authorization. Discoveries. 2021;9:e122.
Hui EK. Reasons for the increase in emerging and re-emerging viral infectious diseases. Microbes Infect. 2006;8:905–16.
Neiderud CJ. How urbanization affects the epidemiology of emerging infectious diseases. Infect Ecol Epidemiol. 2015;24:27060.
Baker RE, Mahmud AS, Miller IF, Rajeev M, Rasambainarivo F, Rice BL, et al. Infectious disease in an era of global change. Nat Rev Microbiol. 2022;20:193–205.
Campbell-Lendrum D, Manga L, Bagayoko M, Sommerfeld J. Climate change and vector-borne diseases: what are the implications for public health research and policy? Philos Trans R Soc Lond B Biol Sci. 2015;5:370.
Caminade C, McIntyre KM, Jones AE. Impact of recent and future climate change on vector-borne diseases. Ann N Y Acad Sci. 2019;1436:157–73.
Tompa DR, Immanuel A, Srikanth S, Kadhirvel S. Trends and strategies to combat viral infections: a review on FDA approved antiviral drugs. Int J Biol Macromol. 2021;172:524–41.
Kausar S, Said Khan F, Ishaq Mujeeb Ur Rehman M, Akram M, Riaz M, Rasool G, et al. A review: mechanism of action of antiviral drugs. Int J Immunopathol Pharmacol. 2021;35:33726557.
Strasfeld L, Chou S. Antiviral drug resistance: mechanisms and clinical implications. Infect Dis Clin North Am. 2010;24:413–37.
Seley-Radtke KL, Yates MK. The evolution of nucleoside analogue antivirals: a review for chemists and non-chemists. Part 1: early structural modifications to the nucleoside scaffold. Antiviral Res. 2018;154:66–86.
Broder S. The development of antiretroviral therapy and its impact on the HIV-1/AIDS pandemic. Antiviral Res. 2010;85:1–18.
Rampersad S, Tennant P. Replication and expression strategies of viruses. Viruses. 2018;30:55–82.
V'kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. 2021;19:155–70.
Razonable RR. Antiviral drugs for viruses other than human immunodeficiency virus. Mayo Clin Proc. 2011;86:1009–26.
Strasfeld L, Chou S. Antiviral drug resistance: mechanisms and clinical implications. Infect Dis Clin North Am. 2010;24:413–37.
Brower ET, Bacha UM, Kawasaki Y, Freire E. Inhibition of HIV-2 protease by HIV-1 protease inhibitors in clinical use. Chem Biol Drug Des. 2008;71:298–305.
Kumar A, Rajput MK, Paliwal D, Yadav A, Chhabra R, Singh S. Genotyping & diagnostic methods for hepatitis C virus: a need of low-resource countries. Indian J Med Res. 2018;147:445–55.
Aissa Larousse J, Trimoulet P, Recordon Pinson P, Tauzin B, Azzouz MM, Ben Mami N, et al. Prevalence of hepatitis C virus (HCV) variants resistant to NS5A inhibitors in naïve patients infected with HCV genotype 1 in Tunisia. Virol J. 2015;6:84.
Chaudhuri S, Symons JA, Deval J. Innovation and trends in the development and approval of antiviral medicines: 1987–2017 and beyond. Antiviral Res. 2018;155:76–88.
Anderson PL, Rower JE. Zidovudine and lamivudine for HIV infection. Clin Med Rev Ther. 2010;2:a2004.
Portsmouth SD, Scott CJ. The renaissance of fixed-dose combinations: combivir. Ther Clin Risk Manag. 2007;3:579–83.
Bhatia HK, Singh H, Grewal N, Natt NK. Sofosbuvir: a novel treatment option for chronic hepatitis C infection. J Pharmacol Pharmacother. 2014;5:278–84.
Asselah T. Sofosbuvir for the treatment of hepatitis C virus. Exp Opin Pharmacother. 2014;15:121–30.
Cha A, Budovich A. Sofosbuvir: a new oral once-daily agent for the treatment of hepatitis C virus infection. P T. 2014;39:345–52.
Bubna AK. Imiquimod—its role in the treatment of cutaneous malignancies. Indian J Pharmacol. 2015;47:354–9.
Jobanputra KS, Rajpal AV, Nagpur NG. Imiquimod. Indian J Dermatol Venereol Leprol. 2006;72:466–9.
Leung DT, Sacks SL. Docosanol: a topical antiviral for herpes labialis. Expert Opin Pharmacother. 2004;5:2567–71.
Paeshuyse J, Dallmeier K, Neyts J. Ribavirin for the treatment of chronic hepatitis C virus infection: a review of the proposed mechanisms of action. Curr Opin Virol. 2011;1:590–8.
Naffouje R, Grover P, Yu H, Sendilnathan A, Wolfe K, Majd N, et al. Anti-tumor potential of IMP dehydrogenase inhibitors: a century-long story. Cancers. 2019;11:1346.
Miyoshi N, Tanabe H, Suzuki T, Saeki K, Hara Y. Applications of a standardized green tea catechin preparation for viral warts and human papilloma virus-related and unrelated cancers. Molecules. 2020;25:2588.
Vardanyan R, Hruby V. Antiviral drugs. Synthesis of best-seller drugs. In: Vardanyan R, Hruby V, editors. Academic Press; 2016. p. 687–736.
Trivella JP, Gutierrez J, Martin P. Dasabuvir: a new direct antiviral agent for the treatment of hepatitis C. Exp Opin Pharmacother. 2015;16:617–24.
James J, Saquinavir S. (Invirase): first protease inhibitor approved–reimbursement, information hotline numbers. AIDS Treat News. 1995;22:237.
Brenner BG, Thomas R, Blanco JL, Ibanescu RI, Oliveira M, Mesplède T, et al. Development of a G118R mutation in HIV-1 integrase following a switch to dolutegravir monotherapy leading to cross-resistance to integrase inhibitors. J Antimicrob Chemother. 2016;71:1948–53.
Anstett K, Brenner B, Mesplede T. HIV drug resistance against strand transfer integrase inhibitors. Retrovirology. 2017;14:36.
Ng TI, Krishnan P, Pilot-Matias T. In vitro antiviral activity and resistance profile of the next-generation hepatitis C virus NS5A inhibitor pibrentasvir. Antimicrob Agents Chemother. 2017;24:61.
Chaudhuri S, Symons JA, Deval J. Innovation and trends in the development and approval of antiviral medicines: 1987–2017 and beyond. Antiviral Res. 2018;155:76–88.
Ismail AA, Kajal A, Bala S, Kamboj S, Sharma N, Saini V. Schiff bases: a versatile pharmacophore. J Catal. 2013;895512:1–14.
Raczuk E, Dmochowska B, Samaszko-Fiertek J, Madaj J. Different Schiff bases-structure, importance, and classification. Molecules. 2022;27:787.
Dhar DN, Taploo CL. Schiff bases and their applications. J Sci Ind Res. 1982;41:501–6.
Przybylski P, Huczynski A, Pyta K, Brzezinski B, Bartl F. Biological properties of Schiff bases and azo derivatives of phenol. Curr Org Chem. 2009;13:124–48.
Aggarwal S, Paliwal D, Kaushik D, Gupta GK, Kumar A. Pyrazole Schiff base hybrids as anti-malarial agents: synthesis, in vitro screening and computational study. Comb Chem High Throughput Screen. 2018;21:194–203.
Bringmann G, Dreyer M, Faber JH, Dalsgaard PW, Staerk D, Jaroszewski JW, et al. Ancistrotanzanine C and related 5,1’and 7,3‘-coupled naphthylisoquinoline alkaloids from ancistrocladus tanzaniensis. J Nat Prod. 2004;67:743–8.
Souza AO, Galetti FCS, Silva CL, Bicalho B, Parma MM, Fonseca SF, et al. Antimycobacterial and cytotoxicity activity of synthetic and natural compounds. Quim Nova. 2007;30:1563–6.
Guo Z, Xing R, Liu S, Zhong Z, Ji X, Wang L, et al. Antifungal properties of Schiff bases of chitosan, N-substituted chitosan and quaternized chitosan. Carbohydr Res. 2007;342:1329–32.
Kalaivani S, Priya NP, Arunachalam S, et al. Schiff bases: facile synthesis, spectral characterization, and biocidal studies. Int J Appl Bio Pharm Tech. 2012;3:219–23.
Radecka-Paryzek W, Pospieszna-Markiewicz I, Kubicki M. Self-assembled two-dimensional salicylaldimine lanthanum (III) nitrate coordination polymer. Inorg Chim Acta. 2007;360:488–96.
Boghaei DM, Askarizadeh E, Bezaatpour A. Synthesis, characterization, spectroscopic and thermodynamic studies of charge transfer interaction of a new water-soluble cobalt (II) Schiff base complex with imidazole. Spectrochim Acta Part A. 2008;69:624–8.
Prashanthi Y, Kiranmai K, Subhashini NJ, Shivaraj. Synthesis, potentiometric and antimicrobial studies on metal complexes of isoxazole Schiff bases. Spectrochim Acta A Mol Biomol Spectrosc. 2008;70:30–5.
Anand P, Patil VM, Sharma VK, et al. Schiff bases: a review on biological insights. Int J Drug Des Discov. 2012;3:851–68.
Deepika P, Vinusha HM, Begum M, Ramu R, Shirahatti PS, Nagendra Prasad MN. 2-methoxy-4-(5-nitropyridin-2-yl)imino)methyl)phenol Schiff base ligand and its Cu(II) and Zn(II) complexes: synthesis, characterization and biological investigations. Heliyon. 2022;8:09648.
Majid SA, Mir JM, Bhat MA, Shalla AH, Pandey A, Hadda TB, et al. A pair of carbazate derivatives as novel Schiff base ligands: DFT and POM theory supported spectroscopic and biological evaluation. J Biomol Struct Dyn. 2022;35751130:1–17.
Ramezani S, Nakhaei A. Synthesis, absorption, and adsorption properties, and DFT calculations of two new palladium (II) complexes of new fluorescence imidazo[4′,5′:3,4]benzo[1,2-c]isoxazole-based Schiff-bases. Inorg Nano Metal Chem. 2021;51:560–8.
Milosavljevic V, Haddad Y, Merlos Rodrigo MA, Moulick A, Polanska H, Hynek D, et al. The zinc-Schiff base-novicidin complex as a potential prostate cancer therapy. PLoS ONE. 2016;11:e0163983.
Li D, Stovall DB, Wang W, Sui G. Advances of zinc signaling studies in prostate cancer. Int J Mol Sci. 2020;21:667.
Alam A, Ali M, Rehman NU, Ullah S, Halim SA, Latif A, et al. Bio-oriented synthesis of novel (S)-flurbiprofen clubbed hydrazone Schiff’s bases for diabetic management: in vitro and in silico studies. Pharmaceuticals. 2022;15:672.
Mahato RK, Debnath A, Das A, Sarkar D, Bhattacharyya S, Biswas B. Supramolecular encapsulation of nanocrystalline Schiff bases into β-cyclodextrin for multifold enrichment of bio-potency. Carbohydr Polym. 2022;291:119614.
Banerjee A, Banerjee K, Sinha A, Das S, Majumder S, Majumdar S, et al. A zinc Schiff base complex inhibits cancer progression both in vivo and in vitro by inducing apoptosis. Env Toxicol Pharmacol. 2017;56:383–92.
Gowdhami B, Manojkumar Y, Vimala RTV, Ramya V, Karthiyayini B, Kadalmani B, et al. Cytotoxic cobalt (III) Schiff base complexes: in vitro anti-proliferative, oxidative stress and gene expression studies in human breast and lung cancer cells. Biometals. 2022;35:67–85.
Elsaman T, Mohamed MS, Eltayib EM, Abdel-Aziz HA, Abdalla AE, Munir MU, et al. Isatin derivatives as broad-spectrum antiviral agents: the current landscape. Med Chem Res. 2022;31:244–73.
Pandeya SN, Sriram D, Nath G, De Clercq E. Synthesis, antibacterial, antifungal and anti-HIV activities of norfloxacin Mannich bases. Eur J Med Chem. 2000;35:249–55.
Pandeya SN, Yogeeswari P, Sriram D, de Clercq E, Pannecouque C, Witvrouw M. Synthesis and screening for the anti-HIV activity of some N-Mannich bases of isatin derivatives. Chemotherapy. 1999;45:192–6.
Pandeya SN, Smitha S, Jyoti M, Sridhar SK. Biological activities of isatin and its derivatives. Acta Pharm. 2005;55:27–46.
Sevinçli ZŞ, Duran GN, Özbil M, Karalı N. Synthesis, molecular modeling and antiviral activity of novel 5-fluoro-1H-indole-2,3-dione 3-thiosemicarbazones. Bioorg Chem. 2020;104:104202.
Francesconi V, Cichero E, Schenone S, Naesens L, Tonelli M. Synthesis and biological evaluation of novel (thio)semicarbazone-based benzimidazoles as antiviral agents against human respiratory viruses. Molecules. 2020;25:1487.
Padmanabhan P, Khaleefathullah S, Kaveri K, Palani G, Ramanathan G, Thennarasu S, et al. Antiviral activity of thiosemicarbazones derived from α-amino acids against Dengue virus. J Med Virol. 2017;89:546–52.
Soraires Santacruz MC, Fabiani M, Castro EF, Cavallaro LV, Finkielsztein LM. Synthesis, antiviral evaluation and molecular docking studies of N4-aryl substituted/unsubstituted thiosemicarbazones derived from 1-indanones as potent anti-bovine viral diarrhea virus agents. Bioorg Med Chem. 2017;25:4055–63.
Pacca CC, Marques RE, Espindola JWP, Filho GBOO, Leite ACL, Teixeira MM, et al. Thiosemicarbazones and phthalyl-thiazoles compounds exert antiviral activity against the yellow fever virus and Saint Louis encephalitis virus. Biomed Pharmacother. 2017;87:381–7.
Glisoni RJ, Castro EF, Cavallaro LV, Moglioni AG, Sosnik A. Complexation of a 1-indanone thiosemicarbazone with hydroxypropyl-β-cyclodextrin enhances its activity against a hepatitis C virus surrogate model. J Nanosci Nanotechnol. 2015;15:4224–8.
Rogolino D, Bacchi A, De Luca L, Rispoli G, Sechi M, Stevaert A, et al. Investigation of the salicylaldehyde thiosemicarbazone scaffold for inhibition of influenza virus PA endonuclease. J Biol Inorg Chem. 2015;20:1109–21.
Pandeya SN, Sriram D, Nath G, DeClercq E. Synthesis, antibacterial, antifungal and anti-HIV activities of Schiff and Mannich bases derived from isatin derivatives and N-[4-(4′-chlorophenyl) thiazol-2-yl] thiosemicarbazide. Eur J Pharm Sci. 1999;9:25–31.
Banerjee D, Yogeeswari P, Bhat P, Thomas A, Srividya M, Sriram D. Novel isatinyl thiosemicarbazones derivatives as potential molecule to combat HIV-TB co-infection. Eur J Med Chem. 2011;46:106–21.
Kaur R, Kumar K. Synthetic and medicinal perspective of quinolines as antiviral agents. Eur J Med Chem. 2021;215:113220.
Patil VM, Singhal S, Masand N. A systematic review on use of aminoquinolines for the therapeutic management of COVID-19: efficacy, safety and clinical trials. Life Sci. 2020;254:117775.
Wang M, Zhang G, Zhao J, Cheng N, Wang Y, Fu Y, et al. Synthesis and antiviral activity of a series of novel quinoline derivatives as anti-RSV or anti-IAV agents. Eur J Med Chem. 2021;214:113208.
Abuelizz HA, Bakheit AH, Marzouk M, El-Senousy WM, Abdellatif MM, Mostafa GAE, et al. Evaluation of some Benzo[g]Quinazoline derivatives as antiviral agents against human rotavirus Wa strain: biological screening and docking study. Curr Issues Mol Biol. 2023;45:2409–21.
Gillis EP, Parcella K, Bowsher M, Cook JH, Iwuagwu C, Naidu BN, et al. Potent long-acting inhibitors targeting the HIV-1 capsid based on a versatile quinazolin-4-one scaffold. J Med Chem. 2023;66:1941–54.
Zhao J, Zhang Y, Wang M, Liu Q, Lei X, Wu M, et al. Quinoline and quinazoline derivatives inhibit viral RNA synthesis by SARS-CoV-2 RdRp. ACS Infect Dis. 2021;7:1535–44.
Kumar KS, Ganguly S, Veerasamy R, De Clercq E. Synthesis, antiviral activity and cytotoxicity evaluation of Schiff bases of some 2-phenyl quinazoline-4(3)H-ones. Eur J Med Chem. 2010;45:5474–9.
Krishnan SK, Ganguly S, Veerasamy R, Jan B. Synthesis, antiviral and cytotoxic investigation of 2-phenyl-3-substituted quinazoline-4(3H)-ones. Eur Rev Med Pharmacol Sci. 2011;15:673–81.
Grenier D, Chen H, Ben Lagha A, Fournier-Larente J, Morin MP. Dual action of myricetin on porphyromonas gingivalis and the inflammatory response of host cells: a promising therapeutic molecule for periodontal diseases. PLoS ONE. 2015;10:e0131758.
Rashed K, Ćirić A, Glamočlija J, Soković M. Antibacterial and antifungal activities of methanol extract and phenolic compounds from Diospyros virginiana L. Ind Crops Prod. 2014;59:210–5.
Ortega JT, Suárez AI, Serrano ML, Baptista J, Pujol FH, Rangel HR. The role of the glycosyl moiety of myricetin derivatives in anti-HIV-1 activity in vitro. AIDS Res Ther. 2017;14:57.
Park S, Kim JI, Lee I, Lee S, Hwang MW, Bae JY, et al. Aronia melanocarpa and its components demonstrate antiviral activity against influenza viruses. Biochem Biophys Res Commun. 2013;440:14–19.
Xianghui R, Hongju Z, Cheng Z, et al. Syntheses and bioactivities of myricetin derivatives containing piperazine acidamide moiety. Chem J Chin Univ. 2018;39:1197–204.
Chobot V, Hadace F. Exploration of pro-oxidant and antioxidant activities of the flavonoid myricetin. Redox Rep. 2011;16:243–7.
Wang X, Wang Z, Sidhu PS, Desai UR, Zhou Q. 6-hydroxyflavone and derivatives exhibit potent anti-inflammatory activity among mono-, di- and polyhydroxylated flavones in kidney mesangial cells. PLoS ONE. 2015;10:e0116409.
Chen Y, Li P, Su S, Chen M, He J, Liu L, et al. Synthesis and antibacterial and antiviral activities of myricetin derivatives containing a 1,2,4-triazole Schiff base. RSC Adv. 2019;40:23045–52.
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–13.
Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, et al. Transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med. 2020;382:1199–207.
van Doremalen N, Bushmaker T, Morris DH, Holbrook MG, Gamble A, Williamson BN, et al. Aerosol and surface stability of SARS-CoV-2 as compared with SARS-CoV-1. N Engl J Med. 2020;382:1564–7.
Das P, Majumder R, Mandal M, Basak P. In-Silico approach for identification of effective and stable inhibitors for COVID-19 main protease (Mpro) from flavonoid-based phytochemical constituents of Calendula officinalis. J Biomol Struct Dyn. 2021;39:6265–80.
Mohan B, Muhammad S, Al‐Sehemi AG, Bharti S, Kumar S, Choudhary M. Synthesis of copper (II) coordination complex, its molecular docking and computational exploration for novel functional properties: a dual approach. Chem Sel. 2021;6:738–45.
Mohan B, Choudhary M. Synthesis, crystal structure, computational study, and anti-virus effect of mixed ligand copper (II) complex with ONS donor Schiff base and 1, 10-phenanthroline. J Mol Struct. 2021;1246:131246.
El-ajaily MM, Sarangi AK, Mohapatra RK, et al. Transition metal complexes of (E)-2((2-hydroxy benzylidene) amino-3-mercapto propanoic acid: XRD, anticancer, molecular modeling and molecular docking studies. Chem Select. 2019;4:9999–10005.
Mohapatra RK, Perekhoda L, Azam M, Suleiman M, Sarangi AK, Semenets A, et al. Computational investigations of three main drugs and their comparison with synthesized compounds as potent inhibitors of SARS-CoV-2 main protease (Mpro): DFT, QSAR, molecular docking, and in silico toxicity analysis. J King Saud Univ. 2021;33:101315.
Xu Y, Meng X. Molecular simulation elaborating the mechanism of 1β-hydroxy alantolactone inhibiting ubiquitin-conjugating enzyme UbcH5s. Sci Rep. 2020;10:141.
Duru Kamaci U, Kamaci M, Peksel A. Thermally stable Schiff base, and its metal complexes: molecular docking and protein binding studies. J Fluoresc. 2017;27:805–17.
Mohamed GG, Omar MM, Ahmed YM. Metal complexes of tridentate Schiff base: synthesis, characterization, biological activity and molecular docking studies with COVID-19 protein receptor. J Inorg Gen Chem. 2021;647:2201–18.
Wang PH, Keck JG, Lien EJ, Lai MM. Design, synthesis, testing, and quantitative structure-activity relationship analysis of substituted salicylaldehyde Schiff bases of 1-amino-3-hydroxy guanidine tosylate as new antiviral agents against coronavirus. J Med Chem. 1990;33:608–14.
Sriram D, Yogeeswari P, Myneedu NS, Saraswat V. Abacavir prodrugs: microwave-assisted synthesis and their evaluation of anti-HIV activities. Bioorg Med Chem Lett. 2006;16:2127–9.
Luthra P, Liang J, Pietzsch CA, Khadka S, Edwards MR, Wei S, et al. A high throughput screen identifies benzoquinoline compounds as inhibitors of Ebola virus replication. Antivir Res. 2018;150:193–201.
Loregian A, Mercorelli B, Muratore G, Sinigalia E, Pagni S, Massari S, et al. The 6-aminoquinolone WC5 inhibits human cytomegalovirus replication at an early stage by interfering with the transactivating activity of viral immediate-early protein. Antimicrob Agents Chemother. 2010;54:1930–40.
Plantone D, Koudriavtseva T. Current and future use of chloroquine and hydroxychloroquine in infectious, immune, neoplastic, and neurological diseases: a mini-review. Clin Drug Investig. 2018;38:653–71.
Barbosa-Lima G, Moraes AM, Araujo ADS, da Silva ET, de Freitas CS, Vieira YR, et al. 2,8-bis(trifluoromethyl)quinoline analogs show improved anti-Zika virus activity, compared to mefloquine. Eur J Med Chem.2017;127:334–40.
Al-Bari MA. Chloroquine analogues in drug discovery: new directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J Antimicrob Chemother. 2015;70:1608–21.
Delvecchio R, Higa LM, Pezzuto P, Valadao AL, Garcez PP, Monteiro FL, et al. Chloroquine, an endocytosis blocking agent, inhibits Zika virus infection in different cell models. Viruses.2016;8:322
Richter S, Parolin C, Palumbo M, Palù G. Antiviral properties of quinolone-based drugs. Curr Drug Target Infect Disord. 2004;4:111–6.
Alshammari MB, Ramadan M, Aly AA, El-Sheref EM, Bakht MA, Ibrahim M, et al. Synthesis of potentially new schiff bases of N-substituted-2-quinolonylacetohydrazides as anti-COVID-19 agents. J Mol Struc. 2021;1230:129649.
da Silva CM, da Silva DL, Modolo LV, Alves RB, de Resende MA, Martins CVB, et al. Schiff bases: a short review of their antimicrobial activities. J Adv Res. 2011;2:1–8.
Du YX, Chen XP. Favipiravir: pharmacokinetics and concerns about clinical trials for 2019-nCoV. Infection. 2020;108:242–7.
Sheahan TP, Sims AC, Graham RL, Menachery VD, Gralinski LE, Case JB, et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med. 2017;9:396.
Mulangu S, Dodd LE, Davey RT Jr, Tshiani Mbaya O, Proschan M, Mukadi D, et al. A randomized, controlled trial of Ebola virus disease therapeutics. N Engl J Med. 2019;381:2293–303.
Dhama K, Khan S, Tiwari R, Sircar S, Bhat S, Malik YS, et al. Coronavirus disease 2019-COVID-19. Clin Microbiol Rev. 2020;33:e00028–20.
Cirri D, Pratesi A, Marzo T, Messori L. Metallo therapeutics for COVID-19. Exploiting metal-based compounds for the discovery of new antiviral drugs. Expert Opin Drug Discov. 2020;16:39–46.
Mjos KD, Orvig C. Metallodrugs in medicinal inorganic chemistry. Chem Rev. 2014;114:4540–63.
Alexander J, Tinkov A, Strand TA, Alehagen U, Skalny A, Aaseth J. Early nutritional interventions with zinc, selenium and vitamin D for raising anti-viral resistance Against progressive COVID-19. Nutrients. 2020;12:2358.
Pechlivanidou E, Vlachakis D, Tsarouhas K, Panidis D, Tsitsimpikou C, Darviri C, et al. The prognostic role of micronutrient status and supplements in COVID-19 outcomes: a systematic review. Food Chem Toxicol. 2022;162:112901.
Skalny AV, Rink L, Ajsuvakova OP, Aschner M, Gritsenko VA, Alekseenko SI, et al. Zinc and respiratory tract infections: perspectives for COVID-19 (Review). Int J Mol Med. 2020;46:17–26.
Mir JM, Maurya RC. Nitric oxide as a therapeutic option for COVID-19 treatment: a concise perspective. New J Chem. 2021;45:1774–84.
Mir JM, Maurya RC. Nitric oxide boosters as defensive agents against COVID-19 infection: an opinion. J Biomol Struct Dyn. 2022;40:4285–91.
Boopathi S, Poma AB, Kolandaivel P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J Biomol Struct Dyn. 2021;39:3409–18.
Mir JM, Majid SA, Shalla AH. Enhancement of Schiff base biological efficacy by metal coordination and introduction of metallic compounds as anticovid candidates: a simple overview. Rev Inorg Chem. 2021;41:199–211.
Abd El-Lateef HM, El-Dabea T, Khalaf MM, Abu-Dief AM. Development of metal complexes for treatment of coronaviruses. Int J Mol Sci. 2022;23:6418.
Chohan ZH, Hernandes MZ, Sensato FR, Moreira DR, Pereira VR, Neves JK, et al. Sulfonamide–metal complexes endowed with potent anti-Trypanosoma cruzi activity. J Enz Inh Med Chem. 2014;29:230–6.
Chohan ZH, Shad HA, Supuran CT. Synthesis, characterization and biological studies of sulfonamide Schiff’s bases and some of their metal derivatives. J Enz Inh Med Chem. 2012;27:58–68.
Chohan ZH, Shad HA. Sulfonamide-derived compounds and their transition metal complexes: synthesis, biological evaluation and X-ray structure of 4-bromo-2-[(E)-{4-[(3,4-dimethyl isoxazole-5 yl)sulfamoyl]phenyl} iminiomethyl] phenolate. Appl Organomet Chem. 2011;25:591–600.
Chohan ZH, Shad HA. Metal-based new sulfonamides: design, synthesis, antibacterial, antifungal, and cytotoxic properties. J Enzyme Inhib Med Chem. 2012;27:403–12.
Chohan ZH, Youssoufi MH, Jarrahpour A. Identification of antibacterial and antifungal pharmacophore sites for potent bacteria and fungi inhibition: indolenyl sulfonamide derivatives. Eur J Med Chem. 2010;45:1189–99.
Eroglu E. Some QSAR studies for a group of sulfonamide Schiff base as carbonic anhydrase CA II inhibitors. Int J Mol Sci. 2008;9:181–97.
Malcolm X. (EN) Schiff-based aldehydic hyaluronic acid-chitosan hydrogel compositions and uses thereof (FR). 2014;WO/2014/161085:PCT/CA2014/050311.
Narine A, Dickhaut J, Kaiser F. Substituted mesoionic imine compounds for combating animal pests. 2014;WO2014033244A3.
Hameed A, Rashida MA, Uroos M. Schiff bases in medicinal chemistry: a patent review (2010-2015). Expert Opin Ther Pat. 2017;27:63–79.
Login CC, Bâldea I, Tiperciuc B. A novel thiazolyl Schiff base: antibacterial and antifungal effects and in vitro oxidative stress modulation on human endothelial cells. Oxid Med Cell Longev. 2019;10:1607903.
Acknowledgements
The authors (SK and VMP) recognize the substantial assistance of the KIET Group of Institutions, India for this study. One of the authors (MRI) is funded by the intramural research program of the National Institute on Alcohol Abuse and Alcoholism (NIAAA), National Institute of Health (NIH), Bethesda, USA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kaushik, S., Paliwal, S.K., Iyer, M.R. et al. Promising Schiff bases in antiviral drug design and discovery. Med Chem Res 32, 1063–1076 (2023). https://doi.org/10.1007/s00044-023-03068-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-023-03068-0