
Abstract
Integrase strand transfer inhibitors (INSTIs) are the newest class of antiretroviral drugs to be approved for treatment and act by inhibiting the essential HIV protein integrase from inserting the viral DNA genome into the host cell’s chromatin. Three drugs of this class are currently approved for use in HIV-positive individuals: raltegravir (RAL), elvitegravir (EVG), and dolutegravir (DTG), while cabotegravir (CAB) and bictegravir (BIC) are currently in clinical trials. RAL and EVG have been successful in clinical settings but have relatively low genetic barriers to resistance. Furthermore, they share a high degree of cross-resistance, which necessitated the development of so-called second-generation drugs of this class (DTG, CAB, and BIC) that could retain activity against these resistant variants. In vitro selection experiments have been instrumental to the clinical development of INSTIs, however they cannot completely recapitulate the situation in an HIV-positive individual. This review summarizes and compares all the currently available information as it pertains to both in vitro and in vivo selections with all five INSTIs, and the measured fold-changes in resistance of resistant variants in in vitro assays. While the selection of resistance substitutions in response to RAL and EVG bears high similarity in patients as compared to laboratory studies, there is less concurrence regarding the “second-generation” drugs of this class. This highlights the unpredictability of HIV resistance to these inhibitors, which is of concern as CAB and BIC proceed in their clinical development.
Similar content being viewed by others

Background
Since the beginning of the pandemic, HIV/AIDS has claimed the lives of over 35 million people, and approximately 35 million individuals are currently infected [1]. Highly active antiretroviral therapy (HAART) has transformed a positive HIV diagnosis from a former death sentence into a chronic, manageable disease. However, no cure yet exists for HIV and patients must remain on therapy for the entirety of their lives which makes the development of drug resistance in the virus a real concern. In fact, drug resistance has been documented for every currently available drug class in patients [2]. This makes the continued study of the mechanisms of HIV drug resistance and novel therapeutics a top priority for HIV scientists worldwide.
The reverse transcriptase (RT) enzyme of HIV is highly error-prone, introducing mutations into the genome at a rate of 1.4 × 10−5 mutation per base pair, per replication cycle [3]. This high mutation rate allows for the generation of multiple different viruses within an infected individual, sometimes referred to as “quasi-species.” If one of these quasi-species has a mutation that provides a selective advantage for replication in the presence of antiretrovirals (ARVs), it will out-compete other viral forms to become the dominant species [4].
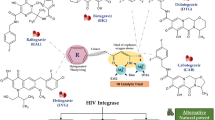
The integrase (IN) enzyme catalyses the insertion of the viral DNA (vDNA) into the host’s genome through two catalytic actions: 3′ processing and strand transfer. In the cytoplasm, IN self-associates into tetramers on the newly reverse transcribed vDNA, where it catalyzes the removal of the last two nucleotides from the 3′ ends of both strands [5]. In addition, IN can spontaneously form larger multimers that are stabilized by the addition of allosteric integrase inhibitors, and reciprocally destabilized in the presence of DNA [6,7,8,9,10]. After nuclear translocation, IN associates with lens epithelium-derived growth factor (LEDGF)/p75 and is directed to sites of open chromatin, where it will initiate strand transfer, i.e. the nucleophilic attack of the 3′ hydroxyl groups on the viral DNA on the nucleotide backbone of the host DNA. The integration process is completed by host gap-repair machinery, resulting in a 5 base-pair repeat that flanks each end of the viral DNA [11].
The integrase strand transfer inhibitor (INSTI) class of antiretroviral drugs is the latest to be approved for treatment of HIV-positive individuals. As their name suggest, INSTIs inhibit the second step catalyzed by IN, i.e. strand transfer, through competitive binding to the enzyme’s active site. INSTIs not only displace the 3′ end of the vDNA from the active site, but also chelate the divalent cation (Mg2+ or Mn2+) that is required for IN enzymatic activity [12]. There are currently three INSTIs approved for the treatment of HIV infection: raltegravir (RAL), elvitegravir (EVG), and dolutegravir (DTG) [13]. Cabotegravir (CAB) and bictegravir (BIC) are newer INSTIs currently in clinical trials [14, 15].
Although highly efficacious in the management of HIV, both RAL and EVG are susceptible to virological failure through the development of resistance mutations. What is more, most of the changes that cause resistance to RAL also cause resistance to EVG, and vice versa [16]. This is, however, not the case with DTG. Not only does DTG appear to have a higher genetic barrier to resistance than either of the other two drugs, it has not yet been shown to definitively select for any resistance-associated changes in treatment-naïve patients [17]. Although two reports of potential emergence of resistance in individuals treated with DTG in first line therapy recently appeared, baseline IN was not sequenced in one of these cases, nor did the supposed-emergent mutation lead to persistent virological failure while DTG was still being used together with an optimized background regimen containing rilpivirine (RPV), an NNRTI with a modest genetic barrier to resistance [18]. Specifically, initial TDF/FTC/DTG treatment was supplemented with ritonavir-boosted darunavir following failure; the latter drug was subsequently substituted with RPV for reason of diffuse erythoderma. The second case reported transient emergence of a T97A substitution that did not confer any resistance on its own against DTG in vitro and was not observed at subsequent time points [19]. Although it cannot be excluded that unambiguously documented cases of emergent resistance mutations against first-line DTG will eventually be reported, it is expected that this will be rare. This is supported by the fact that despite dolutegravir being used by tens of thousands treatment-naïve individuals in Europe and the USA, the abovementioned two cases are the only known reports of potential primary de novo resistance against this drug. There have also been rare cases of treatment failure with resistance mutations in treatment-experienced but INSTI-naïve patients, and, in this setting, DTG has most often selected for the novel resistance substitution R263K [20]. Other substitutions at residues E92, Q148 and N155, have been reported when DTG monotherapy was used in treatment-experienced patients.
Primary resistance substitutions arise first in response to INSTI drug pressure and cause a decrease in susceptibility at the expense of viral fitness, most often through alterations to the enzyme’s active site where the inhibitors bind [16, 21]. Secondary resistance substitutions arise after continued drug pressure and usually act to alleviate the negative effects of primary mutations, and may also increase levels of INSTI resistance [22, 23]. Some of these secondary changes are specific to a certain primary resistance pathway, but many may be selected after several different primary mutations.
Pre-clinical and in vitro studies have been instrumental in the evaluation of novel therapeutic agents for the treatment of HIV infection, however they do not always accurately predict clinical outcomes for patients. Laboratory viral strains and cell lines, although excellent scientific tools, can never recapitulate in vivo human infections with 100% accuracy. In this review, we compare both the in vitro selection and antiviral activity reported for drugs of the INSTI class with the analogous data available from treated patients to assess the predictive power of in vitro studies for INSTI clinical outcomes.
Raltegravir
In 2004 a group of researchers at Merck & Co. reported on the efficacy of the diketo acid (DKA)-based lead compound L-870812 against simian immunodeficiency virus (SIV) in infected rhesus macaques [24]. This led to the approval of the first INSTI, raltegravir, in 2007 for treatment-experienced AIDS patients with multidrug resistance, and two years later for treatment-naïve individuals as well [25, 26]. In the 10 years since its first approval, RAL has been shown to be well tolerated in the vast majority of patients, although it is does require twice daily dosing. It displays a modest genetic barrier to resistance, with the most common mutational pathways consisting of changes at positions Y143, Q148, and N155 [27].
The substitutions in IN that have been selected both in cell culture and in treated patients with RAL are summarized in Tables 1, 2, 3 and 4, and the measured fold-changes in resistance to INSTIs for the different combinations of substitutions in each of the major pathways are displayed in Tables 5, 6, 7 and 8. There were sporadic reports of changes at positions T66 and E92 with RAL, mostly in vitro (Table 1). However, the main resistance pathways that have been reported as selected both in vitro and in vivo with RAL are Y143, Q148, and N155 (Tables 2 and 3). As shown in Table 5, these pathways only provide low to moderate changes in RAL susceptibility, which helps to explain their limited selection. Broadly, there is a high concurrence between the resistance pathways selected both in tissue culture under RAL pressure and in patients undergoing therapy with RAL.
The Y143 pathway is specific to RAL; it is not selected by any other INSTI (Table 2). This specificity was explained when the crystal structure of prototype foamy virus IN in complex with RAL was solved to show that residue 143 interacts directly with the oxadiazole ring of RAL, forming a π − π stacking interaction that is abrogated when this position is mutated [70, 71]. This is in contrast to changes at positions 148 and 155, which disturb the geometry of the IN active site, thereby disrupting the binding of INSTIs [23]. Interestingly, levels of resistance conferred by changes at position 143 are variable depending on the specific amino acid change involved (Table 6). This phenomenon has been extensively studied and been shown to be true also for the fitness of these variants [46, 53]. The most common substitutions at this position are Y143C and Y143R. They cause low to moderate reductions in RAL susceptibility on their own, but the addition of secondary mutations leads to the levels of resistance being greatly increased (Table 6; see also [53]). Although the Y143R pathway provides the highest levels of RAL resistance, it also has a higher genetic barrier to selection, as the amino acid change requires two nucleotide mutations whereas Y143C/H/S only require one change. Both Y143C and Y143H may transition to Y143R and so these substitutions may reflect an intermediary rather than a final selection [53].
There were numerous reports of emergence of substitutions involving position Q148 in response to RAL pressure both in tissue culture and in patient-derived samples, and this is in line with previous studies (Table 3 and [38, 72, 73]). Early in treatment, substitutions at position Q148 may be seen in isolation, but as they impart a severe fitness cost, they are rapidly compensated for by various secondary resistance mutations as shown in Table 3. In growth competition assays, single Q148X mutants showed a significant reduction in fitness compared with WT viruses in the absence of RAL, whereas viruses containing these same mutations outcompeted WT in the presence of the INSTI. Likewise, as secondary mutations are added to Q148X-containing viruses, they outcompete the single mutants whether RAL is present or not [74]. We can see in Table 7 that as secondary resistance mutations accumulate, the fold-changes in susceptibility to RAL greatly increase as well. This helps to explain why the Q148 pathway dominates in RAL selections.
A N155H pathway is also selected with moderate frequency both in vitro and in vivo in response to RAL (Table 3; see also [16, 75]). This single substitution appears to have a less deleterious effect in terms of the replication of the virus, and as such is mostly only co-reported with one or infrequently two additional secondary substitutions [74, 75]. This observation is also supported by the data collected in Table 7: only one or two additional substitutions are required to provide high levels of RAL resistance. In a study examining the evolution of INSTI resistance substitutions in treated patients over time, it was found that mutations at position 155 were often selected earlier during therapy, and then gradually replaced by changes at position 143 or 148 [76]. This may be due to higher levels of resistance conferred by the 143/148 pathways as compared to N155H.
Elvitegravir
EVG is a monoketo acid derivative that also demonstrated high specificity for inhibition of HIV IN strand transfer reactions [77]. EVG was developed by Gilead Sciences and approved for use in HIV infected individuals in 2012 [26]. Because EVG is processed by the cytochrome p450 enzyme CYP3A4/5, it needs to be co-formulated with cobicistat to boost plasma concentrations. This permits once daily dosing of EVG [78].
It is evident from both Tables 3 and 7 that RAL and EVG share both the Q148 and N155H major resistance pathways, although from our literature review it appears that the latter is most often reported for RAL. The data compiled in Table 7 clearly show that the levels of resistance conferred by the various mutations of the N155 pathway for EVG are at or above those for RAL and the selection of a greater number of secondary resistance mutations in addition to N155H in patients treated with RAL may be a reflection of this difference. This pathway has also been extensively characterized in terms of EVG resistance by several groups [28, 62, 75]. Although some mutants containing the Y143 pathway displayed moderate levels of resistance against EVG (Table 6), this is most likely due to the secondary resistance mutations present.
The T66 and E92 pathways are predominately selected by EVG, although they do display increased likelihood of selection in vitro as opposed to in vivo (Table 1). As was reported with RAL, there is a dynamism to the temporal selection of EVG resistance mutations that may help to explain these differences. The T66 and E92 pathways are selected earlier under EVG pressure, and are gradually replaced by other pathways, such as Q148X [36, 49, 69]. As can be seen in Tables 5 and 7, substitutions in the T66 and E92 pathways provide moderate levels of resistance to EVG, while the Q148 and N155 pathways provide larger fold-changes in resistance. Many more secondary mutations were also reported for T66X and E92X in vivo than in vitro. While this could be a reporting bias, it could also be reflective of the different requirements for replication under EVG pressure in tissue culture versus in a human host. The S147G pathway is also sometimes selected by EVG in tissue culture and in the clinic but only confers moderate to high levels of INSTI resistance when combined with two or more other resistance substitutions (Tables 4 and 8).
One of the major limitations for EVG has been that it shares a clinically significant resistance pathway at position 148 with RAL [28]. Just as is the case for RAL, significant selection both in vitro and in vivo of the Q148 pathway in response to EVG was observed (Table 3), and this pathway also conferred significant reductions in EVG susceptibility (Table 7). Thus, substitutions at position 148, and the accompanying secondary changes, predominate selections with RAL and EVG.
Second-generation INSTIs
The relatively low genetic barrier and high degree of cross-resistance among the so called “first-generation” INSTIs RAL and EVG spurred research into the chase for “second-generation” drugs of this class, aimed at retaining efficacy against RAL/EVG resistant variants. There have been four candidate second-generation INSTIs to date. DTG, manufactured by ViiV-Healthcare and GlaxoSmithKline, was approved in 2013 for both treatment-naïve and—experienced patients and is the only second-generation INSTI to be approved to date [79]. MK-2048 showed potent activity against most RAL/EVG resistant variants and did not select for the same substitutions in tissue culture studies but its clinical development was halted due to poor pharmacokinetics. Both CAB and BIC are promising and both are currently in advanced clinical trials [15, 19, 50, 80].
The resistance profile of DTG has been extensively characterized during the past few years (reviewed in [16, 26, 81]). DTG has been shown to have a longer binding half-life to HIV IN than either RAL or EVG, which may help to explain why it maintains activity against most first-generation INSTI resistant variants [61].
It can be seen in Tables 2, 3 and 4 that DTG only sporadically selects for common first-generation INSTI resistance substitutions and that resistance to this compound most often derives from the DTG-specific R263K substitution. Although R263K was seen rarely as a secondary EVG resistance substitution prior to the approval of DTG, it has since been selected by the latter in tissue culture selection studies, and in four INSTI-naïve but treatment-experienced patients undergoing DTG therapy [14, 17, 33, 35, 69, 75, 82]. What is notable about this substitution is that, unlike those discussed for RAL and EVG, the R263K substitution only results in low levels of resistance to DTG. It also has a significant impact on the fitness of the virus, and has yet to be compensated by secondary resistance mutations in tissue culture selections [33, 83]. It has been reported that patients with non-B subtype viruses selected for N155 pathway mutations in response to DTG (Table 3; see also [35]). In vitro, subtype B viruses harbouring this mutation are sensitive to DTG, so these selections may reflect a subtype-specific effect [57].
There are fewer reports on the resistance patterns of CAB, a novel INSTI under development at GlaxoSmithKline. CAB was developed concomitantly with DTG and shares most of its structure; it has the potential to be formulated as a long acting injectable for both pre-exposure prophylaxis and treatment of HIV infection [84]. In the LATTE clinical trial, one patient in the CAB arm did develop a mutation in the Q148 pathway, which suggests that this second-generation INSTI may select for the same mutations as RAL and EVG [15]. In in vitro selection studies, CAB has selected for changes at positions 146 and 153 that could also be selected in the presence of EVG and DTG, respectively (Table 4).
BIC is a more recent second-generation INSTI and as such there is less information available in regard to resistance against this drug. Tissue culture selection studies with BIC performed by Gilead Sciences selected for the R263K substitution in IN, and at an earlier week than occurred with DTG in parallel studies [14]. The fact that BIC selected for R263K may be related to structural similarities between this drug and DTG. So far, the results of a phase II trial of BIC at 48 weeks in HIV infected individuals have been reported and, as yet, there has been no detection of resistance-associated changes in IN [19].
As is shown in Table 7 and been reported previously, the Q148 pathway seems to confer the highest fold-changes in resistance to second-generation INSTIs upon the addition of at least two secondary mutations, and has been selected in a patient failing CAB-based therapy [14, 15, 29, 60]. However, the fold-changes in susceptibility to second-generation INSTIs are almost always below those that have been observed with RAL and EVG. Table 3 notes that the Q148 pathway has yet to be selected for in vitro or in vivo by DTG or BIC, suggesting that decreases in susceptibility with Q148 may only be worrisome in INSTI-experienced patients with Q148 mutations; neither compound appears to select for this pathway on their own when they are used in combination with RTI. Of the other first-generation INSTI resistance pathways, only changes associated with position 155 appear to have any effect on the susceptibility of HIV to DTG, CAB, or BIC, and these changes are relatively low-level (Table 7).
Experienced patients and selections using resistant viruses
Due to the high degree of cross-resistance between RAL and EVG, neither may be used as salvage therapy for patients failing the other. DTG, however, has been used in select cases in patients failing RAL- or EVG-based therapies. The results of these studies are summarized in Table 9. This strategy was first explored in the VIKING phase II clinical trial, in which 27 highly treatment-experienced patients with drug resistant viruses were switched from their RAL-containing regimens to DTG. At week 24 69% of participants achieved undetectable viral loads, as compared to 88% in treatment-naïve patients in the SPRING-2 trial [17, 43]. Patients with Q148X+ two additional secondary mutations fared the worst in this study, which is in line with the in vitro susceptibility assays reported in Table 7. Interestingly, patients who experienced failure with DTG in this study did so through the accumulation of several RAL/EVG resistance mutations in addition to those present at baseline, and not through the selection of DTG-specific mutations such as R263K. It has been reported that the presence of various first-generation INSTI resistance mutations may be incompatible with R263K [57].
There have been other, infrequent reports of RAL-experienced patients failing DTG salvage therapy. One such patient failed RAL and subsequently DTG with the only known resistance-associated change being E157Q [85]. Although the authors found that the IN derived from this patient was highly resistant to both INSTIs, others showed that the laboratory viral strain NL4.3 containing E157Q was hyper-sensitive to DTG, highlighting the variability that background changes and polymorphisms may introduce into analyses [59]. A different patient failed DTG with a combination of the Q148 and N155 pathways, which is reminiscent of the combinations found in some VIKING patients [45, 86]. A RAL-experienced patient was also reported to have failed DTG with N155 pathway mutations [64].
A G118R mutation was selected by MK-2048 drug pressure, as well as by DTG in certain non-B subtypes of HIV-1 [33, 80, 87]. G118R was also shown to be present in two patients, one previously treated with EVG and the other with RAL, during failure on DTG monotherapy [34]. The selection of G118R in certain settings and not others is most likely due to codon usage at position 118; although rare in certain subtypes of HIV-1, the presence of the GGA (G) codon is favourable to a transition to AGA (R).
Analogous to these sporadic reports of INSTI-experienced patients subsequently failing DTG-based therapies, tissue culture selection studies have used HIV with INSTI resistance mutations to mimic the situation seen in some patient populations. The results of these studies are summarized in Table 10. When viruses with mutations of the Q148 pathway are placed under DTG pressure, they select for additional secondary resistance mutations, similar to what was reported in the patients in the VIKING clinical trial. As expected, viruses containing other primary INSTI resistance substitutions can acquire secondary INSTI resistance mutations under continued selection with either RAL or EVG (Table 10; see also [52]). After 30 weeks of DTG selection, viruses that contained E92Q or N155H at baseline selected for R263K [57]. In another study that lasted only 6 weeks, additional substitutions to E92Q, Y143R or N155H were not detected [52].
Attempts to select further changes to DTG-specific resistance pathways have yielded more nuanced results. R263K-containing viruses are sensitive to RAL and unable to select for additional changes under pressure by this compound unless secondary mutations such as H51Y or E138K are also present. R263K, however, readily selects for EVG resistance (Table 10). This is in line with previous data that identified R263K as a secondary EVG resistance mutation [69]. G118R readily selects both primary and secondary resistance mutations under pressure with all three INSTIs, with or without the initial presence of additional secondary mutations. Lastly, the DTG resistance mutation H51Y facilitates the emergence of other resistance-associated substitutions in selections with all three INSTIs, in agreement with its previously characterized role as a secondary change [67].
Discussion and conclusions
For the first-generation INSTIs RAL and EVG, the resistance pathways that were selected in vitro were generally predictive of the mutations that would arise in patients failing therapy with these drugs, although the frequencies of primary and/or secondary mutations selected may vary depending on whether in vitro or in vivo results are considered. The picture is not so straight-forward for the newer INSTIs. We have very limited information on resistance against newer INSTIs such as CAB and BIC. Since CAB has selected for the Q148 pathway in vivo, it is possible that the clinical resistance profile of this INSTI will resemble that of the first-generation INSTIs. However, since CAB did not select RAL or EVG resistance pathways in tissue culture, the situation may be more complex. BIC has so far selected for the same substitutions in vitro as DTG and this suggests that this compound might also select for similar pathways as DTG in patients.
Although the most common substitution selected in vitro by DTG, R263K, has also been the most common pathway seen in patients failing DTG, other aspects of tissue culture selection studies with DTG have not been as predictive. One-third of all INSTI-naïve patients reported to have failed DTG to date have done so with the N155 pathway (2/6), even though the N155H substitution alone does not cause large fold-changes in DTG resistance in in vitro assays (Table 7). Part of the explanation may be that the majority of selection studies and in vitro INSTI resistance testing has been performed with subtype B HIV-1. Indeed, the two patients who developed N155H in response to DTG both had non-B viruses. In culture selection studies, non-B viruses predominantly selected the G118R substitution and N155H was not observed [33]. This shows a divergence between the in vivo and in vitro resistance profile of DTG.
In the case of the two INSTI-experienced patients who failed DTG monotherapy with the G118R mutation, the effect of polymorphisms and subtype differences on the selection of INSTI resistance may be important, as the GGA glycine codon can more easily transition to AGA, explaining why arginine is present at a higher frequency in non-B subtypes [34]. Many of the secondary resistance mutations listed in Tables 1, 2, 3, 4 and 9 occur at positions that are considered polymorphic, i.e. dependent on subtype and geographical distribution; this complicates the nature of the selection of these polymorphic changes as a function of INSTI exposure. Any description of transmitted INSTI drug resistance (for a review of the effect of subtype diversity and polymorphisms on HIV-1 INSTI resistance see [37]) must also be thereby complicated. A more accurate portrait of patterns of second-generation INSTI resistance mutations in non-B subtypes, which is increasingly important as access to these medications increases in developing countries, will require that selection studies be conducted more frequently with non-B primary isolates [90].
The Q148 pathway remains the dominant route to INSTI resistance, regardless of the individual compound used. All second-generation INSTIs show lower activity against HIV as secondary mutations of this pathway accumulate, and the results of INSTI-experienced patients on DTG therapy suggest that once present, the sequential selection of further mutations in this pathway will result in greatly diminished susceptibilities to this compound (Table 9; see also [86]). Even though the Q148 substitutions may not be selected by either DTG or BIC in vivo, they remain important for the future of these compounds.
Predictions of which pathways will be important for resistance to second-generation INSTIs, unlike first-generation INSTIs, may not easily follow from in vitro studies. If resistance to these compounds turns out to be due to random genetic changes that are not easily identified, genotyping of patient-derived viruses may not be able to predict treatment outcome when these compounds are employed [37, 86]. Perhaps, the high genetic barrier to resistance of DTG will force HIV to evolve along different mutational pathways in vitro versus in vivo, depending on subtype and baseline polymorphisms.
References
World Health Organisation t. HIV/AIDS fact sheet 2016. 2016.
Wainberg MA, Zaharatos GJ, Brenner BG. Development of antiretroviral drug resistance. N Engl J Med. 2011;365(7):637–46.
Smyth RP, Davenport MP, Mak J. The origin of genetic diversity in HIV-1. Virus Res. 2012;169(2):415–29.
Larder BA, Kemp SD. Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT). Science. 1989;246(4934):1155–8.
Maertens GN, Hare S, Cherepanov P. The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature. 2010;468(7321):326–9.
Borrenberghs D, Dirix L, De Wit F, Rocha S, Blokken J, De Houwer S, et al. Dynamic oligomerization of integrase orchestrates HIV nuclear entry. Sci Rep. 2016;6:36485.
Deng N, Hoyte A, Mansour YE, Mohamed MS, Fuchs JR, Engelman AN, et al. Allosteric HIV-1 integrase inhibitors promote aberrant protein multimerization by directly mediating inter-subunit interactions: structural and thermodynamic modeling studies. Protein Sci. 2016;25(11):1911–7.
Deprez E, Tauc P, Leh H, Mouscadet JF, Auclair C, Brochon JC. Oligomeric states of the HIV-1 integrase as measured by time-resolved fluorescence anisotropy. Biochemistry. 2000;39(31):9275–84.
Guiot E, Carayon K, Delelis O, Simon F, Tauc P, Zubin E, et al. Relationship between the oligomeric status of HIV-1 integrase on DNA and enzymatic activity. J Biol Chem. 2006;281(32):22707–19.
Passos DO, Li M, Yang R, Rebensburg SV, Ghirlando R, Jeon Y, et al. Cryo-EM structures and atomic model of the HIV-1 strand transfer complex intasome. Science. 2017;355(6320):89–92.
Van Maele B, Busschots K, Vandekerckhove L, Christ F, Debyser Z. Cellular co-factors of HIV-1 integration. Trends Biochem Sci. 2006;31(2):98–105.
Delelis O, Carayon K, Saib A, Deprez E, Mouscadet JF. Integrase and integration: biochemical activities of HIV-1 integrase. Retrovirology. 2008;5:114.
Quashie PK, Mesplede T, Wainberg MA. HIV drug resistance and the advent of integrase inhibitors. Curr Infect Dis Rep. 2013;15(1):85–100.
Tsiang M, Jones GS, Goldsmith J, Mulato A, Hansen D, Kan E, et al. Antiviral activity of bictegravir (GS-9883), a novel potent HIV-1 integrase strand transfer inhibitor with an improved resistance profile. Antimicrob Agents Chemother. 2016;60(12):7086–97.
Margolis DA, Brinson CC, Smith GH, de Vente J, Hagins DP, Eron JJ, et al. Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral-naive adults with HIV-1 infection (LATTE): a randomised, phase 2b, dose-ranging trial. Lancet Infect Dis. 2015;15(10):1145–55.
Quashie PK, Mesplede T, Wainberg MA. Evolution of HIV integrase resistance mutations. Curr Opin Infect Dis. 2013;26(1):43–9.
Raffi F, Jaeger H, Quiros-Roldan E, Albrecht H, Belonosova E, Gatell JM, et al. Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naive adults with HIV-1 infection (SPRING-2 study): 96 week results from a randomised, double-blind, non-inferiority trial. Lancet Infect Dis. 2013;13(11):927–35.
Fulcher AJ, Du Y, Sun R, Landovitz RJ. Emergence of integrase resistance mutations during initial therapy with TDF/FTC/DTG. In: Conference on retroviruses and opportunistic infections; February 13–16. Seattle; 2017.
Sax PE, DeJesus E, Crofoot G, Ward D, Benson P, Dretler R, et al. Bictegravir versus dolutegravir, each with emtricitabine and tenofovir alafenamide, for initial treatment of HIV-1 infection: a randomised, double-blind, phase 2 trial. Lancet HIV. 2017;4(4):e154–60.
Cahn P, Pozniak AL, Mingrone H, Shuldyakov A, Brites C, Andrade-Villanueva JF, et al. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV: week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet. 2013;382(9893):700–8.
Shafer RW. Rationale and uses of a public HIV drug-resistance database. J Infect Dis. 2006;194(Suppl 1):S51–8.
Delelis O, Malet I, Na L, Tchertanov L, Calvez V, Marcelin AG, et al. The G140S mutation in HIV integrases from raltegravir-resistant patients rescues catalytic defect due to the resistance Q148H mutation. Nucleic Acids Res. 2009;37(4):1193–201.
Hare S, Vos AM, Clayton RF, Thuring JW, Cummings MD, Cherepanov P. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc Natl Acad Sci USA. 2010;107(46):20057–62.
Hazuda DJ, Young SD, Guare JP, Anthony NJ, Gomez RP, Wai JS, et al. Integrase inhibitors and cellular immunity suppress retroviral replication in rhesus macaques. Science. 2004;305(5683):528–32.
FDA notifications. FDA approves raltegravir for HIV-1 treatment-naive patients. AIDS Alert. 2009;24(9):106–7.
Metifiot M, Marchand C, Pommier Y. HIV integrase inhibitors: 20-year landmark and challenges. Adv Pharmacol. 2013;67:75–105.
Malet I, Delelis O, Valantin MA, Montes B, Soulie C, Wirden M, et al. Mutations associated with failure of raltegravir treatment affect integrase sensitivity to the inhibitor in vitro. Antimicrob Agents Chemother. 2008;52(4):1351–8.
Blanco JL, Varghese V, Rhee SY, Gatell JM, Shafer RW. HIV-1 integrase inhibitor resistance and its clinical implications. J Infect Dis. 2011;203(9):1204–14.
Yoshinaga T, Kobayashi M, Seki T, Miki S, Wakasa-Morimoto C, Suyama-Kagitani A, et al. Antiviral characteristics of GSK1265744, an HIV integrase inhibitor dosed orally or by long-acting injection. Antimicrob Agents Chemother. 2015;59(1):397–406.
Lataillade M, Chiarella J, Kozal MJ. Natural polymorphism of the HIV-1 integrase gene and mutations associated with integrase inhibitor resistance. Antivir Ther. 2007;12(4):563–70.
Malet I, Gimferrer Arriaga L, Artese A, Costa G, Parrotta L, Alcaro S, et al. New raltegravir resistance pathways induce broad cross-resistance to all currently used integrase inhibitors. J Antimicrob Chemother. 2014;69(8):2118–22.
Blanco Arevalo JL, Whitlock GG. Dolutegravir: an exciting new kid on the block. Expert Opin Pharmacother. 2014;15(4):573–82.
Quashie PK, Mesplede T, Han YS, Oliveira M, Singhroy DN, Fujiwara T, et al. Characterization of the R263K mutation in HIV-1 integrase that confers low-level resistance to the second-generation integrase strand transfer inhibitor dolutegravir. J Virol. 2012;86(5):2696–705.
Brenner BG, Thomas R, Blanco JL, Ibanescu RI, Oliveira M, Mesplede T, et al. Development of a G118R mutation in HIV-1 integrase following a switch to dolutegravir monotherapy leading to cross-resistance to integrase inhibitors. J Antimicrob Chemother. 2016;71(7):1948–53.
Underwood M, Deanda F, Dorey D, Hightower GK, Wang R, Griffith S, et al. Resistance post week 48 in ART-experienced, integrase inhibitor-naïve subjects with dolutegravir (DTG) vs. raltegravir (RAL) in SAILING (ING111762). In: Proceedings of the 13th European HIV and hepatitis workshop; June 3–5, 2015. Barcelona; 2015.
Shimura K, Kodama E, Sakagami Y, Matsuzaki Y, Watanabe W, Yamataka K, et al. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137). J Virol. 2008;82(2):764–74.
Han YS, Mesplede T, Wainberg MA. Differences among HIV-1 subtypes in drug resistance against integrase inhibitors. Infect Genet Evol. 2016;46:286–91.
Cooper DA, Steigbigel RT, Gatell JM, Rockstroh JK, Katlama C, Yeni P, et al. Subgroup and resistance analyses of raltegravir for resistant HIV-1 infection. N Engl J Med. 2008;359(4):355–65.
Wainberg MA, Mesplede T, Quashie PK. The development of novel HIV integrase inhibitors and the problem of drug resistance. Curr Opin Virol. 2012;2(5):656–62.
Kobayashi M, Nakahara K, Seki T, Miki S, Kawauchi S, Suyama A, et al. Selection of diverse and clinically relevant integrase inhibitor-resistant human immunodeficiency virus type 1 mutants. Antivir Res. 2008;80(2):213–22.
Goethals O, Van Ginderen M, Vos A, Cummings MD, Van Der Borght K, Van Wesenbeeck L, et al. Resistance to raltegravir highlights integrase mutations at codon 148 in conferring cross-resistance to a second-generation HIV-1 integrase inhibitor. Antivir Res. 2011;91(2):167–76.
Codoner FM, Pou C, Thielen A, Garcia F, Delgado R, Dalmau D, et al. Dynamic escape of pre-existing raltegravir-resistant HIV-1 from raltegravir selection pressure. Antivir Res. 2010;88(3):281–6.
Eron JJ, Clotet B, Durant J, Katlama C, Kumar P, Lazzarin A, et al. Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING Study. J Infect Dis. 2013;207(5):740–8.
Varghese V, Pinsky BA, Smith DS, Klein D, Shafer RW. Q148N, a novel integrase inhibitor resistance mutation associated with low-level reduction in elvitegravir susceptibility. AIDS Res Hum Retroviruses. 2016;32(7):702–4.
Malet I, Thierry E, Wirden M, Lebourgeois S, Subra F, Katlama C, et al. Combination of two pathways involved in raltegravir resistance confers dolutegravir resistance. J Antimicrob Chemother. 2015;70(10):2870–80.
Delelis O, Thierry S, Subra F, Simon F, Malet I, Alloui C, et al. Impact of Y143 HIV-1 integrase mutations on resistance to raltegravir in vitro and in vivo. Antimicrob Agents Chemother. 2010;54(1):491–501.
Wirden M, Simon A, Schneider L, Tubiana R, Malet I, Ait-Mohand H, et al. Raltegravir has no residual antiviral activity in vivo against HIV-1 with resistance-associated mutations to this drug. J Antimicrob Chemother. 2009;64(5):1087–90.
Santos JR, Blanco JL, Masia M, Gutierrez F, Perez-Elias MJ, Iribarren JA, et al. Virological failure to raltegravir in Spain: incidence, prevalence and clinical consequences. J Antimicrob Chemother. 2015;70(11):3087–95.
Goethals O, Clayton R, Van Ginderen M, Vereycken I, Wagemans E, Geluykens P, et al. Resistance mutations in human immunodeficiency virus type 1 integrase selected with elvitegravir confer reduced susceptibility to a wide range of integrase inhibitors. J Virol. 2008;82(21):10366–74.
Van Wesenbeeck L, Rondelez E, Feyaerts M, Verheyen A, Van der Borght K, Smits V, et al. Cross-resistance profile determination of two second-generation HIV-1 integrase inhibitors using a panel of recombinant viruses derived from raltegravir-treated clinical isolates. Antimicrob Agents Chemother. 2011;55(1):321–5.
Canducci F, Marinozzi MC, Sampaolo M, Boeri E, Spagnuolo V, Gianotti N, et al. Genotypic/phenotypic patterns of HIV-1 integrase resistance to raltegravir. J Antimicrob Chemother. 2010;65(3):425–33.
Seki T, Suyama-Kagitani A, Kawauchi-Miki S, Miki S, Wakasa-Morimoto C, Akihisa E, et al. Effects of raltegravir or elvitegravir resistance signature mutations on the barrier to dolutegravir resistance in vitro. Antimicrob Agents Chemother. 2015;59(5):2596–606.
Huang W, Frantzell A, Fransen S, Petropoulos CJ. Multiple genetic pathways involving amino acid position 143 of HIV-1 integrase are preferentially associated with specific secondary amino acid substitutions and confer resistance to raltegravir and cross-resistance to elvitegravir. Antimicrob Agents Chemother. 2013;57(9):4105–13.
Munir S, Thierry E, Malet I, Subra F, Calvez V, Marcelin AG, et al. G118R and F121Y mutations identified in patients failing raltegravir treatment confer dolutegravir resistance. J Antimicrob Chemother. 2015;70(3):739–49.
Quashie PK, Oliviera M, Veres T, Osman N, Han YS, Hassounah S, et al. Differential effects of the G118R, H51Y, and E138K resistance substitutions in different subtypes of HIV integrase. J Virol. 2015;89(6):3163–75.
Liang J, Mesplede T, Oliveira M, Anstett K, Wainberg MA. The combination of the R263K and T66I resistance substitutions in HIV-1 integrase is incompatible with high level viral replication and the development of high-level drug resistance. J Virol. 2015;85(22):11269–74.
Anstett K, Mesplede T, Oliveira M, Cutillas V, Wainberg MA. Dolutegravir resistance mutation R263K cannot coexist in combination with many classical integrase inhibitor resistance substitutions. J Virol. 2015;89(8):4681–4.
Anstett K, Fusco R, Cutillas V, Mesplede T, Wainberg MA. Dolutegravir-selected HIV-1 containing the N155H and R263K resistance substitutions does not acquire additional compensatory mutations under drug pressure that lead to higher-level resistance and increased replicative capacity. J Virol. 2015;89(20):10482–8.
Anstett K, Cutillas V, Fusco R, Mesplede T, Wainberg MA. Polymorphic substitution E157Q in HIV-1 integrase increases R263K-mediated dolutegravir resistance and decreases DNA binding activity. J Antimicrob Chemother. 2016;71(8):2083–8.
Kobayashi M, Yoshinaga T, Seki T, Wakasa-Morimoto C, Brown KW, Ferris R, et al. In Vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob Agents Chemother. 2011;55(2):813–21.
Hightower KE, Wang R, Deanda F, Johns BA, Weaver K, Shen Y, et al. Dolutegravir (S/GSK1349572) exhibits significantly slower dissociation than raltegravir and elvitegravir from wild-type and integrase inhibitor-resistant HIV-1 integrase-DNA complexes. Antimicrob Agents Chemother. 2011;55(10):4552–9.
Abram ME, Hluhanich RM, Goodman DD, Andreatta KN, Margot NA, Ye L, et al. Impact of primary elvitegravir resistance-associated mutations in HIV-1 integrase on drug susceptibility and viral replication fitness. Antimicrob Agents Chemother. 2013;57(6):2654–63.
Cutillas V, Mesplede T, Anstett K, Hassounah S, Wainberg MA. The R262K substitution combined with H51Y in HIV-1 subtype B integrase confers low-level resistance against dolutegravir. Antimicrob Agents Chemother. 2015;59(1):310–6.
Hardy I, Brenner B, Quashie P, Thomas R, Petropoulos C, Huang W, et al. Evolution of a novel pathway leading to dolutegravir resistance in a patient harbouring N155H and multiclass drug resistance. J Antimicrob Chemother. 2015;70(2):405–11.
Mesplede T, Osman N, Wares M, Quashie PK, Hassounah S, Anstett K, et al. Addition of E138K to R263K in HIV integrase increases resistance to dolutegravir, but fails to restore activity of the HIV integrase enzyme and viral replication capacity. J Antimicrob Chemother. 2014;69(10):2733–40.
Wares M, Mesplede T, Quashie PK, Osman N, Han Y, Wainberg MA. The M50I polymorphic substitution in association with the R263K mutation in HIV-1 subtype B integrase increases drug resistance but does not restore viral replicative fitness. Retrovirology. 2014;11(1):7–14.
Mesplede T, Quashie PK, Osman N, Han Y, Singhroy DN, Lie Y, et al. Viral fitness cost prevents HIV-1 from evading dolutegravir drug pressure. Retrovirology. 2013;10:22–8.
Mesplede T, Quashie PK, Wainberg MA. Resistance to HIV integrase inhibitors. Curr Opin HIV AIDS. 2012;7(5):401–8.
Margot NA, Hluhanich RM, Jones GS, Andreatta KN, Tsiang M, McColl DJ, et al. In vitro resistance selections using elvitegravir, raltegravir, and two metabolites of elvitegravir M1 and M4. Antivir Res. 2012;93(2):288–96.
Krishnan L, Li X, Naraharisetty HL, Hare S, Cherepanov P, Engelman A. Structure-based modeling of the functional HIV-1 intasome and its inhibition. Proc Natl Acad Sci USA. 2010;107(36):15910–5.
Metifiot M, Vandegraaff N, Maddali K, Naumova A, Zhang X, Rhodes D, et al. Elvitegravir overcomes resistance to raltegravir induced by integrase mutation Y143. Aids. 2011;25(9):1175–8.
Malet I, Delelis O, Soulie C, Wirden M, Tchertanov L, Mottaz P, et al. Quasispecies variant dynamics during emergence of resistance to raltegravir in HIV-1-infected patients. J Antimicrob Chemother. 2009;63(4):795–804.
Fransen S, Gupta S, Danovich R, Hazuda D, Miller M, Witmer M, et al. Loss of raltegravir susceptibility by human immunodeficiency virus type 1 is conferred via multiple nonoverlapping genetic pathways. J Virol. 2009;83(22):11440–6.
Hu Z, Kuritzkes DR. Effect of raltegravir resistance mutations in HIV-1 integrase on viral fitness. J Acquir Immune Defic Syndr. 2010;55(2):148–55.
Hurt CB, Sebastian J, Hicks CB, Eron JJ. Resistance to HIV integrase strand transfer inhibitors among clinical specimens in the United States, 2009–2012. Clin Infect Dis. 2014;58(3):423–31.
Fransen S, Gupta S, Frantzell A, Petropoulos CJ, Huang W. Substitutions at amino acid positions 143, 148, and 155 of HIV-1 integrase define distinct genetic barriers to raltegravir resistance in vivo. J Virol. 2012;86(13):7249–55.
Sato M, Motomura T, Aramaki H, Matsuda T, Yamashita M, Ito Y, et al. Novel HIV-1 integrase inhibitors derived from quinolone antibiotics. J Med Chem. 2006;49(5):1506–8.
Zolopa AR, Berger DS, Lampiris H, Zhong L, Chuck SL, Enejosa JV, et al. Activity of elvitegravir, a once-daily integrase inhibitor, against resistant HIV Type 1: results of a phase 2, randomized, controlled, dose-ranging clinical trial. J Infect Dis. 2010;201(6):814–22.
Greener BN, Patterson KB, Prince HM, Sykes CS, Adams JL, Dumond JB, et al. Dolutegravir pharmacokinetics in the genital tract and colorectum of HIV-negative men after single and multiple dosing. J Acquir Immune Defic Syndr. 2013;64(1):39–44.
Bar-Magen T, Sloan RD, Donahue DA, Kuhl BD, Zabeida A, Xu H, et al. Identification of novel mutations responsible for resistance to MK-2048, a second-generation HIV-1 integrase inhibitor. J Virol. 2010;84(18):9210–6.
Thierry E, Deprez E, Delelis O. Different pathways leading to integrase inhibitors resistance. Front Microbiol. 2016;7:2165.
Vavro C, Palumbo P, Wiznia A, Alvero C, Graham B, Fenton T, et al. Evolution of HIV-1 integrase following selection of R263K with further dolutegravir treatment: a case report from the P1093 study. In: Proceedings of the 8th international AIDS society (IAS) conference. Vancouver; 2015.
Wainberg MA, Han YS, Mesplede T. Might dolutegravir be part of a functional cure for HIV? Can J Microbiol. 2016;62(5):375–82.
Whitfield T, Torkington A, van Halsema C. Profile of cabotegravir and its potential in the treatment and prevention of HIV-1 infection: evidence to date. HIV AIDS (Auckl). 2016;8:157–64.
Danion F, Belissa E, Peytavin G, Thierry E, Lanternier F, Scemla A, et al. Non-virological response to a dolutegravir-containing regimen in a patient harbouring a E157Q-mutated virus in the integrase region. J Antimicrob Chemother. 2015;70(6):1921–3.
Castagna A, Maggiolo F, Penco G, Wright D, Mills A, Grossberg R, et al. Dolutegravir in antiretroviral-experienced patients with raltegravir- and/or elvitegravir-resistant HIV-1: 24-week results of the phase III VIKING-3 study. J Infect Dis. 2014;210(3):354–62.
Quashie PK, Mesplede T, Han YS, Veres T, Osman N, Hassounah S, et al. Biochemical analysis of the role of G118R-linked dolutegravir drug resistance substitutions in HIV-1 integrase. Antimicrob Agents Chemother. 2014;58(1):633.
Oliveira M, Mesplede T, Moisi D, Ibanescu RI, Brenner B, Wainberg MA. The dolutegravir R263K resistance mutation in HIV-1 integrase is incompatible with the emergence of resistance against raltegravir. AIDS. 2015;29(17):2255–60.
Oliveira M, Mesplede T, Quashie PK, Moisi D, Wainberg MA. Resistance mutations against dolutegravir in HIV integrase impair the emergence of resistance against reverse transcriptase inhibitors. AIDS. 2014;28(6):813–9.
Brenner B, Wainberg MA. We need to use the best antiretroviral drugs worldwide to prevent HIV drug resistance. AIDS. 2016;30(17):2725–7.
Authors’ contributions
KA performed the literature review, prepared the tables, and wrote the first draft of the manuscript, all under the guidance of TM. BB, TM and MAW reviewed and edited the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This review is dedicated to the memory of Dr. Mark Wainberg, our friend and mentor, whose dedication to the cause of those affected by HIV will continue to inspire us and elicit our admiration.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Funding
MAW was supported by grants from the Canadian Institutes of Health Research (CIHR). KA is the recipient of a doctoral studentship from CIHR.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Anstett, K., Brenner, B., Mesplede, T. et al. HIV drug resistance against strand transfer integrase inhibitors. Retrovirology 14, 36 (2017). https://doi.org/10.1186/s12977-017-0360-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12977-017-0360-7