
Abstract
Huntington’s disease (HD) is a hereditary and fatal disease causing profound neurodegeneration. Deficits in cerebral energy and neurotransmitter metabolism have been suggested to play a central role in the neuronal dysfunction and death associated with HD. The branched-chain amino acids (BCAAs), leucine, isoleucine and valine, are important for cerebral nitrogen homeostasis, neurotransmitter recycling and can be utilized as energy substrates in the tricarboxylic acid (TCA) cycle. Reduced levels of BCAAs in HD have been validated by several reports. However, it is still unknown how cerebral BCAA metabolism is regulated in HD. Here we investigate the metabolism of leucine and isoleucine in the R6/2 mouse model of HD. Acutely isolated cerebral cortical and striatal slices of control and R6/2 mice were incubated in media containing 15N- or 13C-labeled leucine or isoleucine and slice extracts were analyzed by gas chromatography–mass spectrometry (GC–MS) to determine isotopic enrichment of derived metabolites. Elevated BCAA transamination was found from incubations with [15N]leucine and [15N]isoleucine, in both cerebral cortical and striatal slices of R6/2 mice compared to controls. Metabolism of [U-13C]leucine and [U-13C]isoleucine, entering oxidative metabolism as acetyl CoA, was maintained in R6/2 mice. However, metabolism of [U-13C]isoleucine, entering the TCA cycle as succinyl CoA, was elevated in both cerebral cortical and striatal slices of R6/2 mice, suggesting enhanced metabolic flux via this anaplerotic pathway. To support the metabolic studies, expression of enzymes in the BCAA metabolic pathway was assessed from a proteomic resource. Several enzymes related to BCAA metabolism were found to exhibit augmented expression in the R6/2 brain, particularly related to isoleucine metabolism, suggesting an increase in the BCAA metabolic machinery. Our results show that the capacity for cerebral BCAA metabolism, predominantly of isoleucine, is amplified in the R6/2 brain and indicates that perturbations in cerebral BCAA homeostasis could have functional consequences for HD pathology.






Similar content being viewed by others
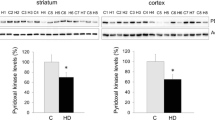
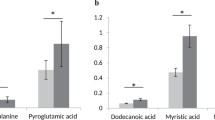

References
Bates GP et al (2015) Huntington disease. Nat Rev Dis Primers 1:15005
Ross CA, Tabrizi SJ (2011) Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10(1):83–98
MacDonald ME et al (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72(6):971–983
Browne SE, Beal MF (2004) The energetics of Huntington’s disease. Neurochem Res 29(3):531–546
Mochel F, Haller RG (2011) Energy deficit in Huntington disease: why it matters. J Clin Invest 121(2):493–499
Acuna AI et al (2013) A failure in energy metabolism and antioxidant uptake precede symptoms of Huntington’s disease in mice. Nat Commun 4:2917
Kim J et al (2010) Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum Mol Genet 19(20):3919–3935
Mochel F et al (2012) Early alterations of brain cellular energy homeostasis in Huntington disease models. J Biol Chem 287(2):1361–1370
Powers WJ et al (2007) Selective defect of in vivo glycolysis in early Huntington’s disease striatum. Proc Natl Acad Sci U S A 104(8):2945–2949
Camandola S, Mattson MP (2017) Brain metabolism in health, aging, and neurodegeneration. EMBO J 36(11):1474–1492
Yudkoff M (1997) Brain metabolism of branched-chain amino acids. Glia 21(1):92–98
Sperringer JE, Addington A, Hutson SM (2017) Branched-chain amino acids and brain metabolism. Neurochem Res 42(6):1697–1709
Conway ME, Hutson SM (2016) BCAA metabolism and NH3 homeostasis. Adv Neurobiol 13:99–132
Wang TJ et al (2011) Metabolite profiles and the risk of developing diabetes. Nat Med 17(4):448–453
Ruiz HH et al (2016) Increased susceptibility to metabolic dysregulation in a mouse model of Alzheimer’s disease is associated with impaired hypothalamic insulin signaling and elevated BCAA levels. Alzheimers Dement 12(8):851–861
Novarino G et al (2012) Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science 338(6105):394–397
Tarlungeanu DC et al (2016) Impaired amino acid transport at the blood brain barrier is a cause of autism spectrum disorder. Cell 167(6):1481.e18–1494.e18
Perry TL et al (1969) Plasma-aminoacid levels in Huntington’s chorea. Lancet 1(7599):806–808
Perry TL, Hansen S, Lesk D (1972) Plasma amino acid levels in children of patients with Huntington’s chorea. Neurology 22(1):68–70
Mochel F et al (2007) Early energy deficit in Huntington disease: identification of a plasma biomarker traceable during disease progression. PLoS One 2(7):e647
Mochel F et al (2011) Validation of plasma branched chain amino acids as biomarkers in Huntington disease. Arch Neurol 68(2):265–267
Graham SF et al (2016) Metabolic signatures of Huntington’s disease (HD): (1)H NMR analysis of the polar metabolome in post-mortem human brain. Biochim Biophys Acta 1862(9):1675–1684
Cheng ML et al (2016) Metabolic disturbances in plasma as biomarkers for Huntington’s disease. J Nutr Biochem 31:38–44
Adanyeguh IM et al (2015) Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology 84(5):490–495
Mochel F (2017) Triheptanoin for the treatment of brain energy deficit: a 14-year experience. J Neurosci Res 95(11):2236–2243
Sonnewald U (2014) Glutamate synthesis has to be matched by its degradation—where do all the carbons go? J Neurochem 131(4):399–406
Skotte NH et al (2018) Integrative characterization of the R6/2 mouse model of Huntington’s disease reveals dysfunctional astrocyte metabolism. Cell Rep 23(7):2211–2224
Mangiarini L et al (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87(3):493–506
Andrew SE et al (1994) A CCG repeat polymorphism adjacent to the CAG repeat in the Huntington disease gene: implications for diagnostic accuracy and predictive testing. Hum Mol Genet 3(1):65–67
Andersen JV et al (2017) Improved cerebral energetics and ketone body metabolism in db/db mice. J Cereb Blood Flow Metab 37(3):1137–1147
McNair LF et al (2017) Metabolic characterization of acutely isolated hippocampal and cerebral cortical slices using [U-13C]glucose and [1,2-13C]acetate as substrates. Neurochem Res 42(3):810–826
Smith PK et al (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150(1):76–85
Walls AB et al (2014) Metabolic mapping of astrocytes and neurons in culture using stable isotopes and gas chromatography-mass spectrometry (GC–MS). In: Hirrlinger J, Waagepetersen HS (eds) Brain energy metabolism. Neuromethods, vol 90. Humana Press, New York
Andersen JV et al (2017) Alterations in cerebral cortical glucose and glutamine metabolism precedes amyloid plaques in the APPswe/PSEN1dE9 mouse model of Alzheimer’s disease. Neurochem Res 42(6):1589–1598
Aldana BI et al (2017) Characterization of energy and neurotransmitter metabolism in cortical glutamatergic neurons derived from human induced pluripotent stem cells: a novel approach to study metabolism in human neurons. Neurochem Int 106:48–61
Behrens PF et al (2002) Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain 125(Pt 8):1908–1922
Tkac I et al (2007) Neurochemical changes in Huntington R6/2 mouse striatum detected by in vivo 1H NMR spectroscopy. J Neurochem 100(5):1397–1406
Tsang TM et al (2006) Metabolic characterization of the R6/2 transgenic mouse model of Huntington’s disease by high-resolution MAS 1H NMR spectroscopy. J Proteome Res 5(3):483–492
Zacharoff L et al (2012) Cortical metabolites as biomarkers in the R6/2 model of Huntington’s disease. J Cereb Blood Flow Metab 32(3):502–514
Patassini S et al (2015) Identification of elevated urea as a severe, ubiquitous metabolic defect in the brain of patients with Huntington’s disease. Biochem Biophys Res Commun 468(1–2):161–166
Yudkoff M et al (1983) [15 N] leucine as a source of [15 N] glutamate in organotypic cerebellar explants. Biochem Biophys Res Commun 115(1):174–179
Kanamori K, Ross BD, Kondrat RW (1998) Rate of glutamate synthesis from leucine in rat brain measured in vivo by 15 N NMR. J Neurochem 70(3):1304–1315
Skotte NH et al (2017) Palmitoylation of caspase-6 by HIP14 regulates its activation. Cell Death Differ 24(3):433–444
Huang K et al (2010) Palmitoylation and function of glial glutamate transporter-1 is reduced in the YAC128 mouse model of Huntington disease. Neurobiol Dis 40(1):207–215
Perluigi M et al (2005) Proteomic analysis of protein expression and oxidative modification in r6/2 transgenic mice: a model of Huntington disease. Mol Cell Proteomics 4(12):1849–1861
Islam MM et al (2007) A novel branched-chain amino acid metabolon. Protein-protein interactions in a supramolecular complex. J Biol Chem 282(16):11893–11903
Cole JT (2015) Metabolism of BCAAs. In: Rajendram R, Preedy VR, Patel VB (eds) Branched chain amino acids in clinical nutrition. Humana Press, New York
Brosnan JT, Brosnan ME (2006) Branched-chain amino acids: enzyme and substrate regulation. J Nutr 136(1 Suppl):207s–211s
Karasinska JM, Hayden MR (2011) Cholesterol metabolism in Huntington disease. Nat Rev Neurol 7(10):561–572
Valenza M et al (2007) Progressive dysfunction of the cholesterol biosynthesis pathway in the R6/2 mouse model of Huntington’s disease. Neurobiol Dis 28(1):133–142
Hull J et al (2012) Distribution of the branched chain aminotransferase proteins in the human brain and their role in glutamate regulation. J Neurochem 123(6):997–1009
Bak LK, Schousboe A, Waagepetersen HS (2006) The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem 98(3):641–653
Walls AB et al (2015) The glutamine-glutamate/GABA cycle: function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem Res 40(2):402–409
Lim RG, Salazar LL, Wilton DK, King AR, Stocksdale JT, Sharifabad D, Lau AL, Stevens B, Reidling JC, Winokur ST, Casale MS (2017) Developmental alterations in Huntington’s disease neural cells and pharmacological rescue in cells and mice. Nat Neurosci 20(5):648–660
Johansen ML et al (2007) The metabolic role of isoleucine in detoxification of ammonia in cultured mouse neurons and astrocytes. Neurochem Int 50(7–8):1042–1051
Gluud LL et al (2017) Branched-chain amino acids for people with hepatic encephalopathy. Cochrane Database Syst Rev 5:cd001939
Bak LK et al (2013) Role of branched chain amino acids in cerebral ammonia homeostasis related to hepatic encephalopathy. Metab Brain Dis 28(2):209–215
Skene DJ et al (2017) Metabolic profiling of presymptomatic Huntington’s disease sheep reveals novel biomarkers. Sci Rep 7:43030
Chiang MC et al (2007) Dysregulation of C/EBPalpha by mutant Huntingtin causes the urea cycle deficiency in Huntington’s disease. Hum Mol Genet 16(5):483–498
Ott P, Clemmesen O, Larsen FS (2005) Cerebral metabolic disturbances in the brain during acute liver failure: from hyperammonemia to energy failure and proteolysis. Neurochem Int 47(1–2):13–18
Schousboe A et al (2014) Glutamate metabolism in the brain focusing on astrocytes. Adv Neurobiol 11:13–30
Lerchundi R et al (2015) NH4(+) triggers the release of astrocytic lactate via mitochondrial pyruvate shunting. Proc Natl Acad Sci USA 112(35):11090–11095
Leke R et al (2011) Detoxification of ammonia in mouse cortical GABAergic cell cultures increases neuronal oxidative metabolism and reveals an emerging role for release of glucose-derived alanine. Neurotox Res 19(3):496–510
Zwingmann C (2007) The anaplerotic flux and ammonia detoxification in hepatic encephalopathy. Metab Brain Dis 22(3–4):235–249
Tefera TW et al (2016) Triheptanoin protects motor neurons and delays the onset of motor symptoms in a mouse model of amyotrophic lateral sclerosis. PLoS One 11(8):e0161816
Acknowledgements
JVA would like to express his sincere gratitude to The Scholarship of Peter & Emma Thomsen for personal financial support (JVA). Furthermore, this project is financially supported by The Foundation of Aase and Ejnar Danielsen (10-002028) and the Augustinus Foundation (17-4115) (both Grants to JVA).
Author information
Authors and Affiliations
Contributions
JVA performed the slice incubation experiments and GC–MS analysis. NHS analyzed the proteomic resource. BIAG performed the HPLC analysis. AN bred and genotyped the mice. JVA, NHS and HSW wrote the manuscript. All authors have contributed critical intellectual content and have approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no potential conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Andersen, J.V., Skotte, N.H., Aldana, B.I. et al. Enhanced cerebral branched-chain amino acid metabolism in R6/2 mouse model of Huntington’s disease. Cell. Mol. Life Sci. 76, 2449–2461 (2019). https://doi.org/10.1007/s00018-019-03051-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03051-2