
Abstract
Tau neuronal and glial pathologies drive the clinical presentation of Alzheimer’s disease and related human tauopathies. There is a growing body of evidence indicating that pathological tau species can travel from cell to cell and spread the pathology through the brain. Throughout the last decade, physiological and pathological tau have become attractive targets for AD therapies. Several therapeutic approaches have been proposed, including the inhibition of protein kinases or protein-3-O-(N-acetyl-beta-D-glucosaminyl)-L-serine/threonine Nacetylglucosaminyl hydrolase, the inhibition of tau aggregation, active and passive immunotherapies, and tau silencing by antisense oligonucleotides. New tau therapeutics, across the board, have demonstrated the ability to prevent or reduce tau lesions and improve either cognitive or motor impairment in a variety of animal models developing neurofibrillary pathology. The most advanced strategy for the treatment of human tauopathies remains immunotherapy, which has already reached the clinical stage of drug development. Tau vaccines or humanised antibodies target a variety of tau species either in the intracellular or extracellular spaces. Some of them recognise the amino-terminus or carboxy-terminus, while others display binding abilities to the proline-rich area or microtubule binding domains. The main therapeutic foci in existing clinical trials are on Alzheimer’s disease, progressive supranuclear palsy and non-fluent primary progressive aphasia. Tau therapy offers a new hope for the treatment of many fatal brain disorders. First efficacy data from clinical trials will be available by the end of this decade.
Similar content being viewed by others
Introduction
Tau protein is considered to be one of the most peculiar proteins in the central nervous system. It is located in several cell compartments, including the axon, dendrites, nucleus, nucleolus, cell membrane and synapses [310]. However, tau is also present in the interstitial fluid [284, 370], and can pass into cerebrospinal fluid (CSF), where it is found at concentrations of 10-25 pg/ml (pT181-tau) or 300-400 pg/ml (tau) [28, 29, 248]. In physiological conditions, extracellular tau may enter neurons either via a dynamin-mediated endocytic mechanism or by classical endocytosis [95]. In neurodegenerative tauopathy, diseased modified tau can propagate along neuroanatomically connected brain areas via multiple mechanisms and spread tau pathology throughout the brain [231].
Tau belongs to the group of natively disordered proteins, which exist in a highly flexible, unfolded structural state, largely devoid of well-defined secondary and tertiary structure, although they are able to fold after binding to targets [329]. The highly flexible structure of tau protein allows interaction with multiple partners, suggesting its involvement in numerous signalling pathways [308]. The dark side of its structural repertoire is its ability to interact with other tau molecules to form oligomers and filaments [298, 338, 339]. These complexes cause degeneration of neurons and glial cells [97], manifesting as a group of neurodegenerative disorders termed ‘tauopathies’ [312].
The most prominent tauopathy is Alzheimer’s disease (AD), the common cause of dementia in older adults. AD is an incurable, progressive degenerative disease of the brain, characterized by the presence of tau and ß- amyloid (Aß) pathology [286]. There are no disease-modifying drugs available for AD; only symptomatic treatments trying to counterbalance the neurotransmitter disturbance exist. No significant new drug for AD has been approved in the last 14 years, despite extensive clinical trials. The pipeline has been plagued with significant failures, with more than 400 failed clinical trials since the last symptomatic Alzheimer’s drug was approved [71].
Despite the field being aware that tau pathology correlates well with the onset and progression of AD for almost 40 years [39], it is only now that tau targetted therapy has become attractive for clinical trials. A multitude of tau antibodies and vaccines have been tested in preclinical studies in the last two decades. Currently, eight humanised tau antibodies and two tau vaccines have entered clinical trials either for AD or frontotemporal dementia (FTD) [65, 71](www.alzforum.org). In light of the failure of the clinical trials with amyloid targeting drugs, tau therapy is manifesting as the frontrunner in the search for an effective treatment for AD.
Tour de tau - tau as a protein with multiple faces
In contrast to amyloid precursor protein (APP), the function of tau protein was already known at the time of the discovery of it as a constituent of neurofibrillary degeneration. Tau is a microtubule-associated protein (MAP), promoting the polymerization and assembly of microtubules [351]. In the adult human brain, there are six isoforms of tau protein generated by alternative splicing from a single gene located on chromosome 17 [120, 238]. At the N-terminal end, they differ by the addition of a 29 amino-acid sequence (1 N) or as replicates (2 N - total of 58 amino acids) coded by exons 2 and 3. The sequence coded by exon 3 is only present if the sequence encoded by exon 2 is inserted. Interestingly, the 2 N tau isoforms are weakly expressed in the human brain [119, 214, 295]. The microtubule binding region (MTBR), has three (3R: R1, R3, R4) or four repeat domains (4R: R1-R4). The sequence encoded by exon 10 allows the insertion of a 31 amino acid microtubule binding domain (R2) which is inserted after the first repeat R1. Tau isoforms with 3R and 4R are equally expressed, since their ratio is about 1:1 in the human brain [295]. However, some neurons do not express 4R tau isoforms. For instance, granular cells of the dentate gyrus only express mRNAs of 3R-tau isoforms [119]. Thus, tau isoforms have different cellular and laminar distribution in the human brain [46].
The strict classification of tau protein as a MAP may have delayed research on its other biological functions. If sequence homology (70-90%) with other MAPs is evident in the microtubule binding domains, the N-terminal portion of tau is unique. It must therefore have other unique functions [194]. Logically, as a MAP, tau has functions in cell trafficking, but it also interacts with dynactin and synaptogyrin-3, suggesting specific related-functions, such as synaptic vesicle control [213, 224].
The first unexpected functions of tau may be related to its nuclear localization [201]. These initial findings were widely discussed, but nowadays, it is clearly established that tau binds to nucleic acids, and may be involved in chromatin remodelling [53, 104, 146, 252, 266, 267]. The binding of tau to DNA may allow protection against reactive oxygen species [316, 349], and binding to RNA may contribute to ribosome stability and miRNA activity [35]. Altogether, these data strongly suggest that tau may modulate gene expression and RNA stability. Such observations are also supported by tau loss-of-function in pathological conditions. For instance, formation of tau oligomers leads to DNA/RNA damage [337], RNA and ribosome instability [225] and changes in nuclear organization and protein expression [103]. Binding of tau to tRNAs may also initiate tau aggregation by forming droplets through complex coacervation [378]. Moreover, pathological tau can interact with nucleoporins of the nuclear pore complex (NPC) and affect their structural and functional integrity [93] (Fig. 1).

Yin and Yang of Tau protein
Secondly, tau may also play a role in cell signalling. The longest brain tau isoform with 441 amino acids (aa) has 85 putative sites of phosphorylation. Thus, tau may act as a buffer for cell signalling. For instance, tau may serve as a ‘phosphorylation sink’ for the p25-Cdk5 complex, hence sequestering it away from other death-inducing substrates [130]. Tau may also interfere with tyrosine kinase family Src/Fyn signalling at dendrites [49, 152]. Tau also interacts with phosphatase and tensin homolog (PTEN) and modulates insulin signalling. Recent data suggest that loss of tau function leads to an impaired hippocampal response to insulin, caused by altered insulin receptor substrate 1 (IRS-1) and PTEN activities [218].
Finally, the cytosolic tau protein may also be secreted. This secretion is stimulated by neuronal activity [263]. Such secretion is likely to occur through non-conventional secretory pathways [44]. Recent data suggest that such secretion may be similar to that of fibroblast growth factor 2 (FGF-2), including oligomerization, binding to phospho-inositol, and extracellular capture by heparan sulphate proteoglycans [164]. An alternative pathway is the secretion of pro-interleukin 1, which requires proteolysis. Interestingly, C-terminal-tau fragment Δ422–441 was significantly more secreted than full length tau [261]. Tau is also secreted within extracellular vesicles such as exosomes [346] and ectosomes [89]. In pathological conditions, secreted tau may participate to tau seeding and spread (discussed later).
To sum up, tau has multiple functions in addition to axonal microtubule assembly. All of these recently discovered tau functions may contribute to the development of tau pathology and related events (Fig. 1). These discoveries further strengthen the case for tau as the therapeutic target for AD and tauopathies.
Tau as a driver of neurodegeneration
AD is a double proteinopathy, characterized by the presence of both tau-reactive neurofibrillary lesions and β-amyloid (Aβ) depositions (senile plaques; SPs). The importance of both proteins, which are present also under physiological circumstances, in the development of AD is extensively debated. Numerous clinicopathological studies were published, favouring both histological lesions, i.e. NFTs and SPs. However, since the early nineties, most studies found a strong correlation between neocortical NFT load and cognitive impairment [94].
The progression of neurofibrillary pathology begins in the entorhinal cortex, in contrast to the spreading of Aβ, where the presence of neocortical SPs precedes the appearance of hippocampal SPs [39, 91, 320, 327]. Aβ pathology is present even in cognitively intact persons, so amyloid deposition is not sufficient to explain the clinical phenotype of AD [77]. In contrast, NFT burden in associative neocortical areas is strongly related with clinically overt dementia. The Braak staging [39] for NFTs, used to define the neuropathological severity of AD in the general neuropathological practice, reveals a strong correlation with cognitive decline [92, 121]. In a study of an oldest-old population, Gold and colleagues [121] found that unlike younger cohorts, Braak stages did not precisely reflect the severity of dementia. Braak stage III correlates poorly with cognitive decline, while Braak stages IV or greater are consistently associated with at least mild dementia. This discrepancy is most likely due to the increasing prevalence of mixed neuropathologies in the oldest-old, such as a combination of vascular lesions and AD pathology [156].
As in all neurodegenerative diseases, AD is characterised by selective vulnerability of specific brain regions, cortical layers, and neuronal populations. The anatomical distribution of tau and neuronal loss reflects the different clinical signs of AD well. Anterograde memory problems at the beginning of the symptomatology are related to tau-burden in the medial temporal lobe [94]. During the progression of the clinical presentation, other signs, such as agnosia, apraxia or speech and behavioural problems will add to the memory problems, corresponding to the involvement of different associative or limbic regions. The neuropathological background for acalculia and visuospatial dysfunction is related to the involvement of tau pathology in the parietal lobe [94]. Ideomotor and dressing apraxia is linked to NFT densities in the anterior cingulate cortex, while constructional apraxia relate to NFT densities in the superior parietal, posterior cingulate and occipital cortex [113]. A significant relationship exists between associative visual agnosia and tau burden in the secondary visual cortex (Brodmann area 18) and the occipitotemporal visual association cortex (Brodmann area 37 and ventral 19) [114]. The high NFT density in the superior parietal cortex (Brodmann area 7), posterior cingulate cortex (Brodmann area 23), and CA1 subfield of the hippocampus plays a role in developing temporo-spatial disorientation [115]. Cases with atypical AD, such as posterior cortical atrophy, also underline the importance of tau pathology in developing clinical signs. Patients presenting mainly with visual symptomatology have a high NFT burden in the occipito-parieto-temporal junction and posterior cingulate cortex [138]. The anterior brain regions are less involved as compared to the “classic” form of AD.
Behavioural problems or speech disorders, more suggestive of other neurodegenerative diseases such as frontotemporal dementia, could also be present in neuropathologically confirmed AD. In contrast, prefrontal syndromes are correlated with atypical distribution of NFTs in the dorsolateral, median and orbitofrontal areas [340]. These clinicopathological observations underline the importance of the tau protein in the pathogenesis of AD and its subtypes (amnestic, dysexecutive/behavioural, visuo-spatial, and language presentation).
Tauopathies are clinically, biochemically and morphologically heterogeneous neurodegenerative diseases characterized by the deposition of abnormal tau (microtubule associated protein tau; MAPT) in the brain. Neuropathological phenotypes are distinguished based on the distinct involvement of anatomical areas, cell type, and presence of distinct isoforms of tau in the pathological deposits [172]. If tau protein deposition is the predominant feature, the term primary tauopathy is used. The nomenclature overlaps with the classification of frontotemporal lobar degeneration (FTLD). Disorders characterized by tau pathologies considered having other (possibly diverse) driving forces (e.g. Creutzfeldt–Jakob disease, Down’s syndrome) are called secondary tauopathies [108].
Tauopathies are distinguished based on the ratio of 3 repeat (3R)- and 4R-tau and two or three major bands (60, 64, and 68 kDa) in Western blot of sarkosyl-insoluble fractions [184, 296, 312]. FTLD-tau is grouped based on the tau isoform predominating the morphology. Pick’s disease (PiD) is a 3R tauopathy (60 and 64 kDa bands). 4R tauopathies (64 and 68 kDa bands) is comprised of progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), argyrophilic grain disease (AGD), and globular glial tauopathy (GGT) [172]. Mixed 3R and 4R tauopathy (60, 64 and 68 kDa bands) is the neurofibrillary tangle (NFT)-dementia (discussed also in the frame of primary age-related tauopathy, PART), and this type of tau pathology is seen in Alzheimer diseased (AD) brains.
Hyperphosphorylated tau is the major constituent of neuronal and glial inclusions, although there are further biochemical modifications (N- and C-terminal truncation, glycosylation, glycation, nitration of tyrosine residues, transglutamination, deamidation; acetylation; oligomer forms) [173] which are not examined routinely in diagnostic practice. Using phospho-dependent tau antibodies several morphologies of cellular tau immunoreactivity can be detected [172]. Tau immunoreactivity in neurons comprises pre-tangles (diffuse cytoplasmic neuronal tau immunoreactivity), NFTs, Pick bodies (3R-tau immunoreactive), spherical inclusions (usually 4R immunoreactive), dystrophic neurites, neuropil threads (axonal), and grains (dendritic). Astrocytic tau pathology includes tufted astrocytes (PSP), astrocytic plaques (CBD), ramified astrocytes (PiD), globular astroglial inclusions (GGT), thorn-shaped astrocytes, and granular-fuzzy astrocytes (the latter two seen mostly in age-related tau astrogliopathy, ARTAG). In oligodendrocytes, coiled bodies (PSP, CBD, AGD) and globular inclusions (PiD, GGT) can be detected (Fig. 2). The constellation of these morphologies and their anatomical distribution characterize primary tauopathies, e.g. NFTs in the medial temporal lobe is characteristic for PART [68] and NFTs in subcortical structures together with tufted astrocytes are pathognomonic for PSP [172]. Neuropathologic hallmarks of CBD comprise neuronal inclusions, threads in the white and grey matter, coiled bodies and astrocytic plaques [85]. AGD is characterized by the presence of argyrophilic and 4R tau immunoreactive grains in medial temporal lobe structures together with pre-tangles, oligodendroglial coiled bodies, and astrocytic tau pathology [324]. Globular oligodendroglial and astroglial inclusions characterize the GGTs [7]. PiD is a 3R tauopathy with Pick bodies, with less glial tau pathology and prominent FTLD [172]. In addition, neuronal tau pathology in the form of NFTs, threads and dystrophic neurites associated with Aß plaques is a hallmark of AD [39, 46, 91]. Finally, hereditary frontotemporal dementia (FTD) associated with mutations in the MAPT gene shows 3R-, 4R- and 3R/4R-tau pathologies overlapping with the neuropathologic features of primary tauopathies [101, 111]. However, in hereditary FTD, tau mutations lead to conformational changes before tau hyperphosphorylation [90].

Tau pathologies in diverse tauopathies. Tau pathology in AD and PART comprise dystrophic neurites (a), axonal threads (b), pretangles (c) and NFTs (d). PSP is characterized by pretangles and threads (e), subcortical tangles (f), tufted astrocytes (g), and oligodendroglial coiled bodies (h). In CBD cases pretangles and threads (i), globose neuronal CBD-bodies (j), astrocytic plaques (k), and oligodendroglial coiled bodies (l) can be seen. AGD is characterized by 4R-tau positive neuronal dendritic grains (m), pretangles (n), granular/fuzzy astrocytes (o), and oligodendroglial coiled bodies (p). In GGT cases neuronal pretangles (q), spherical cytoplasmic inclusions (r), globular astroglial (s) and oligodendroglial (t) inclusions are detected. In Pick’s disease neuronal Pick bodies are frequent in the dentate gyrus (u) and show 3R immunoreactivity (v; here CA1 subregion is shown), furthermore, ramified astrocytes (w) and small globular oligodendroglial inclusions (x) can be noticed as well. Finally ARTAG comprises thorn shaped astrocytes and granular fuzzy astrocytes here demonstrated in the subependymal (y1), subpial (y2), perivascular (upper part of image 4) and white matter (lower part of image) (y3), and grey matter (y4) areas. All images show immunostaining for the AT8 antibody except (m) and (v) where immunostaining for 4R- and 3R-tau isoform, respectively, was performed. The bar in (a) represents 50 μm for a, e, f, g, h, l, m, t, u, v, y1, and y4; 35 μm for b, c, d, j, k, o, p, x; 30 μm for q and r; 40 μm for w and y2; 100 μm for i; 25 μm for s; and 150 μm for y3
Tau pathologies show hierarchical involvement of anatomical regions. This is exemplified by the six stages of NFT pathology in AD [38] and PART (usually only up to stage IV) [68] and the three stages of AGD-associated pathology [277]. For PSP and CBD hierarchical involvement is being studied; this is hindered by the heterogeneity of these diseases. A recent study described sequential distribution patterns of astroglial tau pathologies in CBD, PSP and in ARTAG types [175]. These observations on various stages complement experimental observations in cell culture and animal models, suggesting spreading of tau pathologies along neuronal connections and provide a basis for the concept of tau-strains as a background for disease heterogeneity [31, 60, 236, 280]. In fact, 3R, 4R and mutated tau species are likely to display different spreading behaviors [90]. Recent studies suggest that astrocytes might play a previously underappreciated role in the disease process. Indeed, astroglial tau pathology may precede neuronal tau immunoreactivities in primary FTLD-tauopathies [174, 193]. Astroglial tau pathologies might reflect their contribution to disease spreading or clearance of disease-associated proteins, and might lead to astroglial dysfunction contributing to neuronal degeneration [174].
Pet imaging of tau pathology
Recently, the development of positron emission tomography (PET) radioligands presumably binding to tau has enabled the in vivo mapping and quantification of tau pathology, hitherto largely confirming autopsy findings. The radioligand [18F] Flortaucipir (FTP, previously AV1451 or T807), a benzimidazole pyrimidine derivative, is by far the most widely employed to date. It has been shown to bind with high affinity to mixed 3R- and 4R-tau isoforms in paired-helical filaments (PHF) of AD patients [26, 309, 361]. A recent study furthermore showed that in vivo FTP-binding and post mortem PHF load were highly correlated in a subject with a MAPT R406W mutation, which causes AD-like 3R/4R tau pathology [309]. However, large inter- and intra-individual differences were observed in a recent autopsy study of several tauopathies [361], calling for further investigation of FTP binding characteristics.
Off-target binding of tau PET ligands is another major limitation and challenge to be addressed in novel tracer development [26, 187, 200]. For example, the alleged tau PET ligand [18F]THK5351 demonstrated strong binding to monoaminoxidase B (MAO-B) in and ex vivo [133, 239], with ligand uptake being reduced by up to 50% in selected brain regions by the MAO-B inhibitor selegiline, preventing accurate quantification of tau [239]. Among the currently available tracers, the binding characteristics of FTP have been characterized best. FTP off-target binding has been observed in the caudate, putamen, and pallidum in elderly individuals regardless of their clinical diagnosis [20, 42, 205, 333, 354], and has been attributed to, amongst others, iron binding [59]. Its pronounced binding to the substantia nigra, also in cases with no apparent tau pathology, has been related to neuromelanin [219,220,221], as has elevated FTP binding in the pituitary gland, retinal pigment epithelial cells, leptomeninges, and malignant melanocytes in metastatic melanoma [205, 219, 221]. High FTP signal in the choroid plexus has been attributed to calcification/mineralization [205], binding to tangle-like structures corresponding to so-called Biondi ring tangles [150], or melanocyte binding [180, 219, 221] and constitutes an issue for the quantification of hippocampal ligand uptake due to their close proximity. Here, partial volume correction (PVC) might reduce bias from choroid plexus signal on hippocampal signal [180, 211, 212, 288]. FTP has also been shown to bind to MAO-A and B in vitro [335], however, no significant differences were observed in vivo between FTP scans of patients with and without MAO-B inhibitors [133].
A second generation of tau radioligands is supposed to be affected less by off-target binding issues, however, in vivo data are thus far limited for these ligands, which include, amongst others, [18F]RO6958948 (Roche) [142, 359], [18F]MK-6240 (Merck/Cerveau) [24, 199, 255], [18F]GTP-1 (Genentech) [278, 279, 350], [18F]PI2620 (Life Molecular Imaging, formerly Piramal Imaging) [314] and [18F]PM-PBB3 [249, 299].
For [18F] FTP, tracer uptake in physiological aging and AD appears to follow a particular spatial and temporal pattern. Although longitudinal data are limited to this date [153, 311], the distribution appears to begin in the entorhinal cortex, to spread into inferolateral temporal lobes and medial parietal lobes, and to eventually cover most of the neocortex in disease cases. To capture this high regionality, which is significantly different from e.g. PET imaging of Aβ pathology (often found throughout the neocortex), several approaches have been suggested for A) binary categorization of tau “positivity” [154, 212, 229, 344], and B) topographical staging approaches that recapitulate post mortem findings of tau distribution [211, 288, 290]. This regionality of tau PET ligand uptake in the brain is further emphasized by studies employing data-driven approaches without prior definition of anatomical regions [293, 352]. However, a few studies have suggested that ligand uptake assessment based on larger composite regions may be sufficient to capture AD-related tau PET signal and the longitudinal accumulation of tau [153, 211]. On a group level, FTP demonstrated clinical usefulness when its discriminative accuracy between AD dementia and non-AD neurodegenerative disorders was examined in a large multisite study, yielding very high sensitivity and specificity based on medial-basal and lateral temporal cortex ligand uptake [250].
In general, elevated tau tracer binding in the medial temporal lobe (MTL) can be observed in cognitively healthy older adults, whereas widespread binding in neocortical regions of any individual commonly is associated with the presence of cortical Aβ [58, 124, 161, 198, 211, 262, 288, 291, 294]. However, despite an overall correlation between brain Aβ and tau [161], the spatial distributions of these two aggregated proteins are discordant [161, 198, 294]. Interestingly, the strongest association can be observed between global Aβ and entorhinal tau PET signal [333], rendering this region important for the detection of AD-related tau PET signal.
Tau deposition outside the MTL is more common in individuals with AD; however, elevated tau tracer uptake has been reported for in neocortical areas in cognitively normal and even Aβ negative individuals [204]. While AD patients commonly have more widespread and pronounced tracer uptake than controls, exceptions have been found in AD patients who are Aβ-positive and show relatively low levels of tau deposition [262, 344]. Longitudinal studies have also demonstrated that increasing levels of Aβ are associated with more tau deposition in limbic and neocortical Braak regions several years later, even in nominally Aβ-negative individuals [179, 325]. Despite the limited availability of longitudinal data, it appears that tau accumulates over time in the temporal lobes of cognitively healthy individuals and AD patients, albeit this seems to be limited to Aβ-positive individuals [153, 311].
Compared to associations with Aβ, correlations between tau PET measures and age across healthy elderly seem to be weaker and confined to MTL regions [212, 289]. Greatest differences in FTP uptake between healthy young and elderly subjects are commonly observed in the choroid plexus and basal ganglia; however, tracer uptake in these regions most likely represents off-target binding [205, 206]. The age of symptom onset among AD patients clearly affects tau PET uptake patterns. Sporadic early-onset AD patients (EOAD) exhibit distinctly greater parietotemporal and frontal ligand uptake when compared with late onset AD (LOAD) which exhibits rather confined temporal lobe uptake [289]. Data from studies in early-onset familial/autosomal-dominant AD are limited, suggesting earliest FTP uptake in the medial temporal lobes of Aβ-positive presymptomatic mutation carriers but high cortical uptake, spatially comparable to sporadic EOAD cases in later symptomatic stages [268, 289].
Tau has, in contrast to Aβ, long been known to be much stronger associated with measures of cognitive decline and neurodegeneration [86, 88, 136, 155, 237]. In fact, greater FTP uptake has been shown to be related to both poorer cognitive function cross-sectionally and retrospective longitudinal decline in cognition functioning [13, 212]. In cognitively healthy elderly, associations are strongest between episodic memory performance and MTL, namely entorhinal cortical tracer uptake, whereas associations with global cognition are either absent or found for wider, less specific neocortical regions. Interestingly, the effect of MTL tau on episodic memory seems to be independent of global Aβ load [211, 288] both in these individuals and in individuals experiencing subjective cognitive decline [45]. Moreover, MTL tau accumulation in cognitively normal elderly is associated with patterns of neurodegeneration as assessed by both structural magnetic resonance imaging (MRI) and [18F] Fluorodeoxyglucose (FDG) PET that are topographically similar to the patterns seen in AD patients [2, 74, 125, 132, 176], suggesting that early-stage MTL tau might have a pathogenic role even in cognitively healthy individuals.
The relationship between tau, cognition, and neurodegeneration is even more pronounced in AD patients, especially in cases of EOAD who frequently exhibit language, visuospatial, or executive dysfunction rather than memory impairment and where the spatial distribution of tau deposition strongly reflects the clinical phenotype [250, 368]. In these patients, tau deposition is also strongly associated with the neurodegeneration markers of atrophy and glucose hypometabolism [27, 148, 250, 344], a relationship that cannot be explained by measures of or the distribution of Aβ [269]. Statistically, cognitive impairment can be related to both brain atrophy and tau, however, tau remains solely correlated with cognitive dysfunction, even when controlling for atrophy [23].
Generally, FTP uptake might be helpful in distinguishing clinical variants of AD, e.g. a recent study employing a data-driven clustering approach demonstrated that the majority of patients with relatively low entorhinal FTP uptake, compared to overall neocortical uptake, have an atypical clinical EOAD presentations, while most patients with high FTP uptake in both entorhinal and neocortex present with EOAD and a typical amnestic phenotype, and most with low FTP uptake in both entorhinal and neocortex present with typical LOAD [352].
In summary, the assessment of tau accumulation with PET has revealed a pattern of aggregation on a continuum from normal aging through AD that parallels neuropathological data and now offers the possibility of longitudinal studies. The strong relationship between tau PET measures and measures of neurodegeneration and cognition, taking in account the relationship between tau and Aβ, will elucidate how Aβ and tau pathology interact in the development of the processes that are linked to cognitive decline and clinical dementia.
Extracellular and intracellular tau – Two sides of one coin
In pathological conditions, tau protein undergoes post-translational modifications, such as, truncation [241, 242, 357, 358], phosphorylation [127], ubiquitination [32, 181], glycation [283, 373], glycosylation [196, 343], nitration [144, 271, 272] and sumoylation [87, 209]. Among them, phosphorylation and truncation are the most studied. Many laboratories suggest that tau hyperphosphorylation on Ser and Thr residues facilitates tau aggregation. Tau is posttranslationally modified at Ser/Thr residies by O-linked N-acetylglucosamine (O-GlcNAc), and thus increasing tau O-GlcNAcylation may protect against tau aggregation. In tau transgenic mouse models, inhibition of β-N-acetyl-glucosaminidase, the enzyme responsible for O-GlcNAc removal, is protective [33].
It has been shown that truncated tau proteins are contained in the core of the paired helical filaments. Expression of the tau protein in the brain of transgenic rats and mice induced the formation of extensive neurofibrillary pathology, suggesting that truncated tau is a driving force of neurofibrillary degeneration [98, 381, 382, 384, 385].
Therapeutic approaches against tau pathology target either intracellular or extracellular tau or eventually both. It has been demonstrated that an increase in the level of intracellular tau could result in tau secretion into the extracellular space or in cell death [122, 304]. Toxic extracellular tau could interact with neuronal cell receptors such as M1/M3 muscarinic receptors [122, 123], or with heparin sulfate linked to cell membrane [372]. The result of that interaction could be again the onset of neuron toxicity and intracellular tau secretion. In this way, tau pathology could be propagated. Thus, possible therapies involving the use of muscarinic antagonists [131, 334, 336], or agents decreasing heparin sulphation [372], are under discussion for AD therapy.
Extracellular tau is found at significant levels in the interstitial fluid of the central nervous system (CNS), and can pass into cerebrospinal fluid (CSF) [370]. Initially, extracellular tau was thought to be only passively released by dying neurons, with selective vulnerability of neuronal types and cellular signals contributing to the disease progression [285]. However, there is now growing evidence that tau is actively transferred between neurons under pathological and physiological conditions. Aggregated and soluble tau variants have been shown to transfer between anatomically connected regions of the brain [75, 149, 197], and trans-synaptically between cells in culture [280, 363]. How tau is actively transferred between neurons is a major focus of dementia research, as attenuating the pathological spread may limit the progression of disease. Active tau transfer is thought to involve discrete steps including post translational modification (PTM), extracellular release and subsequent tau internalization.
Intracellular tau undergoes various PTMs including phosphorylation and proteolytic cleavage. Levels of total and phosphorylated tau detected in the CSF are important biomarkers for dementia [28]. Several tau modifications are detected at proportionally higher levels in extracellular compared with intercellular fractions, implicating specific tau modifications in active neuronal export [248]. Higher levels of extracellular aberrantly hyperphosphorylated tau are detected in patients with dementia [79]. Hyperphosphorylated tau has a lower binding affinity to microtubules (MT) [192] and mislocalizes to somatic and dendritic cell compartments [106, 143, 323]; these factors may contribute to active export as dissociation from MTs would allow a greater opportunity for tau to interact with components that facilitate protein export. C-terminally truncated tau (lacking approximately the last 50 amino acids) is detected at proportionally higher levels in the CSF samples of healthy individuals and dementia patients [284], and in neurons in culture [43, 163]. These tau species may be more readily detectable or resistant to degradation. Post-translational modifications and exon splicing events influence intra- and extracellular tau stability. Phosphorylated and 4R-tau isoform peptides have faster turnover rates than unphosphorylated and 3R-tau isoform peptides, respectively. Peptides from the N-terminal to mid-domain tau are more stable and have similar half-lives both inside and outside of the cell [284]. Notwithstanding these differences in stability, the proportionally higher levels of extracellular truncated tau suggest that physiological active tau release may be regulated by proteolytic cleavage.
Distinguishing between active tau release mechanisms and passive tau release, due to cell death, is challenging. The process of active tau release has been linked with several cellular mechanisms. In cell culture, monomeric tau can directly interact with the plasma membrane and proteoglycans, leading to unconventional secretion of tau [55, 164, 304], or the release of ectosomes containing tau [89]. Active tau release is also proposed to be regulated by neuronal activity. Depolarization of neurons promotes tau release [102, 263, 371] and release of exosomes containing hypophosphorylated tau [346]. These mechanisms aren’t mutually exclusive. However, it is unclear how they are associated and whether they relate to all forms of tau.
Following release into extracellular space, pathogenic tau can be taken up by healthy neurons, and promote seeded aggregation [165]. There have been conflicting reports regarding the forms of tau and route of entry of tau into various cell types. Studies suggest that aggregated tau is the predominant form internalized into cells [105, 362]. However, monomeric full-length tau can also be efficiently taken up by neurons [95]. These reports show that tau is taken up by endocytosis. Levels of the clathrin-mediated endocytosis component myc box-dependent-interacting protein 1 (BIN1) negatively correlate with tau uptake [52]. Different forms of tau enter neurons via distinct but overlapping pathways. Monomeric tau can enter neurons via dynamin-dependent endocytosis that is saturable, suggesting uptake is dependent on carrier proteins or receptors [95]. Entry of aggregated tau is attenuated by heparin in cell culture, indicating that heparan sulphate proteoglycans serve as receptor for tau uptake [140].
Hyperphosphorylated tau isolated from AD brain tissue is also recognized by the CNS immune system; microglia internalize and degrade tau in an Fc-dependent manner [210], and the cytosolic Fc receptor – tripartite motif-containing protein 21 (TRIM21), inhibits seeded tau aggregation [223]. Conversely, it is also suggested that the migration of microglia through the CNS transfers pathogenic species of tau to new areas of the brain [216].
It is currently unknown if tau transfer is a disease-specific phenomenon or physiological process appropriated during disease. Physiological tau transfer may be involved in network signaling or neuronal maintenance. Independently of the ability of pathological tau to seed aggregation, extracellular tau itself has been shown to be neurotoxic [84] and extracellular tau from individuals harboring amyloid precursor protein (APP) gene duplication can also cause synaptic dysfunction [145]. Tau immunotherapies that attenuate transfer of tau with the aim of limiting disease progression are under development [43, 64]. Tau antibodies have been shown to attenuate intracellular tau aggregation [375], while tau-antibody complexes can be internalized and targeted for degradation [56, 129, 215]. Identifying epitopes and conformations that distinguish between physiological and pathological tau transfer are important considerations when developing immunotherapies that target extracellular tau.
Tau passive immunotherapy
In Alzheimer’s disease, tau protein is burdened by numerous post-translational modifications resulting in aggregation and tangle formation. Therefore, a number of passive vaccines for tau immunotherapy raised against various epitopes or conformation/s of tau have been developed, showing varied degrees of efficacy in attenuating tau pathology in animals, along with improvement in cognitive or motor functions. Several animal models have been used for testing of the therapeutic efficacy of monoclonal antibodies. Tau pathology is localized in various brain areas including hippocampus and brainstem. The location of tau pathology is mostly determined by the gene promotor. The clinical presentation is driven by topographic distribution of tau pathology, some of rodent models demonstrated cognitive decline while others suffer from impairment of sensori-motor functions [383]. The majority of preclinical studies have been performed on transgenic mice expressing mutant tau proteins (Table 1). However, tau mutations are not linked to familial forms of AD, but can cause frontotemporal dementia.
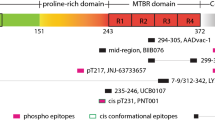
In general, tau therapeutic antibodies target, neutralize and/or eliminate either monomeric [36, 374, 375], aggregated forms [54], phospho-specific, or conformationally altered forms of tau protein [36, 56, 72, 129, 167, 342] (Table 1) and thus preventing formation neurofibrillary lesions. Anti-tau antibodies also differ in their binding site on tau. They recognise either the N-terminus [4, 73, 374, 375], the proline rich region [73, 342], the microtubule binding region [167, 375] or C-terminus [36, 56, 151].
The N-terminus of the tau protein has become attractive for preclinical development of tau therapeutic antibodies [4, 73, 374, 375]. This can be attributed to following reasons. Firstly, the conformational changes in the N-terminal region of tau occur very early in the disease pathogenesis in AD, which affects the function of the protein [62]. Furthermore, the exposure of the N-terminal is associated with early pathological event in human tauopathies [63]. The N-terminal fragment containing Gln124 displayed stronger ability to stabilize microtubules [78]. In addition, only N-terminal fragments were detected in the CSF from AD subjects [160, 284]. Similar results were also obtained from cortical neurons cultured from AD brains [43]. Moreover, the N-terminal fragment of tau protein was shown to increase amyloid beta production [43], and impair mitochondrial function, synaptic plasticity, and in turn was detrimental to neurons [9, 10, 34, 100]. Several studies focusing on antibodies targeting N-terminal sequences of tau have reported varied degree yet promising efficacy in reducing tau pathology and improveing cognitive or motor deficits during preclinical trials [4, 14, 73, 374, 375].
On the other hand, it has been shown that the majority of tau in the AD brain is truncated, mostly at the N-terminus [384]. A recent study showed that high molecular weight tau species from AD brain extract demonstrated strong immuno-positivity to C-terminal specific antibodies, and were weakly stained with N-terminal specific antibodies, indicating substantial lack of N-terminal sequences in oligomers and fibrils from the AD brain [380]. In concordance with this study, two recent papers demonstrated that N-terminal tau antibodies do not recognise truncated tau and the whole spectrum of aggregated forms of tau in Alzheimer’s disease brain. They mainly decorate a triplet of hyperphosporylated full-length tau – A68 [183]. This means that a large portion of pathological tau is not recognised by N-terminal tau antibodies [67, 331, 380]. By using a seeded aggregation cell model, N-terminal antibodies (PT26, aa 23-26; PT93, aa27-32; hTau10, aa29-36) showed incomplete depletion of human-derived seeds even at the concentration, which was sufficient for complete depletion of tau seeds from P301S transgenic model (300 nM) [331]. Similarly, two tested N-terminal antibodies (aa15-24, aa 25-30) and MC1 (which recognises both N-terminus and microtubule binding domain) failed to fully prevent seeding of AD tau in a seeded aggregation cell model [67] and in vivo [8]. In contrast, Nobuhara and colleagues [240] demonstrated that N-terminal antibody C13 (aa2-18) efficiently removed tau from rTg4510 brain extracts and human AD high molecular weight tau (HMW). Moreover, the antibody reduced tau uptake of pathological mouse and human AD HMW tau in a sensitive FRET-based in mouse primary neurons. It is important to note that the antibodies targeting the N-terminus on tau are not specific to diseased tau, and they possibly reduce the level of physiological tau.
While beneficial effects of N-terminal antibodies on reduction of tau uptake or inhibition of seeding activity are still a matter of discussion, the development of novel therapeutic tau antibodies has shifted to the mid domain of tau protein. In the mid region, phosphorylation of tau at the position pS202 and pT205 was reported as an intracellular and extracellular marker for tau pathology in AD [39], and is potentially involved in neuronal apoptosis [166]. Moreover, phosphorylation of tau at T231 was also reported as an early event in AD [207, 208]. Several mid-domain tau antibodies (PT51, aa153-158, PT79, aa131-140, PT89, aa173-178) demonstrated complete depletion of mouse transgenic tau P301S-derived tau seeds. However, incomplete depletion of human derived seeds even at maximal concentration of 300 nM [331], suggests the different composition of mouse and human tau seeds. On the other hand, the antibody 6C5 (aa125-131) efficiently removed tau (> 85% reduction) from both mouse transgenic (Tg4510) brain extracts and human AD HMW tau (82% reduction). Furthermore, the antibody was the most effective in reducing tau uptake of pathological mouse tau (> 90% reduction) and human AD HMW tau (> 75% reduction) as well in a sensitive FRET-based assay in mouse primary neurons [240]. Similarly, the antibody recognising aa235-250, fully neutralised seeding activity of AD and PSP tau in a seeded aggregation cell model with an IC50 of 2.9 nM and 5.6 nM, respectively [67]. These results demonstrate that antibodies recognising the mid region of tau can be effective in the reduction of tau uptake and neutralisation of tau seeding activity. In contrast to in vitro experiments, studies using tau antibodies raised against this region of tau showed inconsistent results in preclinical in vivo experiments [72, 73, 342].
The third class of antibodies target the microtubule binding region (MTBR), which plays a crucial role in polymerization and stability of microtubules [36, 168, 328]. On the other hand, this region is responsible for the pathological tau-tau interaction. It was reported that the C-terminal fragments were more prone to filament formation than the N-terminal sequences [257, 258]. Specifically, the region spanning aa244-372 corresponds to the amyloid-forming region on tau protein [315]. This property is attributed to the hexapeptide sequence 306VQIVYK311 on the 2nd repeat of MTBR which was shown to promote tau aggregation by a nucleation dependent mechanism [338]. Recent cryo-electron microscopy study demonstrated that this hexapeptide packed through a heterotypic, non-staggered interface with the opposing residues 373–378 [99]. Moreover, the hexapeptide on the 3rd MTBR also caused formation of fibrils in vitro [315]. Currently, only two preclinical studies on passive immunotherapies targeting the MTBR were performed, both showing promising results [168, 375]. More specifically, the antibody DC8E8 [168] binds to the four highly homologous and yet independent hexapeptides localised in each microtubule binding domain, while mAb HJ9.3 (epitope 306-321) recognises the hexapeptide sequence 306VQIVYK311 [375]. Both antibodies were effective in reduction of neurofibrillary pathology in the brain of transgenic rodent models.
It has been shown that the C-terminus enhanced the microtubule binding capacity of tau protein and also influenced pathological tau aggregation [177, 232]. More specifically, the C-terminal region of tau harbors several phosphorylation sites which regulate microtubule binding of tau and hyperphosphorylation of phospho-sites in this region, such as pS413, pS396, pS404, are observed in early and late stages of AD progression [15, 300]. Therefore, several studies are devoted to investigating the effect of C-terminal specific tau antibodies in animal models [36, 56, 129, 151, 182, 328].
Finally, conformational changes and oligomer formation of tau protein represent early events in the pathogenesis of tau lesions in AD [39, 256, 348]. For example, with MC1 (aa7–9 and aa313–322), conformational epitope specific reactivity is observed in Braak stages I and II in AD [348]. In addition, MC1 immuno-purified soluble tau species readily assembled into paired helical filaments in vitro [348]. Therefore, antibodies against these unique species of tau are also being investigated in preclinical studies to attenuate tau pathogenesis. MC1 therapy slightly reduced insoluble tau and number of tangles in the brain of experimental mice [54, 56, 72].
Currently, only a handful of humanised tau antibodies are being investigated at various stages of clinical development (Clinicaltrials.gov). Humanized versions of N-terminal specific antibodies 8E12 [374, 375], and BIIB092 (also known as BMS-986168 or IPN007) [43] are being currently investigated at various phases of trials for treatment of PSP and AD. Another N-terminal antibody RO 7105705 (RG 6100) has already entered Phase 2 clinical trials, targeting Alzheimer’s disease. Janssen is also starting phase 1 clinical trials in mild AD with the antibody JNJ-63733657 which is effective at eliminating pathological tau seeds. Antibody UCB0107 that targets the mid region of tau is currently in Phase I (healthy volunteers). Antibody LY3303560 (modified MC1 antibody) recognising both N-terminus and microtubule binding domain is in Phase 2 trial in MCI-to-AD or mild to moderate AD patients. Finally, antibody BIIB076 that has the ability to bind monomeric and fibrillar forms of tau is being tested in the Phase I clinical trial in AD [65, 71](www.alzforum.org).
There are several advantages of passive immunotherapy. In terms of pharmacology definition, antibodies are precisely characterised both in vitro and in vivo (avidity, affinity, target specificity, half-life, concentration, single isotype). Passive immunotherapy does not require the immune system to generate an immune response. The main disadvantages are expensive production, the short half-life of antibodies and chronic systemic administration (i.v.). Chronic administration may lead to formation of anti-antibodies, which could result in neutralization and/or have other unwanted immunological side effects [128].
Importance of binding mechanism and affinity of therapeutic anti-tau antibodies
The binding of antigen by an antibody is effectuated by direct contacts between the antigen epitope and antibody complementarity determining regions (CDRs). The three-dimensional structure of the CDRs and its temporal fluctuations conditioned by the flexibility of the antibody molecule determine (1) the specificity for an epitope, (2) the binding selectivity between various presentations of the epitope and (3) the strength of interaction (the stability of the antibody-antigen complex), where the strength is quantified as association (equilibrium) constant, Ka, or its reciprocal quantity, the dissociation constant Kd. All these aspects are interconnected, where the latter, quantified strength of interaction, is being used for the determination of previous two, i.e. specificity and selectivity.
According to the available data, not all three of the abovementioned aspects were evaluated for all the anti-tau therapeutic antibodies. The specific epitopes are the best characterized and thoroughly described in a recent review [189, 244]. They comprise linear, conformational or phosphorylation-dependent sites on tau [302]. The second aspect, selectivity towards pathogenic presentation of the epitope, is important for both the safety and the efficacy of the anti-tau therapy. This avoids the side effects caused by knocking out healthy tau and focuses the antibody action towards the initial and/or the most toxic pathological tau forms. In this respect, some of the antibodies have had claims for their selectivity for pathological tau at various stages of tau neurodegeneration, e.g., MC1 for a conformation associated with tau filaments [99, 159], ACI-5400 for a phospho-epitope inducing a pathological conformation [321] or DC8E8 for multiple epitopes selectively presented on conformational ensemble of pathogenic truncated tau [168, 243]. The third aspect, interaction strength, has been frequently evaluated by relative quantification on western blot, or, more precisely, by ELISA. For an absolute quantification the surface plasmon resonance (SPR) technique has been used.
A confusing aspect of quantification of binding strength arises in the distinction between monovalent and multivalent arrangement of the quantification protocol. A full-length monoclonal antibody of IgG class contains two binding sites for the antigen. For determination of binding strength, one has to measure KA or KD of interaction of one binding site with one epitope on the antigen molecule, e.g. using monovalent antibody Fab. This quantity is commonly called as antibody affinity. The affinity is a constant value, characteristic for the given antibody binding site – antigen epitope pair, and may be used for an unbiased comparison of antibody binding strength. Affinity is independent of the spatial arrangement of antigen. If performed properly, it is independent on the design of the measurement.
The strength of binding of a whole IgG molecule, which is bivalent, may be expressed equally as a KA or KD, but with this we measure the avidity of antibody. The avidity is not a constant and depends on the availability of the antigenic epitopes in the vicinity of both IgG antibody binding sites simultaneously. When an epitope is present at a high local concentration (that is, at a high areal/spatial density), e.g. on a surface (during Western blotting, on the ELISA plate/SPR sensorchip with a high density of immobilized protein etc.) or on the polymerized antigen (tau filaments), the overall level of bound antibody may be very high with the probability that at least one of the antibody binding sites can at any one moment be bound to the antigen.
Antibody avidity is effective in situ (in the inter-neuronal space) towards protein particles with a high spatial density of its epitopes (e.g. oligomerized, aggregated and filamentous tau, but not monomeric tau). Generally, the avidity of a mature, functional antibody can reach extreme values, ranging from 10− 12 to 10− 15 M (picomolar to femtomolar), whereas the affinity of a single antibody binding site is proportionally lower, in the range of 10− 8 to 10− 10 M (nanomolar to subnanomolar). It is of note that the immune system employs an affinity ceiling at ~ 10− 10 M during antibody maturation, eliminating the antibodies with excessively high affinities, that are not beneficial for the organism [22]. It was postulated that for therapeutic antibodies for tauopathies, a strong selectivity towards pathological tau may be more important than high affinity [72, 301].
Whereas affinity, the constant measure characteristic for a given antibody-antigen pair can be quantified reproducibly on different SPR instruments in different laboratories, using various immobilization chemistries and a range of time kinetic protocols, the avidities are more difficult to reproduce with a new sensorchip or with different arrangement of measurement, because they are intrinsically dependent on the conditions of measurements. It is known that a low flow rate used in SPR could artificially decrease the dissociation rate constant and therefore increase the affinity due to rebinding events [234]. Equally, the amount of protein on the chip could also increase rebinding and mass transport artefacts [235].
Reactivity of anti-tau antibodies HJ8.5, HJ9.4 and HJ9.3 were measured at conditions where the avidity was effective due to the use of bivalent full-length antibodies, and a very high density of tau epitopes on the surface of sensorchip [375]. Therefore, determined values represent avidity rather than affinity. Reactivity of antibody ACI-5400 was also measured with bivalent full-length antibody, but with a low density of epitopes on the sensorchip [321]. Therefore, the determined value likely corresponds to the affinity; although a correction for a bivalent analyte has to be performed. Antibody DC8E8 was measured with low densities of antibody on the sensorchip, therefore, strictly under conditions measuring affinity, and thus, the values represent affinities [167] (Table 2).
For unbiased comparison of binding strength and specificity of candidate therapeutic anti-tau antibodies, the affinity should be strictly used. Binding of therapeutic antibody to oligomerized tau protein species in the interstitial brain space would benefit from increased avidity of a bivalent antibody, assuming that the antibody epitope is present on the polymerized tau in sufficiently high spatial density. The latter requirement might be fulfilled for repeat region-directed antibodies, as the repeat region is the constitutive component of the core structure of assembled tau [99, 242]. The avidity enhancement for binding of N-terminal anti-tau antibodies like HJ9.4 and HJ8.5 is compromised from two reasons: (1) the N-terminal part of tau is not regularly arranged in the tau polymers, but rather forms a fuzzy coat [99] and (2) a significant portion of high molecular weight tau species in the Alzheimer’s brain is N-terminally truncated [384] and may be lacking the antibody epitopes.
Tau therapeutic vaccines
Like their passive immunotherapy counterparts, active vaccines targeting the mid-region, microtubule binding domain and C-terminus have been extensively investigated in preclinical studies (Table 2). Most of these studies demonstrated reduction in tau pathology [14, 30, 167, 270, 274, 322] along with improvement in cognitive or sensorimotor abilities in animals [36, 37, 167, 322, 326] (Table 3).
Interestingly, the majority of preclinical studies with tau active vaccines have paid only marginal attention to the characterization of the antibody response induced by the vaccines. It should be emphasized, that the primary goal of all designed tau vaccines is antibody-mediated protection. The quantity and quality of the vaccine antibodies may represent a critical correlate of the efficacy of tau vaccines. In general, the measurement of titer or concentration by ELISA is the widely accepted approach for quantification of antibody response in body fluids [66, 369]. Unfortunately, there is still no agreement on the optimal methods for measurement of anti-tau antibodies, or how the results of such assays should be reported [3]. Many preclinical studies of the tau vaccines have analysed the antibody response in a rather descriptive manner as “good, robust, high or low”, and did not elaborate on its quantitative aspect [14, 37, 270, 322]. Only two studies published so far, have defined the titer of the antibody response [167, 274]. There is an urgent need for development of common standards for the measurement of antibody response with the most sensitive and reproducible methods. This will allow us to perform a direct comparison of antibody responses between different assays and different clinical trials [3]. Another determining factor of vaccine efficacy is quality of vaccine-induced antibodies (e.g., their isotypes, affinity/avidity, target epitope, functional activity). For example, antibody isotype already more or less indicates antibody affinity. Moreover, to some extent, affinity reflects therapeutic effectivity of antibody.
In comparison with passive tau immunotherapy, there are only two tau active vaccines that have been tested in human clinical trials, AADvac1 for Alzheimer’s disease and non-fluent primary progressive aphasia (Axon Neuroscience SE), and ACI-35 vaccine for Alzheimer’s disease (AC Immune SA, Janssen). Active vaccine AADvac1 consists of tau peptide (aa 294-305/4R) that was coupled to keyhole limpet haemocyanin (KLH) in order to stimulate production of specific antibodies. The 24-week first-in-man study on AADvac1 in patients with mild to moderate AD dementia demonstrated encouraging results in both safety and immunogenicity. Twenty nine of 30 patients developed an IgG response against the tau peptide component of AADvac1 and against recombinant pathological tau (aa151-391/4R) [381]. The serum antibodies showed a pronounced preference for pathological truncated tau over healthy full-length tau protein [245]. Similarly, a 72-week open-label single arm interventional follow-up trial (FUNDAMANT) displayed a benign safety profile of the vaccine. No cases of meningoencephalitis or vasogenic oedema were observed. There was a tendency towards slower atrophy in MRI and less decline in cognitive assessment in patients with high titres [243]. Currently, a phase II clinical trial in AD and a phase I trial in non-fluent primary progressive aphasia are underway (alzforum.org) (Fig. 3).
Much less is known about the ACI35 clinical trial. ACI-35 is a liposome-based vaccine consisting of a synthetic peptide to mimic the phospho-epitope of tau at residues pS396/pS404 anchored into a lipid bilayer. A phase 1b multi-centre double-blind randomized placebo-controlled trial in 24 patients with mild to moderate Alzheimer’s disease compared low, medium, and high doses of the vaccine to placebo.
Active immunization is long lasting because it induces immunological memory. Active vaccines are easy to administer (different routes) and the production is cost-effective. Immunisation generates polyclonal response; antibodies can recognize multiple epitopes on the target protein with different affinity and avidity. On the other hand, the immune response depends on the host immune system, there is a variability in the antibody response across patients [128, 353].
Antisense therapies for tauopathies
Direct targeting of tau gene (MAPT) expression is gaining currency as a therapeutic approach with an antisense oligonucleotide (ASO) therapy already in Phase I clinical trials. Several in vivo and cell studies have demonstrated the benefit of tau reduction in slowing pathological progression and improving functional deficits in tauopathy models both dependent and independent of ß-amyloid pathology. Tau reduction also results in significant improvements in seizures associated with AD pathology and in a model for Dravet syndrome [112].
The fibrillar tau pathology in tauopathy brains consists of abnormally hyperphosphorylated tau protein [169, 360]. Normal phosphorylation and dephosphorylation of residues within and flanking the microtubule (MT)-binding repeat domain (MTBR) mediates the dynamic binding and release of tau from MTs [303]. Hyperphosphorylation could cause or be the result of aberrant release of tau from MTs, with hyperphosphorylated tau unable to bind to MTs [41]. The resulting surplus of unbound tau together with localised concentrations, could lead to the triggering of pathological conformational conversion of tau to a seed-competent form [228] and the initiation of the aggregation cascade that leads to the fibrillar tau accumulations.
The genetics of tau have informed us on the role of tau defects as directly contributing to neurodegeneration. The early dominance of Aß and the amyloid hypothesis [292] subsumed tau to a consequence or bystander in the AD pathogenesis cascade. However, it was clear that the spread and severity of tau pathology better correlated with clinical progression of AD [40, 116, 126]. The identification of mutations in the tau gene (MAPT) that cause familial forms of FTLD with tau pathology (FTLD-tau) [147, 313] cemented the primary role of defective tau as a neurodegenerative agent. From these genetic studies, the identification of common genetic variation in MAPT emerged, defining the H1 haplotype, that is a strong risk factor for primary tauopathies with dominant 4R-tau pathology, progressive supranuclear palsy (PSP; OR = 5.46) [19, 139, 260] and corticobasal degeneration (CBD; OR = 3.7) [139, 147, 171] and, more surprisingly, Parkinson’s disease (OR = 0.77) [306].
The FTLD-tau mutations in MAPT fall into two broad classes; missense mutations that chiefly affect residues within the MTBR that impair microtubule binding capacity and/or increase fibrillogenicity of tau, and splicing mutations in intronic sequences flanking the alternatively spliced exon 10 and in splicing regulatory motifs within exon 10 [147]. The latter cause increased inclusion of exon 10 and ensuing increased ratio of tau isoforms with four MTBRs (4R-tau) over those containing three MTBRs (3R-tau) [118]. The splicing of MAPT exons 2, 3 and 10 is developmentally regulated, and in the healthy adult brain, there are about equal amounts of 3R- and 4R-tau [117, 170]. The basis of the increased risk conferred by the H1 haplotype of MAPT and its defining common polymorphisms, spanning the entire gene and beyond, could be the demonstrated allele-specific differences in transcription [233] and of splicing of exons 3 and 10 of the MAPT pre-mRNA [50, 233]. The result is an overall increase in tau levels, particularly the more fibrillogenic 4R-tau, leading to the 4R-tau dominated pathology seen in PSP and CBD [195]. Furthermore, it was shown that 17q21.31 duplication leads to early-onset dementia with an AD clinical phenotype [178].
Therapeutic reduction of tau
Surplus availability of unbound tau, particularly of the more fibrillogenic mutants or 4R-tau could, with abnormal hyperphosphorylation, lead to mislocalisation and aberrant interaction with other cellular components and milieux. This leads to conformational conversion of tau from its highly soluble, intrinsically disordered characteristic to the seed-competent aggregation-prone form [228]. This has led to the notion that reduction of total tau (or surplus 4R-tau) could be therapeutically beneficial. Although the recent stable of passive immunotherapy approaches targeting tau could be blockading intercellular transmission of pathological tau seeds, a plausible mechanism could also be a reduction of pathological tau mediated by microglial or neuronal uptake and clearance of extracellular tau-antibody complexes [107, 210, 223].
Several published pre-clinical studies with cell and animal models of AD and tauopathies have persuasively demonstrated the possible therapeutic benefit of tau reduction (Table 4). An ASO-based approach already having entered Phase I of clinical trials [227]. In early work, SantaCruz and colleagues demonstrated recovery of memory function and reduced neuronal loss after conditional repression of tau expression in the rTg4510 mouse [282]. Reduction of endogenous tau levels in AD mouse models overexpressing human amyloid precursor protein (hAPP) with familial AD mutations dose-dependently ameliorated Aß-related learning and memory deficits and protected the mice from early mortality [152, 275]. The benefit of tau reduction occurred without influencing Aß burden suggesting that tau reduction uncouples Aß from downstream pathogenic mechanisms [275] including the prevention of Aß-induced defects in axonal transport [341]. Other mouse studies have also shown tau reduction-mediated mitigation of cognitive deficits as a consequence of mild repetitive brain injury [57], or type-1 diabetes [1].
With excitotoxicity implicated in AD, and increased incidence of seizures in AD patients [11], tau reduction also prevented increased susceptibility of hAPP mice to evoked seizures [275]. This protection extended to seizures independent of AD pathology with ASO-mediated knockdown of endogenous tau in adult non-transgenic mice [81] and in mouse (Kcna1−/−) and Drosophila (kcc and eas) models of hyperexcitability [141] as well as a mouse model for Dravet syndrome [112].
Antisense therapies
This is an exciting juncture in the hunt for therapies against neurodegenerative disorders by directly targeting those causative genes. The efficacy and safety of ASO therapy has been demonstrated in clinical trials for nusinersen (Spinraza®; ClinicalTrials.gov Identifier: NCT02193074) for the treatment of spinal muscular atrophy (SMA) and eteplirsen (Exondys51®; NCT00844597, NCT01396239/NCT01540409, NCT02255552) to treat Duchenne muscular dystrophy (DMD). More recently, IONIS-HTTRx (RG6042; NCT02519036) was tested for the treatment of Huntington’s disease (HD) [317]. This specifically targets the mutant, expanded huntingtin gene (HTT) mRNA and supresses its expression. A recent Phase 1/2a clinical trial with intrathecal delivery of the ASO has had no adverse drug-related incidents and showed promising reduction of mutant HTT mRNA levels in CSF [317].
ASOs are short, single-stranded oligonucleotides (8-50 nucleotides) that are designed to bind with complete specificity to complementary sense pre-messenger RNA (mRNA) or mature mRNA sequences. Depending on design and binding site, they could mediate degradation of the target mRNA or prevent translation and thus attenuate protein production. Gene down-regulation by ASOs exploits cellular mechanisms either via RNA interference (RNAi) and the degradation of the target mRNA by RNA-induced silencing complex (RISC), or by recruitment RNase H1 to degrade mRNA at the site of the DNA-RNA duplex. Owing to their size and highly charged nature, ASOs present challenges in terms of cellular uptake, stability and susceptibility to degradation by nucleases and, particularly with CNS targeted therapies, overcoming the blood-brain barrier (BBB). These can in part be overcome by chemical modifications of the DNA or RNA phosphodiester backbone or ribose sugar [190] and the use of the likes of viral vectors, liposomes, polyplexes, or cell-penetrating peptides to enhance delivery [96, 222, 367].
Based on the striking success and safety profile of recent ASO-based clinical trials and, and the recent in vivo ASO-based tau reduction work by de Vos and colleagues [80], a clinical trial of IONIS-MAPTRx (BIIB080, ISIS 814907), the first ASO targeting tau in mild AD patients, is currently under way [ClinicalTrials.gov Identifier: NCT03186989]. Via repeated intrathecal delivery, it appears that this ASO can overcome the BBB in non-human primates with about 75% reduction of MAPT mRNA in both hippocampus and cortex and no dose-limiting side-effects [227].
As shown with nusinersen in SMA and eteplirsen in DMD, ASOs could also be used to target splice acceptor or donor sites or splicing enhancers or repressors to block or enhance splicing of alternatively spliced exons [69, 190]. SMA is caused by survival motor neuron 1 (SMN1) gene mutation causing loss of SMN1 protein, resulting in loss of motor neuron function [202]. The intrathecally administered ASO targets the paralogous SMN2 pre-mRNA, promoting inclusion of exon 7 and production of active SMN in place of the depleted SMN1 product [307]. DMD is a fatal X-linked recessive neuromuscular disorder characterised by progressive muscle weakening and wasting caused by disruptive mutations throughout the large (79 exon) DMD gene [203]. ASO approaches for DMD, including eteplirsen, are designed to induce exon skipping, thereby excluding dispensable downstream exons and avoiding exons with disruptive loss-of-function frame-shift or splice site mutations, while still producing an internally truncated, partially functional protein [190].
Noting the pathogenic role of increased availability of 4R-tau due to exon 10 mutations in FTLD-tau and the MAPT H1 haplotype in PSP and CBD, rebalancing exon 10 is also being tested [276, 287]. This includes ASO-based targeting of exon 10 splice motifs leading to exon-skipping and reduced 4R-tau [287], or reprogramming using a spliceosome-mediated trans-splicing (SMaRT) technique that acts by creating a hybrid mRNA through a trans-splicing reaction between the MAPT pre-mRNA and a pre-trans-splicing molecule, comprised of a binding domain that hybridises with the 3′ end of intron 9 and exons 11–13, designed to exclude exon 10 [276].
MAPT-AS1 natural antisense transcript as a physiological repressor of tau expression
In addition to ASOs, we have seen recent upsurge in our understanding of natural antisense transcripts (NATs). These are endogenous RNA molecules formed by antisense transcription at coding genes and play multi-layered role(s) in regulation of expression of their paired coding gene [347]. The MAPT-AS1 long non-coding RNA (lncRNA) gene partially overlaps head-to-head with the promoter and 5′ untranslated region (5’-UTR) of MAPT and by alternative splicing and use of alternate exons and splice sites, expresses multiple NATs (tau-NATs) [305]. Both in vitro and in vivo, some of the tau-NATs potently repress tau translation [305]. This presents a novel, physiological repressor of tau protein acting in the cytoplasm that, unlike synthetic ASOs, does not rely on RISC or RNAseH and is amenable to adeno-associated virus (AAV) vector-based delivery. Several clinical trials using AAV vectors, including intracranial delivery, have been shown to be safe [137]. Widespread CNS distribution and persistence for up to 10 years and no adverse effects [188] could imply treatment limited to a single delivery unlike ASOs where in ongoing clinical trials, involve repeat intrathecal injection of large doses, every few weeks over several months.
Consequences of tau reduction
Given the importance of tau in multiple facets of neuronal function, mainly by its role in axonal MT assembly and stabilisation and mediation of axonal transport, deficits in tau could have undesirable consequences. Mice that completely lack tau have normal learning and memory and cognition [191, 230, 275], with a minor, variable motor phenotype in later life [186, 191, 230, 330]. On the other hand, it is important to note, that tau deletion was shown to be associated with brain iron accumulation, brain insulin resistance and deficits in synaptic plasticity and cognition [6, 185, 218]. However, observations in knockout models could be hampered by developmental compensation by other MT-associated proteins such as MAP1B [134, 318] and it is thus crucial to understand the consequences of tau knockdown, post-development, in the adult brain. In one recent study, bilateral hippocampal shRNA mediated knockdown of tau in adult mice caused significantly impaired motor coordination and spatial memory accompanied by reduced synaptic markers and dendritic spine density. Behavioural deficits were restored once tau repression was removed [332]. However, in other studies, generalised knockdown of CNS tau in adult mice caused no deviations in normal sensory, motor or cognitive tasks [82]. Based on these mixed findings, it would be important to ascertain the tolerability of different levels of tau knockdown – it is perceivable that partial knockdown of tau in the adult brain could be beneficial, and yet minimising any undesirable effects.
Anti-aggregation agents
Hyperphosphorylated and truncated tau protein is susceptible to aggregation and loss of cytoskeletal microtubule-stabilizing properties, leading to neuronal damage and cell death. Compounds able to prevent aggregation may represent a promising strategy for effective treatment of Alzheimer’s disease [162, 356]. Two major approaches focus on phosphorylation of tau and prevention of tau oligomerization. The former involves the search for inhibitors of kinases which phosphorylate tau or phosphatase activators which dephosphorylate the protein [5, 189]. The latter seeks direct inhibitors of the tau aggregation process.
Regulation of tau phosphorylation
Phosphorylation of tau is under tight control of various protein kinases and phosphatases [5, 189]. Among them, glycogen synthase kinase 3β (GSK-3β) and phosphatase 2A (PP2A) are two key enzymes involved in regulation of the phosphorylation state of tau. GSK-3β is a multitasking serine/threonine kinase largely expressed in CNS that phosphorylates tau mainly at the Ser199, Ser396 and Ser413 sites [16]. Furthermore, it has been shown that an increase in GSK-3β activity induces Αβ formation and is also implicated in other processes, including neuroinflammation and apoptosis [51]. Therefore, GSK-3β is validated as a therapeutic target for AD, and several chemical classes of GSK-3β inhibitors have been discovered and developed in preclinical [217, 253] or even clinical trials. Tideglusib (NP031112, NP-12), is an ATP non-competitive GSK-3β inhibitor demonstrated to reduce spatial memory deficits in transgenic mice in preclinical studies [76]. While it has reached clinical trials, no satisfactory therapeutic results were obtained during phase II.
Tau phosphorylation is also regulated by O-GlcNAcylation, a non-canonical glycosylation involving the attachment of single O-linked N-acetylglucosamine (O-GlcNAc) moieties to serine and threonine residues [376]. O-GlcNAcylation is regulated by two enzymes, O-GlcNAc transferase catalyzing the transfer of GlcNAc to proteins, and N-acetylglucosaminidase (OGA) catalyzing the removal of GlcNAc from proteins [377]. Thiamet-G – a potent OGA inhibitor, that can influence O-GlcNAc levels in the brain, reduced tau phosphorylation in the brain after intraventricular administration [377]. This finding was successfully replicated in additional study, where Thiamet-G prevented the progression of hyperactivity, slowed brain atrophy, and reduced brain hyperphosphorylated tau in tau transgenic model TG4510 [345]. Similarly, ASN120290 – a small molecule that inhibits O-GlcNAcase reduced tau phosphorylation and the number of neurofibrillary pathology in the brain of transgenic mice P301L. ASN120290 which received Orphan Drug Designation from the Food and Drug Administration (FDA) for PSP, has already initiated Phase I clinical trials.
Inhibition of tau aggregation
The most common direct inhibitor of tau protein aggregation is methylene blue (MB), which belongs to the class of thiazine dyes. Methylene blue, also known as methylthionine chloride (MTC), was originally synthesized at the end of the nineteenth century and used to treat malaria. It later found use as an antibacterial, antiviral and anticancer agent, applied in the treatment of various disorders. It is worth noting that its structure has played an important role in the development of phenothiazine-like compounds, including antipsychotic and antihistamine drugs. The anti-aggregating effect of phenothiazines upon the tau protein, discovered by Wischik and co-workers [355] over twenty years ago, paved the way for a new class of for potential anti-AD agents. However, during this time, most researchers focused on β-amyloid targets, and over the next two decades few achievements concerning tau were reported. Nonetheless, several chemical classes of tau aggregation inhibitors have been synthesized and presented [47, 48].
The tau aggregation inhibitor, methylene blue, occurs in two main forms, which are in equilibrium, depending on the redox potential of the solution. The first oxidized cationic state is characterized by a dark blue color, while the second (reduced form) is colorless and also referred to as leucomethylene blue (leuco-methylthioninum, LMT). Structurally, MTC is an aromatic anthracene compound (polyaromatic), whereas LMT is classified as a phenothiazine. It has been shown that anthracene-type compounds inhibit the tau protein, while phenothiazines, with nonaromatic tricyclic structures are inactive in this respect. MTC acts as a prodrug, and in acidic pH converts into leuco-methylthioninium, which can penetrate the BBB and reach brain tissues [17]. Many studies have demonstrated that MTC has a broad spectrum of pharmacological activity [251, 319]. The inhibition of tau aggregation by MTC has been confirmed by numerous in vitro tests as well as in in vivo models in transgenic mice. The dye has properties which inhibit microtubule assembly, prevent tau interaction, inhibit β-amyloid aggregation as well as α-synuclein aggregation. MTC counteracts mitochondrial damage caused by oxidative stress; it also has a positive effect on regulation of autophagy, acetylcholine E (AchE) inhibition, monoamine oxidases, the glutamatergic system and inhibition of noradrenaline uptake. From the point of view of potential clinical applications, the most important properties of MTC include: inhibition of microtubule formation, improvement of mitochondrial oxidation and inhibition of monoamine oxidase A [239].
In clinical trials, MTC was introduced under the name Rember™ (TauRx Therapeutics) as a potential anti-AD drug candidate. Some improvements in AD-related symptoms have been reported, but the drug failed phase II trials due to undesirable side effects, including diarrhoea, urgency, painful urination, dizziness and others (Clinical Trial Identifier, NCT00515333 and NCT00684944). Results of these studies prompted researchers to develop a new generation of MTC derivatives. These new compounds (LMTX) include leuco-methylthionium bis (hydro-methanesulfonate (LMTM) and leuco-methylthionium dihydrobromide (LMTB) – stable, reduced forms that permit direct absorption of LMT without the need for the aforementioned conversion step (Fig. 3).

Chemical structures of methylene blue derivatives
LMTM (TRx0237) has reached phase III trials, and was better absorbed, with improved safety and tolerability compared to methylene blue (Rember™). Nevertheless, results of Phase III clinical trials involving LMTM in the treatment of AD were disappointing as they did not yield unambiguously positive data. The first phase III trial (NCT01689246) included 891 participants with mild to moderate AD, who received 125 mg of LMTM twice a day, or 75 mg twice a day while the control group received 4 mg twice a day. No significant difference in cognitive faculties or the ability to perform daily activities was observed between the treatment and control groups [110]. Due to the low number of participants (79) in this study, these results require further confirmation. Currently, TauRx has begun a new clinical trial (LUCIDUTY, NCT03446001) using FDG-PET imaging to examine the potential of LMTX in delaying the progression of pathological changes in the brain in AD patients who do not receive cholinesterase inhibitors or memantine. This trial is aimed at patients with early AD, with treatment lasting for 9 months (at doses of 8 mg/day and 16 mg/day). Thus, LMTM is being developed as an anti-AD treatment option based on inhibition of tau aggregation. Moreover, LMTC has demonstrated amelioration of α-synuclein pathology in a transgenic mouse model of synucleinopathy, and may therefore find use as a potential disease modification therapy in Parkinson’s disease (PD) and other synucleinopathies [290].
Since the discovery of the tau aggregation inhibitory activity of methylene blue, several chemical classes of compounds have been identified. These include derivatives of phenothiazines, polyphenols, benzothiazoles and porphyrins [319]. It has been observed that all these tested derivatives inhibited both tau filament formation and Aβ fibril formation. Further research carried out by Bulic and E. Mandelkow [47, 48], based on screening of a random library of 200,000 compounds, led to the identification of new chemical structures for potential tau inhibitors, including rhodamines, phenylthiazolyl-hydrazides, N-phenylamines, anthraquinones, benzothiazoles. Using quantitative high-throughput screening, Crowe and co-workers [70] discovered that aminothienopyrydazines (AZPZs) also inhibit of tau assembly.
Another potential source of anti-aggregation agents is provided by the multi-target-directed ligand approach. This strategy is suitable for complex diseases such as Alzheimer’s disease [18, 83, 264]. Therefore, many multifunctional compounds have been obtained by combining various pharmacophores targeting neurodegenerative processes into a single molecule. Among them multimodal molecules have been discovered that are endowed with tau aggregation inhibitory activity as well as other desirable properties. Selected examples of multifunctional agents are presented below.
Compound AZP2006, an N,N′-disubstituted piperazine [226, 297], reduces the release of Aβ species and targets both amyloid and tau pathologies. It was demonstrated to improve cognitive faculties in various mouse models of both amyloid and tau pathology [21]. AZP2006 underwent phase I clinical trials on AD, and has now been classed as an orphan drug for the treatment of progressive supranuclear palsy (PSP). Another new compound, named RPEL, is a piperazine derivative that contains the pharmacophore fragment of tacrine [226] (Fig. 4). This dual-action compound showed inhibitory potency against cholinesterase (IC50 hAChE = 0.8 nM), reduced the phosphorylation of tau protein and inhibited the release of the Aβ peptide. Moreover, it displayed in vivo potency in transgenic mouse models and reduced memory loss.

Multifunctional derivatives of piperazine
Japanese researchers [246, 247] presented a new tau inhibitor compound, PE859, based on the curcumin structure (Fig. 5). Promising results were obtained in both in vitro and in vivo studies – the compound was shown to counteract tau aggregation and prevent the onset and progression of nerve dysfunction in an in vivo model. Furthermore, it inhibits both tau and Aβ aggregation and alleviates cognitive dysfunction in vivo.

Structure of curcumin derivative PE859 dual tau and β-amyloid inhibitor
Two carbazole-based cyanine compounds named SLM and SLOH were described as strong inhibitors of Aβ aggregation in vitro and were able to alleviate the pathological symptoms and memory deterioration in AD model mice [364,365,366] (Fig. 6). These multifunctional compounds also reduced tau hyperphosphorylation as well as significantly attenuated neuroinflammation through inhibitiion of GSK-3β activity. They showed a good pharmacokinetic profile, with high BBB permeability, which justifies their further development as AD drug candidates [379].

Structure of multifunctional carbazole–based cyanine compounds
Dual inhibitors acting against β-secretase (BACE1) and glycogen synthase kinase 3β (GSK-3β), with well-balanced in vitro activity (in μM range), were synthesized in the class of triazinone derivatives [265]. These compounds displayed strong neuroprotective and neurogenic effects, and also showed good BBB permeability in a pharmacokinetic evaluation in mice. A new multi-target strategy for designing anti-AD agents involves compounds which combine GSK-3β and tau aggregation inhibitors [109]. Derivatives of 2,4-thiazolidinedione showed activity against GSK-3β (at micromolar IC50 values) and were also found to inhibit tau aggregation. Other examples of multifunctional compounds include rhein-huprine hybrids, which showed AChE and BACE1 inhibitory activity, as well as Aβ1-42 and tau anti-aggregating properties [259]. A 1-benzylamino-2-hydroxyalkyl derivative with a diphenylpiperazine fragment, selected form a series of compounds, showed balanced inhibitory activity against both disease-modifying targets, inhibition of BACE1, inhibition of Aβ, inhibition of tau aggregation, as well as inhibition of BuChE as a symptomatic target [254]. Jiang and co-workers [158] described a new class of dual GSK-3β and AChE inhibitors. These multifunctional compounds were designed by incorporating a tacrine fragment at the thiazolyl ring, as the pharmacophore responsible for GSK-3β inhibition. The resulting derivatives were very potent inhibitors of both targets (in the nanomolar range). The most promising compound from this series significantly inhibited tau protein phosphorylation and counteracted self-aggregation of Aβ1-42. In addition, it was not toxic and proved effective in an in vivo assay in mice, by significantly improving memory.
Most of the above-described direct tau inhibitors and multifunctional compounds have shown activity in in vitro tests, but only some of them have been evaluated in vivo in extended pharmacological, preclinical studies. Moreover, it is difficult to predict further development of these compounds. Due to the complex nature of AD, it seems reasonable to pursue development of combination therapies, as well as new alternative approaches which involve multi-target drugs. It is probable that a molecule capable of acting on two recognized targets, with one of them belonging to the tau cascade, might bring clinical benefits compared to drugs which only address only specific target.
Concluding remarks and future directions
Tau is a multifaceted protein with a plethora of physiological functions. In the disease condition, tau protein drives neurodegeneration and causes neurodegenerative disorders such as Alzheimer’s disease. Pathologically modified tau has become an important therapeutic target for AD and related tauopathies. Although no disease-modifying treatments are yet available, many new therapeutic approaches targeting pathological forms of tau are being tested in clinical trials. Disease-modifying therapy is aimed at preventing, slowing or ameliorating the production, oligomerisation, aggregation and deposition of pathological tau protein. The most promising therapeutic strategies include active tau vaccines and therapeutic monoclonal antibodies. Besides immunotherapy, there are many other therapies currently being explored in the treatment of tau neurodegeneration such as modulation of tau phosphorylation, inhibition of tau aggregation or regulation of its expression. While waiting for the results of ongoing clinical trials, we can continue to unravel the complexities of tau proteome and different biological functions of this peculiar brain protein.
Abbreviations
- 1 N:
-
First insert
- 2 N:
-
Second insert
- 3R:
-
Three repeat
- 4R:
-
Four repeat
- Aβ:
-
β-amyloid
- aa:
-
Amino acids
- AAV:
-
Adeno-associated virus
- AChE:
-
Acetylcholine E
- AD:
-
Alzheimer’s disease
- AGD:
-
Argyrophilic grain disease
- ApoE4:
-
Apolipoprotein E4
- APP:
-
Amyloid precursor protein
- ARTAG:
-
Age-related tau astrogliopathy
- ASO:
-
Antisense oligonucleotide
- BBB:
-
Blood-brain barrier
- BIN1:
-
Myc box-dependent-interacting protein 1
- CDB:
-
Corticobasal degeneration
- CDRs:
-
Complementarity determining regions
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- C-terminal:
-
Carboxy-terminal
- DMD:
-
Duchenne muscular dystrophy
- EOAD:
-
Early-onset Alzheimer’s disease
- FDA:
-
Food and Drug Administration
- FDG:
-
Fluorodeoxyglucose
- FGF-2:
-
Fibroblast growth factor 2
- FTD:
-
Frontotemporal dementia
- FTLD:
-
Frontotemporal lobar degeneration
- FTP:
-
[18F] Flortaucipir
- GGT:
-
Globular glial tauopathy
- GSK-3β:
-
Glycogen synthase kinase 3β
- HD:
-
Huntington’s disease
- HMW:
-
High molecular weight tau
- HTT :
-
Huntingtin gene
- IRS-1:
-
Insulin receptor substrate 1
- Ka :
-
Association constant
- Kd :
-
Dissociation constant
- KLH:
-
Keyhole limpet haemocyanin
- LMT:
-
Leuco-methylthioninum
- LMTB:
-
Leuco-methylthionium dihydrobromide
- LMTM:
-
Leuco-methylthionium bis(hydro-methanesulfonate
- lncRNA:
-
Long non-coding RNA
- LOAD:
-
Late- onset Alzheimer’s disease
- MAO-B:
-
Monoaminoxidase B
- MAP:
-
Microtubule-associated protein
- MB:
-
Methylene blue
- MIR:
-
Mammalian interspersed repeat
- MRI:
-
Magnetic resonance imaging
- mRNA:
-
Messenger RNA
- MT:
-
Microtubules
- MTBR:
-
Microtubule binding region
- MTC:
-
Methylthionine chloride
- MTL:
-
Medial temporal lobe
- NATs:
-
Natural antisense transcripts
- NFT:
-
Neurofibrillary tangles
- NPC:
-
Nuclear pore complex
- N-terminal:
-
Amino-terminal
- O-GlcNAc:
-
O-linked N-acetylglucosamine
- PART:
-
Primary age-related tauopathy
- PET:
-
Positron emission tomography
- PHF:
-
Paired-helical filaments
- PiD:
-
Pick’s disease
- PP2A:
-
Phosphatase 2A
- PSP:
-
Progressive supranuclear palsy
- PTEN:
-
Phosphatase and tensin homolog
- PTM:
-
Post translational modification
- RISC:
-
RNA-induced silencing complex
- RNAi:
-
RNA interference
- SMA:
-
Spinal muscular atrophy
- SMaRT:
-
Spliceosome-mediated trans-splicing
- SMN:
-
Survival motor neuron
- SPR:
-
Surface plasmon resonance spectroscopy
- SPs:
-
Senile plaques
- TRIM21:
-
Tripartite motif-containing protein 21
References
Abbondante S, Baglietto-Vargas D, Rodriguez-Ortiz CJ, Estrada-Hernandez T, Medeiros R, Laferla FM (2014) Genetic ablation of tau mitigates cognitive impairment induced by type 1 diabetes. Am J Pathol 184:819–826. https://doi.org/10.1016/j.ajpath.2013.11.021
Adams JN, Lockhart SN, Li L, Jagust WJ (2018) Relationships between tau and glucose metabolism reflect Alzheimer’s disease pathology in cognitively Normal older adults. Cereb Cortex. https://doi.org/10.1093/cercor/bhy078
Agadjanyan MG, Petrovsky N, Ghochikyan A (2015) A fresh perspective from immunologists and vaccine researchers: active vaccination strategies to prevent and reverse Alzheimer's disease. Alzheimers Dement 11:1246–1259. https://doi.org/10.1016/j.jalz.2015.06.1884
Agadjanyan MG, Zagorski K, Petrushina I, Davtyan H, Kazarian K, Antonenko M, Davis J, Bon C, Blurton-Jones M, Cribbs DH, Ghochikyan A (2017) Humanized monoclonal antibody armanezumab specific to N-terminus of pathological tau: characterization and therapeutic potency. Mol Neurodegener 12:33. https://doi.org/10.1186/s13024-017-0172-1
Ahmed T, Blum D, Burnouf S, Demeyer D, Buee-Scherrer V, D'Hooge R, Buee L, Balschun D (2015) Rescue of impaired late-phase long-term depression in a tau transgenic mouse model. Neurobiol Aging 36:730–739. https://doi.org/10.1016/j.neurobiolaging.2014.09.015
Ahmed T, Van der Jeugd A, Blum D, Galas MC, D'Hooge R, Buee L, Balschun D (2014) Cognition and hippocampal synaptic plasticity in mice with a homozygous tau deletion. Neurobiol Aging 35(11):2474-2478. https://doi.org/10.1016/j.neurobiolaging.2014.05.005.
Ahmed Z, Bigio EH, Budka H, Dickson DW, Ferrer I, Ghetti B, Giaccone G, Hatanpaa KJ, Holton JL, Josephs KA, Powers J, Spina S, Takahashi H, White CL 3rd, Revesz T, Kovacs GG (2013) Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol 126:537–544. https://doi.org/10.1007/s00401-013-1171-0
Albert M, Mairet-Coello G, Danis C, Lieger S, Caillierez R, Carrier S, Skrobala E, Landrieu I, Michel A, Schmitt M, Citron M, Downey P, Courade JP, Buée L, Colin M (2018) Prevention of tau seeding and propagation by immunotherapy with a central tau epitope antibody. AAIC2018, Chicago, IL, USA.
Amadoro G, Corsetti V, Florenzano F, Atlante A, Ciotti MT, Mongiardi MP, Bussani R, Nicolin V, Nori SL, Campanella M, Calissano P (2014) AD-linked, toxic NH2 human tau affects the quality control of mitochondria in neurons. Neurobiol Dis 62:489–507. https://doi.org/10.1016/j.nbd.2013.10.018
Amadoro G, Corsetti V, Stringaro A, Colone M, D'Aguanno S, Meli G, Ciotti M, Sancesario G, Cattaneo A, Bussani R, Mercanti D, Calissano P (2010) A NH2 tau fragment targets neuronal mitochondria at AD synapses: possible implications for neurodegeneration. J Alzheimers Dis 21:445–470. https://doi.org/10.3233/jad-2010-100120
Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, Albert M, Brandt J, Stern Y (2006) Incidence and predictors of seizures in patients with Alzheimer's disease. Epilepsia 47:867–872. https://doi.org/10.1111/j.1528-1167.2006.00554.x
Ando K, Kabova A, Stygelbout V, Leroy K, Heraud C, Frederick C, Suain V, Yilmaz Z, Authelet M, Dedecker R, Potier MC, Duyckaerts C, Brion JP (2014) Vaccination with Sarkosyl insoluble PHF-tau decrease neurofibrillary tangles formation in aged tau transgenic mouse model: a pilot study. J Alzheimers Dis 40(Suppl 1):S135–S145. https://doi.org/10.3233/jad-132237
Aschenbrenner AJ, Gordon BA, Benzinger TLS, Morris JC, Hassenstab JJ (2018) Influence of tau PET, amyloid PET, and hippocampal volume on cognition in Alzheimer disease. Neurology 91:e859–e866. https://doi.org/10.1212/wnl.0000000000006075
Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM (2007) Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci 27:9115–9129. https://doi.org/10.1523/jneurosci.2361-07.2007
Augustinack JC, Schneider A, Mandelkow EM, Hyman BT (2002) Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol 103:26–35
Avila J, Wandosell F, Hernandez F (2010) Role of glycogen synthase kinase-3 in Alzheimer's disease pathogenesis and glycogen synthase kinase-3 inhibitors. Expert Rev Neurother 10:703–710. https://doi.org/10.1586/ern.10.40
Baddeley TC, McCaffrey J, Storey JM, Cheung JK, Melis V, Horsley D, Harrington CR, Wischik CM (2015) Complex disposition of methylthioninium redox forms determines efficacy in tau aggregation inhibitor therapy for Alzheimer's disease. J Pharmacol Exp Ther 352:110–118. https://doi.org/10.1124/jpet.114.219352
Bajda M, Guzior N, Ignasik M, Malawska B (2011) Multi-target-directed ligands in Alzheimer's disease treatment. Curr Med Chem 18:4949–4975
Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, Hardy J, Lynch T, Bigio E, Hutton M (1999) Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet 8:711–715
Baker SL, Maass A, Jagust WJ (2017) Considerations and code for partial volume correcting [(18)F]-AV-1451 tau PET data. Data Brief 15:648–657. https://doi.org/10.1016/j.dib.2017.10.024
Barrier MBL, Burlet S, Delacourte A, Estrella C, Melnyk P, Sergeant N, Verwaerde P (2014) Sulfate salts of N-(3-(4-(3-(diisobutylamino)propyl)piperazin-1-yl)propyl)-1H-benzo [d]imidazol-2 amine, preparation thereof and use of the same
Batista FD, Neuberger MS (1998) Affinity dependence of the B cell response to antigen: a threshold, a ceiling, and the importance of off-rate. Immunity 8:751–759
Bejanin A, Schonhaut DR, La Joie R, Kramer JH, Baker SL, Sosa N, Ayakta N, Cantwell A, Janabi M, Lauriola M, O'Neil JP, Gorno-Tempini ML, Miller ZA, Rosen HJ, Miller BL, Jagust WJ, Rabinovici GD (2017) Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer's disease. Brain 140:3286–3300. https://doi.org/10.1093/brain/awx243
Betthauser TJ, Cody KA, Zammit MD, Murali D, Converse AK, Barnhart TE, Stone CK, Rowley HA, Johnson SC, Christian BT (2018) In vivo characterization and quantification of neurofibrillary tau PET radioligand [(18)F]MK-6240 in humans from Alzheimer's disease dementia to young controls. J Nucl Med. https://doi.org/10.2967/jnumed.118.209650
Bi M, Ittner A, Ke YD, Gotz J, Ittner LM (2011) Tau-targeted immunization impedes progression of neurofibrillary histopathology in aged P301L tau transgenic mice. PLoS One 6:e26860. https://doi.org/10.1371/journal.pone.0026860
Bischof GN, Endepols H, van Eimeren T, Drzezga A (2017) Tau-imaging in neurodegeneration. Methods 130:114–123. https://doi.org/10.1016/j.ymeth.2017.08.003
Bischof GN, Jessen F, Fliessbach K, Dronse J, Hammes J, Neumaier B, Onur O, Fink GR, Kukolja J, Drzezga A, van Eimeren T (2016) Impact of tau and amyloid burden on glucose metabolism in Alzheimer's disease. Ann Clin Transl Neurol 3:934–939. https://doi.org/10.1002/acn3.339
Blennow K, Wallin A, Agren H, Spenger C, Siegfried J, Vanmechelen E (1995) Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease? Mol Chem Neuropathol 26:231–245. https://doi.org/10.1007/bf02815140
Blennow K, Zetterberg H (2018) Biomarkers for Alzheimer's disease: current status and prospects for the future. J Intern Med. https://doi.org/10.1111/joim.12816
Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H (2010) Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp Neurol 224:472–485. https://doi.org/10.1016/j.expneurol.2010.05.010
Boluda S, Iba M, Zhang B, Raible KM, Lee VM, Trojanowski JQ (2015) Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer's disease or corticobasal degeneration brains. Acta Neuropathol 129:221–237. https://doi.org/10.1007/s00401-014-1373-0
Bondareff W, Wischik CM, Novak M, Amos WB, Klug A, Roth M (1990) Molecular analysis of neurofibrillary degeneration in Alzheimer's disease. An immunohistochemical study. Am J Pathol 137:711–723
Borghgraef P, Menuet C, Theunis C, Louis JV, Devijver H, Maurin H, Smet-Nocca C, Lippens G, Hilaire G, Gijsen H, Moechars D, Van Leuven F (2013) Increasing brain protein O-GlcNAc-ylation mitigates breathing defects and mortality of tau.P301L mice. PLoS One 8:e84442. https://doi.org/10.1371/journal.pone.0084442
Borreca A, Latina V, Corsetti V, Middei S, Piccinin S, Della Valle F, Bussani R, Ammassari-Teule M, Nistico R, Calissano P, Amadoro G (2018) AD-related N-terminal truncated tau is sufficient to recapitulate in vivo the early perturbations of human neuropathology: implications for immunotherapy. Mol Neurobiol 55:8124–8153. https://doi.org/10.1007/s12035-018-0974-3
Bou Samra E, Buhagiar-Labarchede G, Machon C, Guitton J, Onclercq-Delic R, Green MR, Alibert O, Gazin C, Veaute X, Amor-Gueret M (2017) A role for tau protein in maintaining ribosomal DNA stability and cytidine deaminase-deficient cell survival. Nat Commun 8:693. https://doi.org/10.1038/s41467-017-00633-1
Boutajangout A, Ingadottir J, Davies P, Sigurdsson EM (2011) Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J Neurochem 118:658–667. https://doi.org/10.1111/j.1471-4159.2011.07337.x
Boutajangout A, Quartermain D, Sigurdsson EM (2010) Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J Neurosci 30:16559–16566. https://doi.org/10.1523/jneurosci.4363-10.2010
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404. https://doi.org/10.1007/s00401-006-0127-z
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Braak H, Braak E (1995) Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol Aging 16:271–278 discussion 278-284
Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM (1993) Abnormal tau phosphorylation at Ser396 in Alzheimer's disease recapitulates development and contributes to reduced microtubule binding. Neuron 10:1089–1099
Brier MR, Gordon B, Friedrichsen K, McCarthy J, Stern A, Christensen J, Owen C, Aldea P, Su Y, Hassenstab J, Cairns NJ, Holtzman DM, Fagan AM, Morris JC, Benzinger TL, Ances BM (2016) Tau and Abeta imaging, CSF measures, and cognition in Alzheimer’s disease. Sci Transl Med 8:338ra366. https://doi.org/10.1126/scitranslmed.aaf2362
Bright J, Hussain S, Dang V, Wright S, Cooper B, Byun T, Ramos C, Singh A, Parry G, Stagliano N, Griswold-Prenner I (2015) Human secreted tau increases amyloid-beta production. Neurobiol Aging 36:693–709. https://doi.org/10.1016/j.neurobiolaging.2014.09.007
Brough D, Pelegrin P, Nickel W (2017) An emerging case for membrane pore formation as a common mechanism for the unconventional secretion of FGF2 and IL-1beta. J Cell Sci 130:3197–3202. https://doi.org/10.1242/jcs.204206
Buckley RF, Hanseeuw B, Schultz AP, Vannini P, Aghjayan SL, Properzi MJ, Jackson JD, Mormino EC, Rentz DM, Sperling RA, Johnson KA, Amariglio RE (2017) Region-specific Association of Subjective Cognitive Decline with Tauopathy Independent of global beta-amyloid burden. JAMA Neurol 74:1455–1463. https://doi.org/10.1001/jamaneurol.2017.2216
Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 33:95–130
Bulic B, Pickhardt M, Mandelkow EM, Mandelkow E (2010) Tau protein and tau aggregation inhibitors. Neuropharmacology 59:276–289. https://doi.org/10.1016/j.neuropharm.2010.01.016
Bulic B, Pickhardt M, Schmidt B, Mandelkow EM, Waldmann H, Mandelkow E (2009) Development of tau aggregation inhibitors for Alzheimer's disease. Angew Chem Int Ed Engl 48:1740–1752. https://doi.org/10.1002/anie.200802621
Burnouf S, Martire A, Derisbourg M, Laurent C, Belarbi K, Leboucher A, Fernandez-Gomez FJ, Troquier L, Eddarkaoui S, Grosjean ME, Demeyer D, Muhr-Tailleux A, Buisson A, Sergeant N, Hamdane M, Humez S, Popoli P, Buee L, Blum D (2013) NMDA receptor dysfunction contributes to impaired brain-derived neurotrophic factor-induced facilitation of hippocampal synaptic transmission in a tau transgenic model. Aging Cell 12:11–23. https://doi.org/10.1111/acel.12018
Caffrey TM, Joachim C, Wade-Martins R (2008) Haplotype-specific expression of the N-terminal exons 2 and 3 at the human MAPT locus. Neurobiol Aging 29:1923–1929. https://doi.org/10.1016/j.neurobiolaging.2007.05.002
Cai Z, Zhao Y, Zhao B (2012) Roles of glycogen synthase kinase 3 in Alzheimer's disease. Curr Alzheimer Res 9:864–879
Calafate S, Flavin W, Verstreken P, Moechars D (2016) Loss of Bin1 promotes the propagation of tau pathology. Cell Rep 17:931–940. https://doi.org/10.1016/j.celrep.2016.09.063
Camero S, Benitez MJ, Barrantes A, Ayuso JM, Cuadros R, Avila J, Jimenez JS (2014) Tau protein provides DNA with thermodynamic and structural features which are similar to those found in histone-DNA complex. J Alzheimers Dis 39:649–660. https://doi.org/10.3233/jad-131415
Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Lasagna-Reeves CA, Gerson JE, Singh G, Estes DM, Barrett AD, Dineley KT, Jackson GR, Kayed R (2014) Passive immunization with tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J Neurosci 34:4260–4272. https://doi.org/10.1523/jneurosci.3192-13.2014
Chai X, Dage JL, Citron M (2012) Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol Dis 48:356–366. https://doi.org/10.1016/j.nbd.2012.05.021
Chai X, Wu S, Murray TK, Kinley R, Cella CV, Sims H, Buckner N, Hanmer J, Davies P, O'Neill MJ, Hutton ML, Citron M (2011) Passive immunization with anti-tau antibodies in two transgenic models: reduction of tau pathology and delay of disease progression. J Biol Chem 286:34457–34467. https://doi.org/10.1074/jbc.M111.229633
Cheng JS, Craft R, Yu GQ, Ho K, Wang X, Mohan G, Mangnitsky S, Ponnusamy R, Mucke L (2014) Tau reduction diminishes spatial learning and memory deficits after mild repetitive traumatic brain injury in mice. PLoS One 9:e115765. https://doi.org/10.1371/journal.pone.0115765
Cho H, Choi JY, Hwang MS, Kim YJ, Lee HM, Lee HS, Lee JH, Ryu YH, Lee MS, Lyoo CH (2016) In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum. Ann Neurol 80:247–258. https://doi.org/10.1002/ana.24711
Choi JY, Cho H, Ahn SJ, Lee JH, Ryu YH, Lee MS, Lyoo CH (2018) Off-target (18)F-AV-1451 binding in the basal ganglia correlates with age-related Iron accumulation. J Nucl Med 59:117–120. https://doi.org/10.2967/jnumed.117.195248
Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, Probst A, Winkler DT, Reichwald J, Staufenbiel M, Ghetti B, Goedert M, Tolnay M (2013) Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A 110:9535–9540. https://doi.org/10.1073/pnas.1301175110
Collin L, Bohrmann B, Gopfert U, Oroszlan-Szovik K, Ozmen L, Gruninger F (2014) Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer's disease. Brain 137:2834–2846. https://doi.org/10.1093/brain/awu213
Combs B, Hamel C, Kanaan NM (2016) Pathological conformations involving the amino terminus of tau occur early in Alzheimer's disease and are differentially detected by monoclonal antibodies. Neurobiol Dis 94:18–31. https://doi.org/10.1016/j.nbd.2016.05.016
Combs B, Kanaan NM (2017) Exposure of the amino terminus of tau is a pathological event in multiple Tauopathies. Am J Pathol 187:1222–1229. https://doi.org/10.1016/j.ajpath.2017.01.019
Congdon EE, Lin Y, Rajamohamedsait HB, Shamir DB, Krishnaswamy S, Rajamohamedsait WJ, Rasool S, Gonzalez V, Levenga J, Gu J, Hoeffer C, Sigurdsson EM (2016) Affinity of tau antibodies for solubilized pathological tau species but not their immunogen or insoluble tau aggregates predicts in vivo and ex vivo efficacy. Mol Neurodegener 11:62. https://doi.org/10.1186/s13024-016-0126-z
Congdon EE, Sigurdsson EM (2018) Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol 14:399–415. https://doi.org/10.1038/s41582-018-0013-z
Cooper H M and Paterson Y. Determination of specific antibody titer. Current protocols in Molecular Biology. 2000, 11.17.1-11.17.13, John Wiley & Sons, Inc.
Courade J-P, Angers R, Mairet-Coello G, Pacico N, Tyson K, Lightwood D, Munro R, McMillan D, Griffin R, Baker T, Starkie D, Nan R, Westwood M, Mushikiwabo M-L, Jung S, Odede G, Sweeney B, Popplewell A, Burgess G, Downey P, Citron MJAN (2018) Epitope determines efficacy of therapeutic anti-tau antibodies in a functional assay with human Alzheimer tau. 136:729–745. https://doi.org/10.1007/s00401-018-1911-2
Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, Arnold SE, Attems J, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Gearing M, Grinberg LT, Hof PR, Hyman BT, Jellinger K, Jicha GA, Kovacs GG, Knopman DS, Kofler J, Kukull WA, Mackenzie IR, Masliah E, McKee A, Montine TJ, Murray ME, Neltner JH, Santa-Maria I, Seeley WW, Serrano-Pozo A, Shelanski ML, Stein T, Takao M, Thal DR, Toledo JB, Troncoso JC, Vonsattel JP, White CL 3rd, Wisniewski T, Woltjer RL, Yamada M, Nelson PT (2014) Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128:755–766. https://doi.org/10.1007/s00401-014-1349-0
Crooke ST (2017) Molecular mechanisms of antisense oligonucleotides. Nucleic Acid Ther 27:70–77. https://doi.org/10.1089/nat.2016.0656
Crowe A, Huang W, Ballatore C, Johnson RL, Hogan AM, Huang R, Wichterman J, McCoy J, Huryn D, Auld DS, Smith AB 3rd, Inglese J, Trojanowski JQ, Austin CP, Brunden KR, Lee VM (2009) Identification of aminothienopyridazine inhibitors of tau assembly by quantitative high-throughput screening. Biochemistry 48:7732–7745. https://doi.org/10.1021/bi9006435
Cummings J, Lee G, Ritter A, Zhong K (2018) Alzheimer's disease drug development pipeline: 2018. Alzheimers Dement (N Y) 4:195–214. https://doi.org/10.1016/j.trci.2018.03.009
d'Abramo C, Acker CM, Jimenez HT, Davies P (2013) Tau passive immunotherapy in mutant P301L mice: antibody affinity versus specificity. PLoS One 8:e62402. https://doi.org/10.1371/journal.pone.0062402
Dai CL, Chen X, Kazim SF, Liu F, Gong CX, Grundke-Iqbal I, Iqbal K (2015) Passive immunization targeting the N-terminal projection domain of tau decreases tau pathology and improves cognition in a transgenic mouse model of Alzheimer disease and tauopathies. J Neural Transm (Vienna) 122:607–617. https://doi.org/10.1007/s00702-014-1315-y
Das SR, Xie L, Wisse LEM, Ittyerah R, Tustison NJ, Dickerson BC, Yushkevich PA, Wolk DA (2018) Longitudinal and cross-sectional structural magnetic resonance imaging correlates of AV-1451 uptake. Neurobiol Aging 66:49–58. https://doi.org/10.1016/j.neurobiolaging.2018.01.024
de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT (2012) Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73:685–697. https://doi.org/10.1016/j.neuron.2011.11.033
del Ser T, Steinwachs KC, Gertz HJ, Andres MV, Gomez-Carrillo B, Medina M, Vericat JA, Redondo P, Fleet D, Leon T (2013) Treatment of Alzheimer's disease with the GSK-3 inhibitor tideglusib: a pilot study. J Alzheimers Dis 33:205–215. https://doi.org/10.3233/jad-2012-120805
Delaere P, He Y, Fayet G, Duyckaerts C, Hauw JJ (1993) Beta A4 deposits are constant in the brain of the oldest old: an immunocytochemical study of 20 French centenarians. Neurobiol Aging 14:191–194
Derisbourg M, Leghay C, Chiappetta G, Fernandez-Gomez FJ, Laurent C, Demeyer D, Carrier S, Buee-Scherrer V, Blum D, Vinh J, Sergeant N, Verdier Y, Buee L, Hamdane M (2015) Role of the tau N-terminal region in microtubule stabilization revealed by new endogenous truncated forms. Sci Rep 5:9659. https://doi.org/10.1038/srep09659
Derkinderen P, Scales TM, Hanger DP, Leung KY, Byers HL, Ward MA, Lenz C, Price C, Bird IN, Perera T, Kellie S, Williamson R, Noble W, Van Etten RA, Leroy K, Brion JP, Reynolds CH, Anderton BH (2005) Tyrosine 394 is phosphorylated in Alzheimer’s paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase. J Neurosci 25:6584–6593. https://doi.org/10.1523/jneurosci.1487-05.2005
DeVos SL, Corjuc BT, Commins C, Dujardin S, Bannon RN, Corjuc D, Moore BD, Bennett RE, Jorfi M, Gonzales JA, Dooley PM, Roe AD, Pitstick R, Irimia D, Frosch MP, Carlson GA, Hyman BT (2018) Tau reduction in the presence of amyloid-beta prevents tau pathology and neuronal death in vivo. Brain 141:2194–2212. https://doi.org/10.1093/brain/awy117
DeVos SL, Goncharoff DK, Chen G, Kebodeaux CS, Yamada K, Stewart FR, Schuler DR, Maloney SE, Wozniak DF, Rigo F, Bennett CF, Cirrito JR, Holtzman DM, Miller TM (2013) Antisense reduction of tau in adult mice protects against seizures. J Neurosci 33:12887–12897. https://doi.org/10.1523/jneurosci.2107-13.2013
DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, Wegener AJ, Chen G, Shen T, Tran H, Nichols B, Zanardi TA, Kordasiewicz HB, Swayze EE, Bennett CF, Diamond MI, Miller TM (2017) Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med 9. https://doi.org/10.1126/scitranslmed.aag0481
Dias KS, Viegas C Jr (2014) Multi-target directed drugs: a modern approach for Design of new Drugs for the treatment of Alzheimer's disease. Curr Neuropharmacol 12:239–255. https://doi.org/10.2174/1570159x1203140511153200
Díaz-Hernández M, Gomez-Ramos A, Rubio A, Gomez-Villafuertes R, Naranjo JR, Miras-Portugal MT, Avila J (2010) Tissue-nonspecific alkaline phosphatase promotes the neurotoxicity effect of extracellular tau. J Biol Chem 285:32539–32548. https://doi.org/10.1074/jbc.M110.145003
Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, Jellinger K, Lantos PL, Lippa CF, Mirra SS, Tabaton M, Vonsattel JP, Wakabayashi K, Litvan I (2002) Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 61:935–946
Donohue MC, Sperling RA, Petersen R, Sun CK, Weiner MW, Aisen PS (2017) Association between elevated brain amyloid and subsequent cognitive decline among cognitively Normal persons. Jama 317:2305–2316. https://doi.org/10.1001/jama.2017.6669
Dorval V, Fraser PE (2006) Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J Biol Chem 281:9919–9924. https://doi.org/10.1074/jbc.M510127200
Dubois B, Epelbaum S, Nyasse F, Bakardjian H, Gagliardi G, Uspenskaya O, Houot M, Lista S, Cacciamani F, Potier MC, Bertrand A, Lamari F, Benali H, Mangin JF, Colliot O, Genthon R, Habert MO, Hampel H (2018) Cognitive and neuroimaging features and brain beta-amyloidosis in individuals at risk of Alzheimer's disease (INSIGHT-preAD): a longitudinal observational study. Lancet Neurol 17:335–346. https://doi.org/10.1016/s1474-4422(18)30029-2
Dujardin S, Begard S, Caillierez R, Lachaud C, Delattre L, Carrier S, Loyens A, Galas MC, Bousset L, Melki R, Auregan G, Hantraye P, Brouillet E, Buee L, Colin M (2014) Ectosomes: a new mechanism for non-exosomal secretion of tau protein. PLoS One 9:e100760. https://doi.org/10.1371/journal.pone.0100760
Dujardin S, Lachaud C, Caillierez R, Bégard S, Carrier S, Lieger S, Gonzales JA, Moore BD, Deramecourt V, Deglon N, Maurage CA, Frosch MP, Hyman BT, Colin M, Buée L (2018) Different tau species lead to heterogeneous tau pathology propagation and misfolding. Acta Neuropathol Commun 6:132.
Duyckaerts C, Hauw JJ (1997) Diagnosis and staging of Alzheimer disease. Neurobiol Aging 18:S33–S42
Duyckaerts C, Hauw JJ (1997) Prevalence, incidence and duration of Braak's stages in the general population: can we know? Neurobiol Aging 18:362–369 discussion 389-392
Eftekharzadeh B, Daigle JG, Kapinos LE, Coyne A, Schiantarelli J, Carlomagno Y, Cook C, Miller SJ, Dujardin S, Amaral AS, Grima JC, Bennett RE, Tepper K, DeTure M, Vanderburgh CR, Corjuc BT, DeVos SL, Gonzalez JA, Chew J, Vidensky S, Gage FH, Mertens J, Troncoso J, Mandelkow E, Salvatella X, Lim RYH, Petrucelli L, Wegmann S, Rothstein JD, Hyman BT (2018) Tau protein disrupts nucleocytoplasmic transport in Alzheimer's disease. Neuron 99:925–940.e927. https://doi.org/10.1016/j.neuron.2018.07.039
Elahi FM, Miller BL (2017) A clinicopathological approach to the diagnosis of dementia. Nat Rev Neurol 13:457–476. https://doi.org/10.1038/nrneurol.2017.96
Evans LD, Wassmer T, Fraser G, Smith J, Perkinton M, Billinton A, Livesey FJ (2018) Extracellular monomeric and aggregated tau efficiently enter human neurons through overlapping but distinct pathways. Cell Rep 22:3612–3624. https://doi.org/10.1016/j.celrep.2018.03.021
Evers MM, Toonen LJ, van Roon-Mom WM (2015) Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv Drug Deliv Rev 87:90–103. https://doi.org/10.1016/j.addr.2015.03.008
Ferrer I, Lopez-Gonzalez I, Carmona M, Arregui L, Dalfo E, Torrejon-Escribano B, Diehl R, Kovacs GG (2014) Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol 73:81–97. https://doi.org/10.1097/nen.0000000000000030
Filipcik P, Zilka N, Bugos O, Kucerak J, Koson P, Novak P, Novak M (2012) First transgenic rat model developing progressive cortical neurofibrillary tangles. Neurobiol Aging 33:1448–1456. https://doi.org/10.1016/j.neurobiolaging.2010.10.015
Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, Scheres SHW (2017) Cryo-EM structures of tau filaments from Alzheimer's disease. Nature 547:185–190. https://doi.org/10.1038/nature23002
Florenzano F, Veronica C, Ciasca G, Ciotti MT, Pittaluga A, Olivero G, Feligioni M, Iannuzzi F, Latina V, Maria Sciacca MF, Sinopoli A, Milardi D, Pappalardo G, Marco S, Papi M, Atlante A, Bobba A, Borreca A, Calissano P, Amadoro G (2017) Extracellular truncated tau causes early presynaptic dysfunction associated with Alzheimer's disease and other tauopathies. Oncotarget 8:64745–64778. https://doi.org/10.18632/oncotarget.17371
Forrest SL, Kril JJ, Stevens CH, Kwok JB, Hallupp M, Kim WS, Huang Y, McGinley CV, Werka H, Kiernan MC, Gotz J, Spillantini MG, Hodges JR, Ittner LM, Halliday GM (2018) Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain 141:521–534. https://doi.org/10.1093/brain/awx328
Frandemiche ML, De Seranno S, Rush T, Borel E, Elie A, Arnal I, Lante F, Buisson A (2014) Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J Neurosci 34:6084–6097. https://doi.org/10.1523/jneurosci.4261-13.2014
Frost B, Bardai FH, Feany MB (2016) Lamin dysfunction mediates neurodegeneration in Tauopathies. Curr Biol 26:129–136. https://doi.org/10.1016/j.cub.2015.11.039
Frost B, Hemberg M, Lewis J, Feany MB (2014) Tau promotes neurodegeneration through global chromatin relaxation. Nat Neurosci 17:357–366. https://doi.org/10.1038/nn.3639
Frost B, Jacks RL, Diamond MI (2009) Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 284:12845–12852. https://doi.org/10.1074/jbc.M808759200
Fu H, Hussaini SA, Wegmann S, Profaci C, Daniels JD, Herman M, Emrani S, Figueroa HY, Hyman BT, Davies P, Duff KE (2016) 3D visualization of the temporal and spatial spread of tau pathology reveals extensive sites of tau accumulation associated with neuronal loss and recognition memory deficit in aged tau transgenic mice. PLoS One 11:e0159463. https://doi.org/10.1371/journal.pone.0159463
Funk KE, Mirbaha H, Jiang H, Holtzman DM, Diamond MI (2015) Distinct therapeutic mechanisms of tau antibodies: promoting microglial clearance versus blocking neuronal uptake. J Biol Chem 290:21652–21662. https://doi.org/10.1074/jbc.M115.657924
Fuster-Matanzo A, Hernandez F, Avila J (2018) Tau spreading mechanisms; implications for dysfunctional Tauopathies. Int J Mol Sci 19. https://doi.org/10.3390/ijms19030645
Gandini A, Bartolini M, Tedesco D, Martinez-Gonzalez L, Roca C, Campillo NE, Zaldivar-Diez J, Perez C, Zuccheri G, Miti A, Feoli A, Castellano S, Petralla S, Monti B, Rossi M, Moda F, Legname G, Martinez A, Bolognesi ML (2018) Tau-centric multitarget approach for Alzheimer's disease: development of first-in-class dual glycogen synthase kinase 3beta and tau-aggregation inhibitors. J Med Chem 61:7640–7656. https://doi.org/10.1021/acs.jmedchem.8b00610
Gauthier S, Feldman HH, Schneider LS, Wilcock GK, Frisoni GB, Hardlund JH, Moebius HJ, Bentham P, Kook KA, Wischik DJ, Schelter BO, Davis CS, Staff RT, Bracoud L, Shamsi K, Storey JM, Harrington CR, Wischik CM (2016) Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer's disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 388:2873–2884. https://doi.org/10.1016/s0140-6736(16)31275-2
Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M (2015) Invited review: frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol 41:24–46. https://doi.org/10.1111/nan.12213
Gheyara AL, Ponnusamy R, Djukic B, Craft RJ, Ho K, Guo W, Finucane MM, Sanchez PE, Mucke L (2014) Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann Neurol 76:443–456. https://doi.org/10.1002/ana.24230
Giannakopoulos P, Duc M, Gold G, Hof PR, Michel JP, Bouras C (1998) Pathologic correlates of apraxia in Alzheimer disease. Arch Neurol 55:689–695
Giannakopoulos P, Gold G, Duc M, Michel JP, Hof PR, Bouras C (1999) Neuroanatomic correlates of visual agnosia in Alzheimer's disease: a clinicopathologic study. Neurology 52:71–77
Giannakopoulos P, Gold G, Duc M, Michel JP, Hof PR, Bouras C (2000) Neural substrates of spatial and temporal disorientation in Alzheimer's disease. Acta Neuropathol 100:189–195
Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, Morrison JH, Gold G, Hof PR (2003) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology 60:1495–1500
Goedert M, Jakes R (1990) Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J 9:4225–4230
Goedert M, Spillantini MG (2001) Tau gene mutations and neurodegeneration. Biochem Soc Symp 67:59–71
Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA (1989) Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J 8:393–399
Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A (1988) Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A 85:4051–4055
Gold G, Bouras C, Kovari E, Canuto A, Glaria BG, Malky A, Hof PR, Michel JP, Giannakopoulos P (2000) Clinical validity of Braak neuropathological staging in the oldest-old. Acta Neuropathol 99:579–582 discussion 583-574
Gomez-Ramos A, Diaz-Hernandez M, Cuadros R, Hernandez F, Avila J (2006) Extracellular tau is toxic to neuronal cells. FEBS Lett 580:4842–4850. https://doi.org/10.1016/j.febslet.2006.07.078
Gomez-Ramos A, Diaz-Hernandez M, Rubio A, Miras-Portugal MT, Avila J (2008) Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol Cell Neurosci 37:673–681. https://doi.org/10.1016/j.mcn.2007.12.010
Gordon BA, Friedrichsen K, Brier M, Blazey T, Su Y, Christensen J, Aldea P, McConathy J, Holtzman DM, Cairns NJ, Morris JC, Fagan AM, Ances BM, Benzinger TL (2016) The relationship between cerebrospinal fluid markers of Alzheimer pathology and positron emission tomography tau imaging. Brain 139:2249–2260. https://doi.org/10.1093/brain/aww139
Gordon BA, McCullough A, Mishra S, Blazey TM, Su Y, Christensen J, Dincer A, Jackson K, Hornbeck RC, Morris JC, Ances BM, Benzinger TLS (2018) Cross-sectional and longitudinal atrophy is preferentially associated with tau rather than amyloid beta positron emission tomography pathology. Alzheimers Dement (Amst) 10:245–252. https://doi.org/10.1016/j.dadm.2018.02.003
Grober E, Dickson D, Sliwinski MJ, Buschke H, Katz M, Crystal H, Lipton RB (1999) Memory and mental status correlates of modified Braak staging. Neurobiol Aging 20:573–579
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI (1986) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83:4913–4917
Gu H, Dodel R, Farlow M, Du Y (2014) Advances in the development of antibody-based immunotherapy against prion disease. https://doi.org/10.2147/ANTI.S53336
Gu J, Congdon EE, Sigurdsson EM (2013) Two novel tau antibodies targeting the 396/404 region are primarily taken up by neurons and reduce tau protein pathology. J Biol Chem 288:33081–33095. https://doi.org/10.1074/jbc.M113.494922
Hamdane M, Bretteville A, Sambo AV, Schindowski K, Begard S, Delacourte A, Bertrand P, Buee L (2005) p25/Cdk5-mediated retinoblastoma phosphorylation is an early event in neuronal cell death. J Cell Sci 118:1291–1298. https://doi.org/10.1242/jcs.01724
Hampel H, Toschi N, Baldacci F, Zetterberg H, Blennow K, Kilimann I, Teipel SJ, Cavedo E, Melo Dos Santos A, Epelbaum S, Lamari F, Genthon R, Dubois B, Floris R, Garaci F, Lista S (2018) Alzheimer's disease biomarker-guided diagnostic workflow using the added value of six combined cerebrospinal fluid candidates: Abeta1-42, total-tau, phosphorylated-tau, NFL, neurogranin, and YKL-40. Alzheimers Dement 14:492–501. https://doi.org/10.1016/j.jalz.2017.11.015
Hanseeuw BJ, Betensky RA, Schultz AP, Papp KV, Mormino EC, Sepulcre J, Bark JS, Cosio DM, LaPoint M, Chhatwal JP, Rentz DM, Sperling RA, Johnson KA (2017) Fluorodeoxyglucose metabolism associated with tau-amyloid interaction predicts memory decline. Ann Neurol 81:583–596. https://doi.org/10.1002/ana.24910
Hansen AK, Brooks DJ, Borghammer P (2018) MAO-B inhibitors do not block in vivo Flortaucipir([(18)F]-AV-1451) binding. Mol Imaging Biol 20:356–360. https://doi.org/10.1007/s11307-017-1143-1
Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, Hirokawa N (1994) Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 369:488–491. https://doi.org/10.1038/369488a0
Hayashi ML, Lu J, Driver D, Alvarado A. (2016) Antibodies to tau and uses thereof. Eli Lilly and Company
Hedden T, Oh H, Younger AP, Patel TA (2013) Meta-analysis of amyloid-cognition relations in cognitively normal older adults. Neurology 80:1341–1348. https://doi.org/10.1212/WNL.0b013e31828ab35d
Hocquemiller M, Giersch L, Audrain M, Parker S, Cartier N (2016) Adeno-associated virus-based gene therapy for CNS diseases. Hum Gene Ther 27:478–496. https://doi.org/10.1089/hum.2016.087
Hof PR, Vogt BA, Bouras C, Morrison JH (1997) Atypical form of Alzheimer's disease with prominent posterior cortical atrophy: a review of lesion distribution and circuit disconnection in cortical visual pathways. Vis Res 37:3609–3625. https://doi.org/10.1016/s0042-6989(96)00240-4
Hoglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, van Swieten JC, Heutink P, Wszolek ZK, Uitti RJ, Vandrovcova J, Hurtig HI, Gross RG, Maetzler W, Goldwurm S, Tolosa E, Borroni B, Pastor P, Cantwell LB, Han MR, Dillman A, van der Brug MP, Gibbs JR, Cookson MR, Hernandez DG, Singleton AB, Farrer MJ, Yu CE, Golbe LI, Revesz T, Hardy J, Lees AJ, Devlin B, Hakonarson H, Muller U, Schellenberg GD (2011) Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet 43:699–705. https://doi.org/10.1038/ng.859
Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, Kotzbauer PT, Miller TM, Papy-Garcia D, Diamond MI (2013) Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A 110:E3138–E3147. https://doi.org/10.1073/pnas.1301440110
Holth JK, Bomben VC, Reed JG, Inoue T, Younkin L, Younkin SG, Pautler RG, Botas J, Noebels JL (2013) Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J Neurosci 33:1651–1659. https://doi.org/10.1523/jneurosci.3191-12.2013
Honer M, Gobbi L, Knust H, Kuwabara H, Muri D, Koerner M, Valentine H, Dannals RF, Wong DF, Borroni E (2018) Preclinical evaluation of (18)F-RO6958948, (11)C-RO6931643, and (11)C-RO6924963 as novel PET radiotracers for imaging tau aggregates in Alzheimer disease. J Nucl Med 59:675–681. https://doi.org/10.2967/jnumed.117.196741
Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D (2010) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68:1067–1081. https://doi.org/10.1016/j.neuron.2010.11.030
Horiguchi T, Uryu K, Giasson BI, Ischiropoulos H, LightFoot R, Bellmann C et al (2003) Nitration of tau protein is linked to neurodegeneration in tauopathies. Am J Pathol 163:1021–1031. https://doi.org/10.1016/s0002-9440(10)63462-1
Hu NW, Corbett GT, Moore S, Klyubin I, O'Malley TT, Walsh DM, Livesey FJ, Rowan MJ (2018) Extracellular forms of Abeta and tau from iPSC models of Alzheimer's disease disrupt synaptic plasticity. Cell Rep 23:1932–1938. https://doi.org/10.1016/j.celrep.2018.04.040
Hua Q, He RQ (2003) Tau could protect DNA double helix structure. Biochim Biophys Acta 1645:205–211
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705. https://doi.org/10.1038/31508
Iaccarino L, Tammewar G, Ayakta N, Baker SL, Bejanin A, Boxer AL, Gorno-Tempini ML, Janabi M, Kramer JH, Lazaris A, Lockhart SN, Miller BL, Miller ZA, O'Neil JP, Ossenkoppele R, Rosen HJ, Schonhaut DR, Jagust WJ, Rabinovici GD (2018) Local and distant relationships between amyloid, tau and neurodegeneration in Alzheimer's disease. Neuroimage Clin 17:452–464. https://doi.org/10.1016/j.nicl.2017.09.016
Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM (2013) Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's-like tauopathy. J Neurosci 33:1024–1037. https://doi.org/10.1523/jneurosci.2642-12.2013
Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, Clark RS, Marion DW, Wisniewski SR, DeKosky ST (2004) Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol 190:192–203. https://doi.org/10.1016/j.expneurol.2004.06.011
Ittner A, Bertz J, Suh LS, Stevens CH, Gotz J, Ittner LM (2015) Tau-targeting passive immunization modulates aspects of pathology in tau transgenic mice. J Neurochem 132:135–145. https://doi.org/10.1111/jnc.12821
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Gotz J (2010) Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell 142:387–397. https://doi.org/10.1016/j.cell.2010.06.036
Jack CR Jr, Wiste HJ, Schwarz CG, Lowe VJ, Senjem ML, Vemuri P, Weigand SD, Therneau TM, Knopman DS, Gunter JL, Jones DT, Graff-Radford J, Kantarci K, Roberts RO, Mielke MM, Machulda MM, Petersen RC (2018) Longitudinal tau PET in ageing and Alzheimer's disease. Brain 141:1517–1528. https://doi.org/10.1093/brain/awy059
Jack CR Jr, Wiste HJ, Weigand SD, Therneau TM, Lowe VJ, Knopman DS, Gunter JL, Senjem ML, Jones DT, Kantarci K, Machulda MM, Mielke MM, Roberts RO, Vemuri P, Reyes DA, Petersen RC (2017) Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement 13:205–216. https://doi.org/10.1016/j.jalz.2016.08.005
Jansen WJ, Ossenkoppele R, Tijms BM, Fagan AM, Hansson O, Klunk WE, van der Flier WM, Villemagne VL, Frisoni GB, Fleisher AS, Lleo A, Mintun MA, Wallin A, Engelborghs S, Na DL, Chetelat G, Molinuevo JL, Landau SM, Mattsson N, Kornhuber J, Sabri O, Rowe CC, Parnetti L, Popp J, Fladby T, Jagust WJ, Aalten P, Lee DY, Vandenberghe R, Resende de Oliveira C, Kapaki E, Froelich L, Ivanoiu A, Gabryelewicz T, Verbeek MM, Sanchez-Juan P, Hildebrandt H, Camus V, Zboch M, Brooks DJ, Drzezga A, Rinne JO, Newberg A, de Mendonca A, Sarazin M, Rabinovici GD, Madsen K, Kramberger MG, Nordberg A, Mok V, Mroczko B, Wolk DA, Meyer PT, Tsolaki M, Scheltens P, Verhey FRJ, Visser PJ, Aarsland D, Alcolea D, Alexander M, Almdahl IS, Arnold SE, Baldeiras I, Barthel H, van Berckel BNM, Blennow K, van Buchem MA, Cavedo E, Chen K, Chipi E, Cohen AD, Forster S, Fortea J, Frederiksen KS, Freund-Levi Y, Gkatzima O, Gordon MF, Grimmer T, Hampel H, Hausner L, Hellwig S, Herukka SK, Johannsen P, Klimkowicz-Mrowiec A, Kohler S, Koglin N, van Laere K, de Leon M, Lisetti V, Maier W, Marcusson J, Meulenbroek O, Mollergard HM, Morris JC, Nordlund A, Novak GP, Paraskevas GP, Perera G, Peters O, Ramakers I, Rami L, Rodriguez-Rodriguez E, Roe CM, Rot U, Ruther E, Santana I, Schroder J, Seo SW, Soininen H, Spiru L, Stomrud E, Struyfs H, Teunissen CE, Vos SJB, van Waalwijk van Doorn LJC, Waldemar G, Wallin AK, Wiltfang J, Zetterberg H (2018) Association of Cerebral Amyloid-beta Aggregation with Cognitive Functioning in persons without dementia. JAMA Psychiatry 75:84–95. https://doi.org/10.1001/jamapsychiatry.2017.3391
Jellinger KA, Attems J (2010) Prevalence of dementia disorders in the oldest-old: an autopsy study. Acta Neuropathol 119:421–433. https://doi.org/10.1007/s00401-010-0654-5
Ji M, Xie XX, Liu DQ, Yu XL, Zhang Y, Zhang LX, Wang SW, Huang YR, Liu RT (2018) Hepatitis B core VLP-based mis-disordered tau vaccine elicits strong immune response and alleviates cognitive deficits and neuropathology progression in tau.P301S mouse model of Alzheimer's disease and frontotemporal dementia. Alzheimers Res Ther 10:55. https://doi.org/10.1186/s13195-018-0378-7
Jiang XY, Chen TK, Zhou JT, He SY, Yang HY, Chen Y, Qu W, Feng F, Sun HP (2018) Dual GSK-3beta/AChE inhibitors as a new strategy for multitargeting anti-Alzheimer's disease drug discovery. ACS Med Chem Lett 9:171–176. https://doi.org/10.1021/acsmedchemlett.7b00463
Jicha GA, Bowser R, Kazam IG, Davies P (1997) Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res 48:128–132
Johnson GV, Seubert P, Cox TM, Motter R, Brown JP, Galasko D (1997) The tau protein in human cerebrospinal fluid in Alzheimer's disease consists of proteolytically derived fragments. J Neurochem 68:430–433
Johnson KA, Schultz A, Betensky RA, Becker JA, Sepulcre J, Rentz D, Mormino E, Chhatwal J, Amariglio R, Papp K, Marshall G, Albers M, Mauro S, Pepin L, Alverio J, Judge K, Philiossaint M, Shoup T, Yokell D, Dickerson B, Gomez-Isla T, Hyman B, Vasdev N, Sperling R (2016) Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol 79:110–119. https://doi.org/10.1002/ana.24546
Jouanne M, Rault S, Voisin-Chiret AS (2017) Tau protein aggregation in Alzheimer's disease: an attractive target for the development of novel therapeutic agents. Eur J Med Chem 139:153–167. https://doi.org/10.1016/j.ejmech.2017.07.070
Kanmert D, Cantlon A, Muratore CR, Jin M, O'Malley TT, Lee G, Young-Pearse TL, Selkoe DJ, Walsh DM (2015) C-terminally truncated forms of tau, but not full-length tau or its C-terminal fragments, are released from neurons independently of cell death. J Neurosci 35:10851–10865. https://doi.org/10.1523/jneurosci.0387-15.2015
Katsinelos T, Zeitler M, Dimou E, Karakatsani A, Muller HM, Nachman E, Steringer JP, Ruiz de Almodovar C, Nickel W, Jahn TR (2018) Unconventional secretion mediates the trans-cellular spreading of tau. Cell Rep 23:2039–2055. https://doi.org/10.1016/j.celrep.2018.04.056
Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI (2012) Trans-cellular propagation of tau aggregation by fibrillar species. J Biol Chem 287:19440–19451. https://doi.org/10.1074/jbc.M112.346072
Kobayashi K, Nakano H, Hayashi M, Shimazaki M, Fukutani Y, Sasaki K, Sugimori K, Koshino Y (2003) Association of phosphorylation site of tau protein with neuronal apoptosis in Alzheimer's disease. J Neurol Sci 208:17–24
Kontsekova E, Zilka N, Kovacech B, Novak P, Novak M (2014) First-in-man tau vaccine targeting structural determinants essential for pathological tau-tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer's disease model. Alzheimers Res Ther 6:44. https://doi.org/10.1186/alzrt278
Kontsekova E, Zilka N, Kovacech B, Skrabana R, Novak M (2014) Identification of structural determinants on tau protein essential for its pathological function: novel therapeutic target for tau immunotherapy in Alzheimer's disease. Alzheimers Res Ther 6:45. https://doi.org/10.1186/alzrt277
Kosik KS, Joachim CL, Selkoe DJ (1986) Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A 83:4044–4048
Kosik KS, Orecchio LD, Bakalis S, Neve RL (1989) Developmentally regulated expression of specific tau sequences. Neuron 2:1389–1397
Kouri N, Ross OA, Dombroski B, Younkin CS, Serie DJ, Soto-Ortolaza A, Baker M, Finch NC, Yoon H, Kim J, Fujioka S, McLean CA, Ghetti B, Spina S, Cantwell LB, Farlow MR, Grafman J, Huey ED, Ryung Han M, Beecher S, Geller ET, Kretzschmar HA, Roeber S, Gearing M, Juncos JL, Vonsattel JP, Van Deerlin VM, Grossman M, Hurtig HI, Gross RG, Arnold SE, Trojanowski JQ, Lee VM, Wenning GK, White CL, Hoglinger GU, Muller U, Devlin B, Golbe LI, Crook J, Parisi JE, Boeve BF, Josephs KA, Wszolek ZK, Uitti RJ, Graff-Radford NR, Litvan I, Younkin SG, Wang LS, Ertekin-Taner N, Rademakers R, Hakonarsen H, Schellenberg GD, Dickson DW (2015) Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun 6:7247. https://doi.org/10.1038/ncomms8247
Kovacs GG (2015) Invited review: neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol 41:3–23. https://doi.org/10.1111/nan.12208
Kovacs GG (2016) Molecular pathological classification of neurodegenerative diseases: turning towards precision medicine. Int J Mol Sci 17. https://doi.org/10.3390/ijms17020189
Kovacs GG, Lee VM, Trojanowski JQ (2017) Protein astrogliopathies in human neurodegenerative diseases and aging. Brain pathol 27:675–690. https://doi.org/10.1111/bpa.12536
Kovacs GG, Xie SX, Robinson JL, Lee EB, Smith DH, Schuck T, Lee VM, Trojanowski JQ (2018) Sequential stages and distribution patterns of aging-related tau astrogliopathy (ARTAG) in the human brain. Acta Neuropathol Commun 6:50. https://doi.org/10.1186/s40478-018-0552-y
LaPoint MR, Chhatwal JP, Sepulcre J, Johnson KA, Sperling RA, Schultz AP (2017) The association between tau PET and retrospective cortical thinning in clinically normal elderly. Neuroimage 157:612–622. https://doi.org/10.1016/j.neuroimage.2017.05.049
Lathuiliere A, Valdes P, Papin S, Cacquevel M, Maclachlan C, Knott GW, Muhs A, Paganetti P, Schneider BL (2017) Motifs in the tau protein that control binding to microtubules and aggregation determine pathological effects. Sci Rep 7:13556. https://doi.org/10.1038/s41598-017-13786-2
Le Guennec K, Quenez O, Nicolas G, Wallon D, Rousseau S, Richard AC, Alexander J, Paschou P, Charbonnier C, Bellenguez C, Grenier-Boley B, Lechner D, Bihoreau MT, Olaso R, Boland A, Meyer V, Deleuze JF, Amouyel P, Munter HM, Bourque G, Lathrop M, Frebourg T, Redon R, Letenneur L, Dartigues JF, Martinaud O, Kalev O, Mehrabian S, Traykov L, Strobel T, Le Ber I, Caroppo P, Epelbaum S, Jonveaux T, Pasquier F, Rollin-Sillaire A, Genin E, Guyant-Marechal L, Kovacs GG, Lambert JC, Hannequin D, Campion D, Rovelet-Lecrux A (2017) 17q21.31 duplication causes prominent tau-related dementia with increased MAPT expression. Mol Psychiatry 22:1119–1125. https://doi.org/10.1038/mp.2016.226
Leal SL, Lockhart SN, Maass A, Bell RK, Jagust WJ (2018) Subthreshold amyloid predicts tau deposition in aging. J Neurosci 38:4482–4489. https://doi.org/10.1523/jneurosci.0485-18.2018
Lee CM, Jacobs HIL, Marquie M, Becker JA, Andrea NV, Jin DS, Schultz AP, Frosch MP, Gomez-Isla T, Sperling RA, Johnson KA (2018) 18F-Flortaucipir binding in choroid plexus: related to race and Hippocampus signal. J Alzheimers Dis 62:1691–1702. https://doi.org/10.3233/jad-170840
Lee MJ, Lee JH, Rubinsztein DC (2013) Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol 105:49–59. https://doi.org/10.1016/j.pneurobio.2013.03.001
Lee SH, Le Pichon CE, Adolfsson O, Gafner V, Pihlgren M, Lin H, Solanoy H, Brendza R, Ngu H, Foreman O, Chan R, Ernst JA, DiCara D, Hotzel I, Srinivasan K, Hansen DV, Atwal J, Lu Y, Bumbaca D, Pfeifer A, Watts RJ, Muhs A, Scearce-Levie K, Ayalon G (2016) Antibody-mediated targeting of tau in vivo does not require effector function and microglial engagement. Cell Rep 16:1690–1700. https://doi.org/10.1016/j.celrep.2016.06.099
Lee VM, Balin BJ, Otvos L Jr, Trojanowski JQ (1991) A68: a major subunit of paired helical filaments and derivatized forms of normal tau. Science 251:675–678
Lee VM, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159. https://doi.org/10.1146/annurev.neuro.24.1.1121
Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK et al (2012) Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med 18:291–295. https://doi.org/10.1038/nm.2613
Lei P, Ayton S, Moon S, Zhang Q, Volitakis I, Finkelstein DI, Bush AI (2014) Motor and cognitive deficits in aged tau knockout mice in two background strains. Mol Neurodegener 9:29. https://doi.org/10.1186/1750-1326-9-29
Lemoine L, Leuzy A, Chiotis K, Rodriguez-Vieitez E, Nordberg A (2018) Tau positron emission tomography imaging in tauopathies: the added hurdle of off-target binding. Alzheimers Dement (Amst) 10:232–236. https://doi.org/10.1016/j.dadm.2018.01.007
Leone P, Shera D, McPhee SW, Francis JS, Kolodny EH, Bilaniuk LT, Wang DJ, Assadi M, Goldfarb O, Goldman HW, Freese A, Young D, During MJ, Samulski RJ, Janson CG (2012) Long-term follow-up after gene therapy for canavan disease. Sci Transl Med 4:165ra163. https://doi.org/10.1126/scitranslmed.3003454
Li C, Gotz J (2017) Tau-based therapies in neurodegeneration: opportunities and challenges. Nat Rev Drug Discov 16:863–883. https://doi.org/10.1038/nrd.2017.155
Li D, Mastaglia FL, Fletcher S, Wilton SD (2018) Precision medicine through antisense oligonucleotide-mediated exon skipping. Trends Pharmacol Sci 39:982–994. https://doi.org/10.1016/j.tips.2018.09.001
Li Z, Hall AM, Kelinske M, Roberson ED (2014) Seizure resistance without parkinsonism in aged mice after tau reduction. Neurobiol Aging 35:2617–2624. https://doi.org/10.1016/j.neurobiolaging.2014.05.001
Lindwall G, Cole RD (1984) The purification of tau protein and the occurrence of two phosphorylation states of tau in brain. J Biol Chem 259:12241–12245
Ling H, Kovacs GG, Vonsattel JP, Davey K, Mok KY, Hardy J, Morris HR, Warner TT, Holton JL, Revesz T (2016) Astrogliopathy predominates the earliest stage of corticobasal degeneration pathology. Brain 139:3237–3252. https://doi.org/10.1093/brain/aww256
Liu C, Song X, Nisbet R, Gotz J (2016) Co-immunoprecipitation with tau isoform-specific antibodies reveals distinct protein interactions and highlights a putative role for 2N tau in disease. J Biol Chem 291:8173–8188. https://doi.org/10.1074/jbc.M115.641902
Liu F, Gong CX (2008) Tau exon 10 alternative splicing and tauopathies. Mol Neurodegener 3:8. https://doi.org/10.1186/1750-1326-3-8
Liu F, Zaidi T, Iqbal K, Grundke-Iqbal I, Merkle RK, Gong CX (2002) Role of glycosylation in hyperphosphorylation of tau in Alzheimer's disease. FEBS Lett 512:101–106
Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K (2012) Trans-synaptic spread of tau pathology in vivo. PLoS One 7:e31302. https://doi.org/10.1371/journal.pone.0031302
Lockhart SN, Schöll M, Baker SL, Ayakta N, Swinnerton KN, Bell RK, Mellinger TJ, Shah VD, O'Neil JP, Janabi M, Jagust WJ (2017) Amyloid and tau PET demonstrate region-specific associations in normal older people. Neuroimage 150:191–199. https://doi.org/10.1016/j.neuroimage.2017.02.051
Lohith TG, Bennacef I, Vandenberghe R, Vandenbulcke M, Salinas-Valenzuela C, Declercq R, Reynders T, Telan-Choing F, Riffel K, Celen S, Serdons K, Bormans G, Tsai K, Walji A, Hostetler ED, Evelhoch JL, Van Laere K, Forman M, Stoch A, Sur C, Struyk A (2018) First-in-human brain imaging of Alzheimer dementia patients and elderly controls with (18)F-MK-6240, a PET tracer targeting neurofibrillary tangle pathology. J Nucl Med. https://doi.org/10.2967/jnumed.118.208215
Lois C, Gonzalez I, Johnson KA, Price JC (2018) PET imaging of tau protein targets: a methodology perspective. Brain Imaging Behav. https://doi.org/10.1007/s11682-018-9847-7
Loomis PA, Howard TH, Castleberry RP, Binder LI (1990) Identification of nuclear tau isoforms in human neuroblastoma cells. Proc Natl Acad Sci U S A 87:8422–8426
Lorson CL, Rindt H, Shababi M (2010) Spinal muscular atrophy: mechanisms and therapeutic strategies. Hum Mol Genet 19:R111–R118. https://doi.org/10.1093/hmg/ddq147
Love DR, Davies KE (1989) Duchenne muscular dystrophy: the gene and the protein. Mol Biol Med 6:7–17
Lowe VJ, Bruinsma TJ, Min HK, Lundt ES, Fang P, Senjem ML, Boeve BF, Josephs KA, Pandey MK, Murray ME, Kantarci K, Jones DT, Schwarz CG, Knopman DS, Petersen RC, Jack CR Jr (2018) Elevated medial temporal lobe and pervasive brain tau-PET signal in normal participants. Alzheimers Dement (Amst) 10:210–216. https://doi.org/10.1016/j.dadm.2018.01.005
Lowe VJ, Curran G, Fang P, Liesinger AM, Josephs KA, Parisi JE, Kantarci K, Boeve BF, Pandey MK, Bruinsma T, Knopman DS, Jones DT, Petrucelli L, Cook CN, Graff-Radford NR, Dickson DW, Petersen RC, Jack CR Jr, Murray ME (2016) An autoradiographic evaluation of AV-1451 tau PET in dementia. Acta Neuropathol Commun 4:58. https://doi.org/10.1186/s40478-016-0315-6
Lowe VJ, Wiste HJ, Senjem ML, Weigand SD, Therneau TM, Boeve BF, Josephs KA, Fang P, Pandey MK, Murray ME, Kantarci K, Jones DT, Vemuri P, Graff-Radford J, Schwarz CG, Machulda MM, Mielke MM, Roberts RO, Knopman DS, Petersen RC, Jack CR Jr (2018) Widespread brain tau and its association with ageing, Braak stage and Alzheimer's dementia. Brain 141:271–287. https://doi.org/10.1093/brain/awx320
Luna-Munoz J, Chavez-Macias L, Garcia-Sierra F, Mena R (2007) Earliest stages of tau conformational changes are related to the appearance of a sequence of specific phospho-dependent tau epitopes in Alzheimer's disease. J Alzheimers Dis 12:365–375
Luna-Munoz J, Garcia-Sierra F, Falcon V, Menendez I, Chavez-Macias L, Mena R (2005) Regional conformational change involving phosphorylation of tau protein at the Thr231, precedes the structural change detected by Alz-50 antibody in Alzheimer's disease. J Alzheimers Dis 8:29–41
Luo HB, Xia YY, Shu XJ, Liu ZC, Feng Y, Liu XH et al (2014) SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc Natl Acad Sci U S A 111:16586–16591. https://doi.org/10.1073/pnas.1417548111
Luo W, Liu W, Hu X, Hanna M, Caravaca A, Paul SM (2015) Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci Rep 5:11161. https://doi.org/10.1038/srep11161
Maass A, Landau S, Baker SL, Horng A, Lockhart SN, La Joie R, Rabinovici GD, Jagust WJ (2017) Comparison of multiple tau-PET measures as biomarkers in aging and Alzheimer's disease. Neuroimage 157:448–463. https://doi.org/10.1016/j.neuroimage.2017.05.058
Maass A, Lockhart SN, Harrison TM, Bell RK, Mellinger T, Swinnerton K, Baker SL, Rabinovici GD, Jagust WJ (2018) Entorhinal tau pathology, episodic memory decline, and neurodegeneration in aging. J Neurosci 38:530–543. https://doi.org/10.1523/jneurosci.2028-17.2017
Magnani E, Fan J, Gasparini L, Golding M, Williams M, Schiavo G, Goedert M, Amos LA, Spillantini MG (2007) Interaction of tau protein with the dynactin complex. EMBO J 26:4546–4554. https://doi.org/10.1038/sj.emboj.7601878
Mailliot C, Sergeant N, Bussiere T, Caillet-Boudin ML, Delacourte A, Buee L (1998) Phosphorylation of specific sets of tau isoforms reflects different neurofibrillary degeneration processes. FEBS Lett 433:201–204
Mallery DL, McEwan WA, Bidgood SR, Towers GJ, Johnson CM, James LC (2010) Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc Natl Acad Sci U S A 107:19985–19990. https://doi.org/10.1073/pnas.1014074107
Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, Bhaskar K (2015) Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 138:1738–1755. https://doi.org/10.1093/brain/awv081
Maqbool M, Mobashir M, Hoda N (2016) Pivotal role of glycogen synthase kinase-3: a therapeutic target for Alzheimer's disease. Eur J Med Chem 107:63–81. https://doi.org/10.1016/j.ejmech.2015.10.018
Marciniak E, Leboucher A, Caron E, Ahmed T, Tailleux A, Dumont J, Issad T, Gerhardt E, Pagesy P, Vileno M, Bournonville C, Hamdane M, Bantubungi K, Lancel S, Demeyer D, Eddarkaoui S, Vallez E, Vieau D, Humez S, Faivre E, Grenier-Boley B, Outeiro TF, Staels B, Amouyel P, Balschun D, Buee L, Blum D (2017) Tau deletion promotes brain insulin resistance. J Exp Med 214:2257–2269. https://doi.org/10.1084/jem.20161731
Marquie M, Normandin MD, Vanderburg CR, Costantino IM, Bien EA, Rycyna LG, Klunk WE, Mathis CA, Ikonomovic MD, Debnath ML, Vasdev N, Dickerson BC, Gomperts SN, Growdon JH, Johnson KA, Frosch MP, Hyman BT, Gomez-Isla T (2015) Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol 78:787–800. https://doi.org/10.1002/ana.24517
Marquie M, Siao Tick Chong M, Anton-Fernandez A, Verwer EE, Saez-Calveras N, Meltzer AC, Ramanan P, Amaral AC, Gonzalez J, Normandin MD, Frosch MP, Gomez-Isla T (2017) [F-18]-AV-1451 binding correlates with postmortem neurofibrillary tangle Braak staging. Acta Neuropathol 134:619–628. https://doi.org/10.1007/s00401-017-1740-8
Marquie M, Verwer EE, Meltzer AC, Kim SJW, Aguero C, Gonzalez J, Makaretz SJ, Siao Tick Chong M, Ramanan P, Amaral AC, Normandin MD, Vanderburg CR, Gomperts SN, Johnson KA, Frosch MP, Gomez-Isla T (2017) Lessons learned about [F-18]-AV-1451 off-target binding from an autopsy-confirmed Parkinson's case. Acta Neuropathol Commun 5:75. https://doi.org/10.1186/s40478-017-0482-0
McClorey G, Banerjee S (2018) Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics. Biomedicines 6. https://doi.org/10.3390/biomedicines6020051
McEwan WA, Falcon B, Vaysburd M, Clift D, Oblak AL, Ghetti B, Goedert M, James LC (2017) Cytosolic fc receptor TRIM21 inhibits seeded tau aggregation. Proc Natl Acad Sci U S A 114:574–579. https://doi.org/10.1073/pnas.1607215114
McInnes J, Wierda K, Snellinx A, Bounti L, Wang YC, Stancu IC, Apostolo N, Gevaert K, Dewachter I, Spires-Jones TL, De Strooper B, De Wit J, Zhou L, Verstreken P (2018) Synaptogyrin-3 mediates presynaptic dysfunction induced by tau. Neuron 97:823–835.e828. https://doi.org/10.1016/j.neuron.2018.01.022
Meier S, Bell M, Lyons DN, Rodriguez-Rivera J, Ingram A, Fontaine SN, Mechas E, Chen J, Wolozin B, LeVine H 3rd, Zhu H, Abisambra JF (2016) Pathological tau promotes neuronal damage by impairing ribosomal function and decreasing protein synthesis. J Neurosci 36:1001–1007. https://doi.org/10.1523/jneurosci.3029-15.2016
Melnyk P, Vingtdeux V, Burlet S, Eddarkaoui S, Grosjean ME, Larchanche PE, Hochart G, Sergheraert C, Estrella C, Barrier M, Poix V, Plancq P, Lannoo C, Hamdane M, Delacourte A, Verwaerde P, Buee L, Sergeant N (2015) Chloroquine and chloroquinoline derivatives as models for the design of modulators of amyloid peptide precursor metabolism. ACS Chem Neurosci 6:559–569. https://doi.org/10.1021/cn5003013
Mignon L, Kordasiewicz H, Lane R, Smith A, Miller T, Narayanan P, Swayze EN, D., Fitzsimmons B, Bennett F (2018) Design of the First-in-Human Study of IONIS-MAPTRx, a Tau-lowering Antisense Oligonucleotide, in Patients With Alzheimer Disease (S2.006). Neurology 90; S2.006.
Mirbaha H, Chen D, Morazova OA, Ruff KM, Sharma AM, Liu X, Goodarzi M, Pappu RV, Colby DW, Mirzaei H, Joachimiak LA, Diamond MI (2018) Inert and seed-competent tau monomers suggest structural origins of aggregation. Elife 7. https://doi.org/10.7554/eLife.36584
Mishra S, Gordon BA, Su Y, Christensen J, Friedrichsen K, Jackson K, Hornbeck R, Balota DA, Cairns NJ, Morris JC, Ances BM, Benzinger TLS (2017) AV-1451 PET imaging of tau pathology in preclinical Alzheimer disease: defining a summary measure. NeuroImage 161:171–178. https://doi.org/10.1016/j.neuroimage.2017.07.050
Morris M, Hamto P, Adame A, Devidze N, Masliah E, Mucke L (2013) Age-appropriate cognition and subtle dopamine-independent motor deficits in aged tau knockout mice. Neurobiol Aging 34:1523–1529. https://doi.org/10.1016/j.neurobiolaging.2012.12.003
Mudher A, Colin M, Dujardin S, Medina M, Dewachter I, Alavi Naini SM, Mandelkow EM, Mandelkow E, Buee L, Goedert M, Brion JP (2017) What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol Commun 5:99. https://doi.org/10.1186/s40478-017-0488-7
Mukrasch MD, von Bergen M, Biernat J, Fischer D, Griesinger C, Mandelkow E, Zweckstetter M (2007) The “jaws” of the tau-microtubule interaction. J Biol Chem 282:12230–12239. https://doi.org/10.1074/jbc.M607159200
Myers AJ, Pittman AM, Zhao AS, Rohrer K, Kaleem M, Marlowe L, Lees A, Leung D, McKeith IG, Perry RH, Morris CM, Trojanowski JQ, Clark C, Karlawish J, Arnold S, Forman MS, Van Deerlin V, de Silva R, Hardy J (2007) The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol Dis 25:561–570. https://doi.org/10.1016/j.nbd.2006.10.018
Myszka DG (1999) Improving biosensor analysis. J Mol Recognit 12:279–284. https://doi.org/10.1002/(sici)1099-1352(199909/10)12:5<279::Aid-jmr473>3.0.Co;2-3
Myszka DG, He X, Dembo M, Morton TA, Goldstein B (1998) Extending the range of rate constants available from BIACORE: interpreting mass transport-influenced binding data. Biophys J 75:583–594. https://doi.org/10.1016/s0006-3495(98)77549-6
Narasimhan S, Guo JL, Changolkar L, Stieber A, McBride JD, Silva LV, He Z, Zhang B, Gathagan RJ, Trojanowski JQ, Lee VMY (2017) Pathological tau strains from human brains recapitulate the diversity of Tauopathies in nontransgenic mouse brain. J Neurosci 37:11406–11423. https://doi.org/10.1523/jneurosci.1230-17.2017
Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, Duyckaerts C, Frosch MP, Haroutunian V, Hof PR, Hulette CM, Hyman BT, Iwatsubo T, Jellinger KA, Jicha GA, Kovari E, Kukull WA, Leverenz JB, Love S, Mackenzie IR, Mann DM, Masliah E, McKee AC, Montine TJ, Morris JC, Schneider JA, Sonnen JA, Thal DR, Trojanowski JQ, Troncoso JC, Wisniewski T, Woltjer RL, Beach TG (2012) Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 71:362–381. https://doi.org/10.1097/NEN.0b013e31825018f7
Neve RL, Harris P, Kosik KS, Kurnit DM, Donlon TA (1986) Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res 387:271–280
Ng KP, Pascoal TA, Mathotaarachchi S, Therriault J, Kang MS, Shin M, Guiot MC, Guo Q, Harada R, Comley RA, Massarweh G, Soucy JP, Okamura N, Gauthier S, Rosa-Neto P (2017) Monoamine oxidase B inhibitor, selegiline, reduces (18)F-THK5351 uptake in the human brain. Alzheimers Res Ther 9:25. https://doi.org/10.1186/s13195-017-0253-y
Nobuhara CK, DeVos SL, Commins C, Wegmann S, Moore BD, Roe AD, Costantino I, Frosch MP, Pitstick R, Carlson GA, Hock C, Nitsch RM, Montrasio F, Grimm J, Cheung AE, Dunah AW, Wittmann M, Bussiere T, Weinreb PH, Hyman BT, Takeda S (2017) Tau antibody targeting pathological species blocks neuronal uptake and interneuron propagation of tau in vitro. Am J Pathol 187:1399–1412. https://doi.org/10.1016/j.ajpath.2017.01.022
Novak M (1994) Truncated tau protein as a new marker for Alzheimer's disease. Acta Virol 38:173–189
Novak M, Kabat J, Wischik CM (1993) Molecular characterization of the minimal protease resistant tau unit of the Alzheimer's disease paired helical filament. EMBO J 12:365–370
Novak P, Cehlar O, Skrabana R, Novak M (2018) Tau conformation as a target for disease-modifying therapy: the role of truncation. J Alzheimers Dis 64:S535–s546. https://doi.org/10.3233/jad-179942
Novak P, Kontsekova E, Zilka N, Novak M (2018) Ten years of tau-targeted immunotherapy: the path walked and the roads ahead. Front Neurosci 12:798. https://doi.org/10.3389/fnins.2018.00798
Novak P, Schmidt R, Kontsekova E, Zilka N, Kovacech B, Skrabana R, Vince-Kazmerova Z, Katina S, Fialova L, Prcina M, Parrak V, Dal-Bianco P, Brunner M, Staffen W, Rainer M, Ondrus M, Ropele S, Smisek M, Sivak R, Winblad B, Novak M (2017) Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer's disease: a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol 16:123–134. https://doi.org/10.1016/s1474-4422(16)30331-3
Okuda M, Hijikuro I, Fujita Y, Teruya T, Kawakami H, Takahashi T, Sugimoto H (2016) Design and synthesis of curcumin derivatives as tau and amyloid beta dual aggregation inhibitors. Bioorg Med Chem Lett 26:5024–5028. https://doi.org/10.1016/j.bmcl.2016.08.092
Okuda M, Hijikuro I, Fujita Y, Wu X, Nakayama S, Sakata Y, Noguchi Y, Ogo M, Akasofu S, Ito Y, Soeda Y, Tsuchiya N, Tanaka N, Takahashi T, Sugimoto H (2015) PE859, a novel tau aggregation inhibitor, reduces aggregated tau and prevents onset and progression of neural dysfunction in vivo. PLoS One 10:e0117511. https://doi.org/10.1371/journal.pone.0117511
Olsson A, Vanderstichele H, Andreasen N, De Meyer G, Wallin A, Holmberg B, Rosengren L, Vanmechelen E, Blennow K (2005) Simultaneous measurement of beta-amyloid(1-42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin Chem 51:336–345. https://doi.org/10.1373/clinchem.2004.039347
Ono Mea., et al. Development of novel tau PET tracers, [18F]AM-PPB3 and [18F]PM-PPB3. In: Human Amyloid Imaging Meeting, 2017. p 34
Ossenkoppele R, Schonhaut DR, Schöll M, Lockhart SN, Ayakta N, Baker SL, O'Neil JP, Janabi M, Lazaris A, Cantwell A, Vogel J, Santos M, Miller ZA, Bettcher BM, Vossel KA, Kramer JH, Gorno-Tempini ML, Miller BL, Jagust WJ, Rabinovici GD (2016) Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain 139:1551–1567. https://doi.org/10.1093/brain/aww027
Oz M, Lorke DE, Petroianu GA (2009) Methylene blue and Alzheimer's disease. Biochem Pharmacol 78:927–932. https://doi.org/10.1016/j.bcp.2009.04.034
Padmaraju V, Indi SS, Rao KS (2010) New evidences on tau-DNA interactions and relevance to neurodegeneration. Neurochem Int 57:51–57. https://doi.org/10.1016/j.neuint.2010.04.013
Palomo V, Perez DI, Roca C, Anderson C, Rodriguez-Muela N, Perez C, Morales-Garcia JA, Reyes JA, Campillo NE, Perez-Castillo AM, Rubin LL, Timchenko L, Gil C, Martinez A (2017) Subtly modulating glycogen synthase kinase 3 beta: allosteric inhibitor development and their potential for the treatment of chronic diseases. J Med Chem 60:4983–5001. https://doi.org/10.1021/acs.jmedchem.7b00395
Panek D, Wieckowska A, Jonczyk J, Godyn J, Bajda M, Wichur T, Pasieka A, Knez D, Pislar A, Korabecny J, Soukup O, Sepsova V, Sabate R, Kos J, Gobec S, Malawska B (2018) Design, synthesis, and biological evaluation of 1-Benzylamino-2-hydroxyalkyl derivatives as new potential disease-modifying multifunctional anti-Alzheimer's agents. ACS Chem Neurosci 9:1074–1094. https://doi.org/10.1021/acschemneuro.7b00461
Pascoal TA, Shin M, Kang MS, Chamoun M, Chartrand D, Mathotaarachchi S, Bennacef I, Therriault J, Ng KP, Hopewell R, Bouhachi R, Hsiao HH, Benedet AL, Soucy JP, Massarweh G, Gauthier S, Rosa-Neto P (2018) In vivo quantification of neurofibrillary tangles with [(18)F]MK-6240. Alzheimers Res Ther 10:74. https://doi.org/10.1186/s13195-018-0402-y
Patterson KR, Remmers C, Fu Y, Brooker S, Kanaan NM, Vana L, Ward S, Reyes JF, Philibert K, Glucksman MJ, Binder LI (2011) Characterization of prefibrillar tau oligomers in vitro and in Alzheimer disease. J Biol Chem 286:23063–23076. https://doi.org/10.1074/jbc.M111.237974
Pérez M, Arrasate M, Montejo De Garcini E, Munoz V, Avila J (2001) In vitro assembly of tau protein: mapping the regions involved in filament formation. Biochemistry 40:5983–5991
Pérez M, Valpuesta JM, Medina M, Montejo de Garcini E, Avila J (1996) Polymerization of tau into filaments in the presence of heparin: the minimal sequence required for tau-tau interaction. J Neurochem 67:1183–1190
Perez-Areales FJ, Betari N, Viayna A, Pont C, Espargaro A, Bartolini M, De Simone A, Rinaldi Alvarenga JF, Perez B, Sabate R, Lamuela-Raventos RM, Andrisano V, Luque FJ, Munoz-Torrero D (2017) Design, synthesis and multitarget biological profiling of second-generation anti-Alzheimer rhein-huprine hybrids. Future Med Chem 9:965–981. https://doi.org/10.4155/fmc-2017-0049
Pittman AM, Myers AJ, Abou-Sleiman P, Fung HC, Kaleem M, Marlowe L, Duckworth J, Leung D, Williams D, Kilford L, Thomas N, Morris CM, Dickson D, Wood NW, Hardy J, Lees AJ, de Silva R (2005) Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J Med Genet 42:837–846. https://doi.org/10.1136/jmg.2005.031377
Plouffe V, Mohamed NV, Rivest-McGraw J, Bertrand J, Lauzon M, Leclerc N (2012) Hyperphosphorylation and cleavage at D421 enhance tau secretion. PLoS One 7:e36873. https://doi.org/10.1371/journal.pone.0036873
Pontecorvo MJ, Devous MD Sr, Navitsky M, Lu M, Salloway S, Schaerf FW, Jennings D, Arora AK, McGeehan A, Lim NC, Xiong H, Joshi AD, Siderowf A, Mintun MA (2017) Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain 140:748–763. https://doi.org/10.1093/brain/aww334
Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP (2013) Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep 14:389–394. https://doi.org/10.1038/embor.2013.15
Prati F, Cavalli A, Bolognesi ML (2016) Navigating the chemical space of multitarget-directed ligands: from hybrids to fragments in Alzheimer's disease. Molecules 21:466. https://doi.org/10.3390/molecules21040466
Prati F, De Simone A, Bisignano P, Armirotti A, Summa M, Pizzirani D, Scarpelli R, Perez DI, Andrisano V, Perez-Castillo A, Monti B, Massenzio F, Polito L, Racchi M, Favia AD, Bottegoni G, Martinez A, Bolognesi ML, Cavalli A (2015) Multitarget drug discovery for Alzheimer's disease: triazinones as BACE-1 and GSK-3beta inhibitors. Angew Chem Int Ed Engl 54:1578–1582. https://doi.org/10.1002/anie.201410456
Qi H, Cantrelle FX, Benhelli-Mokrani H, Smet-Nocca C, Buee L, Lippens G, Bonnefoy E, Galas MC, Landrieu I (2015) Nuclear magnetic resonance spectroscopy characterization of interaction of tau with DNA and its regulation by phosphorylation. Biochemistry 54:1525–1533. https://doi.org/10.1021/bi5014613
Qu MH, Li H, Tian R, Nie CL, Liu Y, Han BS, He RQ (2004) Neuronal tau induces DNA conformational changes observed by atomic force microscopy. Neuroreport 15:2723–2727
Quiroz YT, Sperling RA, Norton DJ, Baena A, Arboleda-Velasquez JF, Cosio D, Schultz A, Lapoint M, Guzman-Velez E, Miller JB, Kim LA, Chen K, Tariot PN, Lopera F, Reiman EM, Johnson KA (2018) Association between amyloid and tau accumulation in Young adults with autosomal dominant Alzheimer disease. JAMA Neurol 75:548–556. https://doi.org/10.1001/jamaneurol.2017.4907
Rabinovici GD, Furst AJ, Alkalay A, Racine CA, O'Neil JP, Janabi M, Baker SL, Agarwal N, Bonasera SJ, Mormino EC, Weiner MW, Gorno-Tempini ML, Rosen HJ, Miller BL, Jagust WJ (2010) Increased metabolic vulnerability in early-onset Alzheimer's disease is not related to amyloid burden. Brain 133:512–528. https://doi.org/10.1093/brain/awp326
Rajamohamedsait H, Rasool S, Rajamohamedsait W, Lin Y, Sigurdsson EM (2017) Prophylactic active tau immunization leads to sustained reduction in both tau and amyloid-beta pathologies in 3xTg mice. Sci Rep 7:17034. https://doi.org/10.1038/s41598-017-17313-1
Reyes JF, Fu Y, Vana L, Kanaan NM, Binder LI (2011) Tyrosine nitration within the proline-rich region of tau in Alzheimer's disease. Am J Pathol 178:2275–2285. https://doi.org/10.1016/j.ajpath.2011.01.030
Reynolds MR, Reyes JF, Fu Y, Bigio EH, Guillozet-Bongaarts AL, Berry RW, Binder LI (2006) Tau nitration occurs at tyrosine 29 in the fibrillar lesions of Alzheimer's disease and other tauopathies. J Neurosci 26:10636–10645. https://doi.org/10.1523/jneurosci.2143-06.2006
Richter M, Hoffmann R, Singer D (2013) T-cell epitope-dependent immune response in inbred (C57BL/6J, SJL/J, and C3H/HeN) and transgenic P301S and Tg2576 mice. J Pept Sci 19:441–451. https://doi.org/10.1002/psc.2518
Richter M, Mewes A, Fritsch M, Krugel U, Hoffmann R, Singer D (2014) Doubly phosphorylated peptide vaccines to protect transgenic P301S mice against Alzheimer's disease like tau aggregation. Vaccines 2:601–623. https://doi.org/10.3390/vaccines2030601
Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L (2007) Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science 316:750–754. https://doi.org/10.1126/science.1141736
Rodriguez-Martin T, Anthony K, Garcia-Blanco MA, Mansfield SG, Anderton BH, Gallo JM (2009) Correction of tau mis-splicing caused by FTDP-17 MAPT mutations by spliceosome-mediated RNA trans-splicing. Hum Mol Genet 18:3266–3273. https://doi.org/10.1093/hmg/ddp264
Saito Y, Ruberu NN, Sawabe M, Arai T, Tanaka N, Kakuta Y, Yamanouchi H, Murayama S (2004) Staging of argyrophilic grains: an age-associated tauopathy. J Neuropathol Exp Neurol 63:911–918
Sanabria Bohorquez S, Barret O, Tamagnan G, Alagille D, Marik J, Ayalon G, Bengtsson T, de Crespigny A, Jennings D, Seibyl JP, Marek K, Weimer R, Kerchner GA (2016) Evaluation of tau burden in a cross-sectional cohort of Alzheimer’s disease subjects using [18F]GTP1 (Genentech tau probe 1). Alzheimers Dement 12:P1172. https://doi.org/10.1016/j.jalz.2016.07.096
Sanabria Bohorquez, S., et al., Kinetics of [18F]GTP (Genentech tau probe 1) in the basal ganglia of Alzheimer's disease patients and healthy controls, in Human Amyloid Imaging Meeting. 2017. p. 32
Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC, Miller TM, Grinberg LT, Seeley WW, Diamond MI (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82:1271–1288. https://doi.org/10.1016/j.neuron.2014.04.047
Sankaranarayanan S, Barten DM, Vana L, Devidze N, Yang L, Cadelina G, Hoque N, DeCarr L, Keenan S, Lin A, Cao Y, Snyder B, Zhang B, Nitla M, Hirschfeld G, Barrezueta N, Polson C, Wes P, Rangan VS, Cacace A, Albright CF, Meredith J Jr, Trojanowski JQ, Lee VM, Brunden KR, Ahlijanian M (2015) Passive immunization with phospho-tau antibodies reduces tau pathology and functional deficits in two distinct mouse tauopathy models. PLoS One 10:e0125614. https://doi.org/10.1371/journal.pone.0125614
Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309:476–481. https://doi.org/10.1126/science.1113694
Sasaki N, Fukatsu R, Tsuzuki K, Hayashi Y, Yoshida T, Fujii N et al (1998) Advanced glycation end products in Alzheimer's disease and other neurodegenerative diseases. Am J Pathol 153:1149–1155. https://doi.org/10.1016/s0002-9440(10)65659-3
Sato C, Barthelemy NR, Mawuenyega KG, Patterson BW, Gordon BA, Jockel-Balsarotti J, Sullivan M, Crisp MJ, Kasten T, Kirmess KM, Kanaan NM, Yarasheski KE, Baker-Nigh A, Benzinger TLS, Miller TM, Karch CM, Bateman RJ (2018) Tau kinetics in neurons and the human central nervous system. Neuron 98:861–864. https://doi.org/10.1016/j.neuron.2018.04.035
Saxena S, Caroni P (2011) Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron 71:35–48. https://doi.org/10.1016/j.neuron.2011.06.031
Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, Van der Flier WM (2016) Alzheimer's disease. Lancet 388:505–517. https://doi.org/10.1016/s0140-6736(15)01124-1
Schoch KM, DeVos SL, Miller RL, Chun SJ, Norrbom M, Wozniak DF, Dawson HN, Bennett CF, Rigo F, Miller TM (2016) Increased 4R-tau induces pathological changes in a human-tau mouse model. Neuron 90:941–947. https://doi.org/10.1016/j.neuron.2016.04.042
Schöll M, Lockhart SN, Schonhaut DR, O'Neil JP, Janabi M, Ossenkoppele R, Baker SL, Vogel JW, Faria J, Schwimmer HD, Rabinovici GD, Jagust WJ (2016) PET imaging of tau deposition in the aging human brain. Neuron 89:971–982. https://doi.org/10.1016/j.neuron.2016.01.028
Schöll M, Ossenkoppele R, Strandberg O, Palmqvist S, Jogi J, Ohlsson T, Smith R, Hansson O (2017) Distinct 18F-AV-1451 tau PET retention patterns in early- and late-onset Alzheimer's disease. Brain 140:2286–2294. https://doi.org/10.1093/brain/awx171
Schwab K, Frahm S, Horsley D, Rickard JE, Melis V, Goatman EA, Magbagbeolu M, Douglas M, Leith MG, Baddeley TC, Storey JMD, Riedel G, Wischik CM, Harrington CR, Theuring F (2017) A protein aggregation inhibitor, Leuco-Methylthioninium Bis (Hydromethanesulfonate), decreases alpha-Synuclein inclusions in a transgenic mouse model of Synucleinopathy. Front Mol Neurosci 10:447. https://doi.org/10.3389/fnmol.2017.00447
Schwarz AJ, Yu P, Miller BB, Shcherbinin S, Dickson J, Navitsky M, Joshi AD, Devous MD Sr, Mintun MS (2016) Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 139:1539–1550. https://doi.org/10.1093/brain/aww023
Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 8:595–608. https://doi.org/10.15252/emmm.201606210
Sepulcre J, Grothe MJ, Sabuncu M, Chhatwal J, Schultz AP, Hanseeuw B, El Fakhri G, Sperling R, Johnson KA (2017) Hierarchical Organization of tau and Amyloid Deposits in the cerebral cortex. JAMA Neurol 74:813–820. https://doi.org/10.1001/jamaneurol.2017.0263
Sepulcre J, Schultz AP, Sabuncu M, Gomez-Isla T, Chhatwal J, Becker A, Sperling R, Johnson KA (2016) In vivo tau, amyloid, and gray matter profiles in the aging brain. J Neurosci 36:7364–7374. https://doi.org/10.1523/jneurosci.0639-16.2016
Sergeant N, David JP, Goedert M, Jakes R, Vermersch P, Buee L, Lefranc D, Wattez A, Delacourte A (1997) Two-dimensional characterization of paired helical filament-tau from Alzheimer's disease: demonstration of an additional 74-kDa component and age-related biochemical modifications. J Neurochem 69:834–844
Sergeant N, Delacourte A, Buee L (2005) Tau protein as a differential biomarker of tauopathies. Biochim Biophys Acta 1739:179–197. https://doi.org/10.1016/j.bbadis.2004.06.020
Sergeant N, Delacourte A, Melnyk P, Buée L (2006) Use of 1,4-bis (3-aminoalkyl) piperazine derivatives in the treatment of neurodegenerative diseases
Shammas SL, Garcia GA, Kumar S, Kjaergaard M, Horrocks MH, Shivji N, Mandelkow E, Knowles TP, Mandelkow E, Klenerman D (2015) A mechanistic model of tau amyloid aggregation based on direct observation of oligomers. Nat Commun 6:7025. https://doi.org/10.1038/ncomms8025
Shimada H, Kitamura S, Ono M, Kimura Y, Ichise M, Takahata K, Moriguchi S, Kubota M, Ishii T, Takado Y, Seki C, Hirano S, Shinotoh H, Sahara N, Tempest P, Tamagnan G, Seibyl J, Barret O, Alagille D, Zhang M-R, Kuwabara S, Jang M-K, Marek K, Suhara T, Higuchi M (2017) FIRST-IN-HUMAN PET STUDY WITH <sup>18</sup>F-AM-PBB3 AND <sup>18</sup>F-PM-PBB3. Alzheimers Dement 13:P146. https://doi.org/10.1016/j.jalz.2017.06.2573
Shiurba RA, Ishiguro K, Takahashi M, Sato K, Spooner ET, Mercken M, Yoshida R, Wheelock TR, Yanagawa H, Imahori K, Nixon RA (1996) Immunocytochemistry of tau phosphoserine 413 and tau protein kinase I in Alzheimer pathology. Brain Res 737:119–132
Sigurdsson EM (2016) Tau immunotherapy. Neurodegener Dis 16:34–38. https://doi.org/10.1159/000440842
Sigurdsson EM (2018) Tau immunotherapies for Alzheimer's disease and related Tauopathies: Progress and potential pitfalls. J Alzheimers Dis 64:S555–s565. https://doi.org/10.3233/jad-179937
Simic G, Babic Leko M, Wray S, Harrington C, Delalle I, Jovanov-Milosevic N, Bazadona D, Buee L, de Silva R, Di Giovanni G, Wischik C, Hof PR (2016) Tau protein hyperphosphorylation and aggregation in Alzheimer's disease and other Tauopathies, and possible neuroprotective strategies. Biomolecules 6:6. https://doi.org/10.3390/biom6010006
Simon D, Garcia-Garcia E, Gomez-Ramos A, Falcon-Perez JM, Diaz-Hernandez M, Hernandez F, Avila J (2012) Tau overexpression results in its secretion via membrane vesicles. Neurodegener Dis 10:73–75. https://doi.org/10.1159/000334915
Simone R (2017) Antisense long non-coding RNA represses MAPT translation through an embedded MIR repeat. Alzheimers Dement 13:918
Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T (2009) Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet 41:1308–1312. https://doi.org/10.1038/ng.487
Singh NK, Singh NN, Androphy EJ, Singh RN (2006) Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol 26:1333–1346. https://doi.org/10.1128/mcb.26.4.1333-1346.2006
Skrabana R, Sevcik J, Novak M (2006) Intrinsically disordered proteins in the neurodegenerative processes: formation of tau protein paired helical filaments and their analysis. Cell Mol Neurobiol 26:1085–1097. https://doi.org/10.1007/s10571-006-9083-3
Smith R, Puschmann A, Schöll M, Ohlsson T, van Swieten J, Honer M, Englund E, Hansson O (2016) 18F-AV-1451 tau PET imaging correlates strongly with tau neuropathology in MAPT mutation carriers. Brain 139:2372–2379. https://doi.org/10.1093/brain/aww163
Sotiropoulos I, Galas MC, Silva JM, Skoulakis E, Wegmann S, Maina MB, Blum D, Sayas CL, Mandelkow EM, Mandelkow E, Spillantini MG, Sousa N, Avila J, Medina M, Mudher A, Buee L (2017) Atypical, non-standard functions of the microtubule associated tau protein. Acta Neuropathol Commun 5:91. https://doi.org/10.1186/s40478-017-0489-6
Southekal S, Devous MD Sr, Kennedy I, Navitsky M, Lu M, Joshi AD, Pontecorvo MJ, Mintun MA (2018) Flortaucipir F 18 quantitation using parametric estimation of Reference signal intensity. J Nucl Med 59:944–951. https://doi.org/10.2967/jnumed.117.200006
Spillantini MG, Goedert M (2013) Tau pathology and neurodegeneration. Lancet Neurol 12:609–622. https://doi.org/10.1016/s1474-4422(13)70090-5
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A 95:7737–7741
Stephens A, Seibyl JP, Mueller A, Barret O, Berndt M, Madonia J, et al. Clinical Update: [18F]PI-2620, a next generation Tau PET agent evaluated in subjects with Alzheimer's disease and progressive supranuclear palsy. Alzheimer's Association International Conference (2018), p. 24827
Stöhr J, Wu H, Nick M, Wu Y, Bhate M, Condello C, Johnson N, Rodgers J, Lemmin T, Acharya S, Becker J, Robinson K, Kelly MJS, Gai F, Stubbs G, Prusiner SB, DeGrado WF (2017) A 31-residue peptide induces aggregation of tau's microtubule-binding region in cells. Nat Chem 9:874–881. https://doi.org/10.1038/nchem.2754
Sultan A, Nesslany F, Violet M, Begard S, Loyens A, Talahari S, Mansuroglu Z, Marzin D, Sergeant N, Humez S, Colin M, Bonnefoy E, Buee L, Galas MC (2011) Nuclear tau, a key player in neuronal DNA protection. J Biol Chem 286:4566–4575. https://doi.org/10.1074/jbc.M110.199976
Tabrizi S, Leavitt B, Kordasiewicz H, Czech C, Swayze EE, Norris DA, Baumann T, Gerlach I, Schobel S, Smith A et al (2018) Effects of IONIS-HTTRx in Patients with Early Huntington’s Disease, Results of the First HTT-Lowering Drug Trial (CT.002). Neurology 90
Takei Y, Teng J, Harada A, Hirokawa N (2000) Defects in axonal elongation and neuronal migration in mice with disrupted tau and map 1b genes. J Cell Biol 150:989–1000
Taniguchi S, Suzuki N, Masuda M, Hisanaga S, Iwatsubo T, Goedert M, Hasegawa M (2005) Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J Biol Chem 280:7614–7623. https://doi.org/10.1074/jbc.M408714200
Thal DR, Rub U, Orantes M, Braak H (2002) Phases of a beta-deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800
Theunis C, Adolfsson O, Crespo-Biel N, Piorkowska K, Pihlgren M, Hickman DT, Gafner V, Borghgraef P, Devijver H, Pfeifer A, Van Leuven F, Muhs A (2017) Novel Phospho-tau monoclonal antibody generated using a liposomal vaccine, with enhanced recognition of a conformational Tauopathy epitope. J Alzheimers Dis 56:585–599. https://doi.org/10.3233/JAD-160695
Theunis C, Crespo-Biel N, Gafner V, Pihlgren M, Lopez-Deber MP, Reis P, Hickman DT, Adolfsson O, Chuard N, Ndao DM, Borghgraef P, Devijver H, Van Leuven F, Pfeifer A, Muhs A (2013) Efficacy and safety of a liposome-based vaccine against protein tau, assessed in tau. P301L mice that model tauopathy. PLoS One 8:e72301. https://doi.org/10.1371/journal.pone.0072301
Thies E, Mandelkow EM (2007) Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci 27:2896–2907. https://doi.org/10.1523/jneurosci.4674-06.2007
Tolnay M, Probst A (2008) Argyrophilic grain disease. Handb Clin Neurol 89:553–563. https://doi.org/10.1016/s0072-9752(07)01251-1
Tosun D, Landau S, Aisen PS, Petersen RC, Mintun M, Jagust W, Weiner MW (2017) Association between tau deposition and antecedent amyloid-beta accumulation rates in normal and early symptomatic individuals. Brain 140:1499–1512. https://doi.org/10.1093/brain/awx046
Troquier L, Caillierez R, Burnouf S, Fernandez-Gomez FJ, Grosjean ME, Zommer N, Sergeant N, Schraen-Maschke S, Blum D, Buee L (2012) Targeting phospho-Ser422 by active tau immunotherapy in the THYTau22 mouse model: a suitable therapeutic approach. Curr Alzheimer Res 9:397–405
Tsartsalis S, Xekardaki A, Hof PR, Kovari E, Bouras C (2018) Early Alzheimer-type lesions in cognitively normal subjects. Neurobiol Aging 62:34–44. https://doi.org/10.1016/j.neurobiolaging.2017.10.002
Umeda T, Eguchi H, Kunori Y, Matsumoto Y, Taniguchi T, Mori H, Tomiyama T (2015) Passive immunotherapy of tauopathy targeting pSer413-tau: a pilot study in mice. Ann Clin Transl Neurol 2:241–255. https://doi.org/10.1002/acn3.171
Uversky VN (2002) Natively unfolded proteins: a point where biology waits for physics. Protein Sci 11:739–756. https://doi.org/10.1110/ps.4210102
van Hummel A, Bi M, Ippati S, van der Hoven J, Volkerling A, Lee WS, Tan DC, Bongers A, Ittner A, Ke YD, Ittner LM (2016) No overt deficits in aged tau-deficient C57Bl/6.Mapttm1(EGFP) kit GFP Knockin mice. PLoS One 11:e0163236. https://doi.org/10.1371/journal.pone.0163236
Vandermeeren M, Borgers M, Van Kolen K, Theunis C, Vasconcelos B, Bottelbergs A, Wintmolders C, Daneels G, Willems R, Dockx K, Delbroek L, Marreiro A, Ver Donck L, Sousa C, Nanjunda R, Lacy E, Van De Casteele T, Van Dam D, De Deyn PP, Kemp JA, Malia TJ, Mercken MH (2018) Anti-tau monoclonal antibodies derived from soluble and filamentous tau show diverse functional properties in vitro and in vivo. J Alzheimers Dis 65:265–281. https://doi.org/10.3233/jad-180404
Velazquez R, Ferreira E, Tran A, Turner EC, Belfiore R, Branca C, Oddo S (2018) Acute tau knockdown in the hippocampus of adult mice causes learning and memory deficits. Aging Cell:e12775. https://doi.org/10.1111/acel.12775
Vemuri P, Lowe VJ, Knopman DS, Senjem ML, Kemp BJ, Schwarz CG, Przybelski SA, Machulda MM, Petersen RC, Jack CR Jr (2017) Tau-PET uptake: regional variation in average SUVR and impact of amyloid deposition. Alzheimers Dement (Amst) 6:21–30. https://doi.org/10.1016/j.dadm.2016.12.010
Verma S, Kumar A, Tripathi T, Kumar A (2018) Muscarinic and nicotinic acetylcholine receptor agonists: current scenario in Alzheimer's disease therapy. J Pharm Pharmacol 70:985–993. https://doi.org/10.1111/jphp.12919
Vermeiren C, Motte P, Viot D, Mairet-Coello G, Courade JP, Citron M, Mercier J, Hannestad J, Gillard M (2018) The tau positron-emission tomography tracer AV-1451 binds with similar affinities to tau fibrils and monoamine oxidases. Mov Disord 33:273–281. https://doi.org/10.1002/mds.27271
Vijayraghavan S, Major AJ, Everling S (2018) Muscarinic M1 receptor overstimulation disrupts working memory activity for rules in primate prefrontal cortex. Neuron 98:1256–1268.e1254. https://doi.org/10.1016/j.neuron.2018.05.027
Violet M, Delattre L, Tardivel M, Sultan A, Chauderlier A, Caillierez R, Talahari S, Nesslany F, Lefebvre B, Bonnefoy E, Buee L, Galas MC (2014) A major role for tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front Cell Neurosci 8:84. https://doi.org/10.3389/fncel.2014.00084
von Bergen M, Barghorn S, Biernat J, Mandelkow EM, Mandelkow E (2005) Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim Biophys Acta 1739:158–166. https://doi.org/10.1016/j.bbadis.2004.09.010
von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow EM, Mandelkow E (2000) Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc Natl Acad Sci U S A 97:5129–5134
von Gunten A, Miklossy J, Suva ML, Hof PR, Glannakopoulos P (2005) Environmental reduplicative paramnesia in a case of atypical Alzheimer's disease. Neurocase 11:216–226. https://doi.org/10.1080/13554790590944825
Vossel KA, Xu JC, Fomenko V, Miyamoto T, Suberbielle E, Knox JA, Ho K, Kim DH, Yu GQ, Mucke L (2015) Tau reduction prevents Abeta-induced axonal transport deficits by blocking activation of GSK3beta. J Cell Biol 209:419–433. https://doi.org/10.1083/jcb.201407065
Walls KC, Ager RR, Vasilevko V, Cheng D, Medeiros R, LaFerla FM (2014) P-tau immunotherapy reduces soluble and insoluble tau in aged 3xTg-AD mice. Neurosci Lett 575:96–100. https://doi.org/10.1016/j.neulet.2014.05.047
Wang JZ, Grundke-Iqbal I, Iqbal K (1996) Glycosylation of microtubule-associated protein tau: an abnormal posttranslational modification in Alzheimer's disease. Nat Med 2:871–875
Wang L, Benzinger TL, Su Y, Christensen J, Friedrichsen K, Aldea P, McConathy J, Cairns NJ, Fagan AM, Morris JC, Ances BM (2016) Evaluation of tau imaging in staging Alzheimer disease and revealing interactions between beta-amyloid and Tauopathy. JAMA Neurol 73:1070–1077. https://doi.org/10.1001/jamaneurol.2016.2078
Wang X, Smith K, Pearson M, Hughes A, Cosden ML, Marcus J, Hess JF, Savage MJ, Rosahl T, Smith SM, Schachter JB, Uslaner JM (2018) Early intervention of tau pathology prevents behavioral changes in the rTg4510 mouse model of tauopathy. PLoS One 13:e0195486. https://doi.org/10.1371/journal.pone.0195486
Wang Y, Balaji V, Kaniyappan S, Kruger L, Irsen S, Tepper K, Chandupatla R, Maetzler W, Schneider A, Mandelkow E, Mandelkow EM (2017) The release and trans-synaptic transmission of tau via exosomes. Mol Neurodegener 12:5. https://doi.org/10.1186/s13024-016-0143-y
Wanowska E, Kubiak MR, Rosikiewicz W, Makalowska I, Szczesniak MW (2018) Natural antisense transcripts in diseases: from modes of action to targeted therapies. Wiley Interdiscip Rev RNA 9. https://doi.org/10.1002/wrna.1461
Weaver CL, Espinoza M, Kress Y, Davies P (2000) Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol Aging 21:719–727
Wei Y, Qu MH, Wang XS, Chen L, Wang DL, Liu Y, Hua Q, He RQ (2008) Binding to the minor groove of the double-strand, tau protein prevents DNA from damage by peroxidation. PLoS One 3:e2600. https://doi.org/10.1371/journal.pone.0002600
Weimer R et al (2017) Evaluation of baseline and longitudinal tau burden in Alzheimer's disease using [18F]GTP1 (Genentech Tau Probe 1) PET imaging. In: Human Amyloid Imaging Meeting, p 91
Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW (1975) A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 72:1858–1862
Whitwell JL, Graff-Radford J, Tosakulwong N, Weigand SD, Machulda M, Senjem ML, Schwarz CG, Spychalla AJ, Jones DT, Drubach DA, Knopman DS, Boeve BF, Ertekin-Taner N, Petersen RC, Lowe VJ, Jack CR Jr, Josephs KA (2018) [(18) F]AV-1451 clustering of entorhinal and cortical uptake in Alzheimer's disease. Ann Neurol 83:248–257. https://doi.org/10.1002/ana.25142
Winblad B, Graf A, Riviere ME, Andreasen N, Ryan JM (2014) Active immunotherapy options for Alzheimer's disease. Alzheimers Res Ther 6:7. https://doi.org/10.1186/alzrt237
Winer JR, Maass A, Pressman P, Stiver J, Schonhaut DR, Baker SL, Kramer J, Rabinovici GD, Jagust WJ (2018) Associations between tau, beta-amyloid, and cognition in Parkinson disease. JAMA Neurol 75:227–235. https://doi.org/10.1001/jamaneurol.2017.3713
Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR (1996) Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A 93:11213–11218
Wischik CM, Harrington CR, Storey JM (2014) Tau-aggregation inhibitor therapy for Alzheimer's disease. Biochem Pharmacol 88:529–539. https://doi.org/10.1016/j.bcp.2013.12.008
Wischik CM, Novak M, Edwards PC, Klug A, Tichelaar W, Crowther RA (1988) Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A 85:4884–4888
Wischik CM, Novak M, Thogersen HC, Edwards PC, Runswick MJ, Jakes R et al (1988) Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A 85:4506–4510
Wong DF, Comley R, Kuwabara H, Rosenberg PB, Resnick SM, Ostrowitzki S, Vozzi C, Boess F, Oh E, Lyketsos CG, Honer M, Gobbi L, Klein G, George N, Gapasin L, Kitzmiller K, Roberts J, Sevigny J, Nandi A, Brasic JR, Mishra C, Thambisetty M, Moghekar A, Mathur A, Albert M, Dannals RF, Borroni E (2018) First in-human PET study of 3 novel tau radiopharmaceuticals: [(11)C]RO6924963, [(11)C]RO6931643, and [(18)F]RO6958948. J Nucl Med. https://doi.org/10.2967/jnumed.118.209916
Wood JG, Mirra SS, Pollock NJ, Binder LI (1986) Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau). Proc Natl Acad Sci U S A 83:4040–4043
Wren MC, Lashley T, Arstad E, Sander K (2018) Large inter- and intra-case variability of first generation tau PET ligand binding in neurodegenerative dementias. Acta Neuropathol Commun 6:34. https://doi.org/10.1186/s40478-018-0535-z
Wu JW, Herman M, Liu L, Simoes S, Acker CM, Figueroa H, Steinberg JI, Margittai M, Kayed R, Zurzolo C, Di Paolo G, Duff KE (2013) Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem 288:1856–1870. https://doi.org/10.1074/jbc.M112.394528
Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, Sanders DW, Cook C, Fu H, Boonen RA, Herman M, Nahmani E, Emrani S, Figueroa YH, Diamond MI, Clelland CL, Wray S, Duff KE (2016) Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci 19:1085–1092. https://doi.org/10.1038/nn.4328
Wu X, Kosaraju J, Tam KY (2017) SLM, a novel carbazole-based fluorophore attenuates okadaic acid-induced tau hyperphosphorylation via down-regulating GSK-3beta activity in SH-SY5Y cells. Eur J Pharm Sci 110:101–108. https://doi.org/10.1016/j.ejps.2017.03.037
Wu X, Kosaraju J, Tam KY (2018) Anti-neuroinflammatory effects of SLOH in Abeta-induced BV-2 microglial cells and 3xTg-AD mice involve the inhibition of GSK-3beta. Neurosci Lett 687:207–215. https://doi.org/10.1016/j.neulet.2018.09.056
Wu X, Kosaraju J, Zhou W, Tam KY (2018) SLOH, a carbazole-based fluorophore, mitigates neuropathology and behavioral impairment in the triple-transgenic mouse model of Alzheimer's disease. Neuropharmacology 131:351–363. https://doi.org/10.1016/j.neuropharm.2018.01.003
Wurster CD, Ludolph AC (2018) Antisense oligonucleotides in neurological disorders. Ther Adv Neurol Disord 11:1756286418776932. https://doi.org/10.1177/1756286418776932
Xia C, Makaretz SJ, Caso C, McGinnis S, Gomperts SN, Sepulcre J, Gomez-Isla T, Hyman BT, Schultz A, Vasdev N, Johnson KA, Dickerson BC (2017) Association of in Vivo [18F]AV-1451 tau PET imaging results with cortical atrophy and symptoms in typical and atypical Alzheimer disease. JAMA Neurol 74:427–436. https://doi.org/10.1001/jamaneurol.2016.5755
Yagi M, Palacpac NM, Ito K, Oishi Y, Itagaki S, Balikagala B, Ntege EH, Yeka A, Kanoi BN, Katuro O, Shirai H, Fukushima W, Hirota Y, Egwang TG, Horii T (2016) Antibody titres and boosting after natural malaria infection in BK-SE36 vaccine responders during a follow-up study in Uganda. Sci Rep 6:34363. https://doi.org/10.1038/srep34363
Yamada K, Cirrito JR, Stewart FR, Jiang H, Finn MB, Holmes BB, Binder LI, Mandelkow EM, Diamond MI, Lee VM, Holtzman DM (2011) In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci 31:13110–13117. https://doi.org/10.1523/jneurosci.2569-11.2011
Yamada K, Holth JK, Liao F, Stewart FR, Mahan TE, Jiang H, Cirrito JR, Patel TK, Hochgrafe K, Mandelkow EM, Holtzman DM (2014) Neuronal activity regulates extracellular tau in vivo. J Exp Med 211:387–393. https://doi.org/10.1084/jem.20131685
Yamada M, Hamaguchi T (2018) The sulfation code for propagation of neurodegeneration. J Biol Chem 293:10841–10842. https://doi.org/10.1074/jbc.H118.003970
Yan SD, Chen X, Schmidt AM, Brett J, Godman G, Zou YS et al (1994) Glycated tau protein in Alzheimer disease: a mechanism for induction of oxidant stress. Proc Natl Acad Sci U S A 91:7787–7791
Yanamandra K, Jiang H, Mahan TE, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM (2015) Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann Clin Transl Neurol 2:278–288. https://doi.org/10.1002/acn3.176
Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM (2013) Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80:402–414. https://doi.org/10.1016/j.neuron.2013.07.046
Yang X, Qian K (2017) Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol 18:452–465. https://doi.org/10.1038/nrm.2017.22
Yu Y, Zhang L, Li X, Run X, Liang Z, Li Y et al (2012) Differential effects of an O-GlcNAcase inhibitor on tau phosphorylation. PLoS One 7:e35277. https://doi.org/10.1371/journal.pone.0035277
Zhang X, Lin Y, Eschmann NA, Zhou H, Rauch JN, Hernandez I et al (2017) RNA stores tau reversibly in complex coacervates. PLoS Biol 15:e2002183. https://doi.org/10.1371/journal.pbio.2002183
Zhou W, Hu X, Tam KY (2017) Systemic clearance and brain distribution of carbazole-based cyanine compounds as Alzheimer's disease drug candidates. Sci Rep 7:16368. https://doi.org/10.1038/s41598-017-16635-4
Zhou Y, Shi J, Chu D, Hu W, Guan Z, Gong CX et al (2018) Relevance of phosphorylation and truncation of tau to the Etiopathogenesis of Alzheimer's disease. Front Aging Neurosci 10:27. https://doi.org/10.3389/fnagi.2018.00027
Zilka N, Filipcik P, Koson P, Fialova L, Skrabana R, Zilkova M, Rolkova G, Kontsekova E, Novak M (2006) Truncated tau from sporadic Alzheimer's disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett 580:3582–3588. https://doi.org/10.1016/j.febslet.2006.05.029
Zilka N, Kazmerova Z, Jadhav S, Neradil P, Madari A, Obetkova D, Bugos O, Novak M. Who fans the flames of Alzheimer's disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J Neuroinflammation. 2012 Mar 7;9:47. https://doi.org/10.1186/1742-2094-9-47
Zilka N, Korenova M, Novak M (2009) Misfolded tau protein and disease modifying pathways in transgenic rodent models of human tauopathies. Acta Neuropathol 118:71–86. https://doi.org/10.1007/s00401-009-0499-y
Zilka N, Kovacech B, Barath P, Kontsekova E, Novak M (2012) The self-perpetuating tau truncation circle. Biochem Soc Trans 40:681–686. https://doi.org/10.1042/bst20120015
Zimova I, Brezovakova V, Hromadka T, Weisova P, Cubinkova V, Valachova B et al (2016) Human truncated tau induces mature neurofibrillary pathology in a mouse model of human Tauopathy. J Alzheimers Dis 54:831–843. https://doi.org/10.3233/jad-160347
Acknowledgements
We would like to especially acknowledge the pioneering work of professors Heiko Braak and Constantin Bouras. Without their outstanding contribution to the field, the current progress in the development of tau therapy would not have been possible. The Eurotau2018 meeting was sponsored by UCB BioPharma, Janssen Pharmaceutical Companies, Sanofi, Axon Neuroscience, Servier, Quanterix, Proteome Sciences and Proteintech. It was also supported by French and Spanish academic funds from University of Lille, SFR DN2M, LabEx DISTALZ, LiCEND, and Ciberned.
Author information
Authors and Affiliations
Contributions
All authors participated in writing, read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
SJ, RS, EK and NZ are Axon Neuroscience R&D Services employees. LB received research grants from UCB Biopharma. Other authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jadhav, S., Avila, J., Schöll, M. et al. A walk through tau therapeutic strategies. acta neuropathol commun 7, 22 (2019). https://doi.org/10.1186/s40478-019-0664-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-019-0664-z