
Abstract
Since the discovery of the microtubule-associated protein Tau (MAPT) over 40 years ago, most studies have focused on Tau’s role in microtubule stability and regulation, as well as on the neuropathological consequences of Tau hyperphosphorylation and aggregation in Alzheimer’s disease (AD) brains. In recent years, however, research efforts identified new interaction partners and different sub-cellular localizations for Tau suggesting additional roles beyond its standard function as microtubule regulating protein. Moreover, despite the increasing research focus on AD over the last decades, Tau was only recently considered as a promising therapeutic target for the treatment and prevention of AD as well as for neurological pathologies beyond AD e.g. epilepsy, excitotoxicity, and environmental stress. This review will focus on atypical, non-standard roles of Tau on neuronal function and dysfunction in AD and other neurological pathologies providing novel insights about neuroplastic and neuropathological implications of Tau in both the central and the peripheral nervous system.
Similar content being viewed by others


Introduction
Considering the increasing interest of diverse research fields on the role of Tau in brain function and pathology in and beyond Alzheimer’s disease (AD) and the recent focus on Tau-based therapeutic strategies, the 1st EuroTau Meeting was organized in Lille, France on April 27 and 28 April 2017. The meeting attracted many clinical and basic Tau researchers throughout Europe providing a unique forum to discuss and exchange ideas and hypotheses. The meeting facilitated the integration of the diverse findings implicating Tau in neuronal physiology and pathology. During the conference, a round table discussion was held to discuss the emerging various atypical, non-standard functions of Tau protein in the sense of divergence from its cytoskeletal association and beyond AD as it is summarized in this review report.
Atypical/non-standard functions of Tau
Tau protein and brain pathology – From past to present
Tau protein was discovered in 1975 [1] and its original name was given by Marc Kirschner as a “factor” that was “associated” with tubulin promoting its self-assembly into microtubules (MTs). Indeed, Tau was one of the first microtubule-associated proteins (MAPs) to be characterized. Its discovery [2,3,4,5,6,7] was followed by the characterization of Tau as an axonal protein in neurons [7, 8]. In living cells, the bulk of Tau protein is attached to microtubules and stabilizes them; hence its role in the microtubule-based cytoskeleton was accepted as the standard Tau function (see also Fig. 1). Note that a non-standard role for Tau in relation to RNA, DNA, or actin binding was suggested almost four decades ago [9,10,11] (for review see [12, 13]), but did not maintain its impetus [14].
A major new line of Tau research was established after the discovery that Tau is a major component of abnormal protein deposits in the brains of patients suffering from AD, a neurodegenerative disorder presenting brain atrophy and memory loss. Indeed, Tau was the first protein to be identified as the main component of neurofibrillary tangles (NFTs), one of the main histopathological hallmarks of AD [15,16,17,18,19]. In the early 1980’s, amyloid beta (Aβ) was also found to be deposited in extracellular amyloid plaques [20] based on results obtained with Down syndrome brains [21] and these amyloid plaques accepted as the second histopathological characteristic of AD brains. During the 80’s, different pathological Tau modifications such as aberrant hyperphosphorylation, conformation, ubiquitylation, acetylation, truncation and aggregation and others, were also identified in AD brains and other neurodegenerative disorders [18, 22,23,24,25,26], now collectively called Tauopathies. The term Tauopathy was used for the first time to define the family with the +3 MAPT mutation [27] (see also the article “What is the evidence that the spread of tau pathology occurs via a prion-like mechanism?” in this issue). In addition, increasing research efforts have been focused on elucidating the physiological versus pathological properties of Tau, investigating mechanisms of neuronal dysfunction and pathology attributed to loss-of-normal function or gain-of-toxic Tau properties in AD and other neuronal pathologies with diverse etiologies e.g. epilepsy, excitotoxicity, and environmental stress [28,29,30].
Transcriptomic and proteomic profile of tau – What do we miss?
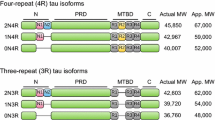
Tau protein in humans is encoded by the MAPT gene, which is located on chromosome 17q21 and comprises 16 exons, where exons 1(E1), E4, E5, E7, E9, E11, E12 and E13 are constitutive, and the others are subjected to alternative splicing. E0 and E1 encode for 5′ untranslated MAPT mRNA sequences, where E0 as part of the promoter, is transcribed but not translated [31, 32]. Alternative mRNA splicing of exons E2, E3 and E10, generates 6 isoforms in the adult human brain. These isoforms differ with regard to the number of 29 residue-long near-amino-terminal inserts, encoded by E2 and E3. Isoforms containing 0, 1 or 2 inserts are known as 0 N, 1 N or 2 N, respectively. Isoforms can also be categorized depending on whether they contain 3 or 4 near carboxyl-terminal repeats (3R and 4R, respectively). The second repeat (R2) is encoded by the alternatively spliced E10, whose inclusion yields the 4R isoform, but it is excluded in mRNA encoding, 3R–Tau [33, 34].
Expression of the six Tau isoforms is developmentally regulated [35], with the smallest and most highly phosphorylated 0N3R (352 a.a) being most abundant in fetal (human or rodent) brains. The Tau expression pattern is modified post-developmentally with a reduction in 0N3R levels and the presence of all six Tau isoforms in the adult human with the levels of 3R and 4R isoforms roughly equal and underrepresentation of the 2 N species [35]. In contrast, there are mainly 4R isoforms in the adult rodent brain [36, 37]. It is unclear at the moment whether such apparent differential regulation of isoform expression of their respective Tau ortholog occurs in invertebrates such as Drosophila or non-mammalian vertebrates [38]. The role of the axon initial segment in the axodendritic sorting of different Tau isoforms has been recently reported in rat cortical neurons [39]. However, these observations raise mostly unanswered questions on whether atypical Tau functions involve particular isoforms exclusively or preferentially. Moreover, the potentially differential distribution of Tau isoforms in the brain and/or their intraneuronal-specific localization remains mostly unanswered.
The round table discussion explored the evidence ascribing atypical Tau functions and debated whether establishment and understanding of these functions would be better unraveled by thorough identification of the intracellular and brain region-specific localization of the different isoforms, or whether its localization alone, disregarding the isoform complexity, can yield expedient understanding of its function(s) in the different locations. The complex nature of the isoform-specific approach in relation to the mouse, rat, human and fruit fly brain was debated. Evidence arguing that a fruitful approach does not necessitate knowledge of isoform-specific subcellular localization was presented from Amrit Mudher suggesting that human Tau isoforms in the Drosophila model present differential phenotypes consistent with unique isoform-specific pathophysiological functions [40]. Consistent with this view, recently published work by Bart Dermaut described a pathological role for the 4R, but not the 3R, Tau during Drosophila development [41], a further demonstration of the utility of this model in addressing such questions in vivo.
A significant point raised in the discussion was the apparent lack of a map detailing Tau isoform-specific or differential localization in a vertebrate brain. However, some published evidence and unpublished work from Maria Spillantini’s lab indicates Tau isoform-specific distribution in the brain, in support of previous studies suggesting considerable regional variation in Tau expression [34]. Hence, Tau mRNA and protein levels in the neocortex are 2~fold higher than those in the white matter and cerebellum [42]. Moreover, splicing of the MAPT primary transcript also presents regional differences. For example, 0N3R Tau is lower in the adult cerebellum than in other regions [42, 43]. Recent findings from Jürgen Götz’s Lab demonstrated that the 1 N tau isoform is highly expressed in the murine pituitary gland, compared to the cortex or hippocampus, but is weaker in the olfactory bulb. The 2 N isoform is enriched in the cerebellum but its levels are also reduced in the olfactory bulb. In contrast, the 0 N isoform presents the highest expression in the olfactory bulb followed by the cortex [44]. These variations may contribute to the well-known differential vulnerability of the distinct brain regions to Tau pathology, while specific disturbances of the normally 1:1 4R/3R ratio are associated with distinct Tauopathies [45, 46]. The regions in which 3R is more abundant could also be associated with higher proliferation or stem cell presence such as the dentate gyrus and olfactory bulb [47].
In terms of intracellular localization, based on immunocytochemical staining, Tau is mainly found in the axons of mature neurons (see Fig. 1). However, it is ubiquitous in immature neurons distributing apparently equally in the cell body and neurites, but becomes primarily axonal during neuronal maturation and emergence of neuronal polarization. This intracellular sorting of Tau is accompanied by a shift towards the higher-molecular-weight 4R isoforms and reduced phosphorylation [4, 48,49,50]. Furthermore, the axonal presence of Tau differs between the ends of the axon, as it is mostly associated with MTs at the distal end of the axon close to the growth cone [51, 52] (see Fig. 1). However, Tau intraneuronal distribution in the human brain is still under debate as nearly equal amounts of Tau were described in the human cerebral gray (somatodendrites) as the underlying white matter (axons) using biochemical assays [53].
Tau phosphorylation is suggested to be involved in this intra-axonal sorting since it was also found to vary along the length of the growing axon. A phosphorylation gradient is evident, with a gradual change from phosphorylated to dephosphorylated Tau going from the soma towards the growth cone [54]. As MTs are more dynamic in the distal regions of growing axons, and dephosphorylation at certain sites increases its affinity towards MTs, these findings suggest that Tau in the growing axon has additional functions to increasing MTs stability. Indeed, a novel function for Tau as a regulator of End Binding proteins 1 and 3 (EB1/3) in extending neurites and axons of developing neurons was presented and discussed by C.L. Sayas [55]. EBs are the core plus-end tracking proteins (+TIPs), which accumulate at the growing ends of MTs, regulating their dynamic state. The current evidence suggests that the interaction between Tau and EBs is direct and dependent on Tau phosphorylation [56] and is dramatically increased by NAP, a neuroprotective peptide, derived from activity-dependent neuroprotective protein [57]. These recent findings offer new insights on the interaction of Tau with other cytoskeletal proteins (e.g. EBs) in mature neurons while future studies should further monitor the role of Tau-EB interaction under pathological conditions e.g. Alzheimer’s disease and other Tauopathies [58].
Multiple studies have provided evidence of low levels of Tau localizing in different intracellular compartments such as the nucleus, nucleolus, plasma membrane, dendrites and dendritic spines (see Fig. 1), as well as in association with various cellular organelles such as the ribosomes, endoplasmic reticulum and the Golgi apparatus [13]. The mechanisms driving this apparent intraneuronal Tau sorting are still not well understood, but evidence suggests that it could occur both at the mRNA or protein level. One of the suggested mechanisms for Tau sorting is based on selective Tau transport into axons or selective degradation in dendrites [59]. An alternative hypothesis suggests that somehow Tau possesses a higher affinity for axons than dendrites [59], consistent with its observed elevation in the axonal compartment. In line with this notion, evidence from Li and colleagues indicated that the axon initial segment (AIS) operates as a barrier against retrograde diffusion of Tau into the dendrites and that Tau phosphorylation and its interaction with MTs is essential for this barrier to be maintained [60]. It has been reported that Tau acetylation destabilizes the AIS cytoskeleton and promote the somatodendritic mislocalization of Tau [61].

A schematic representation of the suggested role(s) of Tau in different subcellular compartments such as neuronal axon, nucleus, post- and pre-synaptic compartments
Furthermore, the projection domain of Tau interacts with membrane complexes and cytoplasmatic components [62], suggesting that it is a differential property of the higher molecular weight isoforms (1 N and 2 N) that possess these domains. It is proposed that Tau interaction with annexin A2, through domains outside those binding MTs [63], contributes to its axon specific distribution and this interaction is modulated by phosphorylation [64], Indeed, Tau mutations leading to aberrant interaction with annexin A2 are likely responsible for the redistribution of Tau away from the axons to the somatodendritic compartment [63].
Interestingly, the intracellular sorting of Tau in different compartments seems to be isoform-dependent [44]. For instance, it has been reported that 1 N isoforms are localized mainly to the nucleus, 0 N isoforms primarily to the cell bodies and axons whereas the 2 N isoforms are elevated in axons and cell bodies [44]. Indeed, Marie Galas and colleagues have recently shown that overexpression of the 0N4R Tau isoform in Tau-knock out (Tau-KO) mouse neurons led to its cytoplasmic localization. Moreover, this Tau isoform goes mostly to the nucleus when tagged with a Nuclear Localization Signal (NLS) [65]. However, such compartment-specific Tau isoform mapping has not been performed in the human brain.
The complexity of using the isoform-specific approach to define other Tau functions was also pointed out, further elaborated because of the existence of Tau species in addition to the six main isoforms [66, 67]. In fact, alternative splicing could yield up to 30 different potential Tau isoforms [32, 66]. In addition, Tau can also be localized in peripheral nervous system (PNS) neurons which express a district high molecular weight (HMW) Tau species [68,69,70]- see also below. This is further complicated by the fact that different Tau transcripts have been described in the literature, including a 2 kb transcript in human cells, that utilize alternate polyadenylation sites on the Tau pre-mRNA, albeit of unknown significance. The 2 kb transcript was found to code for a major nuclear species of Tau [71] and has also been reported in the human frontal cortex by Michel Goedert [19] and in testicular spermatid manchette [72]. The presence of Tau in the sperm and testis has also been reported independently [73, 74]. It is not clear whether the isoform-specific distribution of Tau to either the nucleus, soma and axons reported in the murine brain [44] is dictated by different transcripts (2 kb and 6 kb), or whether analogous transcripts exist in other species e.g. fruit fly. Therefore, unraveling this complexity would provide a better understanding of the isoform-specific localization and function of Tau from the transcript to protein level.
In support of several articles describing a nuclear role for Tau in RNA and DNA protection [50, 75, 76], recent findings from Marie Galas and Eliette Bonnefoy’s teams suggest a structural role in pericentromeric heterochromatin (PCH) architecture, which is impaired in AD brains and a regulatory function for Tau in the expression of PCH lncRNA [65]. Recently, a novel role of Tau in ribosomal DNA transcription and stability has been reported in cells from Bloom’s syndrome patients [77]. Consistent with these findings, data presented by the Serpell Lab provided evidence for a role of Tau in nucleolar transcriptional regulation. Furthermore, extending previous work [78], Alberto Rabano described Tau Nuclear Indentations (TNI) in the entorhinal cortex of early AD patients, which are immune-reactive only to non-phosphorylated Tau epitopes, a potential early marker, and mechanism for the disease. These TNIs may lead to loss of nuclear integrity similar to the effects of lamin invaginations that were reported in the AD brain by the Feany lab [79]. Moreover, the work presented by Bart Dermaut indicated that human Tau expression in Drosophila led to mitotic defects and aneuploidy, similar to the accumulation of aneuploidy observed in splenocytes of Tau-KO mice [80]. This suggests yet another role for Tau in chromosome stability, in agreement with previous studies utilizing peripheral cells from Tauopathy patients [81].
Collectively, the differential distribution of Tau and its isoforms in various cell compartments may reflect distinct subcellularly compartmentalized roles; if so, then disturbances in this Tau sorting and compartmentalization could trigger neuronal dysfunction and neurodegeneration as discussed below. As suggested by different round table participants, future studies should explicitly state the Tau isoform employed in their models, as well as monitor its sub-cellular localization, such that findings can be interpreted taking into consideration that they may not pertain to all Tau isoforms.
Tau splicing and isoform expression in neuronal function and malfunction
Splicing of the MAPT primary transcripts is tightly regulated by several different mechanisms, while its dysregulation and the resulting imbalance of 4R/3R Tau protein and transcripts is causally related to Tau pathology (for review see [24, 82]). The RNA-binding protein Fused in Sarcoma (FUS) may promote skipping of E3 and E10, as FUS knockdown has been reported to increase the expression of 2 N and 4R Tau isoforms [83]. Recently, knockdown of FUS and of Splicing Factor, Proline and Glutamine-rich (SFPQ) was shown to affect E10-related splicing leading to increased 4R/3R ratio, hyperphosphorylation, and neurodegeneration [84]. Small non-coding RNAs (miRNAs) can also influence Tau splicing. For example, miR-132 reduces 4R expression in mouse neuroblastoma cells [85], and miR219 represses Tau protein synthesis by binding to the 3′ untranslated region of the mRNA [86, 87]. Another mechanism that could be linked to the regulation of Tau isoform expression is the formation of ribonucleoprotein granules that results in a shift towards the expression of larger Tau isoforms (see below).
New evidence supports a bi-directional interaction between Tau and the cellular transcriptome. For example, Tau itself can bind to tRNA, a property that may favor Tau fibril formation [88, 89]. Consistent with its role in regulating the cellular transcriptome, unpublished work from Bruno Lefebvre in Luc Buée’s lab provided evidence for an interaction of Tau with the DEAD-box RNA helicase DDX5, supporting a novel role in RNA metabolism and surveillance. Moreover, accumulating evidence from various labs supports a profoundly important role for RNA binding proteins (RBPs) in Tau biology. All RNA is trafficked throughout the neuron in granules composed of RBPs and mRNA. These RBPs appear to spontaneously coalescence into a state resembling lipid droplets or vesicles [90] allowing the RBP/RNA complexes to form granules, which could be considered membraneless organelles. The Tau mRNA-binding proteins RAS GTPase-activating protein-binding protein 1 (G3BP1) and the minor histocompatibility antigen H13 or IMP1 for example, promote the formation of such granules. This leads to a shift towards the production of larger Tau isoforms and therefore, controls axonal sprouting [91] among other functional changes.
Accordingly, a recent study by Akihiko Takashima’s team demonstrated co-localization of Tau mRNA with two RNA binding proteins (RBPs), Stau1 and FMRP, which function as transport proteins. Interestingly, glutamate-driven neuronal activity stimulates local translation of Tau mRNA within mRNP granules in the somatodendritic compartment where the protein accumulates and becomes hyperphosphorylated [92]. Furthermore, another type of RBP/RNA complexes, the Stress Granules (SGs), was recently shown to contribute to Tau pathology and neurodegeneration. SGs normally sequester non-essential mRNA during stressful conditions, allowing the cell to direct protein synthesis towards cytoprotective proteins [93, 94]. However, persistent SG formation seems to be pathological as it directly stimulates Tau aggregation as shown by different studies from the Benjamin Wolozin’s lab [93, 95]. Moreover, Tau was also shown to stimulate the formation of SGs indicating that its interaction with the mRNA trafficking machinery maybe bi-directional [95]. On the other hand, alteration of cytoplasmic eIF2α and reduced SGs formation has been recently reported in the THY-Tau22 tauopathy mouse model under acute hyperthermic stress, raising further questions about the interplay of Tau protein and the cellular transcriptome under physiological and pathological conditions [96].
Novel aspects of physiological functions of tau
Tau hyperphosphorylation and aggregation are well-established key events in AD neuropathology [22]. Although the impact of these disease-associated changes on Tau’s microtubule binding function has been reported [97,98,99,100,101], its effect(s) on atypical Tau functions are not yet known. Thus, the overall contribution of such disease-associated changes to the potential loss or alteration of novel Tau function(s) and AD pathology is still unclear.
Recent experimental evidence from different teams suggests that Tau loss impacts on neuronal function in the CNS and PNS impinging upon different behavioral domains. While deletion of Tau does not precipitate gross behavioral or neurostructural alterations in young/adult mice [28, 102,103,104], previous work has shown that loss of Tau impacts on mechanisms of synaptic plasticity, as Tau-KO animals exhibit deficits in hippocampal LTD [105] and LTP [106]. Moreover, these synaptic changes may be aggravated by aging, as 20-month-old Tau-KO animals also exhibit reduced excitatory synaptic markers and reduced active forms of other MAPs, implicating the cumulative loss of functional MAPs and acetylated tubulin in synaptic deficits and cognitive impairment triggered by aging and loss of Tau [102].
Another age-related phenotype that has been described recently is related to a novel role of Tau in regulated brain insulin signaling [107]. This recent study by David Blum and Luc Buée showed that Tau deletion leads to an impaired hippocampal response to insulin. This could explain the spatial memory deficit upon Tau deletion and peripheral glucose metabolism impairments associated with hypothalamic insulin resistance. In line with this animal evidence, human genetic analyses link the Tau haplotype to glucose homeostasis. The regulatory role of Tau in insulin signaling involves two different nodes. First, Tau-KO mice exhibit higher phosphorylation of IRS-1 at the inhibitory S636 site, known to be linked to insulin resistance in the AD and Tauopathy brain [108, 109], and possibly involve downstream kinase activation. Second, Marininak’s study demonstrates that Tau levels tend to reduce the ability of PTEN lipidphosphatase to dephosphorylate PIP3 into PIP2, an important step in downstream insulin signaling. These findings raise the hypothesis that pathophysiological Tau loss-of-function favors brain insulin resistance, which is likely instrumental for the cognitive and metabolic impairments described in AD patients [107].
Furthermore, Tau involvement in myelination through its interaction with the kinase Fyn and MTs has been also described [110,111,112]. Accordingly, ultrastructural and biochemical analysis of Tau-KO animals demonstrated a hypomyelination phenotype in sciatic nerves of young and adult Tau-KO mice [113] originating in small caliber axons that also exhibit microtubule alterations [114] and altered pain processing [113]. Moreover, these Tau-dependent morphofunctional effects exhibited an age-progressive phenotype with old Tau-KO animals presenting degenerating myelinated fibers and progressive hypomyelination of large-diameter, motor-related axons accompanied by motor deficits [115]. Other studies have also related the age-dependent motor deficits of Tau-KO animals with an age-related loss of substantia nigra (SN) dopaminergic neurons [116] (but also see ref. [103]). Interestingly, similar motor deficits, such as reduced motor strength and coordination, were also found in old animals lacking 4R–Tau, suggesting a potential role for this large isoform in age-dependent development of motor deficits [117]. Note that, although Tau is expressed in both CNS and PNS, the isoforms expressed in adult CNS differ from the HMW Tau isoforms (“big Tau”) found mainly in PNS (e.g., sciatic nerves) but also in optical nerves and retina [70, 118,119,120]. Expression of HMW Tau isoforms may confer increased stabilization and spacing of MTs [121, 122] but to date, our knowledge about Tau function in the PNS is very limited.
Tau protein as key regulator of brain neuroplasticity and neuropathology
In contrast to axons, a small amount of Tau is present in dendrites and dendritic spines under normal, physiological conditions but its function therein has not been well characterized [123, 124]. It is suggested that in this compartment, Tau may regulate synaptic plasticity as pharmacological synaptic activation induces translocation of endogenous Tau from the dendritic shaft to excitatory post-synaptic compartments in cultured mouse neurons and in acute hippocampal slices [125]. Through its interaction with several cellular partners such as tubulin, F-actin, Src family kinases, Tau may play an important role in mediating alterations in the cytoskeletal structure of dendrites and spines as well as synaptic scaffold and signaling [126]. This notion is further supported by the fact that mechanisms of synaptic plasticity are impaired in Tau-KO animals [105, 106] while Tau phosphorylation in specific epitopes is suggested to be critical for synaptic plasticity [127].
Localization of Tau at the synapse has been the focus of several recent reports aiming to determine whether and why Tau is located at the pre-synaptic, the postsynaptic, or both compartments [124]. We now know that Tau interacts directly with filamentous (F) actin [128], localized both in presynaptic boutons and in the head and neck of dendritic spines [129]. Furthermore, using synaptosomes derived from healthy and AD brains, recent studies demonstrated that Tau is present in both pre- and post-synaptic compartments [124], although phosphorylated Tau was found in greater amounts in the postsynaptic sites. Furthermore, using a mouse Tauopathy model expressing the FTDP-17 associated mutation P301L, PHF–Tau was found in both pre- and post-synaptic compartments suggesting that Tau distribution changes in the context of disease [130].
There are several potential mechanisms by which Tau could affect synaptic function and neuronal excitability. It may directly influence synaptic function since, as described above, Tau has been shown to be localized within both pre- and post-synaptic compartments, possibly due to its interaction with other essential synaptic proteins. Further analysis has shown that the phosphorylation status of Tau is modulated through NMDA receptor activation [123]. However, unphosphorylated species are also present in this compartment, suggesting that in synapses, Tau is likely to oscillate between phosphorylated and non-phosphorylated states [123]. Very recently, Kobayachi and colleagues provided evidence that physiological neuronal activity stimulates local translation and phosphorylation of Tau [92]. These data strongly suggest that in dendritic compartments, Tau is involved in physiological synaptic function. However, dendritic localization is more extensively studied in the context of AD pathology, where phosphorylated Tau is missorted into dendrites but also into dendritic spines, causing synaptic dysfunction by suppressing AMPA receptor-mediated synaptic responses, through disruption of post-synaptic targeting and anchoring of glutamate receptors [131].
At the synapse, Tau has been shown to associate with the PSD complex [132], and function in targeting Fyn, a Tyrosine Kinase that belongs to the Src family, to postsynaptic compartments and to be involved in coupling NMDARs to PSD95 [110, 133, 134]. The interaction of Tau with Fyn appears to be essential for targeting Fyn to PSD, where it regulates NMDA receptor function through phosphorylation [135] and the interaction of Fyn with membrane-associated proteins of the plasma membrane [136, 137]. The interaction with Fyn is regulated by the phosphorylation status of Tau, and therefore can be disrupted in disease, when its phosphorylation pattern is altered [133, 136, 138] (see also Fig. 1).
Cumulative evidence from experimental studies using genetic attenuation of Tau levels suggests that it mediates, at least in part, the detrimental effects of Aβ on neuronal function. In fact, Tau ablation has been shown to protect against Aβ-driven AD brain pathology, neurotoxicity and memory impairment [139,140,141,142]. One of the possible mechanisms through which Tau could trigger neuronal and/or synaptic malfunction is based on its Aβ-driven missorting at dendritic spines, a potential early event in AD, preceding the manifestation of detectable neurodegeneration [131, 143]. Recent evidence demonstrated that the intracellular distribution of Tau depends critically on the phosphorylation status of the protein [144]. Accordingly, hyperphosphorylation seems to be necessary for Tau missorting at synapses as mimicking hyperphosphorylation by pseudophosphorylation, mislocalizes it to dendritic spines, an effect not observed with phosphorylation-deficient protein [131]. Importantly, Aβ is a well-known trigger of Tau missorting and dendritic collapse [110, 123, 131, 145,146,147], leading to increased postsynaptic targeting of Fyn [110]. Fyn selectively modulates the function of GluN2B-containing NMDARs, by phosphorylation of the GluN2B on the Y1472 epitope [110, 148]. This phosphorylation is known to stabilize GluN2B at the postsynaptic density linking NMDARs to downstream excitotoxic signaling due to their overexcitation [110, 148].
Recent results from Dr. Sotiropoulos’ team extended the contribution of Tau hyperphosphorylation and missorting to the detrimental effects of exposure to lifetime stress. Stress-dependent Tau missorting may precipitate the dendritic and synaptic malfunctions implicated in the development of neuropsychiatric pathologies such as depression, a known risk factor for AD. These studies demonstrate that chronic stress causes dendritic atrophy, reduced neurogenesis and synaptic deficits in hippocampal integrity leading to cognitive and mood deficits in a Tau-dependent manner [28, 104, 149, 150]. Chronic stress triggers Tau hyperphosphorylation and synaptic missorting of Tau, increased postsynaptic targeting of Fyn and elevation of pGluN2B at the postsynaptic density representing a potential mechanism of stress-driven neurotoxicity. Importantly, all these changes could be abrogated by the ablation of Tau in Tau-KO animals. This, in turn, reveals the protective role of Tau reduction against the establishment of stress-driven hippocampal pathology. This observation is in line with other approaches using Tau-downregulation strategies to tackle neuropathologies with diverse etiology such as AD, epilepsy, Dravet syndrome, excitotoxicity, stress-driven depression [29, 110, 140, 151].
Collectively, these studies highlight Tau protein as a key regulator of neuronal plasticity and pathology in and beyond AD. Indeed, previous studies have shown that Tau hyperphosphorylation and neuronal/synaptic atrophy is also triggered by different intrinsic and extrinsic conditions such as acute stress [152], hypothermia [153], hypometabolism [154], and hibernation [155] in a reversible manner. Thus, future studies are necessary to identify the potential threshold/“point of no return” between Tau-related neuroplasticity and neuropathology during brain aging that may contribute to our understanding of the various precipitating factors of AD as well as of a broader spectrum of brain pathologies.
Future directions
This review further emphasizes the view of Tau as a multifunctional protein. However, it is evident that our knowledge about its atypical/non-standard functions is very limited and could represent only the tip of the Tau “iceberg”. Thus, a main goal of the field is to clarify the exact molecular mechanisms underlying the already-described Tau functions as well as decipher novel Tau physiological roles and their potential involvement in neuropathology. Many participants of this round table discussion suggested that future research efforts should focus on the detailed monitoring of Tau interacting partners, different subcellular locations and post-translational modifications of Tau, as well as the potential implication of various pools of Tau isoforms, aiming to understand their role on Tau action(s) and its role in neuronal (mal)function. Another important issue will be to define the functions of extracellular Tau (see also the article “What is the evidence that the spread of tau pathology occurs via a prion-like mechanism?” in this issue) and their role in the pathophysiological processes.
Conclusions
Although Tau protein was found more than 40 years ago, our knowledge about its role(s) in brain function/malfunction is mainly based on its involvement in AD pathology and other Tauopathies. While we are aware that this review may not cover the entire field (e.g. extracellular Tau –see also above), this short report aimed to summarize recent findings that were presented and discussed in 1st EuroTau meeting related to novel and atypical roles of Tau adding unique insights to our limited knowledge on Tau-related neuronal (mal)function. In light of the accumulating evidence supporting the potential involvement of Tau in neuronal pathologies with diverse etiology, the findings presented and discussed here may trigger novel lines of research that will contribute to better understanding of Tau biology and identify potential therapeutic targets against brain aging and pathology.
Abbreviations
- +TIPs:
-
core plus end tracking proteins
- AD:
-
Alzheimer’s Disease
- AIS:
-
Axonal Initial segment
- AMPA:
-
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- Aβ:
-
amyloid-β
- CNS:
-
Central nervous system
- DDX5:
-
DEAD-box RNA helicase 5
- DNA:
-
Deoxyribonucleic acid
- EBs:
-
End binding proteins
- eIF2a:
-
Eukaryotic translation initiation factor 2A
- FMRP:
-
fragile X mental retardation protein
- FTDP-17:
-
Frontotemporal dementia with parkinsonism linked to chromosome 17
- FUS:
-
RNA-binding protein fused sarcoma
- G3BP1:
-
GTPase-activating protein-bindingprotein 1
- GluN:
-
Glutamate [NMDA] receptor subunit
- H13:
-
Minor histocompatibility antigen
- HMW:
-
High molecular weight
- IMP:
-
Insulin-like growth factor-II mRNA-binding proteins
- IRS-1:
-
Insulin receptor substrate 1
- Kb:
-
Kilo base
- KO:
-
Knockout
- lncRNA:
-
Long non-coding RNA
- LTD:
-
Long-term depression
- LTP:
-
Long-term Potentiation
- MAPs:
-
Microtubule associated proteins
- MAPT:
-
Microtubule Associated Protein Tau
- miRNA:
-
micro RNA.
- mRNA:
-
messenger RNA.
- MTs:
-
Microtubules.
- NAP:
-
Nucleossome assembly protein.
- NFTs:
-
Neurofibrillary Tangle.
- NLS:
-
Nuclear Localization Signal.
- NMD:
-
Nonsense-mediated mRNA decay.
- NMDA:
-
N-methyl-D-aspartate.
- PCH:
-
Pericentromeric heterochromatin.
- PHF:
-
Paired-helical filaments.
- PIP2:
-
Phosphatidylinositol biphosphate.
- PIP3:
-
Phosphatidylinositol triphosphate.
- PNS:
-
Peripheral Nervous System.
- PSD:
-
Post-synaptic Density.
- PTEN:
-
Phosphatase and tensin homolog.
- RBPs:
-
RNA binding protein.
- RNA:
-
Ribonucleic acid.
- SGs:
-
Stress Granules.
- SN:
-
Substantia Nigra.
- TNI:
-
Tau Nuclear Indentations.
References
Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW (1975) A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 72:1858–1862
Cleveland DW, Hwo SY, Kirschner MW (1977) Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol 116:227–247
Cleveland DW, Hwo SY, Kirschner MW (1977) Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J Mol Biol 116:207–225
Drubin DG, Kirschner MW (1986) Tau protein function in living cells. J Cell Biol 103:2739–2746
Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA (1989) Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3:519–526
Himmler A (1989) Structure of the bovine tau gene: alternatively spliced transcripts generate a protein family. Mol Cell Biol 9:1389–1396
Trojanowski JQ, Schuck T, Schmidt ML, Lee VM (1989) Distribution of tau proteins in the normal human central and peripheral nervous system. J Histochem Cytochem 37:209–215
Binder LI, Frankfurter A, Rebhun LI (1895) The distribution of tau in the mammalian central nervous system. J Cell Biol 101:1371–1378
Bryan JB, Nagle BW, Doenges KH (1975) Inhibition of tubulin assembly by RNA and other polyanions: evidence for a required protein. Proc Natl Acad Sci U S A 72:3570–3574
Corces VG, Manso R, De La Torre J, Avila J, Nasr A, Wiche G (1980) Effects of DNA on microtubule assembly. Eur J Biochem 1105:7–16
Corces VG, Salas J, Salas ML, Avila J (1978) Binding of microtubule proteins to DNA: specificity of the interaction. Eur J Biochem 86:473–479
Multhaup G, Huber O, Buée L, Galas M-C (2015) Amyloid precursor protein (APP) metabolites APP intracellular fragment (AICD), Aβ42, and tau in nuclear roles. J Biol Chem 290:23515–23522
Maina MB, Al-Hilaly YK, Serpell LC (2016) Nuclear tau and its potential role in alzheimer’s disease. Biomolecules 6:2–20
Selden SC, Pollard TD (1986) Interaction of actin filaments with microtubules is mediated by microtubule-associated proteins and regulated by phosphorylation. Ann N Y Acad Sci 466:803–812
Brion JP, Couck AM, Passareiro E, Flament-Durand J (1985) Neurofibrillary tangles of Alzheimer’s disease: an immunohistochemical study. J Submicroc Cytol 17:89–96
Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM (1986 May 5) Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem 261(13):6084–6089
Wischik CM, Novak M, Edwards PC, Klug A, Tichelaar W, Crowther RA (1988) Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A 85:4884–4888
Wischik CM, Novak M, Thøgersen HC, Edwards PC, Runswick MJ, Jakes R, Walker JE, Milstein C, Roth M, Klug A (1988) Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A 85:4506–4510
Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A (1988) Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A 85:4051–4055
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (1985) Amyloid plaque core protein in Alzheimer disease and down syndrome. Proc Natl Acad Sci U S A 82:4245–4249
Glenner GG, Wong CW (1984) Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun 122:1131–1135
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI (1986) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83:4913–4917
Derisbourg M, Leghay C, Chiappetta G, Fernandez-Gomez FJ, Laurent C, Demeyer D, Carrier S, Buée-Scherrer V, Blum D, Vinh J, Sergeant N, Verdier Y, Buée L, Hamdane M (2015) Role of the tau N-terminal region in microtubule stabilization revealed by new endogenous truncated forms. Sci Rep 14:9659
Wang Y, Mandelkow E (2015) Tau in physiology and pathology. Nat Rev Neurosci 17:22–35
Iqbal K, Liu F, Gong C-X (2016) Tau and neurodegenerative disease: the story so far. Nat Rev Neurol 12:15–27
Guo T, Noble W, Hanger DP (2017) Roles of tau protein in health and disease. Acta Neuropathol 133:665–704
Spillantini MG, Goedert M, Crowther RA, Murrell JR, Farlow MR, Ghetti B (1997) Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci U S A 94:4113–4118
Lopes S, Vaz-Silva J, Pinto V, Dalla C, Kokras N, Bedenk B et al (2016) Tau protein is essential for stress-induced brain pathology. Proc Natl Acad Sci 113:E3755–E3763
Vossel KA, Tartaglia MC, Nygaard HB, Zeman AZ, Miller BL (2017) Epileptic activity in Alzheimer’s disease: causes and clinical relevance. Lancet Neurol 16:311–322
Li Y, Sun H, Chen Z, Xu H, Bu G, Zheng H (2016) Implications of GABAergic neurotransmission in Alzheimer’s disease. Front Aging Neurosci 23;8:31
Andreadis A, Brown WM, Kosik KS (1992) Structure and novel exons of the human tau gene. Biochemistry 31:10626–10633
Andreadis A (2005) Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim Biophys Acta 1739:91–103
Lee G, Cowan N, Kirschner M (1988) The primary structure and heterogeneity of tau protein from mouse brain. Science 239:285–288
Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA (1989) Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J 8:393–399
Goedert M, Jakes R (1990) Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J 9:4225–4230
Hanes J, Zilka N, Bartkova M, Caletkova M, Dobrota D, Novak M (2009) Rat tau proteome consists of six tau isoforms: implication for animal models of human tauopathies. J Neurochem 108:1167–1176
Bullmann T, Holzer M, Mori H, Arendt T (2009) Pattern of tau isoforms expression during development in vivo. Int J Dev Neurosci 27:591–597
Yoshida H, Goedert M (2002) Molecular cloning and functional characterization of chicken brain tau: isoforms with up to five tandem repeats. Biochemistry 41:15203–15211
Zempel H, Dennissen FJA, Kumar Y, Luedtke J, Biernat J, Mandelkow EM, Mandelkow E (2017) Axodendritic sorting and pathological missorting of tau are isoform-specific and determined by axon initial segment architecture. J Biol Chem 292(29):12192–12207. doi:10.1074/jbc.M117.784702
Sealey MA, Vourkou E, Cowan CM, Bossing T, Quraishe S, Grammenoudi S et al (2017) Distinct phenotypes of three-repeat and four-repeat human tau in a transgenic model of tauopathy. Neurobiol Dis 205:74–83
Malmanche N, Dourlen P, Gistelinck M, Demiautte F, Link N, Dupont C et al (2016) Developmental expression of 4-repeat-tau induces neuronal aneuploidy in drosophila Tauopathy models. Sci Rep 7:1–14
Trabzuni D, Wray S, Vandrovcova J, Ramasamy A, Walker R, Smith C et al (2012) MAPT expression and splicing is differentially regulated by brain region: relation to genotype and implication for tauopathies. Hum Mol Genet 21:4094–4103
Boutajangout A, Boom A, Leroy K, Brion JP (2004) Expression of tau mRNA and soluble tau isoforms in affected and non-affected brain areas in Alzheimer’s disease. FEBS Lett 576:183–189
Liu C, Götz J (2013) Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS One 8
Dickson DW, Kouri N, Murray ME, Josephs KA (2011) Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J Mol Neurosci 45:384–389
Iovino M, Agathou S, Gonzalez-Rueda A, Del Castillo Velasco-Herrera M, Borroni B, Alberici A et al (2015) Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain 138:3345–3359
Fuster-Matanzo A, Llorens-Martín M, Jurado-Arjona J, Avila J, Hernández F (2012) Tau protein and adult hippocampal neurogenesis. Front Neurosci 6:104
Drubin DG, Caput D, Kirschner MW (1984) Studies on the expression of the microtubule-associated protein, tau, during mouse brain development, with newly isolated complementary DNA probes. J Cell Biol 98:1090–1097
Papasozomenos SC, Binder LI (1987) Phosphorylation determines two distinct species of tau in the central nervous system. Cell Motil Cytoskeleton 8:210–226
Sultan A, Nesslany F, Violet M, Bégard S, Loyens A, Talahari S et al (2011) Nuclear tau, a key player in neuronal DNA protection. J Biol Chem 286:4566–4575
Black MM, Slaughter T, Moshiach S, Obrocka M, Fischer I (1996) Tau is enriched on dynamic microtubules in the distal region of growing axons. J Neurosci 16:3601–3619
Hinrichs MH, Jalal A, Brenner B, Mandelkow E, Kumar S, Scholz T (2012) Tau protein diffuses along the microtubule lattice. J Biol Chem 287:38559–38568
Khatoon S, Grundke-Iqbal I, Iqbal K (1994) Levels of normal and abnormally phosphorylated tau in different cellular and regional compartments of Alzheimer disease and control brains. FEBS Lett 351:80–84
Mandell JW, Banker G (1996) A spatial gradient of tau protein phosphorylation in nascent axons. J Neurosci 16:5727–5740
Sayas CL, Tortosa E, Bollati F, Ramírez-Ríos S, Arnal I, Avila J (2015) Tau regulates the localization and function of end-binding proteins 1 and 3 in developing neuronal cells. J Neurochem 133:653–667
Ramirez-Rios S, Denarier E, Prezel E, Vinit A (2016) Tau antagonizes end-binding protein tracking at microtubule ends through a phosphorylation- dependent mechanism. Mol Biol Cell 27:2924–2934
Ivashko-Pachima Y, Sayas CL, Malishkevich A, Gozes I (2017) ADNP/NAP dramatically increase microtubule end-binding protein-tau interaction: a novel avenue for protection against tauopathy. Mol Psychiatry 22:1335–1344
Sayas CL, Ávila J (2014) Crosstalk between axonal classical microtubule-associated proteins and end binding proteins during axon extension: possible implications in neurodegeneration. J Alz Dis 40(Suppl 1):S17–S22
Hirokawa N, Funakoshi T, Sato-Harada R, Kanai Y (1996) Selective stabilization of tau in axons and microtubule-associated protein 2C in cell bodies and dendrites contributes to polarized localization of cytoskeletal proteins in mature neurons. J Cell Biol 132:667–679
Li X, Kumar Y, Zempel H, Mandelkow E-M, Biernat J, Mandelkow E (2011) Novel diffusion barrier for axonal retention of tau in neurons and its failure in neurodegeneration. EMBO J 30:4825–4837
Sohn PD, Tracy TE, Son HI, Zhou Y, Leite RE, Miller BL, Seeley WW, Grinberg LT, Gan L (2016) Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol Neurodegener 11:47
Brandt R, Léger J, Lee (1995). Interaction of tau with the neural plasma membrane mediated by tau's amino-terminal projection domain. J Cell Biol 131:1327–1340
Gauthier-Kemper A, Weissmann C, Golovyashkina N, Sebö-Lemke Z, Drewes G, Gerke V et al (2011) The frontotemporal dementia mutation R406W blocks tau’s interaction with the membrane in an annexin A2-dependent manner. J Cell Biol 192:647–661
Maas T, Eidenmüller J, Brandt R (2000) Interaction of tau with the neural membrane cortex is regulated by phosphorylation at sites that are modified in paired helical filaments. J Biol Chem 275:15733–15740
Mansuroglu Z, Benhelli-Mokrani H, Marcato V, Sultan A, Violet M, Chauderlier A et al (2016) Loss of tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci Rep 6:33047
Luo MH, Tse SW, Memmott J, Andreadis A (2004) Novel isoforms of tau that lack the microtubule-binding domain. J Neurochem 90:340–351
Cross DC, Muñoz JP, Hernández P, Maccioni RB (2000) Nuclear and cytoplasmic tau proteins from human nonneuronal cells share common structural and functional features with brain tau. J Cell Biochem 78:305–317
Georgieff IS, Liem RK, Couchie D, Mavilia C, Nunez J, Shelanski ML (1993) Expression of high molecular weight tau in the central and peripheral nervous systems. J Cell Sci 105:729–737
Nunez J, Fischer I (1997) Microtubule-associated proteins (MAPs) in the peripheral nervous system during development and regeneration. J Mol Neurosci 8:207–222
Goedert M, Spillantini MG, Crowther RA (1992) Proc Natl Acad Sci U S A 89:1983–1987
Wang Y, Loomis PA, Zinkowski RP, Binder LI (1993) A novel tau transcript in cultured human neuroblastoma cells expressing nuclear tau. J Cell Biol 21:257–267
Ashman JB, Hall ES, Eveleth J, Boekelheide K (1992) Tau, the neuronal heat-stable microtubule-associated protein, is also present in the cross-linked microtubule network of the testicular spermatid manchette. Biol Reprod 46(1):120–129
Sigala J, Jumeau F, Caillet-Boudin ML, Sergeant N, Ballot C, Rigot JM, Marcelli F, Tardivel M, Buée L, Mitchell V (2014) Immuno-detection of tau microtubule-associated protein in human sperm and testis. Asian J Androl 16:927–928
Inoue H, Hiradate Y, Shirakata Y, Kanai K, Kosaka K, Gotoh A, Fukuda Y, Nakai Y, Uchida T, Sato E, Tanemura K (2014) Site-specific phosphorylation of tau protein is associated with deacetylation of microtubules in mouse spermato-genic cells during meiosis. FEBS Lett 588:2003–2008
Violet M, Delattre L, Tardivel M, Sultan A, Chauderlier A, Caillierez R et al (2014) A major role for tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front Cell Neurosci 8:84
Violet M, Chauderlier A, Delattre L, Tardivel M, Chouala MS, Sultan A et al (2015) Prefibrillar tau oligomers alter the nucleic acid protective function of tau in hippocampal neurons in vivo. Neurobiol Dis 82:540–551
Bou Samra E (2017) A role for tau protein in maintaining ribosomal DNA stability and cytidine deaminase-deficient cell survival. Nat Commun 8:693
Fernandez-Nogales M, Cabrera JR, Santos-Galindo M, Hoozemans JJ, Ferrer I, Rozemuller AJ et al (2014) Huntington’s disease is a four-repeat tauopathy with tau nuclear rods. Nat Med 20:881–885
Frost B, Bardai FH, Feany MB (2016) Lamin dysfunction mediates neurodegeneration in Tauopathies. Curr Biol 26:129–136
Granic A, Padmanabhan J, Norden M, Potter H (2010) Alzheimer Abeta peptide induces chromosome mis-segregation and aneuploidy, including trisomy 21: requirement for tau and APP. Mol Biol Cell 21:511–520
Rossi G, Conconi D, Panzeri E, Redaelli S, Piccoli E, Paoletta L et al (2013) Mutations in MAPT gene cause chromosome instability and introduce copy number variations widely in the genome. J Alz Dis 33:969–982
Caillet-Boudin M-L, Buée L, Sergeant N, Lefebvre B (2015) Regulation of human MAPT gene expression. Mol Neurodegener 10:28
Orozco D, Tahirovic S, Rentzsch K, Schwenk BM, Haass C, Edbauer D (2012) Loss of fused in sarcoma (FUS) promotes pathological tau splicing. EMBO Rep 13:759–764
Ishigaki S, Fujioka Y, Okada Y, Riku Y, Udagawa T, Honda D et al (2017) Altered tau isoform ratio caused by loss of FUS and SFPQ function leads to FTLD-like phenotypes. Cell Rep 18:1118–1131
Smith PY, Delay C, Girard J, lie PMA, Planel E, Sergeant N et al (2011) MicroRNA-132 loss is associated with tau exon 10 inclusion in progressive supranuclear palsy. Hum Mol Genet 20:4016–4024
Santa-Maria I, Hernandez F, Moreno FJ, Avila J (2007) Taurine, an inducer for tau polymerization and a weak inhibitor for amyloid-beta-peptide aggregation. Neurosci Lett 429:91–94
Santa-Maria I, Alaniz ME, Renwick N, Cela C, Fulga TA, Van Vactor D et al (2015) Dysregulation of microRNA-219 promotes neurodegeneration through post-transcriptional regulation of tau. J Clin Invest 125:681–686
Kampers T, Friedhoff P, Biernat J, Mandelkow EM, Mandelkow E (1996) RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett 399:344–349
Zhang X, Lin Y, Eschmann NA, Zhou H, Rauch JN, Hernandez I, Guzman E, Kosik KS, Han S (2017) RNA stores tau reversibly in complex coacervates. PLoS Biol 15(7):e2002183
Alberti S, Hyman AA (2016) Are aberrant phase transitions a driver of cellular aging? BioEssays 38:959–968
Moschner K, Sündermann F, Meyer H, Da Graca AP, Appel N, Paululat A et al (2014) RNA protein granules modulate tau isoform expression and induce neuronal sprouting. J Biol Chem 289:16814–16825
Kobayashi S, Tanaka T, Soeda Y, Almeida OFX, Takashima A (2017) Local Somatodendritic translation and hyperphosphorylation of tau protein triggered by AMPA and NMDA receptor stimulation. EBioMedicine 20:120–126
Ash PEA, Vanderweyde TE, Youmans KL, Apicco DJ, Wolozin B (2014) Pathological stress granules in Alzheimer’s disease. Brain Res 1584:52–58
Panas MD, Ivanov P, Anderson P (2016) Mechanistic insights into mammalian stress granule dynamics. J Cell Biol:313–323
Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PEA, Cook C, Lummertz da Rocha E et al (2016) Interaction of tau with the RNA-binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep 15:1455–1466
Shelkovnikova TA, Dimasi P, Kukharsky MS, An H, Quintiero A, Schirmer C et al (2017) Chronically stressed or stress-preconditioned neurons fail to maintain stress granule assembly. Cell Death Dis 8:e2788
Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K (1994 Jun 7) Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A 91(12):5562–5566
Alonso AC, Grundke-Iqbal I, Iqbal K (1996 Jul) Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med 2(7):783–787
Alonso AD, Grundke-Iqbal I, Barra HS, Iqbal K (1997 Jan 7) Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci U S A 94(1):298–303
Mudher A, Shepherd D, Newman TA, Mildren P, Jukes JP, Squire A et al (2004) GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in drosophila. Mol Psychiatry 9:522–530
Cowan CM, Chee F, Shepherd D, Mudher A (2010) Disruption of neuronal function by soluble hyperphosphorylated tau in a drosophila model of tauopathy. Biochem Soc Trans 38:564–570
Ma Q-L, Zuo X, Yang F, Ubeda OJ, Gant DJ, Alaverdyan M et al (2014) Loss of MAP function leads to hippocampal synapse loss and deficits in the Morris water maze with aging. J Neurosci 34:7124–7136
Morris M, Hamto P, Adame A, Devidze N, Masliah E, Mucke L (2013) Age-appropriate cognition and subtle dopamine-independent motor deficits in aged tau knockout mice. Neurobiol Aging 34:1523–1529
Lopes S, Teplytska L, Vaz-Silva J, Dioli C, Trindade R, Morais M et al (2016) Tau deletion prevents stress-induced dendritic atrophy in prefrontal cortex: role of synaptic mitochondria. Cereb Cortex 27(4):2580–2591
Kimura T, Whitcomb DJ, Jo J, Regan P, Piers T, Heo S et al (2014) Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos Trans R Soc L. B Biol Sci 369:20130144
Ahmed T, Van der Jeugd A, Blum D, Galas MC, D’Hooge R, Buee L et al (2014) Cognition and hippocampal synaptic plasticity in mice with a homozygous tau deletion. Neurobiol Aging 35:2474–2478
Marciniak E, Leboucher A, Caron E, Ahmed T, Tailleux A, Dumont JIT, Gerhardt E, Pagesy P, Vileno M, Bournonville C, Hamdane MBK, Lancel S, Demeyer D, Eddarkaoui S, Vallez E, Vieau D, Humez SFE, Grenier-Boley B, Outeiro TF, Staels B, Amouyel P, Balschun D, Buée LB, Blum D (2017) Tau deletion promotes brain insulin resistance. J Exp Med 214:2257–2269
Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A et al (2012) Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 122:1316–1338
Yarchoan M, Arnold SE (2014) Repurposing diabetes drugs for brain insulin resistance in Alzheimer disease. Diabetes 63:2253–2261
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J et al (2010) Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142:387–397
Klein C, Kramer E-M, Cardine A-M, Schraven B, Brandt R, Trotter J (2002) Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J Neurosci 22:698–707
Krämer-Albers EM, White R (2011) From axon-glial signalling to myelination: the integrating role of oligodendroglial Fyn kinase. Cell Mol Life Sci 68:2003–2012
Sotiropoulos I, Lopes AT, Pinto V, Lopes S, Carlos S, Duarte-Silva S et al (2014) Selective impact of tau loss on nociceptive primary afferents and pain sensation. Exp Neurol 261:486–493
Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T et al (1994) Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 369:488–491
Lopes S, Lopes A, Pinto V, Guimares MR, Sardinha VM, Duarte-Silva S et al (2016) Absence of tau triggers age-dependent sciatic nerve morphofunctional deficits and motor impairment. Aging Cell 15:208–216
Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK et al (2012) Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med 18:291–295
Gumucio A, Lannfelt L, Nilsson LNG (2013) Lack of exon 10 in the murine tau gene results in mild sensorimotor defects with aging. BMC Neurosci 14:2–25
Sato-Yoshitake R, Shiomura Y, Miyasaka H, Hirokawa N (1989) Microtubule-associated protein 1B: molecular structure, localization, and phosphorylation-dependent expression in developing neurons. Neuron 3:229–238
Georgieff IS, Liem RK, Mellado W, Nunez J, Shelanski ML (1991) High molecular weight tau: preferential localization in the peripheral nervous system. J Cell Sci 100:55–60
Nothias F, Boyne L, Murray M, Tessler A, Fischer I (1995) The expression and distribution of tau proteins and messenger RNA in rat dorsal root ganglion neurons during development and regeneration. Neuroscience 166:707–719
Frappier TF, Georgieff IS, Brown K, Shelanski ML (1994) Regulation of microtubule-microtubule spacing and bundling. J Neurochem 63:2288–2294
Boyne LJ, Martin K, Hockfield S, Fischer I (1995) Expression and distribution of phosphorylated MAP1B in growing axons of cultured hippocampal neurons. J Neurosci Res 140:439–450
Mondragón-Rodríguez S, Trillaud-Doppia E, Dudilot A, Bourgeois C, Lauzon M, Leclerc N et al (2012) Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J Biol Chem 287:32040–32053
Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT (2012) The synaptic accumulation of hyperphosphorylated tau oligomers in alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol 181:1426–1435
Frandemiche ML, De Seranno S, Rush T, Borel E, Elie A, Arnal I et al (2014) Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-Beta oligomers. J Neurosci 34:6084–6097
Morris M, Maeda S, Vossel K, Mucke L (2011) The many faces of tau. Neuron:410–426
Regan P, Piers T, Yi JH, Kim DH, Huh S, Park SJ, Ryu JH, Whitcomb DJ, Cho K (2015) Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J Neurosci 35(12):4804–4812
Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT et al (2007) Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol 9:139–148
Dillon C, Goda Y (2005) The actin cytoskeleton: integrating form and function at the synapse. Annu Rev Neurosci 28:25–55
Harris JA, Koyama A, Maeda S, Ho K, Devidze N, Dubal DB et al (2012) Human P301L-mutant tau expression in mouse entorhinal-hippocampal network causes tau aggregation and presynaptic pathology but no cognitive deficits. PLoS One 7:e45881
Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK et al (2010) Tau Mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68:1067–1081
Kornau H-C, Schenker LT, Kennedy MB, Seeburg PH (1995) Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Source Sci. New Ser 269:1737–1740
Lee G, Newman ST, Gard DL, Band H, Panchamoorthy G (1998) Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci 111:3167–3177
Reynolds C, Garwood C, Wray S, Price C, Kellie S, Perera T et al (2008) Phosphorylation regulates tau interactions with SH3 domains of phosphatidylinositol-3-kinase, phospholipase cgamma 1, GRB2 and SRC-family kinases. J Biol Chem 283(26):18177–18186
Trepanier CH, Jackson MF, MacDonald JF (2012) Regulation of NMDA receptors by the tyrosine kinase Fyn. FEBS J 279:12–19
Usardi A, Pooler AM, Seereeram A, Reynolds CH, Derkinderen P, Anderton B et al (2011) Tyrosine phosphorylation of tau regulates its interactions with Fyn SH2 domains, but not SH3 domains, altering the cellular localization of tau. FEBS J 278:2927–2937
Pooler AM, Usardi A, Evans CJ, Philpott KL, Noble W, Hanger DP (2012) Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol Aging 33:431.e27–431.e38
Bhaskar K, Yen SH, Lee G (2005) Disease-related modifications in tau affect the interaction between Fyn and tau. J Biol Chem 280:35119–35125
Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A (2002) Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A 99:6364–6369
Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T et al (2007) Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316:750–754
Shipton OA, Leitz JR, Dworzak J, Acton CEJ, Tunbridge EM, Denk F et al (2011) Tau protein is required for amyloid β-induced impairment of hippocampal long-term potentiation. J Neurosci 31:1688–1692
Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F et al (2011) Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci 31:700–711
Kimura T, Fukuda T, Park JM, Murayama M, Mizoroki T, Yoshiike Y, Sahara N, Takashima AYS (2007) Hyperphosphorylated tau in parahippocampal cortex impairs place learning in aged mice expressing wild-type human tau. EMBO J 26:5143–5152
Merino-Serrais P, Benavides-Piccione R, Blazquez-Llorca L, Kastanauskaite A, Rábano A, Avila J et al (2013) The influence of phospho-tau on dendritic spines of cortical pyramidal neurons in patients with Alzheimer’s disease. Brain 136:1913–1928
Zempel H, Thies E, Mandelkow E, Mandelkow E-M (2010) Abeta oligomers cause localized ca(2+) elevation, missorting of endogenous tau into dendrites, tau phosphorylation, and destruction of microtubules and spines. J Neurosci 30:11938–11950
Miller EC, Teravskis PJ, Dummer BW, Zhao X, Huganir RL, Liao D (2014) Tau phosphorylation and tau mislocalization mediate soluble a?? Oligomer-induced AMPA glutamate receptor signaling deficits. Eur J Neurosci 39:1214–1224
Tsushima H, Emanuele M, Polenghi A, Esposito A, Vassalli M, Barberis A et al (2015) HDAC6 and RhoA are novel players in Abeta-driven disruption of neuronal polarity. Nat Commun 6:7781
Salter MW, Kalia LV (2004) Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 5:317–328
Pinheiro S, Silva J, Mota C, Vaz-Silva J, Veloso A, Pinto V et al (2015) Tau Mislocation in glucocorticoid-triggered hippocampal pathology. Mol Neurobiol 53:4745–4753
Dioli C, Patrício P, Trindade R, Pinto LG, Silva JM, Morais M, et al (2017) Tau-dependent suppression of adult neurogenesis in the stressed hippocampus. Mol. Psychiatry In press:1–9; 22:1110-1118
Gheyara AL, Ponnusamy R, Djukic B, Craft RJ, Ho K, Guo W et al (2014) Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann Neurol 76:443–456
Rissman RA, Lee K-F, Vale W, Sawchenko PE, Alonso A, Grundke-Iqbal I et al (2007) Corticotropin-releasing factor receptors differentially regulate stress-induced tau phosphorylation. J Neurosci 27:6552–6562
Planel E, Miyasaka T, Launey T, Chui D-H, Tanemura K, Sato S et al (2004) Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer’s disease. J Neurosci 24:2401–2411
van der Harg JM, Nölle A, Zwart R, Boerema AS, van Haastert ES, Strijkstra AM et al (2014) The unfolded protein response mediates reversible tau phosphorylation induced by metabolic stress. Cell Death Dis 5:e1393
Arendt T, Stieler J, Strijkstra AM, R a H, Rüdiger J, E a V d Z et al (2003) Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci 23:6972–6981
Acknowledgements
1st EuroTau meeting has been funded by French academic funds and charities/foundations such as Fondation Plan Alzheimer, LabEx DISTALZ, LiCEND Centre of Excellence, SFR DN2M - University of Lille.
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sotiropoulos, I., Galas, MC., Silva, J.M. et al. Atypical, non-standard functions of the microtubule associated Tau protein. acta neuropathol commun 5, 91 (2017). https://doi.org/10.1186/s40478-017-0489-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-017-0489-6