
Abstract
One major objective for our evolving understanding in the treatment of cancers will be to address how a combination of diagnosis and treatment strategies can be used to integrate patient and tumor variables with an outcome-oriented approach. Such an approach, in a multimodal therapy setting, could identify those patients (1) who should undergo a defined treatment (personalized therapy) (2) in whom modifications of the multimodal therapy due to observed responses might lead to an improvement of the response and/or prognosis (individualized therapy), (3) who might not benefit from a particular toxic treatment regimen, and (4) who could be identified early on and thereby be spared the morbidity associated with such treatments. These strategies could lead in the direction of precision medicine and there is hope of integrating translational molecular data to improve cancer classifications. In order to achieve these goals, it is necessary to understand the key issues in different aspects of biotechnology to anticipate future directions of personalized and individualized diagnosis and multimodal treatment strategies. Providing an overview of translational data in cancers proved to be a challenge as different methods and techniques used to obtain molecular data are used and studies are based on different tumor entities with different tumor biology and prognoses as well as vastly different therapeutic approaches. The pros and cons of the available methodologies and the potential response data in genomics, microRNA, epigenetics and proteomics with a focus on upper gastrointestinal cancers are considered herein to allow for an understanding of where these technologies stand with respect to cancer diagnosis, prognosis and treatment.
Similar content being viewed by others


Introduction
According to the surveillance, epidemiology and end results (SEER) database, the estimated incidence for the USA of male-predominant upper gastrointestinal (GI) cancer (esophagus and gastric) includes 13.6 % of cancers of the digestive system and 2.4 % of all cancer sites with an observed peak in cancer rates in 1991 followed by a decrease of 24 % in men and 16 % in women through 2009, but esophageal cancer remains the 5th leading cause of cancer deaths in males between the ages of 40 and 59 years [1]. Elsewhere, Iranian colleagues reported the high incidence of esophageal cancer as an esophageal cancer belt comparing it with the incidence of laryngeal cancer [2]. The high incidences of upper GI cancers in northern Iran, Kazakhstan, northern central China, (especially Linxian Province), Japan and Singapore [3] can be thought of as an upper GI cancer terrestrial belt.
Despite many variables [4] epidemiologic observations are of value. Since the identification of Helicobacter pylori (H. pylori), the incidences in gastric cancers have decreased worldwide, but there are reports of increases in esophageal squamous cell carcinomas (ESCC) as well as of adenocarcinomas of the esophago-gastral junction [5]. On the other hand, there has been a shift of distal gastric carcinomas to the proximal area of subcardial adenocarcinoma of the esophago-gastral junction (AEG Type III) and cardia localization (AEG Type II) over an 80-year period. This shift in tumor localization and its differences with rising incidences in the US or Asia are not well understood [5, 6] and provides an epidemiological challenge to identify and explain the worldwide differences in order to create effective preventive strategies. Eradication therapy for H. pylori and/or Barrett’s metaplasia alone cannot be the sole explanation as metaplasia of the esophago-gastric mucosa results in just 2 % mortality within 10 years of diagnosis [7]. Currently, ssurveillance seems to be the most important preventive strategy given the implementation in Korea and Japan of early tumor categories through nationwide screening programs with more favourable prognoses and patient outcomes [8]. The absence of an aggressive screening program may be one explanation of why, in Western countries such as the USA, 75 % of patients with upper GI cancers are diagnosed with locally advanced tumor categories and with correspondingly lower survival rates [5]. It is generally accepted that infection with H. pylori results in chronic inflammation, gastritis, and peptic ulcer [9]. The effects of such chronic inflammation are observed in more than 60 % of gastric cancer patients [10]. It is of molecular relevance that E-Cadherin can be observed 48 h after H. pylori infection in small vesicles [11] and that membrane vesicles of bacteria contain lipopolysaccharides, chromosomal deoxyribonucleic acid (DNA), plasmids, and phage DNA [12].
Studies using irradiated human skin fibroblasts have revealed the autocrine function of cyclooxygenase-2 (=Prostaglandin G/H synthetase 2, =COX-2-) dependent prostaglandin (PGE2) and cytokine production in conjunction with nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κβ-) dependent gene expression of cytokines such as Interleukin 1 beta (IL-1B), IL-3, IL-6, IL-8, TNF and PTGS2/COX-2 [13]. Therefore, it appears that chronic inflammation is one of the important sequences in accordance with a recently proposed multistep process of carcinogenesis [14, 15].
The importance of inflammation in carcinogenesis is supported by findings showing that H. pylori induces PTGS2/COX-2 signaling in pre-cancerous lesions and that anti-H. plyori treatment results in a decrease of PGE2 levels with an observed regression of gastric pre-cancerous lesions [16]. This also explains why chronic inflammation plays a pivotal role early in cancer and why, until recently, the origin of less than 15 % of all cancers were shown to be hereditary based on the somatic mutation theory that has been predominant for some 85 years [14, 15]. From genetically derived cancers (estimated to account for some 5 to 10 % of all cancers) only about 1 % represents gastric carcinomas, 3–5 % for colorectal cancers, and about 8 % for breast cancers (breast cancer 1, early onset = BRCA1 or breast cancer 2, early onset = BRCA2) [17–19].
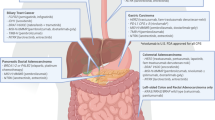
Recently, it has been suggested that in order to more accurately elucidate the origin of cancers, a detailed personalized and individualized conceptual model is needed in terms of both strategy and content [20]. Both seem more difficult than previously assumed as the necessity of providing available and missing evidence is required to bring about the integration of translational molecular biological data to clinical processes such as the cancer classifications proposed by the American Joint Cancer Committee (AJCC). The speed of progress in molecular biology makes such an endeavor difficult and also new technologies e.g. complex nanoparticles might will have significant influence [21]. One aspect involves evaluating available knowledge in a critical manner. Herein, we review translational data in genomics, microRNA, epigenetics and proteomics (Fig. 1) (modified according to [22]) for monitoring responses and, where available, patient outcomes with an emphasis on GI carcinomas.

Schematic drawing of various types of diagnostic and molecular biological options for science and research (modified according to [22])
Review
Genomics
Quantitative RT-PCR, qPCR
General
The invention of the polymerase chain reaction (PCR) in 1955 by Kjell Kleppe and Ian J. Molineux, and its subsequent modification by Kary B. Mullis in 1983 helped realize its potential and in so doing revolutionized biology and medicine [23–25]. PCR allows the amplification of genetic information from a few copies or a single piece of DNA. Various modifications in enzymes, quenchers, primers and protocols are used depending on the specific goal of the researcher and the questions to be addressed. However, the wide spectrum of modifications carries with it the burden of “open-to-interpretation” results and, consequently, their significance or lack thereof. Quantitative real-time reverse-transcription polymerase chain reaction (qRT-PCR) has been widely used to identify gene expression profiles in various cancers and probe differences between the genetic makeup of cancer versus normal tissues [26]. Even though qRT-PCR is perceived as less labor-intensive and a more high-throughput method than conventional RT-PCR (which involves a cDNA synthesis step) two opportunities for variability include template preparation and the dispensing of reagents. Sample collection is a crucial step in the evaluation of the gene expression profiles between tissues.
Up until recently, tissue RNA extraction was carried out from the entire gross resection and/or biopsy. Typically, these tissues consist of a large number of cells with only a small percentage being cells of interest. This is particularly important if qRT-PCR is used to determine small metastases that consist of a small number of cells (10–100 cells/site) and consequently stay undetected because of being masked by the expression level of surrounding cells. This example reveals that tissue sampling is a crucial step in the quality of data obtained and in their subsequent interpretation. Tissue sampling is performed with needles that collect a predetermined volume of the tissue of interest. This method of sample collection became more popular with the utilization of microdissection of the samples by robots that allowed for the collection of small amounts of material (as little as a single cell). However, this increase in dissection capability was not accompanied by a corresponding increase in the capability to perform qRT-PCR. Recently, a single cell RNA expression kit became available [27] but its use in high-output clinical facilities remains to be evaluated. Thus, caution has to be exercised when interpreting qRT-PCR results.
Results from this approach reflect “one” brief moment and only represent the steady state mRNA levels during the time the tissue sample was taken. Therefore, quantification of mRNA levels yields information that is measured during that single time point. No information about mRNA integrity and/or protein level is obtained.
The recently described detection of micro RNA (=miRNA, miR) introduced yet another complexity to the value of qRT-PCR namely post-transcriptional modification of translation in which mRNA level is stable with attenuation of protein synthesis. In addition, there is an influence by mRNA silencing and mRNA splitting such that qRT-PCR no longer represents being a measure of functional expression. RT-PCR data provide less usable information about protein activity or about possible mutations that the target gene might harbor unless it is in the primer binding region. Primer design can partially compensate for the problem of mutations by specifically designing primers to target small point mutations and large deletions but that requires knowing the sequences of these mutations. Therefore, for complete and biologically relevant analysis of gene expression it is necessary to complement qRT-PCR information with data that derived from immunohistochemistry and biochemical assays. Additionally, the qRT-PCR method itself is fraught with issues such as loading of reagents, variability between technicians, normalization between samples and choice of housekeeping genes. This has been reviewed in detail [28, 29].
Tumor heterogeneity and mutations
Intra-patient heterogeneity is a challenge for developing effective therapies. With progress in the human genome project, initial promises such as base pair resolution, genome wide and exon sequencing have become routine. Nowadays, the use of sequencing allows for the collection of enormous amounts of data with the ability to decipher the meaning of the data and attempt to translate these into the treatment of cancer patients. However, 99.9 % of all mutations that occur within the coding regions of the genome are not fully understood nor have they been thoroughly investigated. Additionally, the number of mutated genes and mutations per gene or per cancer in the coding region varies greatly [30]: Some 95–97 % of mutations are single-base substitutions and 3–5 % constitutes insertions and deletions. Furthermore, of the reported single-base mutations, some 90.7 % are missense changes, 7.6 % nonsense, and 1.7 % involves splice sites that are in non-translated regions right after the start or stop codons.
The number of mutated genes varies with smaller number of somatic mutations observed in the younger patient population in comparison with older patients with the same cancer. The number of observed mutations also varies between tissues hosting the primary cancer. Tissues with high rates of cell division, such as the colon or skin, have larger numbers of mutations per cell compared to cancers with slowly dividing tissues i.e., brain [30, 31]. The enormous variability of the mutations combined with the fact that more than 50 % of mutations occur in the cell before the cancer phenotype is established introduces a high noise to signal into genome and/or exon sequencing that confounds the interpretation of data [30, 31]. It has been assumed that mutations occur over long periods of time, in some cases over several decades and this fact alone suggests that sequencing results may vary greatly as a function of the time of sample collection.
Furthermore, it was assumed that mutations occurred after a long latency period of several decades. It has been inferred that this is a main reason for the variability in results. Recently it was demonstrated, using tumor exome sequencing, that acute lymphoblastic B-cell leukemia (B-cell ALL) is not caused by mutations but rather by infection and that mutations of Janus-activated kinase 3 (JAK-3) occur after carcinogenesis is already underway [32]. Thus, somatic mutations are increasingly seen as epiphenomena and subsequent events [14, 15].
An apple found in a car is not synonymous with the proof that apples grow in cars.
The observation of a mutation within a tissue or tumor (i.e., a somatic mutation) is not synonymous with proof that mutations are causally related to the cancer.
In contrast, a recent report suggested that oncogenes, in particular, Kirsten rat sarcoma viral oncogene (KRAS), is causally linked to de novo tumor development in human mammary cells, in vitro and when implanted in immune-deficient mice [33]. Normal human mammary cell types obtained from 37 normal human reduction mammoplasty samples included basal cells (BCs), luminal progenitors (LPs), luminal cells (LCs) and stromal cells (SCs). Such cells were exposed to encoding lentiviral preparations (encoding complementary DNAs) for TP53 and TP53-GFP (green fluorescent protein), phosphatidylinositol-45-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) and PIK3CA-YFP (yellow fluorescent protein), KRAS and mCherry-KRAS, and in some experiments, to a library of biologically neutral, barcoded lentiviral GFP vectors to allow subsequent clonal tracking of their progeny using a DNA sequencing approach. Afterwards these cells were embedded in a collagen gel and the gels transplanted into highly immunodeficient NOD-SCID or NRG female mice. The results showed that BCs and/or LPs isolated from 17 of 27 normal donors and exposed to all three oncogenic vectors produced tumors resembling invasive ductal carcinomas within 8 weeks at similar overall frequencies (46 % of BC isolates and 61 % of LP isolates). However, some major questions were not addressed such as,
-
What happened in the other 54 % of BC isolates and 39 % of LP isolates?
-
Why did identical treatment of LCs and SCs isolated from three of these samples not produce any tumors in the same 8-week period?
Tumors were obtained only when the KRAS oncogene was included and even on its own (64/102 = 63 % for all transductions that included KRAS) compared with 1/12 = 8 % when KRAS was not present—does this allow for the inference that KRAS is the causal factor in the observed tumors in mice? The authors suggest that their studies provide new insights into the earliest phases of malignant transformation in vivo of cells isolated directly from normal human mammary tissue. Five aspects of this study are noteworthy:
-
1.
Rapidity and efficiency (though with high variability) with which this process can be induced using a single transducing oncogene (KRAS).
-
2.
Considerably heterogeneity displayed in the numbers, phenotypes and growth behavior of clonally tracked human cells with tumorigenic activity in vivo within 2–8 weeks.
-
3.
Lack of strong influence of human mammary cell type initially transduced with frequency of clones generated, the histopathology of the tumors produced or their loss of lineage-specific expression profiles. This suggests a greater effect of the potent transforming role of the KRAS oncogene in these cells. What prevents KRAS from being 100 % effective in this mouse model?
-
4.
Frequent delayed activation of clonal growth observed in secondary tumors. This latency could either be biologically determined, reflecting an origin of these late-appearing clones from their normal counterparts with similar features, or simply reflective of a stochastic process, as previously indicated for established human breast cancer cell lines passaged in vivo.
-
5.
If mainly basal cells, which have special location in vivo, produce cancers as proposed, why are the majority of breast cancers found in the epithelial and not in basal cells?
The research by this group provides insights to our understanding of the role of genetics in the origin of cancers, but an old dogma—the somatic mutation theory—is used to explain the results as “important” even though the clinical data tell a different story.
A recent report sheds new light on how enzymes may continuously promote mutations in cancer after the carcinogenesis process has been initiated. High levels of the DNA cytosine deaminase APOBEC3B (A3B) found in breast cancers are associated with poor survival and increased rates of resistance to tamoxifen [34]: A3B changes the microenvironment with observed secondary increases in mutation rates of cytosine within the estrogen receptor positive breast cell line, MCF-7L. Suppression of A3B in a xenograft model was associated with increased responsiveness to tamoxifen. It was suggested that an ongoing stimulus such as a virus, for instance, may affect an increase of A3B and that this would explain why increased mutation rates are found in locally advanced breast cancer.
Furthermore, it has been shown that a liver cancer sample of 2.5 cm (1 inch) contains more than 100 million mutations [35]. The authors showed that this high degree of genetic diversity was independent of whether the tumor sample was a thin or thick one. This observation raises the following questions:
-
Do we know what mutations result in a given percentage of harm to an organism?
-
If a mutation is observed, do we know when it occurred?
-
Does the detection of mutation reveal when it was modified and/or repaired, albeit incorrectly or incompletely?
-
What is the fate of any given mutation in a living organism and how do we know this?
There are other significant biological challenges as well as it has been shown that an identical mutation can result in different phenotypes [36]. We now also know that there is routine processing of mutations within the physiological context of normal growth as an integral part of development and evolution [37]. This raises the issue of how we can assume that any mutations being measured in tissue is necessarily pro-carcinogenic.
Investigating some 17 million single nucleotide variants from genomes of 562 tumors, it was shown that differential DNA repair, and not mutations, is the primary cause of large-scale regional mutation rate variations across the human genome [38].
By examining some 450 somatic mutations accumulated in non-repetitive genome sequences from the blood of a healthy 115-year old woman, the mass of mutations observed were harmless mutations suggesting that “the finite lifespan of hematopoetic stem cells (HSCs), rather than somatic mutation effects, may lead to hematopoietic clonal evolution at extreme ages” [39]. Another report analyzed 4742 tumor-normal pairs across 21 cancer types; the data set consisted out of “3,078,483 somatic single nucleotide variations (SSNVs), 77,270 small insertions and deletions (SINDELs) and 29,837 somatic di-, tri- or oligonucleotide variations (DNVs, TNVs and ONVs, respectively), with an average of 672 per tumour–normal pair. The mutations included 540,831 missense, 207,144 synonymous, 46,264 nonsense, 33,637 splice-site, and 2,294,935 non-coding mutations” [40]. These authors found 145,000 genetic variations per cancer type. Thus, it would appear that somatic mutations are likely an epiphenomena and/or constitute events that occur after carcinogenesis has begun [14, 15, 41, 42], as there are also cancers which are not associated with mutations [43, 44].
The importance of these somatic mutation data is mechanistic in that these serve to trace metastasis and to evaluate entire pathways since proteins with translated mutations interact with the other proteins that may or may not be mutated. In this regard, the influence of activated signaling pathways in the cytoskeleton seems to be particularly relevant. The metastasis of epithelial tumor cells is exemplified by Syndecan-4 as the actin cytoskeleton and cell contractility are modified by a second signal path (PKC) [45].
Another variable which influences our understanding is that proteins with translated mutations can result in interactions with other proteins. For example, point mutations occur at a similar rate in cancer and non-cancerous cells and larger genetic material rearrangements such as translocations and changes in chromosome numbers, occur more frequently in cancer cells than in noncancerous cells which support the importance of the timing of sampling [46]. Despite these important challenges of intratumoral, intermetastatic, intrametastatic and inter-patient variability, some important findings have been obtained by sequencing methods, primarily that more than 138 driver mutations identified to date can be divided into pathways involved in cell survival, cell fate, and genome maintenance [30].
Sequencing
Transcriptome sequencing analysis, also known as RNA-seq, has been used in cancer research for the detection of transcribed mutations and confirmation of known and unknown mutations. RNA-seq is performed on the isolated total RNA from a tumor versus control samples in order to determine the differences between the two [47]. Research in prostate cancer has revealed seven new cancer-specific gene fusions, two involving the E26 transformation-specific or E-twenty-six transcription factor family (ETS) genes, ETS translocation variant 1 (ETV1) and ETS related gene (ERG), and four involving non-ETS genes such as CDKN1A (p21), cell surface glycoprotein encoded by CD9 gene (CD9), and IKBKB (IκK-beta) [48]. Using two different technologies for confirmation provides important fusion proteins like LnCap and VCaP in prostate cancer cell lines followed by identification of those proteins in the patient samples [49]. The downregulation of protein tyrosine kinase 6 (PTK6) in esophageal squamous cell carcinoma (ESCC) has been described [50].
Combining these data using bioinformatics might provide a better understanding of mutations and their role in cancer progression and might also provide insight into how miRNA functions as post-transcriptional regulators of gene expression in cancer progression [51].
MicroRNA
MicroRNA (miR, miRNA) are small (20–24 nucleotides long) well-conserved RNA molecules involved in the control of translation of mRNA in the cell. miRNAs associated with cancers are called oncomirs. Their discovery occurred in 1993 by Ambros, Lee and Feinbaum in the nematode, Caenorhabditis elegans [52]. Since then, the role of miRNAs has been described in several human cancers [53]. There are more than 800 identified miRNAs [54] and their expression patterns vary in different cancers [55]. Altered miRNA expression has been reported for hepatocellular carcinoma [56, 57], pancreatic cancer [58, 59], breast cancer [60, 61] papillary thyroid cancer [62], chronic lymphocytic leukemia [63] and esophageal cancer [64–66]. The results, however, showed that up- and/or down-regulation of the miRNA of interest is usually small when comparing cancerous vs. noncancerous tissues [64, 67]. miRNAs can play different roles in esophageal cancer tumorigenesis exhibiting both pro- and anti- proliferative roles and are differentially expressed in squamous cell carcinoma and in adenocarcinoma, with and without Barrett’s metaplasia [68].
miRNAs have been detected in tissues using qRT-PCR and in situ hybridization [69]. In frozen tissue samples, 509 mature miRNA assay identified several miRNAs distinctively expressed in tumor cells when compared to corresponding normal tissue, hsa-miR-103/107 complex, in particular, showed a strong correlation between low expression and high survival periods [70]. miRNA-21 has been proven to control proliferation in vitro and in humans [69, 71]. Although it was suggested, that miRNA is part of the cancer secretome, miRNA is not a protein and therefore cannot be a part of any protein group. These molecules end up in serum and in exosomes. The term secretome was coined by Tjalsma in 2000 defining the secreted proteins in Bacterium subtilis [72] and, in 2010, Agrawal suggested using this term for ‘the global group of proteins secreted into the extracellular space by a cell, tissue, organ, or organism [73].
In the serum of patients with squamous cell carcinoma vs. patients with benign disease without inflammation, researchers detected exosomes that contain miRNA-21 [74, 75]. Serum samples depleted of exosomes did not have PCR detectable levels of this miRNA. MicroRNA-21 has been detected in the serum of patients with ESCC (100 % in one series) and its level in serum was shown to be dependent on the presence of the tumor. After resection, serum levels of microRNA-21 dropped [74, 75]. More clinical trials with larger patient populations are needed in order to fully explore the significance of microRNA-21 as a marker and predictor of responses to treatment. So far it is not known if free or exosomal miRNA has the most value as a biomarker. However, recently the potential to discriminate between precursor metaplastic Barrett’s mucosa from adenocarcinoma using miRNA was reported [76].
Transfection of colon cancer cell lines with miRNA-21 led to increases in downregulation of programmed cell death protein 4 (PDCD4), transforming growth factor beta receptor 2 levels of beta-catenin, TCF/LEF activity, and expressions of c-Myc, Cyclin-D, which are increased in cancer stem cells (CSCs) and where these are accompanied by an increased sphere-forming ability in vitro and tumor formation in SCID mice [77]. In liver regeneration, it is shown that miRNA-21 regulates rapid translation of G1/S-specific Cyclin D1 (Cyclin D1 and, consequently, increases cell proliferation [78]. MicroRNA-22 is also interesting from a response-to-therapy perspective. Researchers did not find a correlation between levels of miRNA-22 and overall survival but did find a correlation between its levels in tissue and stage of the tumor as well as in the tumor’s response to radiation therapy. Cancers with higher expression of this particular miRNA responded better to radiation therapy than cancers with lower expression of miRNA-22 [79].
Despite the promising aspects of miRNAs in cancer diagnosis and treatment, there are several obstacles for successful implementation of bench findings into the clinic. One such obstacle is detection of miRNA. Initially, Northern blotting techniques were used for miRNA discovery and detection. This method is relatively insensitive to changes in expression of miRNA and requires large amounts of starting material (RNA), usually obtained from tumor resections. Quantification of miRNA is therefore routinely analyzed by RT-PCR methods such as the modified Invader assay [80] and confocal laser-induces fluorescence detection [81], oligo-array based techniques [82] and in situ hybridization [83, 84]. It is of note that none of these techniques have been adequately validated and that their use in laboratory settings is questionable. For example, RT-PCR is semi-quantitative and can provide information on differences between cancers and noncancerous tissues; however, this technique has drawbacks as discussed below in greater detail. Although, miRNA microassays are suitable for large-scale screening, they are semi-quantitative and lack sensitivity to discriminate between small differences. In addition, adjustments of the conditions of the assay have to be balanced for large number of miRNAs which adds to variability in the efficacy of the method. Until now, only certain miRNA species have been preliminarily demonstrated as biomarkers in a clinical setting [85–87].
There are several questions to be answered before miRNAs can be widely used in the clinic. First, we have to fully understand the role of miRNAs and their biological effects. Usually, miRNA binds to multiple mRNA targets [88, 89] and, by such binding, can “label” mRNA to be degraded or translated into protein in an attenuated fashion making it extremely difficult to evaluate the importance of charges in expression [88, 90]. There are efforts underway, using bioinformatics, to model cellular responses to changes in level of particular miRNAs but with no definite conclusions as of this writing. The situation is even more complicated when hyper/hypo-methylation, histone/DNA interactions are included as we have even less mechanistic information to be able to evaluate the resulting data.
For example, in breast cancer cell lines, after 5 h of exposure to a pro-apoptotic dose of LAQ824, a small molecule histone deacetylase inhibitor, changes were measured in 40 % of the >60 different miRNA species expressed in SKBr3 cells with 22 miRNA species shown to be down-regulated and five miRNAs up-regulated [91]. This is a much higher percentage than the 5 % of affected miRNA. What is the significance of this discrepancy between the levels of miRNA and mRNA? How they interact with each other is yet to be understood. Rather than a target for intervention, certain miRNA species may serve as a diagnostic or as a companion diagnostic indicator.
Polymorphisms
The investigation of genetic polymorphisms is important as two or more different phenotypes may exist within the same individual. Biologists investigate certain point mutations in the genotype such as single-nucleotide polymorphisms (SNPs) or variations in homologous DNA by restriction fragment length polymorphisms (RFLPs). Such investigations are performed by chromatography, chromosome cytology, or genetic data. The mechanisms and the distribution of different polymorphisms in different genes are not well understood although these are believed to be a reason for evolutionary disparity for natural selection [92].
Independent from its role in understanding biology and especially tumor biology, the investigation of polymorphisms should not be expected to reveal clinically meaningful data anytime soon. The reasons for this may be understood by considering the following information:
-
1.
We do not understand how polymorphisms reflect a disease and/or respond to a treatment, or if they react in conjunction with polymorphisms of other genes.
-
2.
The number of SNPs, published on 23 July 2013 in the single nucleotide polymorphism database (dbSNP), was 62,676,337 [93].
-
3.
Polymorphisms need to include data on the generation time which is approximately 3 years; the human genome consists of base pairs (6.4 billion, 6.4 × 109 base pairs) and it was assumed, that some 192 mutations (6.4 × 30) per cell generation occur. For example, within the Y chromosome this rate was estimated to be between 100 and 200 [94].
-
4.
Humans have 23 paired chromosomes (46 chromosomes) and the human genome project revealed that humans probably have 21,000 haploid coding genes with approximately 3.3 × 109 base pairs [95].
-
5.
Chromosome 1 alone with its 249,250,621 base pairs has some 4,401,091 variations [96].
-
6.
Mutations occur at an estimated of around 10−6–10−10 in eukaryotes [97]; this allows for an approximation for calculating the possible options and/or combinations.
-
7.
The number of pseudogenes is about 13,000 [95].
-
8.
A wide variation is reported in transposable (mobile) genetic DNA sequences [98, 99]. For example, Alu has about 50,000 active copies while LINE-1 (long interspersed element 1) has approximately 100 active copies per genome.
-
9.
To the best of our knowledge, mobile genetic elements, CLASS I DNA transposons as LTRs (long terminal transposanable retroposons) and non-LTR retrotransposons, as long interspersed elements (LINE), short interspersed elements (SINEs) and CLASS II DNA transposons account for more than 40 % of the total genome [100].
-
10.
We have additional genomic material such as the mitochondrial genome, with little understanding of how these interact with genetic elements in the nucleus.
-
11.
The mitochondrial genome is separated from the nuclear DNA by the nuclear double membrane; however, intranuclear genomic rearrangement takes place frequently by transmission of mitochondrial DNA into the nuclear genome and especially when a normal cell has undergone transformation into a cancer cell [101].
-
12.
Additionally, we have coding and non-coding DNA (98 % of human genome) as well as pseudogenes [95] but are not aware how these interact in any meaningful detail.
Therefore, logically and computationally it seems unlikely to think that a needle in this huge haystack might be found in the near future that could help in treating cancers but there is hope that Big Data might make a difference when the requirements for Big Data projects are addressed [102].
Epigenetics
The main obstacle to sequencing is epigenetic changes which appear to be relevant for understanding cancer occurrence and progression. Epigenetic alterations are, by definition, mitotically and meiotically heritable changes in gene expression that are not caused by changes in the primary DNA sequence. The epigenetic modifications described in the literature generally comprise histone variants, post-translational modifications of amino acids of histone proteins, and changes in the methylation status of cytosine bases (C) in the context of CpG dinucleotides within the DNA itself. Methylation of clusters of CpG dinucleotides (CpGs–called “CpG-islands”) in the promoter region of genes have been associated with heritable gene silencing [103]. Detailed information about modified residues of one or two histones are available at UniProt [104].
A role for epigenetic factors has been shown for several cancers including esophageal cancer [105–107]. Methylation and de-methylation processes are dynamic, and efforts to correlate “methylation fingerprint” with stage of the disease suggest that there are variations in methylation that could be potentially relevant and which correlate to the stage of the disease. Each stage undergoes unique epigenetic changes at different steps of disease progression in esophageal adenocarcinoma, suggesting a step-wise loss of multiple protective barriers against CpG island hypermethylation.
Hyper- and hypo-methylation have distinct roles in the cell making the fingerprinting of cancer cells complex. Hypomethylation usually introduces genome instability and genetic rearrangements while hypermethylation silences various tumor suppressor genes [108]. The aberrant hypomethylation occurs at many different loci suggestive of an overall deregulation of methylation control in tumorigenesis in esophageal adenocarcinoma. However, there is no evidence for correlation of a distinct group of tumors with a CpG island methylator phenotype [109]. Efforts were made to investigate methylation patterns of DNA in the plasma of cancer patients vs. controls such as p16 promoter methylation in ESCC [110]. For example, the beta-catenin signaling pathway Wnt modulator secreted frizzled-related protein 1 (SFRP1) gene is silenced by hypermethylation in ESSC [111] and the COX-2 promoter region is silenced in some ESSC cell lines [112].
The Ras-related protein Rab25 gene implicated in endocytic recycling of integrins and suppressor of invasion and angiogenesis was significantly downregulated in ESCC tumor specimens. Rab25 correlated with decreased overall survival and was also documented in ESCC cell lines as compared to pooled normal tissues [113]. Demethylation treatment and bisulfite genomic sequencing analyses revealed that downregulation of Rab25 expression in both ESCC cell lines and clinical samples was associated with promoter hypermethylation [113]. Further characterization of Rab25 may allow its use as a prognostic biomarker for ESCC and a plausible target in ESCC treatment.
Despite encouraging data, caution must be taken interpreting these results. For example, these studies typically utilized PCRs specifically designed to detect hyper- or hypo- methylation of the DNA with all the downfalls of this method. Also, the origin of the DNA detected by this method is uncertain. There is no evidence whether this DNA originates from dying “normal” cells or from cancer cells or from both.
Alterations in levels of cell-free DNA in plasma or serum as well as increases in the overall level of cell-free DNA is not restricted to any particular tumor site, type or grade. However, there are larger amounts of cell-free DNA in patients with late stage disease and metastasis. Some studies show a correlation between resection of the cancer with diminished levels of cell-free DNA in sera [114]. However, these studies have been conducted on small patient populations and require further investigation to validate their utility [115].
Proteomics
The measurement of proteins in cancerous vs noncancerous tissues termed “cancer profiling” is a promising area for both biomarker discovery and growth in applications for healthcare. Molecular profiling for different biomarkers in a tissue may assist in obtaining more accurate diagnosis for a cancer on a personalized basis providing better information on anti-cancer treatments since every cancer cell appears to have its own pattern of active genes and proteins [116–118]. To achieve this, it will be necessary to collect information on which altered gene expression will result in what kind of final products (proteins) such that the results can be used for diagnostic or for monitoring the efficacy of therapy.
Advances in mass spectrometry (MS) have made it possible for large-scale analysis of the entire proteome of a given tissue to enable the identification of proteins in general and specific marker proteins expressed in that tissue in particular. Multiple platforms and technologies have been utilized including MS-based and antibody-based analyses. Two-dimensional gel electrophoresis (2DE) and liquid chromatography (LC) combined with tandem mass spectrometry (LC/MS/MS) are commonly used as MS-based proteomic approaches. Microarray and immunohistochemistry (IHC) are major platforms in antibody-based analyses.
To be able to find proteins that are differentially expressed in cancerous tissue only, tissue proteomes among normal, precancerous, and cancer patients were analyzed and compared. These tissue samples were obtained via resection and/or biopsies [119–121]. Body fluids including serum and plasma might be better choices not only for discovery studies but also for evaluation of patient’s response to a therapeutic reagent since they can be accessed readily [122, 123]. On the other hand, cancer cell lines were developed and analyzed to identify proteins differentially expressed in vitro, which provides an easier and faster way to discover and develop cancer-specific biomarkers [124, 125].
With respect to the proteomic study of the upper GI tract, a 2DE database for healthy human stomach tissue was reported in 2002 [126]. The authors analyzed both entire homogenate sand soluble fractions of the stomach. Over 600 protein spots were resolved in the 2DE separations. In 2010, Paulo and coworkers identified 134 proteins from normal gastroduodenal fluid using 2DE combined with LC–MS/MS [127]. Such studies can contribute to identifying disease-specific proteins when a diseased sample of the same tissue is analyzed. Likewise, using high-throughput and large scale proteomics technologies for finding upper GI cancer-specific marker proteins have been carried out over the past decade. Hundreds of proteins have been identified from various cancer patients and cell lines [119, 128–130].
A variety of samples from esophageal cancer/esophageal squamous cell carcinoma (ESCC) patients were analyzed and compared with normal tissue. With 2DE combined with MS platform, transglutaminase 3 (TGM3), heat shock protein 70 (Hsp70), TPM4-ALK fusion oncoprotein 2, myosin light polypeptide 6, keratin I and calreticulin were identified as over-expressed in tumor tissues [120, 131]. Using antibody-based technologies, over expression of protein budding uninhibited by benzidazoles 1 homolog beta (BubR1), mitotic arrest deficient-like 1 (Mad2), NF-kappaB-activating kinase, caspase 10, activator protein-1, alpha-actinin 4 (ACTN4), 67 kDa laminin receptor (67LR), COX-2, p53, secret protein acidic and rich in cysteine (SPARC), migration-stimulating factor (MSF), and vascular endothelial growth factor C (VEGF-C) were shown to be associated with ESCC [132–135].
Using an esophageal cancer cell line, proteins over-expressed included hsp70, peroxiredoxin-5, non-muscle myosin light polypeptide 6, keratin 1, annexin A4, keratin 8, tropomyosin 3, stress-induced-phosphoprotein 1, albumin, hsp70 protein 9B precursor, solute carrier family 44 Member 3, heterogeneous nuclear ribonucleoprotein L (hnRNP L), eukaryotic translation initiation factor 4A isoform 2, triosephosphate isomerase1 (TPI), peroxiredoxin1 (PRX1), forminotransferase cyclodeaminase form (FTCD), fibrinogen gamma-A chain precursor, kinesin-like DNA binding protein, lamin A/C, cyclophilin A (CypA), and transcription factor MTSG1 [136–138].
Most of the proteins differentially expressed in upper GI cancers were identified in ESCC cases. Only two proteins, apoC-I and apoC-III, were found to be elevated in the serum of patients with stomach cancer [139]. Serum amyloid A (SAA) was found in rat plasma injected with a stomach cancer cell line SC-M1 and was shown to be up-regulated in human stomach cancer [140]. These findings reinforce the new understanding that chronic inflammation is one of the key sequences in carcinogenesis [14, 15].
Large-scale profiling of cancer targets using a proteomics approach has been recognized and this may be a tool for seeking novel markers in the future. Until recently, different groups applied very different technologies in the mapping of novel upper GI cancer-related proteins and this resulted in a challenge for interpreting the disparate results. A number of proteins were identified as over-expressed in cancers and they may be valuable for future diagnosis and prognosis. For example, ACTN4 and 67LR were found to be up-regulated from stage I to III ESCC, where ACTN4 was associated with advanced tumor stage and lymph node metastasis whereas 67LR was correlated with an advanced tumor stage [133]. It is suggested that both ACTN4 and 67LR may be useful for the classification and evaluation of progression of ESCC and serve as targets for therapy. The levels of VEGF-C expression in gastro-esophageal junction adenocarcinoma were also found to be associated with stages of the tumor and lymph node metastasis. To this end, VEGF-C may be a potential biomarker for diagnosis of lymphatic metastasis and prognosis of survival in cardia carcinoma patients [135].
MSF was identified on the surface of human esophageal cancer endothelial cells (HECECs) and its antibody showed suppression of migration and adhesion of HECECs on a fibronectin matrix first induced by MSF. Furthermore, a biodistribution assay demonstrated that this antibody specifically homed into the xenograft with humanized blood vessels and suppressed tumor growth by inhibition of tumor-related angiogenesis. This observation, if it holds up in vivo, suggests that MSF may be an anti-angiogenic target for treatment of esophageal cancer [134]. TGM3 may be also a prognostic biomarker and may provide strategies to prevent recurrence of ESCC [131]. Excision repair cross complementing group 1 (ERCC1) may also be of prognostic value in multimodal treated upper GI cancer patients [141].
Hsp70 was found to be over-expressed in both cancer tissue samples and cultured cancer cells by several different groups [120, 137, 138] making it a more robust marker. As a known chaperone, Hsp70 plays a crucial role in forming and recycling nucleocytoplasmic transport receptors via direct interaction with the nuclear pore complex. Consequently, it regulates the transport of proteins between nucleus and cytoplasm. Multiple reports revealed its over-expression in esophageal cancers [120, 137, 138]. Thus, Hsp70 appears to be involved in the progression of esophageal cancers. Its impaired expression combined with the inability to transport macromolecules between the nucleus and the cytoplasm, Hsp70 may serve as a biomarker for esophageal cancer [137].
Juan and coworkers introduced five human stomach cancer cell lines (SC-M1, HONE-1, CC-M1, OECM1, GBM 8401) into nude mice separately [140]. After incubation, plasma was collected, analyzed, and compared with plasma from control mice injected with phosphate-buffered saline. In spite of some acute phase proteins found in the plasma of all mice bearing cancer cells, SAA was found over-expressed only in mice with the stomach cancer cell line, SC-M1 [140]. The authors suggested that SAA may be a specific diagnostic marker for patients with gastric cancer.
Using any protein with elevated levels as a therapeutic target may, in most cases, be a overly optimistic approach given the lack of progress in the recent past using such approaches. So far, only Her2 inhibition in Her2-type breast cancer has a positive effect in a fair proportion but not all of these patients. Elevated levels of a protein may serve well as a biomarker but it does not automatically follow that it will also serve as an adequate therapeutic target. The scientific community might put hope over outcome in expecting plausible leads for therapeutics to emerge without an in-depth understanding of the science underpinning the field.
Besides proteins that are up-regulated in esophageal cancers, several down-regulated proteins were also identified. These included subunit alpha type-3 of proteasome, calpain small subunit 1, eIF5A-1, S100-A8 protein, annexin A1, annexin A2, regulatory subunit of dehydrogenase 1calpain, glutamate, histone deacetylase 10 isoform beta, disulfide-isomerase ER-60 precursor, beta-tropomyosin (TMbeta), myosin light chain 2 (and its isoform), myosin regulatory light chain 2, and peroxyredoxin 2 [120, 136–138].
The increase of newly discovered proteins presents both a challenge and an opportunity. In particular, the expression level of two proteins, periplakin and clusterin, were nearly zero in esophageal cancers when compared to healthy tissue [142, 143]. These findings were based on Western Blot and IHC analyses without any quantification data. We may be able to use periplakin and clusterin to assess changes in patients with esophageal cancers. However, their scientific significance can be assessed only when their values in normal tissues are defined. The levels of periplakin were found to have shifted from the cell–cell boundary of normal esophageal epithelial cells to the cytoplasm of epithelial cells in early esophageal cancer, then to have disappeared completely in advanced esophageal cancer [142]. This might be encouraging in that periplakin may not only be useful as a diagnostic biomarker but also a marker for the staging of this cancer [142].
As reviewed above, many groups have investigated different potential biomarkers in upper GI cancers for potential use in making an early diagnosis as well as in providing information about prognosis of these cancers. However, the major challenges for evaluating the clinical significance of any of new biomarkers remain and include:
-
1.
The specificity of occurrence of these proteins,
-
2.
The availability for a drug to get access,
-
3.
Identifying and quantifying a defined usefulness in clinical applications, and
-
4.
A standardization of the different multiple diagnostic tools as well as its investigated biorepository.
Another challenge is that new findings will require more than a few investigations and validation steps although several of them have already been verified by various immunoassays for their presence and sub-cellular locations in cancer tissues. These protein candidates include ACTN4, 67LR, VEGF_C, BubR1, Mad2, SAA, TGM3 and MSF in which both TGM3 and MSF were further investigated in relative functional analysis [131–135, 140]. Beyond the up-regulated proteins, some down-regulated proteins found in upper GI cancer cells or tissues such as annexin A1, keratin 8, annexin A2, and periplakin have also been validated [136, 138, 142].
Due to the availability of antibodies and their costs, not every new protein marker can be tested, validated, or categorized as its biological (tumor) relevance needs to be identified first. Another huge challenge for the possible identification of a protein as a potential biomarker via large-scale protein profiling is the single amino acid mutation, and/or small insertions and deletions in the coding gene. ELISA, western blotting and IHC potentially provide highly specific protein detection but those mutations, insertions and/or deletions are not readily detected. For this, a large number of antibodies will need to be generated, especially to each and every single possible combination of mutations and even conversion of these mutations on 2D or 3D structure and, consequently, the epitope structure. However, an antibody approach is not ideal to detect and to quantify protein modifications and mutations but there is hope that MS might elucidate how best to proceed.
Serum and plasma are under investigation as they are much easier to collect and more practical in clinical applications. The key issue in protein marker discovery in serum/plasma using large-scale and high-throughput proteomics technologies is that approximately 99 % of protein mass in this sample consists of essentially 20 proteins. Nearly all biomarker proteins are present at low levels with a ratio roughly at 1:107, i.e. ng/ml of cancer-related proteins versus mg/ml of physiological levels of albumin [144, 145]. Protein separation prior to sample analysis is required and widely used but with variable levels of contamination or depletion of the protein of interest can occur; and thus, downstream analyses can be biased. As a result, new biomarker discovery in serum/plasma in large-scale proteomics profiling has been limited. Another drawback for use of serum in the search for cancer biomarkers is that even though we possess information on which protein from a tissue can enter the circulation, the half-life of the target protein may not be long enough for it to be detected in serum [146]. There is virtually no published data to suggest that the half-life time of one protein is the same in normal vs. cancer tissue.
Finding novel biomarkers would be an important step for early diagnosis, treatment and prognosis in oncology. The Human Proteome Project with large-scale protein profiling on novel biomarkers discovery and validation may accelerate advancements in human health given in 2005 that more than 3020 proteins exist in the current data base produced by a number of international laboratories [147–149]. This number increased up to 10,546 in 2014 [150] and some 17,000 in 2015 while approximately some 15 % of all proteins have weak proteomic evidence and/or are still missing [151]. The significance how proteomics may influence our understanding of carcinogenesis and its impact on making of clinical decisions will be based on more precise strategies, rather than searching for a needle in a haystack without first knowing what the needle looks like.
Limitations and challenges of reproducibility
The results from in vitro studies in cell lines present a huge challenge in their evaluation and extrapolation to human cancers absent the data to make such a leap. Contamination and incorrect interpretations can result in snowball effects of erroneous secondary research [152]. The HeLa cell line, cultivated in 1951, from Henrietta Lacks, a young cervical cancer patient, is most commonly used [153]. Although HeLa is a robust cell line and has been used in more than 70,000 studies worldwide, it has been known for almost 50 years that approximately 20 % of HeLa cell lines are contaminated and such contamination could impact study results [154]. Thus, the top four cell line repositories in the US, Germany, and Japan should be validated in a standardized manner [155].
The TS gene expression has been under investigation in 25 different human ESCC cell lines, 13 from the Japanese Cancer Research Resources Bank and 12 from the Leibnitz Institute (DSMZ) German Collection of Microorganisms and Cell Cultures [156]. The IC50, a parameter used to determine how much of a substance is needed to inhibit a biological process by 50 %, ranged from 1 to 39.8 µmol/L. This finding by Ando et al. is important as it unequivocally shows that it is not enough to use a cell line for a study given that the range of IC50 s in 25 ESCC cell lines under investigation is quite variable. In keeping with this finding is more recent research comparing molecular details of cell lines to real tumors by genomic profiling [157]. These authors show that common cell lines used for research on ovarian cancer do often not reflect real tumor biology in humans. This means that the results found in cell lines, more often than not, are not representative of the real tumor biology seen in patients.
Furthermore, science faces another challenge which is that a lack of reproducibility of existing cancer research is recognized and frequently discussed. However, we need to remind ourselves that less than 20 % of highly ranked so-called “landmark” publications have been shown to be irreproducible although these are among the most cited references [158]. A recently launched initiative requires that authors provide their raw data for a validation check [159]. This, together with proposed guidelines, could be of helpful to be incorporated into a plan of action thus increasing the value and reliability of cancer research. There is hope overcoming the issues of contamination and poor reproducibility by an U.S. and European initiative, which is on its way replacing its cell lines with patient-derived xenografts (PDX).
Conclusions
There is hope of integrating translational molecular data from genomics, microRNA, epigenetics, and proteomics to improve cancer diagnosis, therapy, and facilitate cancer classifications. Reviewing the available literature for relevance to cancer diagnosis or monitoring of treatment is presently difficult as clearly defined inclusion and exclusion criteria in [1] clinical trials as well as [2] in molecular research are largely absent. Different tumor entities with different tumor biologies, prognoses, and different therapeutic approaches make it difficult to make informed recommendations. As pointed out, many hitherto undervalued criteria such as epidemiology, embryology, and molecular biology will be important to consider when designing future research and clinical trials [4].
In order to achieve these goals, it is necessary to understand the key issues in different aspects of biotechnology so as to anticipate future directions of personalized and individualized diagnosis and multimodal treatment strategies. Providing an overview of translational data in cancers proved to be a challenge as different methods and techniques used to obtain molecular data are based on different tumor entities with different tumor biology and prognoses as well as vastly different therapeutic approaches. The pros and cons of the available methodologies and the potential response data in genomics, microRNA, epigenetics and proteomics as well as their limitations in gastrointestinal cancers are considered to allow for an understanding of where these technologies stand with respect to cancer diagnosis, prognosis and treatment.
A step towards a solution would be if organizations dealing with GI cancers create a framework for biological studies and for clinical trials dealing with tumor entities. This, together with integrating other variables such as providing raw data with a validation check would be helpful. The replacement of cell lines by patient-derived xenografts (PDX) for in vitro studies may significantly enhance the value and rigor of basic and translational research.
Abbreviations
- 2DE:
-
two-dimensional gel electrophoresis
- 67LR:
-
67 kDa laminin receptor
- A3B:
-
apolipoprotein B mRA-editing enzyme, catalytic polypeptide-like 3B (=APOBEC3B)
- ACTN4:
-
activator protein-1, alpha-actinin 4
- AEG:
-
adenocarcinoma of the esophago-gastral junction
- AJCC:
-
American Joint Cancer Committee
- APOBEC3B:
-
apolipoprotein B mRA-editing enzyme, catalytic polypeptide-like 3B (=A3B)
- B cell ALL:
-
acute lymphoblastic B-cell leukemia
- BCs:
-
basal cells
- BRCA1:
-
breast cancer 1, early onset
- BRCA2:
-
breast cancer 2, early onset
- BubR1:
-
benzidazoles 1 homolog beta
- C:
-
cytosine bases
- CpGs:
-
CpG dinucleotides
- CpG-islands:
-
methylation of clusters of CpGs
- CD9:
-
cell surface glycoprotein encoded by CD9 gene
- COX-2:
-
cyclooxygenase-2 (=Prostaglandin G/H synthetase 2)
- CSCs:
-
cancer stem cells
- Cyclin D1:
-
G1/S-specific Cyclin D1
- CypA:
-
cyclophilin A
- dbSNP:
-
single nucleotide polymorphism database
- DNA:
-
deoxyribonucleic acid
- DNVs:
-
somatic di-nucleotide variations
- DSMZ:
-
Leibnitz Institute DSMZ, German Collection of Microorganisms and Cell Cultures
- ERCC1:
-
excision repair cross complementing group 1
- ERG:
-
ETS related gene
- ESCC:
-
esophageal squamous cell carcinomas
- ETS:
-
E26 transformation-specific or E-twenty-six transcription factor family
- ETV1:
-
ETS translocation variant 1
- FTCD:
-
forminotransferase cyclodeaminase form (FTCD)
- GI:
-
gastrointestinal
- HECECs:
-
human esophageal cancer endothelial cells
- hnRNP L:
-
heterogeneous nuclear ribonucleoprotein L
- HSCs:
-
hematopoetic stem cells
- Hsp70:
-
heat shock protein 70
- IC50:
-
half maximal inhibitory concentration
- IHC:
-
immunohistochemistry
- IKBKB:
-
inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase beta (= IκK-beta)
- IκK:
-
inhibitor of κK kinase
- IL-1B:
-
interleukin 1 beta
- JAK-3:
-
Janus-activated kinase
- KRAS:
-
Kirsten rat sarcoma viral oncogene
- LAQ824:
-
a small molecule histone deacetylase inhibitor
- LC:
-
liquid chromatography
- LC/MS/MS:
-
tandem mass spectrometry
- LCs:
-
luminal cells
- LINE-1:
-
long interspersed element 1
- LnCap:
-
cell line: epithelial prostate cancer cell line
- LPs:
-
luminal progenitors
- LTRs:
-
long terminal transposanable retroposons
- LINE:
-
long interspersed elements
- Mad2:
-
mitotic arrest deficient-like 1
- miR:
-
micro RNA (=miRNA)
- miRNA:
-
micro RNA (=miR)
- MCF-7L:
-
human breast cancer adenocarcinoma cell line
- MS:
-
mass spectrometry
- MSF:
-
migration-stimulating factor
- NF-B:
-
nuclear factor kappa-light-chain-enhancer of activated B cells
- NOD-SCID:
-
non-obese diabetic-severe combined immunodeficiency mice, immunodeficient mice with lack of mature T-cells, B-cells and natural killer (NK) cells
- ONVs:
-
somatic oligonucleotide variations
- p21:
-
protein 21, cyclin-dependent kinase inhibitor 1 or CDK-interacting protein 1
- p53:
-
tumor protein p53 (=TP53)
- PCR:
-
polymerase chain reaction
- PDCD4:
-
programmed cell death protein 4
- PDX:
-
patient-derived xenografts
- PGE2:
-
COX-2 dependent prostaglandin E2
- PIK3CA:
-
phosphatidylinositol-45-bisphosphate 3-kinase catalytic subunit alpha
- PIK3CA-YFP:
-
PIK3CA yellow fluorescent protein
- PKC:
-
protein kinase C
- PRX1:
-
peroxiredoxin1
- PTK6:
-
protein tyrosine kinase 6
- qRT-PCR:
-
quantitative real-time reverse-transcription polymerase chain reaction
- Rab25:
-
Ras related protein Rab-25
- RFLPs:
-
restriction fragment length polymorphisms
- SCs:
-
stromal cells
- SEER:
-
surveillance, epidemiology and end results database
- SFRP1:
-
Wnt modulator secreted frizzled-related protein 1
- SINDELs:
-
small insertions and deletions
- SINEs:
-
short interspersed elements
- SNPs:
-
single-nucleotide polymorphisms
- SPARC:
-
secret protein acidic and rich in cysteine
- SSNVs:
-
somatic single nucleotide variations
- TCF/LEF:
-
transcription family group binding to DNA
- TGM3:
-
transglutaminase 3
- TMbeta:
-
beta-tropomyosin
- TNF:
-
tumor necrosis factor
- TNVs:
-
somatic tri-nucleotide variations
- TP53:
-
Tumor protein p53 (= p53)
- TP53-GFP:
-
p53 green fluorescent protein
- TPI:
-
triosephosphate isomerase1
- TPM4-ALK:
-
TPM4-ALK fusion oncoprotein 2
- VCaP:
-
cell line: vertebral cancer of the prostate
- VEGF-C:
-
vascular endothelial growth factor C
- Wnt:
-
beta-catenin signaling pathway
References
Siegel R, Naishadham D, Jemal A (2013) Cancer statistics 2013. CA Cancer J Clin 63:11–30
Saedi B, Razmpa E, Sadeghi M et al (2009) The epidemiology of laryngeal cancer in a country on the esophageal cancer belt. Indian J Otolaryngol Head Neck Surg 61:213–217
Tran GD, Sun XD, Abnet CC et al (2005) Prospective study of risk factors for esophageal and gastric cancers in the linxian general population trial cohort in China. Int J Cancer 113:456–463
Brücher BL, Kitajima M, Siewert JR (2014) Undervalued criteria in the evaluation of multimodal trials for upper GI cancers. Cancer Invest 32:497–506
Liu SZ, Wang B, Zhang F et al (2013) Incidence, survival and prevalence of esophageal and gastric cancer in Linzhou City from 2003 to 2009. Asian Pac J Cancer Prev 14:6031–6034
Jemal A, Murray T, Ward E et al (2005) Cancer statistics, 2005. CA Cancer J Clin 55:10–30
Solaymani-Dodaran M, Card TR, West J (2013) Cause-specific mortality of people with barrett’s esophagus compared with the general population: a population-based cohort study. Gastroenterology 144:1375–1383
Lee JH, Kim KM, Cheong JH et al (2012) Current management and future strategies of gastric cancer. Yonsei Med J 53:248–257
Marshall BJ (1985) The pathogenesis of non-ulcer dyspepsia. Med J Aust 143:319
Pisani P, Parkin DM, Muñoz N et al (1997) Cancer and infection: estimates of the attributable fraction in 1990. Cancer Epidemiol Biomarkers Prev 6:387–400
Conlin VS, Curtis SB, Zhao Y et al (2004) Helicobacter pylori infection targets adherens junction regulatory proteins and results in increased rates of migration in human gastric epithelial cells. Infect Immun 72:5181–5192
Dorward DW, Garon CF et al (1989) DNA-binding proteins in cells and membrane blebs of Neisseria gonorrhoeae. J Bacteriol 171:4196–4201
Aravindan N, Aravindan S, Pandian V et al (2014) Acquired tumor cell radiation resistance at the treatment site is mediated through radiation-orchestrated intercellular communication. Int J Radiat Oncol Biol Phys 88:677–685
Brücher BLDM, Jamall IS (2014) Epistemology of the origin of cancer: a new paradigm. BMC Cancer 14:1–15
Brücher BLDM, Jamall IS (2014) Cell-Cell communication in tumor microenvironment, carcinogenesis and anticancer treatment. Cell Physiol Biochem 34:213–243
Zhang Y, Pan KF, Zhang L et al (2015) Helicobacter pylori, cyclooxygenase-2 and evolution of gastric lesions: results from an intervention trial in China. Carcinogenesis 36:1572–1579
Blattner WA (1999) Human retroviruses: their role in cancer. Proc Assoc Am Physicians 111:563–572
Parkin DM (2006) The global health burden of infection-associated cancers in the year 2002. Int J Cancer 118:3030–3044
Tomlinson IP, Novelli MR, Bodmer WF (1996) The mutation rate and cancer. Proc Natl Acad Sci USA 93:14800–14803
Brücher BLDM, Lyman G, van Hillegersberg R et al (2014) Imagine a world without cancer. BMC Cancer 14:1–8
Balogh LP (2015) Caging cancer. Nanomedicine 11:867–869
Brücher BLDM, Li Y, Schnabel P et al (2016) Biotechnologie und Krebs. CHAZ 2016(17):17–27
Kleppe K, Ohtsuka E, Kleppe R et al (1971) Studies on polynucleotides. XCVI. Repair replications of short synthetic DNA’s as catalyzed by DNA polymerases. J Mol Biol 56:341–361
Mullis K, Faloona F, Scharf S et al (1986) Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol 51:263–273
Saiki RK, Gelfand DH, Stoffel S et al (1988) Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239:487–491
Bernard PS, Wittwer CT (2002) Real-time PCR technology for cancer diagnostics. Clin Chem 48:1178–1185
Kurimoto K, Yabuta Y, Ohinata Y et al (2006) An improved single-cell cDNA amplification method for efficient high-density oligonucleotide microarray analysis. Nucleic Acids Res 34:e42
Bustin SA (2002) Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol 29:23–39
Huggett J, Dheda K, Bustin S et al (2005) Real-time RT-PCR normalisation; strategies and considerations. Genes Immun 6:279–284
Vogelstein B, Papadopoulos N, Velculescu VE et al (2013) Cancer genome landscapes. Science 339:1546–1558
Tomasetti CB, Vogelstein B, Parmigiani G (2013) Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci USA 110:1999–2004
Martin-Lorenzo A, Hauer J, Vicente-Duenas C et al (2015) Infection exposure is a causal factor in B-precursor acute lymphoblastic leukemia as a result of Pax inherited susceptibility. Cancer Discov 5:1328–1343
Nguyen LV, Makarem M, Carles A et al (2014) Clonal analysis via barcoding reveals diverse growth and differentiation of transplanted mouse and human mammary stem cells. Cell Stem Cell 14:253–263
Harris R, Law E, Sieuwerts A et al (2015) Tamoxifen resistance driven by the DNA cytosine deaminase APOBEC3B in recurrent estrogen receptor positive breast cancer. San Antonio Breast Cancer Symposium (SABC) 2015; Abstract S4–07
Ling S, Hu Z, Yang Z et al (2015) Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proc Natl Acad Sci USA 112:E6496
Vu V, Verster AJ, Schertzberg M et al (2015) Natural Variation in gene expression modulates the severity of mutant phenotypes. Cell 162:391–402
Cvijović I, Good BH, Jerison ER et al (2015) Fate of a mutation in a fluctuating environment. Proc Natl Acad Sci USA 112:E5021–E5028
Supek F, Lehner B (2015) Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature 521:81–84
Holstege H, Pfeiffer W, Sie D (2014) Somatic mutations found in the healthy blood compartment of a 115-year-oldwoman demonstrate oligoclonal hematopoiesis. Genome Res 24:733–742
Lawrence MS, Stojanov P, Mermel CH et al (2014) Discovery and saturation analysis of cancer genes across 21 tumor types. Nature 505:495–501
Rosenfeld S (2013) Are the somatic mutation and tissue organization field theories of carcinogenesis incompatible? Cancer Inform 12:221–229
Versteeg R (2014) Cancer: tumours outside the mutation box. Nature 506:438–439
Mack SC, Witt H, Piro RM et al (2014) Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506:445–450
Parker M, Mohankumar KM, Punchihewa C et al (2014) C11orf95-RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature 506:451–455
Huang CP, Cheng CM, Su HL et al (2015) Syndecan-4 promotes epithelial tumor cells spreading and regulates the turnover of PKCα Activity under mechanical stimulation on the elastomeric substrates. Cell Physiol Biochem 36:1291–1304
Beerenwinkel N, Antal T, Dingli D et al (2007) Genetic progression and the waiting time to cancer. PLoS Comput Biol 3:e225
Tuch BB, Laborde RR, Xu X et al (2010) Tumor transcriptome sequencing reveals allelic expression imbalances associated with copy number alterations. PLoS One 5:e9317
Pflueger D, Terry S, Sboner A et al (2011) Discovery of non-ETS gene fusions in human prostate cancer using next-generation RNA sequencing. Genome Res 21:56–67
Maher CA, Kumar-Sinha C, Cao X et al (2009) Transcriptome sequencing to detect gene fusions in cancer. Nature 458:97–101
Ma S, Bao JY, Kwan PS et al (2012) Identification of PTK6, via RNA sequencing analysis, as a suppressor of esophageal squamous cell carcinoma. Gastroenterology 143(675–686):e1–e12
Hu X, MacDonald DM, Huettner PC et al (2009) A miR-200 microRNA cluster as prognostic marker in advanced ovarian cancer. Gynecol Oncol 114:457–464
Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854
Kumar MS, Lu J, Mercer KL et al (2007) Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet 39:673–677
Bentwich I, Avniel A, Karov Y et al (2005) Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet 37:766–770
Volinia S, Calin GA, Liu CG et al (2006) A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA 103:2257–2261
Budhu A, Jia HL, Forgues M et al (2008) Identification of metastasis-related microRNAs in hepatocellular carcinoma. Hepatology 47:897–907
Zhao Y, Jia HL, Zhou HJ et al (2009) Identification of metastasis-related microRNAs of hepatocellular carcinoma in hepatocellular carcinoma cell lines by quantitative real time PCR. Zhonghua Gan Zang Bing Za Zhi 17:526–530
Lee EJ, Gusev Y, Jiang J et al (2007) Expression profiling identifies microRNA signature in pancreatic cancer. Int J Cancer 120:1046–1054
Cheng H, Shi S, Cai X et al (2012) microRNA signature for human pancreatic cancer invasion and metastasis. Exp Ther Med 4:181–187
Brendle A, Lei H, Brandt A et al (2008) Polymorphisms in predicted microRNA-binding sites in integrin genes and breast cancer: iTGB4 as prognostic marker. Carcinogenesis 29:1394–1399
Sung H, Jeon S, Lee KM et al (2012) Common genetic polymorphisms of microRNA biogenesis pathway genes and breast cancer survival. BMC Cancer 28:195
He H, Jazdzewski K, Li W et al (2005) The role of microRNA genes in papillary thyroid carcinoma. Proc Natl Acad Sci USA 102:19075–19080
Calin GA, Pekarsky Y, Croce CM (2007) The role of microRNA and other non-coding RNA in the pathogenesis of chronic lymphocytic leukemia. Best Pract Res Clin Haematol 20:425–437
Feber A, Xi L, Luketich JD et al (2008) MicroRNA expression profiles of esophageal cancer. J Thorac Cardiovasc Surg 135:255–260
Mishima T, Akagi I, Miyashita M et al (2009) Study of MicroRNA expression profiles of esophageal cancer. J Nippon Med Sch 76:43
Hu Y, Correa AM, Hoque A et al (2011) Prognostic significance of differentially expressed miRNAs in esophageal cancer. Int J Cancer 128:132–143
Shinozuka E, Miyashita M, Mizuguchi Y et al (2013) SnoN/SKIL modulates proliferation through control of hsa-miR-720 transcription in esophageal cancer cells. Biochem Biophys Res Commun 430:101–106
Mathé EA, Nguyen GH, Bowman ED et al (2009) MicroRNA expression in squamous cell carcinoma and adenocarcinoma of the esophagus: associations with survival. Clin Cancer Res 215:6192–6200
Hiyoshi Y, Kamohara H, Karashima R et al (2009) MicroRNA-21 regulates the proliferation and invasion in esophageal squamous cell carcinoma. Clin Cancer Res 15:1915–1922
Guo Y, Chen Z, Zhang L et al (2008) Distinctive microRNA profiles relating to patient survival in esophageal squamous cell carcinoma. Cancer Res 68:26–33
Mori Y, Ishiguro H, Kuwabara Y et al (2009) MicroRNA-21 induces cell proliferation and invasion in esophageal squamous cell carcinoma. Mol Med Rep 2:235–239
Tjalsma H, Bolhuis A, Jongbloed JD et al (2000) Signal peptide-dependent protein transport in Bacillus subtilis: a genome-based survey of the secretome. Microbiol Mol Biol Rev 64:515–547
Agrawal GK, Jwa NS, Lebrun MH et al (2010) Plant secretome: unlocking secrets of the secreted proteins. Proteomics 10:799–827
Komatsu S, Ichikawa D, Takeshita H et al (2011) Circulating microRNAs in plasma of patients with oesophageal squamous cell carcinoma. Br J Cancer 105:104–111
Kurashige J, Kamohara H, Watanabe M et al (2012) Serum microRNA-21 is a novel biomarker in patients with esophageal squamous cell carcinoma. J Surg Oncol 106:188–192
Drahos J, Schwameis K et al (2015) MicroRNA profiles of Barrett’s esophagus and esophageal adenocarcinoma: Differences in glandular non-native epithelium. Cancer Epidemiol Biomarkers Prev Nov 24: pii: cebp.0161.2015
Yu Y, Kanwar SS, Patel BB et al (2012) MicroRNA-21 induces stemness by downregulating transforming growth factor beta receptor 2 (TGFbetaR2) in colon cancer cells. Carcinogenesis 33:68–76
Ng R, Song G, Roll GR et al (2012) A microRNA-21 surge facilitates rapid cyclin D1 translation and cell cycle progression in mouse liver regeneration. J Clin Invest 122:1097–1108
Wang XC, Zhang ZB, Wang YY et al (2013) Increased miRNA-22 expression sensitizes esophageal squamous cell carcinoma to irradiation. J Radiat Res 54:401–408
Allawi HT, Dahlberg JE, Olson S et al (2004) Quantitation of microRNAs using a modified Invader assay. RNA 10:1153–1161
Neely LA, Patel S, Garver J et al (2006) A single-molecule method for the quantitation of microRNA gene expression. Nat Methods 3:41–46
Fassan M, Volinia S, Palatini J et al (2011) MicroRNA expression profiling in human Barrett’s carcinogenesis. Int J Cancer 129:1661–1670
Nelson PT, Baldwin DA, Kloosterman WP et al (2006) RAKE and LNA-ISH reveal microRNA expression and localization in archival human brain. RNA 12:187–191
Li J, Li X, Li Y et al (2013) Cell-specific detection of miR-375 downregulation for predicting the prognosis of esophageal squamous cell carcinoma by miRNA in situ hybridization. PLoS One 8:e53582
Schwarzenbach H, Nishida N et al (2014) Clinical relevance of circulating cell-free microRNAs in cancer. Nat Rev Clin Oncol 11:145–156
Anwaar SL, Lehmann U (2015) MicroRNAs: emerging novel clinical biomarkers for hepatocellular carcinomas. J Clin Med 18:1631–1650
Hur K, Toiyama Y et al (2015) Circulating microRNA-203 predicts prognosis and metastasis in human colorectal cancer. Gut, pii: gutjnl-2014-308737
Brennecke J, Stark A, Russell RB et al (2005) Principles of microRNA-target recognition. PLoS Biol 3:e85
Krek A, Grün D, Poy MN et al (2005) Combinatorial microRNA target predictions. Nat Genet 37:495–500
Brodersen P, Voinnet O (2009) Revisiting the principles of microRNA target recognition and mode of action. Nat Rev Mol Cell Biol 10:141–148
Scott GK, Mattie MD, Berger CE et al (2006) Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res 66:1277–1281
Da Cunha AB (1949) Genetic analysis of the polymorphism of color pattern in Drosophila polymorphia. Evolution 3:239–251
National Center for Biotechnology Information, United States National Library of Medicine (2013) NCBI dbSNP build 138 for human: http://www.ncbi.nlm.nih.gov/mailman/pipermail/dbsnp-announce/2013q3/000133.html
Xue Y, Wang Q, Long Q et al (2009) Human Y chromosome base-substitution mutation rate measured by direct sequencing in a deep-rooting pedigree. Curr Biol 19:1453–1457
Human genome project (2013) http://web.ornl.gov/sci/techresources/Human_Genome/project/info.shtml
Ensemble database at the European Bioinformatics Institute (EBI) and Wellcome Trust Sanger (2013) http://useast.ensembl.org/Homo_sapiens/Location/Chromosome?r=1
Watson JD, Baker TA, Bell SP et al (2004) Molecular biology of the gene, 5th edn, In Peason Benjamin Cummings, Cold Spring Harbor, Cold Spring Harbor Laboratory Press
Brouha B (2003) Hot L1 s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci 100:5280–5285
Bennett EA, Keller H, Mills RE et al (2008) Active Alu retrotransposons in the human genome. Genome Res 18:1875–1883
Wicker T, Sabot F, Hua-Van A et al (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8:973–982
Ju YS, Tubio JM, Mifsud W et al (2015) Frequent somatic transfer of mitochondrial DNA into the nuclear genome of human cancer cells. Genome Res 25:814–824
Projectmanagement (2013) Requirements management for big data projects. http://www.projectmanagement.com/articles/279834/Requirements-Management-for-Big-Data-Projects
Dupont C, Armant DR, Brenner CA (2009) Epigenetics: definition, mechanisms and clinical perspective. Semin Reprod Med 27:351–357
UniProt website (2015) UniProtKB–P68432 (H31_Human) http://www.uniprot.org/uniprot/P68431
Herman JG, Baylin SB (2003) Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 349:2042–2054
Nie K, Zhang T, Allawi H et al (2010) Epigenetic down-regulation of the tumor suppressor gene PRDM1/Blimp-1 in diffuse large B cell lymphomas: a potential role of the microRNA let-7. Am J Pathol 177:1470–1479
Kaz AM, Grady WM (2012) Epigenetic biomarkers in esophageal cancer. Cancer Lett 342:193–199
Sharma S, Kelly TK, Jones PA (2010) Epigenetics in cancer. Carcinogenesis 31:27–36
Eads CA, Lord RV, Wickramasinghe K et al (2001) Epigenetic patterns in the progression of esophageal adenocarcinoma. Cancer Res 61:3410–3418
Hibi K, Taguchi M, Nakayama H et al (2001) Molecular detection of p16 promoter methylation in the serum of patients with esophageal squamous cell carcinoma. Clin Cancer Res 7:3135–3138
Meng Y, Wang QG, Wang JX et al (2011) Epigenetic inactivation of the SFRP1 gene in esophageal squamous cell carcinoma. Dig Dis Sci 56:3195–3203
Meng XY, Zhu ST, Zong Y et al (2011) Promoter hypermethylation of cyclooxygenase-2 gene in esophageal squamous cell carcinoma. Dis Esophagus 24:444–449
Tong M, Chan KW, Bao JY et al (2012) Rab25 is a tumor suppressor gene with antiangiogenic and anti-invasive activities in esophageal squamous cell carcinoma. Cancer Res 72:6024–6035
Banki F, Mason RJ, Oh D et al (2007) Plasma DNA as a molecular marker for completeness of resection and recurrent disease in patients with esophageal cancer. Arch Surg 142:533–538
Hauser S, Kogej M, Fechner G et al (2012) Cell-free serum DNA in patients with bladder cancer: results of a prospective multicenter study. Anticancer Res 32:3119–3124
Martini M, Vecchione L, Siena S et al (2011) Targeted therapies: how personal should we go? Nat Rev Clin Oncol 9:87–97
Heng HH, Stevens JB, Bremer SW et al (2011) Evolultionary mechanisms and diversity in cancer. Adv Cancer Res 112:217–253
Stepanenko AA, Vassetzky YS, Kavsan VM (2013) Antagonistic functional duality of cancer genes. Gene 529(2):199–207
Qi Y, Chiu JF, Wang L et al (2005) Comparative proteomic analysis of esophageal squamous cell carcinoma. Proteomics 5:2960–2971
Jazii FR, Najafi Z, Malekzadeh R et al (2006) Identification of squamous cell carcinoma associated proteins by proteomics and loss of beta tropomyosin expression in esophageal cancer. World J Gastroenterol 12:7104–7112
Schaaij-Visser TB, Graveland AP, Gauci S et al (2009) Differential proteomics identifies protein biomarkers that predict local relapse of head and neck squamous cell carcinomas. Clin Cancer Res 15:7666–7675
Wang LD, Wang DC, Zheng S et al (2006) Serum proteomic profiles of the subjects with esophageal precancerous and cancerous lesions from Linzhou, an area with high incidence of esophageal cancer in Henan Province, Northern China. Ai Zheng 25:549–554
Gourin CG, Zhi W, Adam BL (2009) Proteomic identification of serum biomarkers for head and neck cancer surveillance. Laryngoscope 119:1291–1302
Voskuil J (2015) How difficult is the validation of clinical biomarkers? F1000Res 4:101
Ferreira R, Oliviera P, Martins T et al (2015) Comparative proteomic analyses of urine from rat urothelial carcinoma chemically induced by exposure to N-butyl-N-(4-hydroxybutyl)-nitrosamine. Mol BioSyst 11:11594–11602
Ha GH, Lee SU, Kang DG et al (2002) Proteome analysis of human stomach tissue: separation of soluble proteins by two-dimensional polyacrylamide gel electrophoresis and identification by mass spectrometry. Electrophoresis 23:2513–2524
Paulo JA, Lee LS, Wu B et al (2010) Proteomic analysis of endoscopically (endoscopic pancreatic function test) collected gastroduodenal fluid using in-gel tryptic digestion followed by LC-MS/MS. Proteomics Clin Appl 4:715–725
Chung JY, Braunschweig T, Hu N et al (2006) A multiplex tissue immunoblotting assay for proteomic profiling: a pilot study of the normal to tumor transition of esophageal squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev 15:1403–1408
Du XL, Hu H, Lin DC et al (2007) Proteomic profiling of proteins dysregulted in Chinese esophageal squamous cell carcinoma. J Mol Med (Berl) 85:863–875
Kashyap MK, Harsha HC, Renuse S et al (2010) SILAC-based quantitative proteomic approach to identify potential biomarkers from the esophageal squamous cell carcinoma secretome. Cancer Biol Ther 10:796–810
Uemura N, Nakanishi Y, Kato H et al (2009) Transglutaminase 3 as a prognostic biomarker in esophageal cancer revealed by proteomics. Int J Cancer 124:2106–2115
Uemura N, Nakanishi Y, Kato H et al (2009) Antibody-based proteomics for esophageal cancer: identification of proteins in the nuclear factor-kappaB pathway and mitotic checkpoint. Cancer Sci 100:1612–1622
Fu L, Qin YR, Xie D et al (2007) Identification of alpha-actinin 4 and 67 kDa laminin receptor as stage-specific markers in esophageal cancer via proteomic approaches. Cancer 110:2672–2681
Hu H, Ran Y, Zhang Y et al (2009) Antibody library-based tumor endothelial cells surface proteomic functional screen reveals migration-stimulating factor as an anti-angiogenic target. Mol Cell Proteomics 8:816–826
Xie LX, Zhai TT, Yang LP et al (2013) Lymphangiogenesis and prognostic significance of vascular endothelial growth factor C in gastro-oesophageal junction adenocarcinoma. Int J Exp Pathol 94:39–46
Moghanibashi M, Jazii FR, Soheili ZS et al (2012) Proteomics of a new esophageal cancer cell line established from Persian patient. Gene 500:124–133
Moghanibashi M, Rastgar Jazii F, Soheili ZS et al (2013) Esophageal cancer alters the expression of nuclear pore complex binding protein Hsc70 and eIF5A-1. Funct Integr Genomics 13:253–260
Qi YJ, He QY, Ma YF et al (2008) Proteomic identification of malignant transformation-related proteins in esophageal squamous cell carcinoma. J Cell Biochem 104:1625–1635
Cohen M, Yossef R, Erez T et al (2011) () Serum apolipoproteins C-I and C-III are reduced in stomach cancer patients: results from MALDI-based peptidome and immuno-based clinical assays. PLoS One 6(1):e14540
Juan HF, Chen JH, Hsu WT et al (2004) Identification of tumor-associated plasma biomarkers using proteomic techniques: from mouse to human. Proteomics 4:2766–2775
Metzger R, Bollschweiler E, Hölscher AH et al (2010) ERCC1: impact in multimodality treatment of upper gastrointestinal cancer. Future Oncol 6:1735–1749
Nishimori T, Tomonaga T, Matsushita K et al (2006) Proteomic analysis of primary esophageal squamous cell carcinoma reveals downregulation of a cell adhesion protein, periplakin. Proteomics 6:1011–1108
Zhang LY, Ying WT, Mao YS et al (2003) Loss of clusterin both in serum and tissue correlates with the tumorigenesis of esophageal squamous cell carcinoma via proteomics approaches. World J Gastroenterol 9:650–654
Anderson NL, Anderson NG (2002) The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics 1:845–867
Hortin GL, Sviridov D, Anderson NL (2008) High-abundance polypeptides of the human plasma proteome comprising the top 4 logs of polypeptide abundance. Clin Chem 54:1608–1616
Wang P, Whiteaker JR, Paulovich AG (2009) The evolving role of mass spectrometry in cancer biomarker discovery. Cancer Biol Ther 8:1083–1094
Omenn GS, States DJ, Adamski M et al (2005) Overview of the HUPO Plasma Proteome Project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics 5:3226–3245
Haab BB, Geierstanger BH, Michailidis G et al (2005) Immunoassay and antibody microarray analysis of the HUPO Plasma Proteome Project reference specimens: systematic variation between sample types and calibration of mass spectrometry data. Proteomics 5:3278–3291
Rai AJ, Gelfand CA, Haywood BC et al (2005) HUPO Plasma Proteome Project specimen collection and handling: towards the standardization of parameters for plasma proteome samples. Proteomics 5:3262–3277
Nanjappa V, Thomas JK, Marimutu A (2014) Plasma Proteome Database as a resource for proteomics research: 2014 update. Nucleic Acids Res 42:D959–D965 ((Database issue))
Cho JY, Lee HJ, Jeong SK (2015) Combination of multiple spectral libraries improves the current search methods used to identify missing proteins in the chromosome-centric human proteome project. J Proteome Res 14:4959–4966
Neimark J (2014) The dirty little secret in cancer research. 2nd Oct: http://discovermagazine.com/2014/nov/20-trial-and-error
Gey GO, Coffman WD, Kubicek MT (1952) Tissue culture studies of the proliferative capacity of cervical carcinoma and normal epithelium. Cancer Res 12:264–265
Jenkin HM, Hung SC (1967) Effect of vancomycin on the growth of psittacosis-trachoma agents cultivated in eggs and cell culture. Appl Microbiol 15:10–12
Dirks WG, Drexler HG (2011) Online verification of human cell line identity by STR DNA typing. Methods Mol Biol 731:45–55
Ando T, Ishiguro H, Kuwabara Y et al (2008) Relationship between expression of 5-fluorouracil metabolic enzymes and 5-fluorouracil sensitivity in esophageal carcinoma cell lines. Dis Esophagus 21:15–20
Domcke S, Sinha R, Levine DA et al (2013) Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun 4:2126
Begley CG, Ellis LM (2012) Drug development: raise standards for preclinical cancer research. Nature 483:531–533
Van Noorden R (2015) Interdisciplinary research by the numbers. Nature 525:306–307
Authors’ contributions
This manuscript contains original material that has not been previously published. All authors contributed in discussing the contents and approval of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We greatly acknowledge our thanks for the valuable discussions with Vladimir Matveev, Ray Perkins, Peter Shimon as well as the members of the web group of the Theodor-Billroth-Academy® on LinkedIn; this ongoing exchange is of importance to our thinking. Further the authors express their gratitude to the introduction of Vlado Antonic, PhD, University of Maryland, USA at the beginning of the review of the literature for the manuscript.
The manuscript was supported by the Theodor-Billroth-Academy® (TBA®) and INCORE, (International Consortium of Research Excellence) of the (TBA®).
Competing interests
The authors report no conflict of interest. The authors alone are responsible for the content and writing of this paper. Parts of the manuscript were translated into German for a CME accredited article of the CHAZ, Chirurgische Allgemeine Zeitung, Dr. R. Kaden-Verlag, Heidelberg, Germany [22].
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Brücher, B.L.D.M., Li, Y., Schnabel, P. et al. Genomics, microRNA, epigenetics, and proteomics for future diagnosis, treatment and monitoring response in upper GI cancers. Clin Trans Med 5, 13 (2016). https://doi.org/10.1186/s40169-016-0093-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40169-016-0093-6