
Abstract
Mitochondria are the powerhouse of a cell. The structure and function of mitochondria are precisely regulated by multiple signaling pathways. Neddylation, a post-translational modification, plays a crucial role in various cellular processes including cellular metabolism via modulating the activity, function and subcellular localization of its substrates. Recently, accumulated data demonstrated that neddylation is involved in regulation of morphology, trafficking and function of mitochondria. Mechanistic elucidation of how mitochondria is modulated by neddylation would further our understanding of mitochondrial regulation to a new level. In this review, we first briefly introduce mitochondria, then neddylation cascade, and known protein substrates subjected to neddylation modification. Next, we summarize current available data of how neddylation enzymes, its substrates (including cullins/Cullin-RING E3 ligases and non-cullins) and its inhibitor MLN4924 regulate the structure and function of mitochondria. Finally, we propose the future perspectives on this emerging and exciting field of mitochondrial research.
Similar content being viewed by others


Introduction
Mitochondria, the highly dynamic and semi-autonomous organelles wrapped with a double membrane, are deeply integrated into cellular signaling pathways and play the essential role in regulation of a variety of metabolisms such as energy production, calcium homeostasis, and reactive oxidative species (ROS) balance [1, 2]. Moreover, mitochondria regulate various essential cellular physiological processes such as differentiation, cell pluripotency and cell death [3]. Consequently, dysfunctional mitochondria have been observed in many pathological conditions including cancer, cardiovascular disorders, and metabolic diseases [4,5,6,7]. Thus, to maintain structural and functional integrity, and the well-being of a cell, mitochondria are subjected to fine regulations at the multiple levels under various physiological and pathological conditions [8,9,10,11,12,13,14,15].
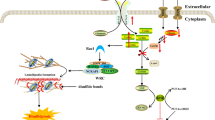
Protein neddylation is an important posttranslational modification in eukaryotes. To date, neddylation modification has been well-established to tag the extremely well conserved neuronal precursor cell-expressed developmentally down-regulated protein8 (NEDD8) onto the substrates to modulate their function, subcellular localization, and activity [16]. Recently, a study also showed that NEDD8 conjugates to SRSF3 on lysine 11 for poly-neddylation and subsequent proteasome-mediated degradation [17]. Similar to ubiquitylation, NEDD8 is first activated in an ATP-dependent manner by one heterodimeric E1 NEDD8-activating enzyme (NAE, NAE1/APPBP1 and NAEβ/UBA3), then transferred to one of the two NEDD8 conjugating enzymes (UBE2M/UBC12 and UBE2F) through a trans-thiolation reaction, and finally conjugated to target substrates catalyzed by one of dozen E3 neddylation ligases [18] (Fig. 1a).

The process of neddylation modification. a Neddylation is a process that tags the ubiquitin-like small molecule NEDD8 onto its substrate through an enzymatic cascade involving NEDD8-activating enzyme E1, NEDD8-conjugating enzyme E2 and substrate-specific NEDD8 E3 ligases. MLN4924 is a NAE inhibitor that blocks the entire neddylation pathway. b Cullin-RING ligase, consisting of a scaffold cullin, a RING protein that binds to NEDD8-loaded E2, an adaptor, and a substrate receptor, promotes ubiquitylation and degradation of their substrates. N8, NEDD8
To date, the best-characterized neddylation substrates are the cullin (CUL) family proteins, including CUL1, CUL2, CUL3, CUL4A, CUL4B, CUL5, CUL7, and CUL9. Cullin is the scaffold component, which complexes with other components, including an adaptor, a substrate receptor and a RING component, to form Cullin-RING ligases (CRLs) [16] (Fig. 1b). The conjugation of NEDD8 to a cullin activates CRL E3 ligase, the largest family of E3 ubiquitin ligases, which is responsible for the ubiquitylation of about 20% cellular proteins for targeted degradation through ubiquitin proteasome system (UPS) [19]. Therefore, neddylation precisely controls many biological processes through CRLs-mediated ubiquitylation and degradation upon neddylation activation [20, 21]. The overactivation of neddylation modification and abnormal expression of CRL components have been found in many human diseases, particularly human cancers [22]. MLN4924, also known as pevonedistat, binds to the active site of NAE catalytic subunit and forms a covalent NEDD8-MLN4924 adduct, which resembles adenylated NEDD8, the first intermediate in the NAE reaction cycle, but cannot be further utilized in subsequent intraenzyme reactions [23]. Therefore, MLN4924 directly inhibits the entire neddylation modification and indirectly inhibits the CRLs [19] (Fig. 1a). As a result, MLN4924 treatment triggers various cellular responses such as cell cycle arrest, apoptosis, senescence, autophagy as well as metabolic reprogramming [19, 24, 25]. Given that preclinical studies both in vitro cell culture settings and in vivo xenograft models showed potent antitumor activity and well-tolerated toxicity, MLN4924 has been advanced into several phase II clinical trials for anticancer therapy as a single agent or in combination with chemotherapeutic drugs [22].
The neddylation has been implicated in various metabolic processes, including adipogenesis, lipid droplet formation, and redox homeostasis [26,27,28,29,30,31]. Interestingly, emerging data revealed that mitochondrial functions are subjected to the regulation by neddylation and CRLs. More importantly, different approaches including mitochondria purification and confocal microscopy image analysis have shown that mitochondria contain various components of neddylation and CRLs, as well as non-cullin substrates, providing molecular basis for such a regulation [28, 32, 33].
Mitochondrial regulation by neddylation enzymes
NEDD8 activating enzyme (NAE)
UBA is the catalytic subunit of NAE [34], and a direct drug target of MLN4924 [19]. Our recent study showed that like MLN4924 (see below), UBA3 knockdown also markedly induces mitochondrial fission-to-fusion conversion to form filamental mitochondria [35]. In addition, one study reported that UBA3 is involved in mitochondrial respiration, since UBA3-deficient neonatal primary hepatocytes manifest reduced basal and maximal respiration, as compared to UBA3-sufficient ones [28].
NEDD8 conjugating enzymes
While there is no direct report to show that mitochondrial structure or function is subjected to regulation by neddylation E2s (Ube2M/UBC12 and Ube2F), our recent study found a negative cross-talk between UBE2M and UBE2F. Specifically, UBE2M complexes with Parkin-DJ1, an E3 localized in mitochondria under stressed conditions to promote ubiquitylation and degradation of UBE2F [21]. However, the biochemical and biological significances as to how this degradation impacts mitochondrial function remain elusive.
NEDD8 ligases
Two well-defined neddylation co-E3s, RBX1 and RBX2 (also known as SAG for Sensitive to Apoptosis Gene) were found to regulate mitochondrial functions [36, 37]. One study showed that upon mitochondrial damage, RBX1 promotes ubiquitylation and degradation of Suppression of Sestrin 2 (SESN2) to trigger the generation of mitochondrial ROS, leading to cell death in neuroblastoma cells [38]. Another recent study showed that in mice hepatocytes with Drp1KO, Rbx1 is recruited to mitochondria in a p62-dependent manner to mediate mitochondrial ubiquitylation and subsequent mitophagy [32].
RBX2/SAG was originally cloned in our laboratory as a redox-inducible antioxidant protein [37], which scavenges oxidant at the expense of forming inter or intra molecular disulfide bond [39]. In mouse embryonic stem cells, Rbx2/Sag disruption increases the steady-state levels of ROS after exposure to ionizing radiation, leading to radiosensitization via enhanced apoptosis [40].
c-CBL is another neddylation E3 [41]. It was reported that the activity of several enzymes involved in mitochondrial fat oxidation and the phosphorylation of acetyl CoA carboxylases are significantly increased in the muscle tissues of c-Cbl deficient mice when fed with the high-fat diet [42]. However, the detailed underlying mechanism of c-CBL action is unknown, nor whether this is through its ubiquitylation or neddylation activity.
Collectively, very limited reports suggest that neddylation enzymes modulate mitochondrial structure and functions. Much more extensive studies with mechanistic elucidation are needed to firmly establish the notion that mitochondria are subjected to neddylation regulation via the enzymatic cascade.
Mitochondrial regulation by neddylation substrates and CRLs
Cullins
Cullins with eight family members are physiological substrates of neddylation [43]. Cullins are scaffold component of the cullin-RING ligases (CRLs), which are the largest family of E3 ubiquitin ligases, consisting of four subunits: a cullin with 8 family members, adaptor proteins with many members, substrate recognition receptors with many members and the RING component with two family members (RBX1/RBX2) [44] (Fig. 1b). Interestingly, RBX1/RBX2 serves as dual E3 for both ubiquitylation and neddylation. Cullin neddylation is required for activation of CRLs [43], and accumulated data have shown that CRLs are actively involved in regulation of morphology, trafficking, functions, and the degradation of mitochondria (Table 1 and Fig. 2).

The modularity of Cullin-RING E3 ligases in mitochondria. Shown are the substrates of CRL1 (a), CRL2 (b), CRL3 (c), CRL4 (d), CRL5 (e) that are involved in the regulation of mitochondrial morphology and function
CRL1
CRL1, also known as SCF (SKP1-Cullin 1-F box protein), is the best studied founding member of CRLs. The F-box protein, the CRL substrate recognition subunit, consists of 69 members in mammalian cells, which are classified into FBXW, FBXL, and FBXO subfamilies [44, 45]. In yeast, F-box protein Mdm30 was shown to target Fzo1 (an ortholog of mammalian mitofusion1/2) or Mdm34, two mitochondrial proteins for their turnover, leading to inhibition of mitochondrial fusion [46,47,48]. The mitochondrial dynamics is also regulated by Mfb1, another F-box protein, although its specific mitochondrial substrate(s) are yet to be identified [49].
In mammalian cells, our recent study showed that SCFβ−TrCP1 E3 ligase, located in mitochondria, targeted MFN1 for ubiquitylation and degradation, and β-TrCP1 knockdown induced mitochondrial fusion due to MFN1 accumulation [35]. FBXW7 is well characterized F-box protein and its knockdown elevated the level of MITF, a lineage-specific master regulator of melanocytes to enhance mitochondrial transcriptional program and oxidative phosphorylation [50]. Furthermore, a recent study showed that SKP2, another well-characterized F-box protein, regulated the stability of IDH1/2, the key TCA cycle enzymes, and SKP2 knockdown promoted the TCA cycle by reducing glycolysis [51]. Other studies revealed that FBXL7 and FBXL4 modulated mitochondrial morphology and function through survivin and other uncharacterized substrates [52,53,54,55,56]. FBXO7 and FBXO11 (also characterized as a NEDD8 E3) were shown to regulate mitochondrial morphology and function, as well as mitochondrial elimination by modulating PARP activation, apoptosis, and mitophagy, respectively [57,58,59]. Finally, FBXO15 was found to modulate mitochondrial function through regulating the stability of CLS1 (cardiolipin synthase), an enzyme for generation of cardiolipin, a mitochondrial-specific lipid [60], while FBXO25 targeted mitochondrial HAX-1 for degradation to induce cellular apoptosis [61]. Thus, CRL1 regulates the structure and function of mitochondria at various aspects via modulating the stability of a variety of mitochondrial substrates.
CRL2
CRL2 is a complex of cullin-2, RBX1, adaptor protein Elongin B/C and substrate recognition receptor VHL-box [62]. HIF1α is the best-known substrate of CRL2 [63]. It was recently reported that pVHL up-regulated (a) CHCHD4, a key component of the disulphide relay system in mitochondrial protein import within the inter-membrane space, and (b) respiratory chain subunits of complexes I and IV, leading to increased rate of oxygen consumption and alterations in glucose and glutamine metabolisms in renal cell carcinoma [64].
CRL3
CRL3 is composed of cullin-3, RBX1, and BTB-containing substrate receptors, such as Kelch-like ECH-associated protein Keap1 and Speckle Type POZ protein SPOP [44, 65]. Nrf2 is the best-characterized substrate of CRL3-Keap1. In non-stress cells, Nrf2 is degraded by Keap1-mediated UPS. The oxidative stress and electrophiles, however, oxidize the cysteine residue in Keap1 to change its conformation, leading to blockage of NRF2 ubiquitylation and degradation. As result, NRF2 translocates to nucleus and transactivates a series of antioxidant genes to scavenge the ROS as the cellular response to the oxidative stress [66, 67]. SPOP, another well-known substrate recognizing component of CRL3, was found to interact with INF2 (inverted formin 2) and promote its atypical ubiquitylation, leading to inhibition of INF2 ER localization instead of degradation. The dissociation of INF2 from ER reduced the formation of mitochondrial associated DRP1 puncta and abrogated mitochondrial fission. Biologically, INF2-mediated mitochondrial fission is involved in migration and invasion, the processes promoted upon mutational inactivation of SPOP in human prostate cancer cells [68].
CRL4
CRL4 consists of cullin-4, RBX1, adaptor protein DDB1 (UV-damaged DNA-binding protein 1), and substrate receptors DCAFs (DDB1-CUL4-associated factor). Cullin-4 has two family members cullin-4A and cullin-4B, which share 82% sequence identity, but target different sets of substrates [69]. Cereblon (CRBN), a Cul-4A substrate receptor, was partially localized within the mitochondrial matrix. Instead of targeting degradation of Cul-4A substrates, CRBN acted as a Lon protease to suppress neuronal cell death upon induced by oxidative stress [70]. Moreover, CC-885, a novel thalidomide derivative that bridges the interaction between CRBN and its neosubstrate BNIP3L (also known as NIX), leading to ubiquitylation and degradation of BNIP3L to abrogate BNIP3L-dependent mitophagy [71]. Furthermore, a recent study showed that muscle-specific deletion of DCAF6, another substrate receptor of Cul-4A, resulted in reduced binding between Z-disc proteins ACTN2 and Cap-Z as well as increased levels of mitochondrial ROS and impaired respiration/ATP production in mouse hearts or cardiomyocytes isolated from these mice. The authors concluded that DCAF6 deficiency contributes to the pathogenesis of limb-girdle muscular dystrophy (LGMD) and heart failure, although detailed molecular mechanism remains elusive [72].
A direct evidence that CRL4B regulates mitochondrial function was shown by a Cul-4b knockout study. Cul-4b deletion in germ cells led to male infertility due to impaired sperm motility, partly caused by reduced mitochondrial activities including lower membrane potential and decreased ATP production. While the authors identified that Insl6, an insulin family member, is a novel substrate of Cul-4b in male germ cells, as possible mechanism, no functional rescue experiment was conducted to demonstrate that accumulation of Insl6 is indeed the cause of infertility derived from impaired mitochondrial function [73]. In another study, Arylhydrocarbon Receptor (AhR), a substrate receptor subunit in the CUL4B-AhR complex [69] was reported to protect melanocytes from oxidative damage via a mechanism that involved the up-regulation of nuclear respiratory factor 1 (NRF1) and its downstream targets to increase mitochondrial DNA synthesis and ATP production [74]. Furthermore, in human lung fibroblasts, genetic ablation of AhR decreased mitochondrial membrane potential and increased mitochondrial ROS to trigger cytochrome c release and subsequent apoptotic cell death when exposed to oxidative stress induced by tobacco extracts. Mechanistically, AhR appears to act as an antioxidant, rather than an E3 ligase for substrate degradation [75].
CRL5
CRL5 consists of cullin-5, a RING finger protein RBX2, adaptor protein Elongin B/C, and SOCS-box containing receptor proteins [62]. Compared to many CRL5 substrates for targeted degradation [76, 77], Cullin-5 bound to TRAF6 and promoted its polyubiquitylation via K63 linkage in response to LPS exposure, which facilitated NF-κB activation to trigger inflammatory response [78]. Interestingly, on depolarized mitochondria, TRAF6 was found to form a complex with PINK1 and SARM1 to promote K63-linked ubiquitylation and stabilization of PINK1. As a result, parkin, a PINK1 partner, was recruited to damaged mitochondria to facilitate mitophagy [79]. DEPTOR, a naturally occurring inhibitor of both mTORC1 and mTORC2, has been identified as the substrate of both CRL1 (SCFβ−TrCP1) and CRL5, respectively [80, 81]. One study showed that knockdown of DEPTOR in cultured term primary human trophoblast (PHT) cells promoted mitochondrial respiration, mtDNA copy number, and citrate synthase activity with mechanism involving activation of mTORC1 and mTORC2 [82].
The CRL7 and CRL9 were seldom studied family members of CRLs, and no reports were found in their involvement of mitochondrial regulation.
Taken together, the CRL regulation of mitochondria occurs by either promoting ubiquitylation and degradation of mitochondrial proteins or their regulators, or in a degradation independent manner via modulating other key molecules that affect mitochondrial functions. The finding that CRLs play an important role in modulating mitochondrial network indeed broadens our understanding of how mitochondria is precisely regulated.
Non-cullin substrates
The neddylation substrates can be broadly classified into two categories: commonly studied cullins and less studied non-cullin substrates. Two studies reported potential regulation of non-cullin substrates in mitochondrial functions. A recent study showed that electron transfer flavoproteins (ETFs: ETFA and ETFB) were subjected to neddylation modification, which resulted in their stabilization by preventing ubiquitylation and degradation in hepatocytes and also partially facilitated fatty acid β-oxidation in neonatal mice. In the same vein, the mutants of ETFA and ETFB with neddylation site abrogated had reduced protein levels or activities which contributed to the pathogenesis of glutaric aciduria type II (GA-II). Furthermore, ETFA knockdown led to substantially decreased basal and maximal respiration [28].
The second study showed that HIF-1α and HIF-2α are subjected to neddylation modification, which led to their stabilization in a manner independent of prolyl hydroxylase (PHD)/VHL oxygen-sensing system, but dependent on mitochondria-generated ROS, although the biological consequence of such stabilization was not determined [83].
Furthermore, some non-cullin substrates were found to regulate mitochondrial function, but whether this is related to neddylation modification is unclear. For example, in response to various DNA damaging agents, p53, a non-cullin substrate of neddylation, was translocated to mitochondria to regulate apoptosis [84, 85]. Although p53 neddylation mediated by MDM2 or SCFFBXO11 inhibited its transcription activity [86, 87], whether its neddylation modification impacts mitochondrial functions remains unclear. One more case is Parkin, an E3 ubiquitin ligase, that promotes ubiquitylation and degradation of MFN1/2 to trigger mitophagy [88]. While Parkin is subjected to neddylation modification, which activated its ligase activity, but not altering its subcellular localization [89], there is no direct biological evidence to show that Parkin neddylation indeed enhanced mitophagy.
Neddylation inhibitor MLN4924 alters mitochondrial morphology and function
Cellular ROS is mainly generated in mitochondrial powerhouse. Abnormal ROS overproduction due to impaired mitochondria alone or in combination with defective antioxidative scavenger systems contributes significantly to the damages to cellular DNA, proteins, and lipids, leading to tumorigenesis [90]. MLN4924 (also known as pevonedistat in the clinical trials), a small molecular inhibitor of catalytic subunit of NEDD8 activating enzyme, blocks the entire neddylation modification and consequently inactivates all CRLs [19]. Given overactivation of neddylation system and CRLs in a variety of human cancers, MLN4924 has been shown impressive anti-cancer activity in many preclinical studies and had advanced to few Phase II clinical trials as an anticancer agent alone or in combination with various chemotherapeutic drugs [22].
Several studies including from our own laboratory showed that MLN4924 regulates mitochondrial ROS production and other mitochondrial functions. In acute myeloid leukemia cells, MLN4924 inactivated CRL1/SCF to inhibit NF-κB via accumulated IκB, leading to increased ROS generation due to downregulation of Mn-SOD (superoxide dismutase), a typical NF-κB downstream gene and a major antioxidant enzyme to scavenge ROS. Thus, MLN4924-induced disruption of cellular redox status was identified as a key event in apoptosis induction [19, 30]. Moreover, other two studies reported that MLN4924 induced ROS generation by impairing mitochondrial membrane potential in both ovarian and live cancer cell lines to cause apoptosis through the upregulation of pro-apoptotic proteins, including PUMA, BIK, NOXA and BIM. Among these proteins, NOXA and BIK were identified as important downstream effectors for apoptosis induced by MLN4924/CQ and MLN4924/cisplatin combination, respectively. Importantly, the ROS scavenger reduced the expression of these pro-apoptotic proteins and attenuated apoptosis, indicating the causal effect of ROS [31, 91].
Recently, we found that MLN4924 induced mitochondrial fission-to-fusion conversion in time-and dose-dependent manners in a variety of human cancer cell lines. Mechanistic studies revealed that MLN4924 inactivated SCFβ−TrCP E3 ligase to cause accumulation of MFN1 [35], which is a protein previously known to trigger the fission-to-fusion conversion [92]. MLN4924 also reduced the level of phospho-DRP1S616 in mitochondria to increase cytoplasmic DRP1 content. The mitochondrial functional assays showed that MLN4924 inhibited TCA cycle but promoted both the basal and maximal oxygen consumption rate (OCR), while reducing the intracellular ATP production. MLN4924 also caused mitochondrial depolarization and increased mitochondrial ROS levels as well as mtDNA copy number. Since MLN4924-induced mitochondrial fusion was coupled with increased oxidative phosphorylation (OXPHOS), we then tested translational implication of this finding by combined treatment of MLN4924 with metformin, an inhibitor of mitochondrial complex I, to determine possible enhanced anticancer efficiency in breast cancer cells. The results indeed showed a synergistic effect both in vitro cell culture and in vivo xenograft models [35]. Thus, our strategy of inhibiting both neddylation modification and OXPHOS provides new avenues for targeted therapy in some types of tumor such as breast cancer. It is interesting to mention that MLN4924 effect on OXPHOS appears to be cell-type dependent. Two other studies showed that MLN4924 decreased both basal and maximal OCR in liver cancer cell line and mouse hepatocytes [28, 93]. Taken together, MLN4924 induces mitochondrial fission-to-fusion conversion, mitochondrial copy number, oxygen consumption and ROS production, but inhibits mitochondrial membrane potential and ATP production (Fig. 3). MLN4924 may, therefore, have unique application against human cancers with dysfunctional mitochondria.

MLN4924 regulates mitochondrial functions. MLN4924 induces mitochondrial fission-to-fusion conversion, increases mitochondrial copy number, oxygen consumption and ROS production, but inhibits mitochondrial membrane potential and ATP production. The proteins and signaling pathways involved in MLN4924-regulated mitochondrial functions are shown [30, 35]
Conclusion and future perspectives
In this review, we summarized neddylation regulation of mitochondrial morphology and functions with potential anticancer application. The fact that several components and substrates of neddylation/CRLs, including NEDD8, UBA3, RBX1, FBXL4 and β-TrCP1, were found in mitochondria [28, 35, 54] provides the physical basis on neddylation regulation of mitochondria. The regulation is likely achieved via altered protein activity after neddylation modification or possible targeted degradation of mitochondrial substrates of CRLs. Nevertheless, neddylation regulation of mitochondria is still an emerging field in the broad area of mitochondria research with many unanswered questions. We propose the following aspects as future perspectives for this exciting field (Fig. 4).

Proposed future directions for neddylation regulation of mitochondria. Five possible directions were proposed. See text for details
Identification of mitochondrial CRL substrates?
Mitochondrial proteins are subjected to degradation by both UPS and proteases. It is generally accepted that the UPS is involved in degradation of the outer membrane proteins, whereas the proteases are responsible for the cleavage and degradation of the proteins of inner mitochondrial compartments [94], although the UPS was also reported to be involved in the degradation of some cysteine-rich proteins in the intermembrane space (e.g. yeast Cox12), the uncoupling protein in the inner membrane, and a subunit of succinate dehydrogenase in the matrix [10, 94]. Several components of neddylation and CRLs were found in the mitochondria [28, 35, 54]. An open question is whether they actually assembly the active CRLs to promote the ubiquitylation of mitochondrial proteins for degradation via the UPS which is mainly localized outside the mitochondria? Given that few mitochondrial proteins were reported as the substrates of CRL, such as MFN1 by SCFβ−TrCP [35] and HAX-1 by SCFFBXO25 [61], and a comprehensive quantitative diGly proteomics analysis identified ~ 150 mitochondria proteins [95], it is likely that some mitochondrial proteins are indeed the substrates of CRLs. Here we propose that a similar analysis could be performed using HEK293 cells with knocked-in ubiquitin tagged with a mitochondrial signal peptide at the N-terminus, followed by mitochondrial purification and diGly proteomics. The approach will validate previous candidates with identification of additional putative mitochondrial substrates of CRLs for further confirmation and functional characterization.
Identification of neddylated mitochondrial proteins
Only one study reporting that mitochondrial protein parkin is subjected to neddylation modification which increased its E3 ligase activity [89]. To identify almost, if not all, mitochondrial proteins subjected to neddylation modification at the whole mitochondria level, an approach similar to serial NEDD8-ubiquitin substrate profiling (sNUSP), reported recently [20] can be employed. Specifically, the approach includes the establishment of a knock-in HEK293 cell line of NEDD8 R74K mutant tagged with a mitochondrial signal peptide at the N-terminus for mitochondria targeting, followed by mitochondrial isolation, Lys-C digestion, K-εGG-peptide enrichment, and finally Mass Spectrometry analysis to identify neddylated mitochondrial proteins. The specificity can be determined by including a MLN4924-treatment control group. Each validated candidate can be further characterized for functional significance of neddylation modification.
Identification of mitochondrial proteins that bind to neddylation enzymes
To mechanistically elucidate neddylation regulation of mitochondrial function, it should be meaningful to isolate mitochondrial proteins that bind to any of neddylation enzymes. Again, a given neddylation enzyme expression construct can be made with the N-terminal tag of mitochondrial signal peptide and the C-terminal FLAG-tag for affinity purification. The construct can then be knocked-in to HEK293 cells, and selected stable clone to be used for routine affinity purification and mass spectrometry, followed by validation of the candidates in an individual basis and biological effect on mitochondrial function determined.
Characterization of mitochondrial metabolic pathways altered by MLN4924
We recently performed Mass-Spectrometry-based metabolic profiling of breast cancer cells to investigate the overall effect of MLN4924 on cell metabolism at a global level, and found that MLN4924 treatment caused significant increase or decrease of many metabolites in cell extracts [35]. Similarly, integrated liquid chromatography-tandem mass spectrometry (LC–MS/MS) analysis of metabolites and mass spectrometry-based proteomic analysis of proteins should be performed using isolated mitochondria upon overall neddylation inhibition (by MLN4924) or manipulation of each individual cullins to define specifically the effect of neddylation/CRLs on mitochondrial metabolism.
The cross-talk between tumor metabolism and tumor microenvironment: involvement of mitochondrial neddylation?
Increasing amount of data has shown that neddylation can modulate both tumor metabolism and tumor microenvironment [18, 35]. Whether and how mitochondria are involved in this cross-talk upon regulation by neddylation/CRLs is an interesting topic for future investigation. As an initial step, mitochondrial proteomics could be performed under the conditions in which tumor metabolism and tumor microenvironment are altered upon MLN4924 treatment to determine potential involvement of mitochondrial neddylation.
Availability of data and materials
Not applicable.
References
Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab TEM. 2016;27(2):105–17. https://doi.org/10.1016/j.tem.2015.12.001.
Pfanner N, Warscheid B, Wiedemann N. Mitochondrial proteins: from biogenesis to functional networks. Nat Rev Mol Cell Biol. 2019;20(5):267–84. https://doi.org/10.1038/s41580-018-0092-0.
Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol. 2020;21(4):204–24. https://doi.org/10.1038/s41580-020-0210-7.
Li S, Pan H, Tan C, Sun Y, Song Y, Zhang X, Yang W, Wang X, Li D, Dai Y, Ma Q, Xu C, Zhu X, Kang L, Fu Y, Xu X, Shu J, Zhou N, Han F, Qin D, Huang W, Liu Z, Yan Q. Mitochondrial dysfunctions contribute to hypertrophic cardiomyopathy in patient iPSC-derived cardiomyocytes with MT-RNR2 mutation. Stem cell Rep. 2018;10(3):808–21. https://doi.org/10.1016/j.stemcr.2018.01.013.
Sabouny R, Shutt TE. Reciprocal regulation of mitochondrial fission and fusion. Trends Biochem Sci. 2020;45(7):564–77. https://doi.org/10.1016/j.tibs.2020.03.009.
Yambire KF, Rostosky C, Watanabe T, Pacheu-Grau D, Torres-Odio S, Sanchez-Guerrero A, Senderovich O, Meyron-Holtz EG, Milosevic I, Frahm J, West AP, Raimundo N. Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo. eLife. 2019. https://doi.org/10.7554/eLife.51031.
Vasan K, Werner M, Chandel NS. Mitochondrial metabolism as a target for cancer therapy. Cell Metab. 2020;32(3):341–52. https://doi.org/10.1016/j.cmet.2020.06.019.
Anand R, Langer T. Baker MJ (2013) Proteolytic control of mitochondrial function and morphogenesis. Biochim Biophys Acta. 1833;1:195–204. https://doi.org/10.1016/j.bbamcr.2012.06.025.
Ross JM, Olson L, Coppotelli G. Mitochondrial and ubiquitin proteasome system dysfunction in ageing and disease: two sides of the same coin? Int J Mol Sci. 2015;16(8):19458–76. https://doi.org/10.3390/ijms160819458.
Lavie J, De Belvalet H, Sonon S, Ion AM, Dumon E, Melser S, Lacombe D, Dupuy JW, Lalou C, Benard G. Ubiquitin-dependent degradation of mitochondrial proteins regulates energy metabolism. Cell Rep. 2018;23(10):2852–63. https://doi.org/10.1016/j.celrep.2018.05.013.
Zhou Q, Wang W, He X, Zhu X, Shen Y, Yu Z, Wang X, Qi X, Zhang X, Fan M, Dai Y, Yang S, Yan Q. Mechanistic study on the nuclear modifier gene MSS1 mutation suppressing neomycin sensitivity of the mitochondrial 15S rRNA C1477G mutation in Saccharomyces cerevisiae. PLoS ONE. 2014;9(3):e90336. https://doi.org/10.1371/journal.pone.0090336.
Zhang X, Zuo X, Yang B, Li Z, Xue Y, Zhou Y, Huang J, Zhao X, Zhou J, Yan Y, Zhang H, Guo P, Sun H, Guo L, Zhang Y, Fu XD. MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell. 2014;158(3):607–19. https://doi.org/10.1016/j.cell.2014.05.047.
Zhou J, Xu L, Duan X, Liu W, Zhao X, Wang X, Shang W, Fang X, Yang H, Jia L, Bai J, Zhao J, Wang L, Tong C. Large-scale RNAi screen identified Dhpr as a regulator of mitochondrial morphology and tissue homeostasis. Sci Adv. 2019;5(9):eaax0365. https://doi.org/10.1126/sciadv.aax0365.
Deshwal S, Fiedler KU, Langer T. Mitochondrial proteases: multifaceted regulators of mitochondrial plasticity. Annu Rev Biochem. 2020;89:501–28. https://doi.org/10.1146/annurev-biochem-062917-012739.
Gao K, Cheng M, Zuo X, Lin J, Hoogewijs K, Murphy MP, Fu XD, Zhang X. Active RNA interference in mitochondria. Cell Res. 2020. https://doi.org/10.1038/s41422-020-00394-5.
Zhao Y, Morgan MA, Sun Y. Targeting Neddylation pathways to inactivate cullin-RING ligases for anticancer therapy. Antioxid Redox Signal. 2014;21(17):2383–400. https://doi.org/10.1089/ars.2013.5795.
Kumar D, Das M, Sauceda C, Ellies LG, Kuo K, Parwal P, Kaur M, Jih L, Bandyopadhyay GK, Burton D, Loomba R, Osborn O, Webster NJ. Degradation of splicing factor SRSF3 contributes to progressive liver disease. J Clin Investig. 2019;129(10):4477–91. https://doi.org/10.1172/JCI127374.
Zhou L, Jiang Y, Luo Q, Li L, Jia L. Neddylation: a novel modulator of the tumor microenvironment. Mol Cancer. 2019;18(1):77. https://doi.org/10.1186/s12943-019-0979-1.
Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–6. https://doi.org/10.1038/nature07884.
Vogl AM, Phu L, Becerra R, Giusti SA, Verschueren E, Hinkle TB, Bordenave MD, Adrian M, Heidersbach A, Yankilevich P, Stefani FD, Wurst W, Hoogenraad CC, Kirkpatrick DS, Refojo D, Sheng M. Global site-specific neddylation profiling reveals that NEDDylated cofilin regulates actin dynamics. Nat Struct Mol Biol. 2020;27(2):210–20. https://doi.org/10.1038/s41594-019-0370-3.
Zhou W, Xu J, Tan M, Li H, Li H, Wei W, Sun Y. UBE2M is a stress-inducible dual E2 for neddylation and ubiquitylation that promotes targeted degradation of UBE2F. Mol Cell. 2018;70(6):1008-1024.e1006. https://doi.org/10.1016/j.molcel.2018.06.002.
Zhou L, Zhang W, Sun Y, Jia L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal. 2018;44:92–102. https://doi.org/10.1016/j.cellsig.2018.01.009.
Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ, Soucy TA, Milhollen MA, Yang X, Burkhardt AL, Ma J, Loke HK, Lingaraj T, Wu D, Hamman KB, Spelman JJ, Cullis CA, Langston SP, Vyskocil S, Sells TB, Mallender WD, Visiers I, Li P, Claiborne CF, Rolfe M, Bolen JB, Dick LR. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell. 2010;37(1):102–11. https://doi.org/10.1016/j.molcel.2009.12.024.
Nawrocki ST, Griffin P, Kelly KR, Carew JS. MLN4924: a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin Investig Drugs. 2012;21(10):1563–73. https://doi.org/10.1517/13543784.2012.707192.
Zhou Q, Sun Y. MLN4924: additional activities beyond neddylation inhibition. Mol Cell Oncol. 2019;6(5):e1618174. https://doi.org/10.1080/23723556.2019.1618174.
Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24(24):10941–53. https://doi.org/10.1128/MCB.24.24.10941-10953.2004.
Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31(6):1121–33. https://doi.org/10.1128/MCB.01204-10.
Zhang X, Zhang YL, Qiu G, Pian L, Guo L, Cao H, Liu J, Zhao Y, Li X, Xu Z, Huang X, Huang J, Dong J, Shen B, Wang HX, Ying X, Zhang WJ, Cao X, Zhang J. Hepatic neddylation targets and stabilizes electron transfer flavoproteins to facilitate fatty acid beta-oxidation. Proc Natl Acad Sci USA. 2020;117(5):2473–83. https://doi.org/10.1073/pnas.1910765117.
Park HS, Ju UI, Park JW, Song JY, Shin DH, Lee KH, Jeong LS, Yu J, Lee HW, Cho JY, Kim SY, Kim SW, Kim JB, Park KS, Chun YS. PPARgamma neddylation essential for adipogenesis is a potential target for treating obesity. Cell Death Differ. 2016;23(8):1296–311. https://doi.org/10.1038/cdd.2016.6.
Swords RT, Kelly KR, Smith PG, Garnsey JJ, Mahalingam D, Medina E, Oberheu K, Padmanabhan S, O’Dwyer M, Nawrocki ST, Giles FJ, Carew JS. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood. 2010;115(18):3796–800. https://doi.org/10.1182/blood-2009-11-254862.
Chen P, Hu T, Liang Y, Jiang Y, Pan Y, Li C, Zhang P, Wei D, Li P, Jeong LS, Chu Y, Qi H, Yang M, Hoffman RM, Dong Z, Jia L. Synergistic inhibition of autophagy and neddylation pathways as a novel therapeutic approach for targeting liver cancer. Oncotarget. 2015;6(11):9002–17. https://doi.org/10.18632/oncotarget.3282.
Yamada T, Murata D, Adachi Y, Itoh K, Kameoka S, Igarashi A, Kato T, Araki Y, Huganir RL, Dawson TM, Yanagawa T, Okamoto K, Iijima M, Sesaki H. Mitochondrial stasis reveals p62-mediated ubiquitination in parkin-independent mitophagy and mitigates nonalcoholic fatty liver disease. Cell Metab. 2018;28(4):588-604.e585. https://doi.org/10.1016/j.cmet.2018.06.014.
Aram L, Braun T, Braverman C, Kaplan Y, Ravid L, Levin-Zaidman S, Arama E. A krebs cycle component limits caspase activation rate through mitochondrial surface restriction of CRL activation. Dev Cell. 2016;37(1):15–33. https://doi.org/10.1016/j.devcel.2016.02.025.
Enchev RI, Schulman BA, Peter M. Protein neddylation: beyond cullin-RING ligases. Nat Rev Mol Cell Biol. 2015;16(1):30–44. https://doi.org/10.1038/nrm3919.
Zhou Q, Li H, Li Y, Tan M, Fan S, Cao C, Meng F, Zhu L, Zhao L, Guan MX, Jin H, Sun Y. Inhibiting neddylation modification alters mitochondrial morphology and reprograms energy metabolism in cancer cells. JCI insight. 2019. https://doi.org/10.1172/jci.insight.121582.
Huang DT, Ayrault O, Hunt HW, Taherbhoy AM, Duda DM, Scott DC, Borg LA, Neale G, Murray PJ, Roussel MF, Schulman BA. E2-RING expansion of the NEDD8 cascade confers specificity to cullin modification. Mol Cell. 2009;33(4):483–95. https://doi.org/10.1016/j.molcel.2009.01.011.
Duan H, Wang Y, Aviram M, Swaroop M, Loo JA, Bian J, Tian Y, Mueller T, Bisgaier CL, Sun Y. SAG, a novel zinc RING finger protein that protects cells from apoptosis induced by redox agents. Mol Cell Biol. 1999;19(4):3145–55. https://doi.org/10.1128/mcb.19.4.3145.
Kumar A, Shaha C. RBX1-mediated ubiquitination of SESN2 promotes cell death upon prolonged mitochondrial damage in SH-SY5Y neuroblastoma cells. Mol Cell Biochem. 2018;446(1–2):1–9. https://doi.org/10.1007/s11010-017-3267-7.
Swaroop M, Bian J, Aviram M, Duan H, Bisgaier CL, Loo JA, Sun Y. Expression, purification, and biochemical characterization of SAG, a ring finger redox-sensitive protein. Free Radical Biol Med. 1999;27(1–2):193–202. https://doi.org/10.1016/s0891-5849(99)00078-7.
Tan M, Zhu Y, Kovacev J, Zhao Y, Pan ZQ, Spitz DR, Sun Y. Disruption of Sag/Rbx2/Roc2 induces radiosensitization by increasing ROS levels and blocking NF-kappaB activation in mouse embryonic stem cells. Free Radical Biol Med. 2010;49(6):976–83. https://doi.org/10.1016/j.freeradbiomed.2010.05.030.
Oved S, Mosesson Y, Zwang Y, Santonico E, Shtiegman K, Marmor MD, Kochupurakkal BS, Katz M, Lavi S, Cesareni G, Yarden Y. Conjugation to Nedd8 instigates ubiquitylation and down-regulation of activated receptor tyrosine kinases. J Biol Chem. 2006;281(31):21640–51. https://doi.org/10.1074/jbc.M513034200.
Molero JC, Waring SG, Cooper A, Turner N, Laybutt R, Cooney GJ, James DE. Casitas b-lineage lymphoma-deficient mice are protected against high-fat diet-induced obesity and insulin resistance. Diabetes. 2006;55(3):708–15. https://doi.org/10.2337/diabetes.55.03.06.db05-0312.
Deshaies RJ. SCF and Cullin/Ring H2-based ubiquitin ligases. Annu Rev Cell Dev Biol. 1999;15:435–67.
Zhao Y, Sun Y. Cullin-RING Ligases as attractive anti-cancer targets. Curr Pharm Des. 2013;19(18):3215–25.
Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 2004;18(21):2573–80.
Fritz S, Weinbach N, Westermann B. Mdm30 is an F-box protein required for maintenance of fusion-competent mitochondria in yeast. Mol Biol Cell. 2003;14(6):2303–13. https://doi.org/10.1091/mbc.e02-12-0831.
Cohen MM, Leboucher GP, Livnat-Levanon N, Glickman MH, Weissman AM. Ubiquitin-proteasome-dependent degradation of a mitofusin, a critical regulator of mitochondrial fusion. Mol Biol Cell. 2008;19(6):2457–64. https://doi.org/10.1091/mbc.E08-02-0227.
Ota K, Kito K, Okada S, Ito T. A proteomic screen reveals the mitochondrial outer membrane protein Mdm34p as an essential target of the F-box protein Mdm30p. Genes Cells Devot Mol Cell Mech. 2008;13(10):1075–85. https://doi.org/10.1111/j.1365-2443.2008.01228.x.
Durr M, Escobar-Henriques M, Merz S, Geimer S, Langer T, Westermann B. Nonredundant roles of mitochondria-associated F-box proteins Mfb1 and Mdm30 in maintenance of mitochondrial morphology in yeast. Mol Biol Cell. 2006;17(9):3745–55. https://doi.org/10.1091/mbc.e06-01-0053.
Abbate F, Badal B, Mendelson K, Aydin IT, Serasinghe MN, Iqbal R, Mohammed JN, Solovyov A, Greenbaum BD, Chipuk JE, Celebi JT. FBXW7 regulates a mitochondrial transcription program by modulating MITF. Pigment Cell Melanoma Res. 2018;31(5):636–40. https://doi.org/10.1111/pcmr.12704.
Liu J, Peng Y, Shi L, Wan L, Inuzuka H, Long J, Guo J, Zhang J, Yuan M, Zhang S, Wang X, Gao J, Dai X, Furumoto S, Jia L, Pandolfi PP, Asara JM, Kaelin WG Jr, Liu J, Wei W. Skp2 dictates cell cycle-dependent metabolic oscillation between glycolysis and TCA cycle. Cell Res. 2021;31(1):80–93. https://doi.org/10.1038/s41422-020-0372-z.
Alsina D, Lytovchenko O, Schab A, Atanassov I, Schober FA, Jiang M, Koolmeister C, Wedell A, Taylor RW, Wredenberg A, Larsson NG. FBXL4 deficiency increases mitochondrial removal by autophagy. EMBO Mol Med. 2020;12(7):e11659. https://doi.org/10.15252/emmm.201911659.
Wang S, Lin L, Wang Y, Wang A, Liu Z, Wu S, Lan X, Jia J, Zhang Y, Yuan F, Wang C, Luo X, Sun X, Avula SK, Tolaymat A, Liu C, Ren Y, Chen Y. Novel homozygous mutation in the FBXL4 gene is associated with mitochondria DNA depletion syndrome-13. J Neurol Sci. 2020;416:116948. https://doi.org/10.1016/j.jns.2020.116948.
Bonnen PE, Yarham JW, Besse A, Wu P, Faqeih EA, Al-Asmari AM, Saleh MA, Eyaid W, Hadeel A, He L, Smith F, Yau S, Simcox EM, Miwa S, Donti T, Abu-Amero KK, Wong LJ, Craigen WJ, Graham BH, Scott KL, McFarland R, Taylor RW. Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am J Hum Genet. 2013;93(3):471–81. https://doi.org/10.1016/j.ajhg.2013.07.017.
Liu Y, Lear T, Iannone O, Shiva S, Corey C, Rajbhandari S, Jerome J, Chen BB, Mallampalli RK. The proapoptotic F-box protein Fbxl7 regulates mitochondrial function by mediating the ubiquitylation and proteasomal degradation of survivin. J Biol Chem. 2015;290(19):11843–52. https://doi.org/10.1074/jbc.M114.629931.
Gai X, Ghezzi D, Johnson MA, Biagosch CA, Shamseldin HE, Haack TB, Reyes A, Tsukikawa M, Sheldon CA, Srinivasan S, Gorza M, Kremer LS, Wieland T, Strom TM, Polyak E, Place E, Consugar M, Ostrovsky J, Vidoni S, Robinson AJ, Wong LJ, Sondheimer N, Salih MA, Al-Jishi E, Raab CP, Bean C, Furlan F, Parini R, Lamperti C, Mayr JA, Konstantopoulou V, Huemer M, Pierce EA, Meitinger T, Freisinger P, Sperl W, Prokisch H, Alkuraya FS, Falk MJ, Zeviani M. Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am J Hum Genet. 2013;93(3):482–95. https://doi.org/10.1016/j.ajhg.2013.07.016.
Li Y, Chen F, Lin F, Guan C, Wei X, Wan Y, Xu A. VIT1/FBXO11 knockdown induces morphological alterations and apoptosis in B10BR mouse melanocytes. Int J Mol Med. 2009;23(5):673–8. https://doi.org/10.3892/ijmm_00000179.
Delgado-Camprubi M, Esteras N, Soutar MP, Plun-Favreau H, Abramov AY. Deficiency of Parkinson’s disease-related gene Fbxo7 is associated with impaired mitochondrial metabolism by PARP activation. Cell Death Differ. 2017;24(1):120–31. https://doi.org/10.1038/cdd.2016.104.
Zhou ZD, Xie SP, Sathiyamoorthy S, Saw WT, Sing TY, Ng SH, Chua HP, Tang AM, Shaffra F, Li Z, Wang H, Ho PG, Lai MK, Angeles DC, Lim TM, Tan EK. F-box protein 7 mutations promote protein aggregation in mitochondria and inhibit mitophagy. Hum Mol Genet. 2015;24(22):6314–30. https://doi.org/10.1093/hmg/ddv340.
Chen BB, Coon TA, Glasser JR, Zou C, Ellis B, Das T, McKelvey AC, Rajbhandari S, Lear T, Kamga C, Shiva S, Li C, Pilewski JM, Callio J, Chu CT, Ray A, Ray P, Tyurina YY, Kagan VE, Mallampalli RK. E3 ligase subunit Fbxo15 and PINK1 kinase regulate cardiolipin synthase 1 stability and mitochondrial function in pneumonia. Cell Rep. 2014;7(2):476–87. https://doi.org/10.1016/j.celrep.2014.02.048.
Baumann U, Fernandez-Saiz V, Rudelius M, Lemeer S, Rad R, Knorn AM, Slawska J, Engel K, Jeremias I, Li Z, Tomiatti V, Illert AL, Targosz BS, Braun M, Perner S, Leitges M, Klapper W, Dreyling M, Miething C, Lenz G, Rosenwald A, Peschel C, Keller U, Kuster B, Bassermann F. Disruption of the PRKCD-FBXO25-HAX-1 axis attenuates the apoptotic response and drives lymphomagenesis. Nat Med. 2014;20(12):1401–9. https://doi.org/10.1038/nm.3740.
Kamura T, Maenaka K, Kotoshiba S, Matsumoto M, Kohda D, Conaway RC, Conaway JW, Nakayama KI. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 2004;18(24):3055–65. https://doi.org/10.1101/gad.1252404.
Pugh CW, Ratcliffe PJ. The von Hippel-Lindau tumor suppressor, hypoxia-inducible factor-1 (HIF-1) degradation, and cancer pathogenesis. Semin Cancer Biol. 2003;13(1):83–9.
Briston T, Stephen JM, Thomas LW, Esposito C, Chung YL, Syafruddin SE, Turmaine M, Maddalena LA, Greef B, Szabadkai G, Maxwell PH, Vanharanta S, Ashcroft M. VHL-mediated regulation of CHCHD4 and mitochondrial function. Front Oncol. 2018;8:388. https://doi.org/10.3389/fonc.2018.00388.
Clark A, Burleson M. SPOP and cancer: a systematic review. Am J Cancer Res. 2020;10(3):704–26.
Itoh K, Ye P, Matsumiya T, Tanji K, Ozaki T. Emerging functional cross-talk between the Keap1-Nrf2 system and mitochondria. J Clin Biochem Nutr. 2015;56(2):91–7. https://doi.org/10.3164/jcbn.14-134.
O’Mealey GB, Plafker KS, Berry WL, Janknecht R, Chan JY, Plafker SM. A PGAM5-KEAP1-Nrf2 complex is required for stress-induced mitochondrial retrograde trafficking. J Cell Sci. 2017;130(20):3467–80. https://doi.org/10.1242/jcs.203216.
Jin X, Wang J, Gao K, Zhang P, Yao L, Tang Y, Tang L, Ma J, Xiao J, Zhang E, Zhu J, Zhang B, Zhao SM, Li Y, Ren S, Huang H, Yu L, Wang C. Dysregulation of INF2-mediated mitochondrial fission in SPOP-mutated prostate cancer. PLoS Genet. 2017;13(4):e1006748. https://doi.org/10.1371/journal.pgen.1006748.
Zhou Z, Song X, Wavelet CM, Wan Y. Cullin 4-DCAF proteins in tumorigenesis. Adv Exp Med Biol. 2020;1217:241–59. https://doi.org/10.1007/978-981-15-1025-0_15.
Kataoka K, Nakamura C, Asahi T, Sawamura N. Mitochondrial cereblon functions as a Lon-type protease. Sci Rep. 2016;6:29986. https://doi.org/10.1038/srep29986.
Hao BB, Li XJ, Jia XL, Wang YX, Zhai LH, Li DZ, Liu J, Zhang D, Chen YL, Xu YH, Lee SK, Xu GF, Chen XH, Dang YJ, Liu B, Tan MJ. The novel cereblon modulator CC-885 inhibits mitophagy via selective degradation of BNIP3L. Acta Pharmacol Sin. 2020. https://doi.org/10.1038/s41401-020-0367-9.
Yang KC, Chuang KW, Yen WS, Lin SY, Chen HH, Chang SW, Lin YS, Wu WL, Tsao YP, Chen WP, Chen SL. Deficiency of nuclear receptor interaction protein leads to cardiomyopathy by disrupting sarcomere structure and mitochondrial respiration. J Mol Cell Cardiol. 2019;137:9–24. https://doi.org/10.1016/j.yjmcc.2019.09.009.
Yin Y, Liu L, Yang C, Lin C, Veith GM, Wang C, Sutovsky P, Zhou P, Ma L. Cell autonomous and nonautonomous function of CUL4B in mouse spermatogenesis. J Biol Chem. 2016;291(13):6923–35. https://doi.org/10.1074/jbc.M115.699660.
Wang X, Li S, Liu L, Jian Z, Cui T, Yang Y, Guo S, Yi X, Wang G, Li C, Gao T, Li K. Role of the aryl hydrocarbon receptor signaling pathway in promoting mitochondrial biogenesis against oxidative damage in human melanocytes. J Dermatol Sci. 2019;96(1):33–41. https://doi.org/10.1016/j.jdermsci.2019.09.001.
Rico de Souza A, Zago M, Pollock SJ, Sime PJ, Phipps RP, Baglole CJ. Genetic ablation of the aryl hydrocarbon receptor causes cigarette smoke-induced mitochondrial dysfunction and apoptosis. J Biol Chem. 2011;286(50):43214–28. https://doi.org/10.1074/jbc.M111.258764.
Zhang S, Sun Y. Cullin RING ligase 5 (CRL-5): neddylation activation and biological functions. Adv Exp Med Biol. 2020;1217:261–83. https://doi.org/10.1007/978-981-15-1025-0_16.
Zhao Y, Xiong X, Sun Y. Cullin-RING ligase 5: functional characterization and its role in human cancers. Semin Cancer Biol. 2020. https://doi.org/10.1016/j.semcancer.2020.04.003.
Zhu Z, Wang L, Hao R, Zhao B, Sun L, Ye RD. Cutting edge: a cullin-5-TRAF6 interaction promotes TRAF6 polyubiquitination and lipopolysaccharide signaling. J Immunol. 2016;197(1):21–6. https://doi.org/10.4049/jimmunol.1600447.
Murata H, Sakaguchi M, Kataoka K, Huh NH. SARM1 and TRAF6 bind to and stabilize PINK1 on depolarized mitochondria. Mol Biol Cell. 2013;24(18):2772–84. https://doi.org/10.1091/mbc.E13-01-0016.
Zhao Y, Xiong X, Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(betaTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell. 2011;44(2):304–16. https://doi.org/10.1016/j.molcel.2011.08.029.
Antonioli M, Albiero F, Nazio F, Vescovo T, Perdomo AB, Corazzari M, Marsella C, Piselli P, Gretzmeier C, Dengjel J, Cecconi F, Piacentini M, Fimia GM. AMBRA1 interplay with cullin E3 ubiquitin ligases regulates autophagy dynamics. Dev Cell. 2014;31(6):734–46. https://doi.org/10.1016/j.devcel.2014.11.013.
Rosario FJ, Gupta MB, Myatt L, Powell TL, Glenn JP, Cox L, Jansson T. Mechanistic target of rapamycin complex 1 promotes the expression of genes encoding electron transport chain proteins and stimulates oxidative phosphorylation in primary human trophoblast cells by regulating mitochondrial biogenesis. Sci Rep. 2019;9(1):246. https://doi.org/10.1038/s41598-018-36265-8.
Ryu JH, Li SH, Park HS, Park JW, Lee B, Chun YS. Hypoxia-inducible factor alpha subunit stabilization by NEDD8 conjugation is reactive oxygen species-dependent. J Biol Chem. 2011;286(9):6963–70. https://doi.org/10.1074/jbc.M110.188706.
Essmann F, Pohlmann S, Gillissen B, Daniel PT, Schulze-Osthoff K, Janicke RU. Irradiation-induced translocation of p53 to mitochondria in the absence of apoptosis. J Biol Chem. 2005;280(44):37169–77. https://doi.org/10.1074/jbc.M502052200.
Zhuang J, Wang PY, Huang X, Chen X, Kang JG, Hwang PM. Mitochondrial disulfide relay mediates translocation of p53 and partitions its subcellular activity. Proc Natl Acad Sci USA. 2013;110(43):17356–61.
Abida WM, Nikolaev A, Zhao W, Zhang W, Gu W. FBXO11 promotes the Neddylation of p53 and inhibits its transcriptional activity. J Biol Chem. 2007;282(3):1797–804. https://doi.org/10.1074/jbc.M609001200.
Xirodimas DP, Saville MK, Bourdon JC, Hay RT, Lane DP. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell. 2004;118(1):83–97. https://doi.org/10.1016/j.cell.2004.06.016.
Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191(7):1367–80. https://doi.org/10.1083/jcb.201007013.
Um JW, Han KA, Im E, Oh Y, Lee K, Chung KC. Neddylation positively regulates the ubiquitin E3 ligase activity of parkin. J Neurosci Res. 2012;90(5):1030–42. https://doi.org/10.1002/jnr.22828.
Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer. 2014;14(11):709–21. https://doi.org/10.1038/nrc3803.
Nawrocki ST, Kelly KR, Smith PG, Espitia CM, Possemato A, Beausoleil SA, Milhollen M, Blakemore S, Thomas M, Berger A, Carew JS. Disrupting protein NEDDylation with MLN4924 Is a novel strategy to target cisplatin resistance in ovarian cancer. Clin Cancer Res. 2013;19(13):3577–90. https://doi.org/10.1158/1078-0432.CCR-12-3212.
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160(2):189–200. https://doi.org/10.1083/jcb.200211046.
Barbier-Torres L, Delgado TC, Garcia-Rodriguez JL, Zubiete-Franco I, Fernandez-Ramos D, Buque X, Cano A, Gutierrez-de Juan V, Fernandez-Dominguez I, Lopitz-Otsoa F, Fernandez-Tussy P, Boix L, Bruix J, Villa E, Castro A, Lu SC, Aspichueta P, Xirodimas D, Varela-Rey M, Mato JM, Beraza N, Martinez-Chantar ML. Stabilization of LKB1 and Akt by neddylation regulates energy metabolism in liver cancer. Oncotarget. 2015;6(4):2509–23. https://doi.org/10.18632/oncotarget.3191.
Song J, Herrmann JM, Becker T. Quality control of the mitochondrial proteome. Nat Rev Mol Cell Biol. 2020. https://doi.org/10.1038/s41580-020-00300-2.
Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, Harper JW, Gygi SP. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44(2):325–40. https://doi.org/10.1016/j.molcel.2011.08.025.
Funding
This work is supported by the National Key R&D Program of China 2016YFA0501800 and Chinese NSFC grant 31701167.
Author information
Authors and Affiliations
Contributions
QZ and YS had the idea for the manuscript. QZ and YZ drafted the manuscript. YS finalized the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have declared that no conflict of interest exists.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, Q., Zheng, Y. & Sun, Y. Neddylation regulation of mitochondrial structure and functions. Cell Biosci 11, 55 (2021). https://doi.org/10.1186/s13578-021-00569-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-021-00569-6