
Abstract
Disulfidptosis is a novel form of cell death that is distinguishable from established programmed cell death pathways such as apoptosis, pyroptosis, autophagy, ferroptosis, and oxeiptosis. This process is characterized by the rapid depletion of nicotinamide adenine dinucleotide phosphate (NADPH) in cells and high expression of solute carrier family 7 member 11 (SLC7A11) during glucose starvation, resulting in abnormal cystine accumulation, which subsequently induces andabnormal disulfide bond formation in actin cytoskeleton proteins, culminating in actin network collapse and disulfidptosis. This review aimed to summarize the underlying mechanisms, influencing factors, comparisons with traditional cell death pathways, associations with related diseases, application prospects, and future research directions related to disulfidptosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cells are the basic organizational units of life. Therefore, cell proliferation, differentiation, and death play important roles in different stages of life. Cell death is a fundamental physiological process in organisms and is crucial for maintaining the stability of the internal environment [1]. In recent years, disulfidptosis, a newly identified form of cell death, has garnered increasing attention. On February 6, 2023, the research group led by Professors Boyi Gan and Junjie Chen published their findings in Nature Cell Biology, unveiling the mechanism of disulfide stress-induced cell death and naming this new mode of cell death disulfidptosis [2].
Although the mechanism of disulfidptosis has not been fully elucidated, studies indicate that increased SLC7A11 protein expression is a critical factor in the occurrence of disulfidptosis. SLC7A11 is a transporter protein that is responsible for translocating cystine from the outside of the cell to the inside. Under conditions of glucose deprivation, increased expression of SLC7A11 results in substantial cystine accumulation, subsequently inducing disulfide stress and leading to cell death [2,3,4].
Recent studies have suggested that disulfidptosis plays a significant role in the onset and progression of various diseases. Associations have been observed with cancer [2, 5, 6], neurodegenerative diseases [7, 8], cardiovascular diseases [9], and liver diseases [10,11,12], and other conditions are closely related to disulfidptosis [13,14,15].
Consequently, comprehensive research on its mechanism holds significant clinical relevance, offering insights into the fundamental nature and principles of life and suggesting novel approaches for disease prevention and treatment.
Differences and relationships between disulfidptosis and traditional cell death modes
Disulfidptosis is a novel form of cell death that is distinguished from traditional cell death modes by its unique mechanism, morphological characteristics, and regulatory networks. Disulfidptosis primarily results from NADPH depletion in cells expressing high levels of SLC7A11 under conditions of glucose deprivation, leading to the abnormal accumulation of cystine and other disulfides and culminating in disulfide stress and rapid cell death [2, 16]. Morphologically, disulfidptosis is characterized by an increase in disulfide bond levels within the cytoskeleton, leading to actin filament contraction, disruption of cytoskeletal integrity, and consequent cell death [2].
The traditional modes of cell death include apoptosis, pyroptosis, autophagy, ferroptosis, and oxeiptosis. Programmed processes are triggered by specific signaling pathways and regulatory networks. For instance, apoptosis caspase activation [17,18,19], pyroptosis is initiated by specific signaling pathways involved in inflammatory responses [20,21,22,23], and autophagy is initiated by autophagosome formation [24,25,26].
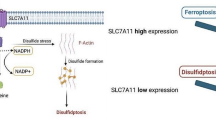
Although disulfidptosis and traditional cell death are distinct, they are not entirely separate. Interactions and connections likely exist between them. For example, disulfidptosis and ferroptosis are connected (Table 1). SLC7A11 is a specific cysteine transporter and a key regulatory protein associated with ferroptosis and disulfidptosis [2]. Downregulation of SLC7A11 indirectly inhibits the activity of glutathione peroxidase 4 (GPX4) by suppressing the cysteine metabolic pathway, leading to reduced intracellular cysteine levels and the depletion of glutathione (GSH) biosynthesis, which in turn leads to the accumulation of lipid peroxides and ultimately induces cell death by ferroptosis [27] and inhibits the occurrence of disulfidptosis [2, 3]. Upregulating SLC7A11 can indirectly promote the activity of GPX4 by promoting the cysteine metabolic pathway, leading to increased intracellular cysteine levels and GSH biosynthesis, which in turn inhibits cell death by ferroptosis. If cystine accumulates at this time, disulfidptosis may occur [13].

The core molecular mechanisms of disulfidptosis. When the NADPH supply is limited under glucose deprivation conditions, high cystine uptake by cells with high SLC7A11 expression results in intracellular NADPH depletion, the excessive accumulation of cystine and other disulfide molecules, and abnormal disulfide bond formation in actin cytoskeleton proteins, culminating in actin network collapse and disulfidptosis. Rac1-WRC-mediated branched actin polymerization and lamellipodia formation likely provide supporting conditions for disulfide bond formation in actin cytoskeleton proteins, thereby facilitating disulfidptosis. Abbreviations: SLC7A11, solute carrier family 7 member 11; SLC3A2, solute carrier family 3 member 2; GLUT1/4, glucose transporter 1/4; PPP, pentose phosphate pathway; NADPH, nicotinamide adenine dinucleotide phosphate; GSH, glutathione; GSSG, oxidized glutathione; GPX4, glutathione peroxidase 4; Arp2/3, actin-related protein 2/3 complex; RAC1, RAS-related C3 botulinum toxin substrate 1; WRC, WAVE regulatory complex; HSPC300, hematopoietic stem/progenitor cell protein 300; NCKAP1, NCK-associated protein 1; CYFIP1, cytoplasmic FMR1-interacting protein 1

The core molecular mechanisms of alkaliptosis. The molecular mechanisms underlying alkaliptosis involve pH-induced alkalization, mainly through the activation of JTC801, and the downregulation of carbonic anhydrase 9 (CA9), which is dependent on the IKBKB-NF-κB pathway and can induce alkaliptosis. Abbreviations: IKBKB, inhibitor of nuclear factor κB kinase subunit-β; CHUK, component of inhibitor of nuclear factor kappa B kinase complex; IKBKG, inhibitor of nuclear factor kappa B kinase regulatory subunit gamma; NF-κBIA, NF-κB inhibitor alpha; RELA, RELA proto-oncogene, NF-κB subunit; NF-κB1, nuclear factor kappa B subunit 1; CA9, carbonic anhydrase 9

The core molecular mechanisms of oxeiptosis. High intracellular ROS levels induce conformational changes in KEAP1, triggering its dissociation from NRF2. KEAP1 translocates into mitochondria and mediates the release of AIFM1 from PGAM5 and AIFM1 translocation to the nucleus, where it dephosphorylates AIFM1 at S116, triggering cell death. Abbreviations: KEAP1, Kelch-like ECH-associated protein-1; NRF2, Nuclear factor erythroid 2-related factor 2; PGAM5, phosphoglycerate mutase family 5; AIFMI, apoptosis-inducing factor mitochondria associated 1

The core molecular mechanisms of autophagy. Autophagy can be divided into three stages: the induction stage, the formation stage of autophagosomes, and the formation of autophagic lysosomes and the degradation of their contents. 1. Autophagy induction stage: The ULK1-Atg13-Atg101-FIP200 complex transmits autophagy signals to the nucleus, and the Class III PI3K-Beclin-1 complex induces the formation of a double-layer membrane, leading to the accumulation of phagocytic vesicles. 2. Formation of the autophagosome: On the one hand, Atg7 and Atg10 activate and transport Atg12, which in turn binds to Atg5 and Atg16 to form the Atg5-Atg12-Atg16 complex. On the other hand, LC3 is decomposed by Atg4 to form LC3-I, which is activated by Atg7 and Atg3 and accumulates on the autophagosome membrane under the induction of the Atg5-Atg12-Atg16 complex to form LC3-II. The membrane extends and surrounds the intracellular degradation substrate to form an autophagosome. 3. Formation of autophagosomes and degradation of their contents: Through the interaction of LAMP-1, LAMP-2, GTPase-RAB-7, and other proteins, autophagosomes fuse with lysosomes to form autophagosomes, which release hydrolytic enzymes to fully degrade substrates. Abbreviations: Atg, Autophagy-related gene; LC3, microtubule-associated protein light-chain 3; PI3K, phosphoinositide 3-kinase; LAMP, lysosome-associated membrane protein; GTPase, GTP hydrolase; RAB-7, ras-related protein Rab-7

The core molecular mechanisms of ferroptosis. PUFAs are oxidized in a stepwise manner into lipid hydroperoxides (PUFAs-OH) by ACSL4, LPCAT3 and PUFA-OOH by LOX. GPX4 uses GSH as a substrate to catalyze the transformation of lipid hydroperoxides into hydroxy derivatives, limiting lipid peroxidation. System Xc- inhibition causes GSH depletion and attenuates GPX4 activity, leading to lipid peroxidation and ferroptosis. Moreover, iron overload generates hydroxyl radicals via the Fenton reaction, which also contributes to lipid peroxidation and ferroptosis. Abbreviations: SLC38A1, solute carrier family 38 member 1; SLC1A5, solute carrier family 1 member 5; SLC3A2, solute carrier family 3 member 2; SLC7A11, solute carrier family 7 member 11; Glucose Transporter 1/4; FPN1, ferroportin-1; DMT1, divalent metal transporter 1; TF, transferrin; CP, ceruloplasmin; TFR1, transferrin receptor 1; NFE2L2, nuclear factor erythroid 2-related factor 2; PPP, pentose phosphate pathway; NADPH, nicotinamide adenine dinucleotide phosphate; GSH, glutathione; GPX4, glutathione peroxidase 4; GSSG, oxidized glutathione; ALOX15, arachidonic acid 15-lipoxygenase; SAT1, spermidine/spermine N1-acetyltransferase 1; P53, tumor protein 53; HO-1, heme oxygenase-1; NRF2, nuclear factor erythroid 2-related factor 2; Keap1, kelch-1ike ECH-associated protein 1; P62, prostacyclin; NCOA4, nuclear receptor coactivator 4; IREB2, iron-responsive element binding protein 2; Atg5/7, autophagy related 5/7

The core molecular mechanisms of parthanatos. The activation of NMDA receptors stimulates NO synthase (nNOS). The abundant levels of NO and superoxide spontaneously generate peroxynitrite (ONOO−). Along with other ROS, this strong prooxidant damages DNA strands and thereby causes the activation of PARP-1. When DNA damage is high, the consequent overactivation of PARP-1 leads to abundant PAR polymer formation in the nucleus; some of the PARy lated carrier proteins exit the nucleus and cause the release of AIF from a pool on the outer mitochondrial membrane. Once in the cytosol, AIF can bind to MIF. Together, these proteins enter the nucleus and cause large-scale DNA degradation and cell death. Abbreviations: ROS, reactive oxygen species; NO, nitric oxide; NMDA, N-methyl-D-aspartic acid receptor; nNOS, nitric oxide synthase; ONOO−, peroxynitrite; PARP-1, poly-ADP-ribosome-polymerase 1; NAD+, nicotinamide adenine dinucleotide; PAR, poly-ADP-ribose; AIF, apoptosis-inducing factor; MIF, macrophage migration inhibitory factor

The core molecular mechanisms of necroptosis. FASL, TRAIL, TNF and IFN-1 activate each of their receptors, and MLKL, RIPK1 and RIPK3 are recruited to assemble the necrosome through phosphorylation. Phosphorylation-mediated activation of MLKL and subsequent MLKL-mediated membrane pore formation result in necroptosis. In response to TNF-α-induced necroptosis, PGAM5 is recruited to the RIPK1/RIPK3 complex on the outer mitochondrial membrane, where it triggers Drp1-mediated mitochondrial fragmentation and the release of large amounts of ROS, thereby activating PARP-1 and resulting in a decrease in NAD+ production and subsequent cycling, which is considered an obligatory step in necroptosis. Abbreviations: FASL, factor-related apoptosis ligand; TRAIL, TNF-related apoptosis-inducing ligand; TNF, tumor necrosis factor; IFN-1, interferon-1; MLKL, mixed lineage kinase domain-like protein; RIPK1, receptor-interacting protein kinase 1; RIPK3, receptor-interacting protein kinase 3; PGAM5, phosphoglycerate mutase family 5; KEAP1, Kelch-like ECH-associated protein 1; NRF2, nuclear factor erythroid 2 related factor 2; PARP-Q, poly-ADP-ribose polymerase; NAD+, nicotinamide adenine dinucleotide

The core molecular mechanisms of NETosis. NETosis is initiated by the activation of neutrophils via PRRs and the subsequent influx of Ca2+. This triggers a cascade of events involving Ca2+−dependent PKC, the PKC-MEK-ERK pathway, and NOX phosphorylation, leading to ROS production. Excessive ROS cause cytoplasmic granule degradation and the release of NE, MPO and PAD4 to the nucleus, leading to chromatin decondensation and ultimately resulting in cell rupture and NET release. PKA-mediated phosphorylation of NOXA1 recruits 14–3-3 proteins, which block the assembly of the NOX1 holoenzyme, ultimately preventing ROS production. Moreover, supraphysiological cAMP concentrations inhibit the formation of NETs and ROS bursts. These findings highlight the role of cAMP signaling in inhibiting NETosis via PKA. Abbreviations: PRRs, pattern recognition receptors; PKC, protein kinase C; MEK, mitogen-activated extracellular signal-regulated kinase; ERK, extracellular regulated protein kinase; NADPH, nicotinamide adenine dinucleotide phosphate; NOX, NADPH oxidase; NE, neutrophil elastase; MPO, myeloperoxidase; PAD4, peptidyl arginine deiminase 4; NETs, neutrophil extracellular traps; PKA, protein kinase A; NOXA1, NADPH oxidase activator 1; NOX1, NADPH oxidase 1; cAMP, cyclic adenosine monophosphate

The core molecular mechanisms of pyroptosis. In response to DAMPs and PAMPs, cytosolic canonical inflammasomes (NLRP3, NLRP1, NLRC4, AIM2, pyrin, etc.) can respond to microbial infection (microbial toxins, etc.) or danger signals (dsDNA and crystals, etc.) to activate caspase-1, while noncanonical inflammasomes directly respond to LPS or other stimuli to activate caspase-4/5/11. After the activation of inflammatory caspases, pro-IL-1β, pro-IL-18, and GSDMD are cleaved to liberate N-terminal GSDMD (GSDMD-N), which forms pores on the plasma membrane and releases inflammatory mediators (IL-1β, IL-18, etc.). Other pathways involved in pyroptosis include the activation of caspase-3, caspase-8 and caspase-9 and the cleavage of gasdermin E, B and C (GSDME, GSDMB, and GSDMC, respectively). GSDMC is cleaved by caspase-8 and transcriptionally upregulated under hypoxic conditions through the interaction of pSTAT3 with programmed death-ligand 1. The amino-terminal PFD of gasdermin N then interacts with the plasma membrane, and 16 monomers oligomerize to form a gasdermin pore. The diameter of these pores is estimated to be in the range of 10–15 nm, which is large enough to release small proteins, including mature IL-1β (4.5 nm diameter), probably at a slow rate. Furthermore, sodium enters the cell, bringing water into the cell, which causes the cell volume to increase. This process can rapidly exceed the capacity of the membrane, resulting in membrane rupture. In response to membrane rupture, all the remaining soluble cytosolic contents are released so rapidly that it is essentially instantaneous, resulting in pyroptosis. Abbreviations: dsDNA, double-stranded DNA; PAMPs, pathogen-associated molecular patterns; DAMPs, damage-associated molecular patterns; NLRP1/3/4, NLR family pyrin domain-containing 1/3/4; AIM2, absent in melanoma 2; ER, endoplasmic reticulum; LPS, lipopolysaccharide; GSDM B/C/D/E, gasdermin B/C/D/E; IRAK-1/4, interleukin receptor associated kinase 1/4; TRAF-6, tumor necrosis factor receptor-associated factor 6; IL-1β, interleukin-1β; MCP-1, monocyte chemoattractant protein-1; ICAM-1, intercellular cell adhesion molecule-1; VCAM-1, vascular cell adhesion molecule-1; pSTAT3, phospho-signal transducer and activator of transcription 3

The core molecular mechanisms of lysosome-dependent cell death. Lysosome-dependent cell death is triggered by ROS or other stimuli. A surge of ROS is one of the main triggers of the increase in calcium, which can occur through hyperactivation of TRPM2 and calcium efflux from lysosomes, leading to LMP and the release of cathepsins into the cytosol. Cathepsins catalyze the formation of multiple substrates, including Bid and apoptotic proteins, and initiate caspase-dependent cell death. Lysosome-dependent cell death occurs through a process involving Ca2+−dependent ADCY1, followed by an increase in cAMP and ultimately the inhibition of lysosomal acid SMase. In addition, ER stress can induce an increase in cytosolic Ca2+. High cytosolic calcium stimulates the activation of calpain, leading to the degradation of lysosomal membrane proteins such as LAMP1/2, which causes lysosomes to rupture, resulting in lysosome-dependent cell death. Abbreviations: TRPM2, transient receptor potential melastatin 2; LMP, lysosomal membrane permeabilization; cAMP, cyclic adenosine monophosphate; ADCY1, adenylate cyclase 1; SMase, sphingomyelinase; ER, endoplasmic reticulum; LAMP1/2, lysosome-associated membrane protein 1/2; Bid, BH3-interacting domain death agonist
Consequently, a comprehensive understanding of the interplay between disulfidptosis and traditional cell death modes is critical for unraveling the complexity and diversity of cell death mechanisms.
The mechanism of disulfidptosis
The fate and function of cells are influenced by environmental and genetic factors. One of the most critical factors that determines cell fate is redox homeostasis. Oxidative stress can produce reactive oxygen species (ROS). Cells experiencing excessive ROS must synthesize protective molecules such as glutathione to mitigate damage [62]. Glutathione synthesis requires cysteine [63], which is typically sourced from the extracellular environment via the cystine/glutamate antiporter (System XC-), which imports cystine [64]. System XC-, which is a Na+-dependent amino acid antiporter embedded in the phospholipid bilayer of cells, is composed of a heterodimer of the light chain SLC7A11 and the heavy chain solute carrier family 3 member 2 (SLC3A2) [65]. Concurrent with the uptake of one cystine molecule, one glutamate molecule is exported. Blocking cystine uptake triggers ferroptosis, an iron-dependent form of cell death characterized by phospholipid peroxidation, especially the peroxidation of polyunsaturated fatty acids, resulting in widespread plasma membrane abnormalities.
The maintenance of proper redox homeostasis is critical for cell survival [66]. During oxidative stress, the body protects cells by upregulating SLC7A11, facilitating the uptake of substantial amounts of cystine [67]. Within the cell, cystine, which has low solubility, is initially reduced to highly soluble cysteine, which involves NADPH [16, 28]. Subsequently, gamma-glutamylcysteine synthetase and glutathione synthetase catalyze the synthesis of glutathione, eliminating excessive ROS and reducing oxidative stress [68, 69]. Glutathione is pivotal for preventing oxidative stress, mitigating lipid peroxidation reactions, and protecting cells [63]. This process requires an adequate glucose supply, enabling sufficient NADPH production via the pentose phosphate pathway (PPP) for the timely reduction of insoluble cystine to soluble cysteine, which is crucial for glutathione biosynthesis.
Glucose starvation and NADPH depletion
Glucose starvation, a condition in which cells experience a scarcity of glucose, disrupts the equilibrium of cellular metabolism with far-reaching consequences. This state not only affects energy production through glycolysis but also has profound implications for anabolic processes, redox balance [70], and antioxidant defense mechanisms, primarily due to its impact on the PPP [71]. The PPP, is a vital metabolic pathway that branches off from glycolysis. It plays a critical role in multiple cellular functions beyond energy generation, including the synthesis of nucleotides, pentoses for nucleic acids, and the provision of reducing equivalents in the form of NADPH [72].
NADPH is crucial for maintaining the cellular redox potential, essential for fatty acid synthesis, cholesterol biosynthesis, and perhaps most importantly, it serves as a cofactor for antioxidants like glutathione, which neutralize ROS and protect cells from oxidative stress [73]. The PPP serves as the primary source of NADPH in animal cells [74,75,76,77]. During glucose starvation, the PPP is inhibited, leading to a decrease in NADPH production and rapid depletion. Reduced NADPH levels compromise the ability of cells to regenerate reduced GSH from its oxidized form (GSSG), thereby weakening the cellular antioxidant defense system [78]. This can lead to an accumulation of ROS, causing oxidative damage to lipids, proteins, and DNA, ultimately compromising cell viability and function [79]. Impaired Lipid and Cholesterol Synthesis.
The balance between oxidants and antioxidants in the cell, crucial for maintaining homeostasis, is disrupted due to decreased NADPH. This redox imbalance can disturb protein function, modulate signaling pathways, and trigger programmed cell death. To cope with glucose starvation, cells may activate alternative metabolic routes to generate ATP and maintain redox balance, such as upregulated fatty acid oxidation, amino acid catabolism, or activating autophagy to recycle intracellular components [80]. However, these adaptive mechanisms may not fully compensate for the loss of PPP activity, especially regarding NADPH production, highlighting the importance of glucose as a fundamental energy and biosynthetic substrate for cellular homeostasis.
SLC7A11 as a dual-edged sword in redox regulation
SLC7A11 plays a crucial role in cellular processes. By serving as a transmembrane protein, it regulates the balance between cystine and glutamate and is intrinsically linked to the maintenance of mitochondrial function [81]. Decreased SLC7A11 expression leads to inadequate cystine uptake, inhibiting timely ROS clearance and culminating in ferroptosis [29, 82]. Conversely, increased SLC7A11 expression results in excessive cystine absorption, which is cytotoxic and can trigger disulfidptosis [2]. Concurrently, extensive glutamate export decreases intracellular glutamate levels, reducing mitochondrial membrane potential, causing mitochondrial swelling and dissolution, and impairing mitochondrial function [83]. Consequently, SLC7A11 expression critically regulates cellular redox status and mitochondrial function, requiring precise modulation to maintain normal cellular physiology.
A decrease in SLC7A11 expression induces ferroptosis
Maintaining proper redox homeostasis is critical for cell survival [66]. Oxidative stress leads to the production of large amounts of ROS, which can hinder normal cell growth and differentiation or even lead to cell death [84]. Therefore, maintaining sufficient glutathione (GSH) levels to neutralize excess ROS and stabilize the intracellular environment is essential [85].
GSH synthesis depends on cysteine, which is derived from cystine and enters the cell from the extracellular milieu through SLC7A11. SLC7A11-mediated GSH synthesis is vital for the cellular antioxidant defense and the maintenance of intracellular stability [86]. Reduced SLC7A11 expression on the cell membrane leads to insufficient cystine uptake, causing a decrease in cysteine availability and, subsequently, a decrease in GSH synthesis [87,88,89]. This results in reduced activity of glutathione peroxidase-4 (GPX4), the only enzyme capable of efficiently reducing lipid peroxides in biological membranes, which requires glutathione as a cofactor [90, 91]. The inability to reduce lipid peroxides in a timely manner leads to the oxidation of intracellular Fe2+, which generates large amounts of lipid radicals and ROS through the Fenton reaction: Fe2+ + H2O2 → Fe3+ + (OH)− + ·OH, Fe3+ + O2− → Fe2+ + O2 [92]. Hydroxyl radicals (·OH) can attack assault polyunsaturated fatty acids embedded in the cellular membrane, instigating a self-amplifying cascade of lipid peroxidation [93]. The concentration of iron ions is meticulously governed by a cohort of iron metabolism-associated proteins, notably the iron export protein (FPN) [94], the iron regulatory hormone (Hepcidin) [95], and the iron storage molecule (Ferritin) [96].
The abundance of polyunsaturated fatty acids in the cell and plasma membranes increases susceptibility to lipid radical-induced cascade reactions, decreasing membrane thickness and compromising barrier function. ROS further accelerate damage, forming protein pores on the cell membrane and destabilizing the intracellular environment. Concurrently, lipid radicals damage the cellular lipid structure, and the resulting peroxidation products (4-hydroxy-nonenal, malondialdehyde) continue to react, perpetually damaging the cell. Ultimately, this leads to irreversible damage to the structure and function of the cell and plasma membrane, culminating in ferroptosis. SIRT1 is a NAD+-dependent deacetylase that plays a central role in cellular responses to metabolic stress and aging. SIRT1 has been shown to positively regulate SLC7A11 expression, thereby enhancing cystine uptake and GSH synthesis, and contributing to ferroptosis resistance [97, 98]. This finding underscores the indispensable role of SLC7A11 in cell survival.
Cells with high SLC7A11 expression under glucose starvation undergo disulfidptosis
SLC7A11 is recognized for its ability to neutralize excess ROS, thereby promoting cell survival under normal conditions [99]. However, research led by Professor Boyi Gan revealed that under glucose starvation conditions, SLC7A11 overexpression paradoxically leads to cell death [16]. This type of cell death has been classified as disulfide stress-induced cell death and is a novel form of programmed cell death defined by stress from disulfide bonds [2]. These findings underscore the complex role of SLC7A11 in the regulation of cellular redox homeostasis and the balance between cell survival and death.
Specifically, cystine, which is an amino acid with very low solubility, can become highly toxic when it accumulates in the cytoplasm [16, 100]. Consequently, cells with high SLC7A11 expression must rapidly convert cystine to the more soluble cysteine in the cytoplasm. This reduction is dependent on NADPH, which is produced via the PPP of glucose metabolism [30, 66]. Under glucose starvation conditions or when glucose uptake is insufficient, NADPH production via the PPP is decreased, inhibiting the prompt conversion of cystine to cysteine and potentially leading to the accumulation of abnormal disulfide bonds, such as cystine, within the cell. This abnormal accumulation can trigger disulfide stress, consequently inducing disulfidptosis [2].
Additionally, studies indicate that 2-deoxy-D-glucose (2-DG) can prevent the death of SLC7A11-overexpressing cells under glucose starvation conditions [101]. 2-DG, which is a glucose analog, inhibits glycolysis and can be diverted to the PPP to produce NADPH. This finding suggested that the protective effect of 2-DG against the death of SLC7A11-overexpressing cells under glucose-deprived conditions may involve its ability to supply NADPH for cysteine reduction.
Formation of disulfide bonds and disulfide stress
Disulfide bonds are common chemical bonds in protein molecules and are crucial in the protein folding process [102]. These bonds significantly impact protein stability and function. Improper formation or disruption of disulfide bonds can lead to abnormal protein structure, thereby affecting protein function. In pathological conditions such as cancer or neurodegenerative diseases, abnormal disulfide bonds can result in protein dysfunction, subsequently impacting cell survival [103].
In cases of glucose starvation, NADPH depletion coupled with high SLC7A11 expression leads to increased cystine uptake but inhibits timely cystine conversion to cysteine, resulting in the accumulation of cystine and other disulfides and triggering disulfide stress [2, 16]. Activation of the Ras-related C3 botulinum toxin substrate 1 (Rac1)-WAVE regulatory complex (WRC)-actin-related protein 2/3 (Arp2/3) signaling pathway occurs, leading to abnormal disulfide bonds in actin cytoskeletal proteins. These disulfide bonds result in F-actin fiber aggregation, causing damage to the cytoskeletal structure, the loss of cell function, and ultimately cell death.
F-actin fibers are protein fibers within the cytoskeleton that provide support and facilitate movement [104]. Aggregation of these fibers damages cytoskeletal structure and leads to the loss of cell function and eventually cell death. This cell death can be mitigated by inhibiting SLC7A11 or by using reducing agents such as dithiothreitol (DTT), β-mercaptoethanol (2ME), and tris-(2-carboxyethyl)-phosphine (TCEP), which prevent disulfide stress, but not by traditional cell death inhibitors, including ferroptosis inhibitors, apoptosis inhibitors, necroptosis inhibitors, autophagy inhibitors, or ROS scavengers. This finding indicates that cell death may be mediated by SLC7A11-induced cystine accumulation and subsequent disulfide stress [2].
F-actin contraction during disulfidptosis
Phalloidin staining was performed by Boyi Gan [2] and revealed significant morphological changes, such as cell and F-actin contraction and F-actin detachment from the plasma membrane, in SLC7A11-overexpressing cells under glucose starvation conditions. Furthermore, glucose starvation-induced actin cytoskeletal remodeling depended on SLC7A11 and could be reversed by cysteine deprivation, 2-DG, or 2ME treatment but not by treatment with ROS scavengers (Tempol or Trolox) [2]. This finding suggests that disulfidptosis is related to the formation of abnormal disulfide bonds in the cytoskeletal protein F-actin and the contraction and detachment of F-actin from the plasma membrane.
The Rac1-WRC-Arp2/3 signaling pathway regulates disulfidptosis
Research indicates that genes and proteins such as NCK-associated protein 1 (NCKAP1), the WRC complex, and Rac1 are crucial for disulfidptosis [2, 105]. The WRC complex, which functions as a downstream effector of the small GTPase Rac, activates Arp2/3, leading to F-actin polymerization and podosome formation. NCKAP1, which is a component of the WRC complex, influences glucose starvation-induced disulfide bond formation and F-actin contraction and detachment from the plasma membrane; its deletion reduces disulfidptosis in UMRC6 cells, and its overexpression promotes disulfidptosis.
Rac1, which is a key GTPase that activates the WRC complex, enhances podosome formation and disulfidptosis in SLC7A11-overexpressing cells [2, 106]. Rac1-WRC-mediated podosome formation can promote disulfidptosis through the F-actin network in podosomes, which is a critical target for disulfide bonding between actin cytoskeletal proteins.
This finding highlights the significant role of the Rac1-WRC-Arp2/3 signaling pathway in disulfidptosis, contributes to a deeper understanding of cell death mechanisms, and suggests new targets for disease treatment.
Other regulatory molecules and pathways related to disulfidptosis
The oxidation‒reduction status and the formation and breakage of disulfide bonds are pivotal factors that regulate disulfidptosis [107]. Various factors, including intracellular and extracellular environments and metabolic states, can affect cellular redox status, thereby influencing disulfide bond dynamics. Disulfidptosis involves sulfur oxidases and sulfatases, which impact the cellular redox state [9]. Additionally, proteins such as glyceraldehyde-3-phosphate dehydrogenase (GAPDH), thioredoxin (Trx), and peroxiredoxin (Prx) regulate disulfidptosis [108–107]. Furthermore, signaling pathways such as the NF-κB pathway [111, 112] and the JNK receptor pathway [113] are instrumental in disulfidptosis. These pathways affect disulfidptosis by regulating intracellular redox levels, protein expression and function. Since SLC7A11 is not essential in normal tissues but is highly expressed in multiple cancers, including lung [114, 115] and kidney [16, 116] cancers, it represents a promising target for novel cancer therapies. Targeting SLC7A11, glucose transport or the PPP, inducing disulfidptosis, and killing cancer cells are potential treatment strategies. Understanding the proteins and signaling pathways involved in disulfidptosis is crucial for developing cancer therapies.
Potential application prospects of disulfidptosis
Following the discovery of disulfidptosis, extensive research has been conducted to determine its role in a range of physiological and pathological conditions. Preliminary studies suggest that disulfidptosis plays a role in the pathogenesis of diseases including cancer [2, 5, 6, 114,115,116], neurodegenerative diseases [7, 8], cardiovascular diseases [9], and liver diseases [10,11,12]. For instance, in cancer cells, altered metabolism often leads to glucose insufficiency, which can trigger disulfidptosis in cells with high SLC7A11 expression [2, 5, 6, 114,115,116]. Similarly, pathological changes related to disulfidptosis have been observed in neurodegenerative diseases such as Alzheimer's disease [7, 8].
Disulfidptosis has also emerged as a focal point in drug development [117]. Certain drugs can induce or inhibit disulfidptosis by modulating SLC7A11 expression or disulfide concentrations. For example, some chemical agents can target tumor cells by inducing disulfidptosis [118, 119]. Additionally, small molecule inhibitors [31] and gene therapy techniques [120] to regulate disulfidptosis are being explored.
In therapeutic contexts, inducing disulfidptosis in cancer cells can effectively eliminate these cells [121]. Modulating immune system functions can prevent autoimmune diseases [13,14,15]. Moreover, targeting disulfidptosis pathways may lead to treatment strategies for other diseases caused by oxidative stress imbalances, such as neurodegenerative [7, 8] and inflammatory diseases [9].
In conclusion, the study of disulfidptosis would not only advance our understanding of cell death mechanisms but also open new avenues for disease prevention and treatment.
Potential application prospects of disulfidptosis in cancer treatment
Some studies have demonstrated that disulfidptosis is intimately related to the occurrence and development of cancer [2, 5, 6, 114,115,116]. Initially, the expression level of SLC7A11 in certain cancer cells was markedly higher than that in normal cells. This could be attributed to the high expression of SLC7A11, which could promote the proliferation and metastasis of cancer cells [122]. Furthermore, research indicates that high SLC7A11 expression may correlate with cancer drug resistance [123,124,125]. This could be due to the high expression of SLC7A11, which enhances the metabolic activity of cancer cells, thus conferring resistance to the toxic effects of certain chemotherapeutic drugs.
Research has revealed that the occurrence of disulfidptosis is closely related to the treatment and prognosis of cancers [121, 126, 127]. For instance, certain anticancer drugs might induce cancer cells to undergo disulfidptosis, thus inhibiting cancer growth and metastasis [121]. Additionally, the occurrence of disulfidptosis may be associated with cancer immune escape [126].
In conclusion, disulfidptosis is fundamentally related to the occurrence and development of cancer, and research on disulfidptosis in cancer treatment holds significant importance and value. By conducting comprehensive research on the regulatory mechanisms and applications of disulfidptosis, it is possible to generate new ideas and methods for cancer treatment.
Potential application prospects of disulfidptosis in the treatment of neurodegenerative diseases
Recent studies indicate that disulfidptosis is also related to neurodegenerative diseases [7, 8]. In certain neurodegenerative diseases, the expression level of SLC7A11 is markedly higher than that in normal cells [118]. This could be attributed to the high expression of SLC7A11, which enhances the metabolic activity of neurons, leading to resistance to certain forms of damage; further research shows that high expression of SLC7A11 may also promote neuronal apoptosis and necrosis [128], contributing to the occurrence and development of neurodegenerative diseases.
Research has revealed that the occurrence of disulfidptosis is related to the treatment and prognosis of neurodegenerative diseases [9]. For instance, certain drugs can inhibit the progression of neurodegenerative diseases by inducing neuronal disulfidptosis. Additionally, research has suggested that the occurrence of disulfidptosis may be linked to immune escape mechanisms in neurodegenerative diseases [7].
Alzheimer's disease, which is a prevalent neurodegenerative condition, has a complex pathogenesis that has not been fully elucidated. Recent studies suggest that disulfidptosis may play a role in the occurrence and development of Alzheimer's disease [7, 8, 32]. Furthermore, a study revealed that high SLC7A11 expression could contribute to neuronal death [129], thus influencing the occurrence and development of Alzheimer's disease. Moreover, evidence indicates that the occurrence of disulfidptosis is related to the treatment and prognosis of Alzheimer's disease [7].
Research on targeting disulfidptosis to treat neurodegenerative diseases has emerged as a pivotal area in neuroscience. Comprehensive investigations of the regulatory mechanisms and applications of disulfidptosis could yield new insights and approaches for treating neurodegenerative diseases.
Potential application prospects of disulfidptosis in cardiovascular disease treatment
The application prospects of disulfidptosis in treating cardiovascular diseases have garnered considerable attention. Recent studies have indicated that the SLC7A11 protein is intimately linked to cardiovascular diseases such as myocardial ischemia‒reperfusion [130, 131], myocardial infarction [132, 133], and myocardial hypertrophy [134] and that its association with disulfidptosis is significant. Consequently, further investigations are needed to determine the relationships between disulfidptosis and cardiovascular diseases, such as myocardial ischemia‒reperfusion, myocardial infarction, and myocardial hypertrophy.
In conclusion, the application prospects of disulfidptosis in treating cardiovascular diseases hold substantial importance and value. Comprehensive investigations of the regulatory mechanisms and applications of disulfidptosis could yield new insights and approaches for treating cardiovascular diseases.
Potential application prospects of disulfidptosis in other diseases
In addition to its role in tumors, neurodegenerative diseases, and cardiovascular diseases, disulfidptosis has also been linked to other conditions, such as diabetes [135] and autoimmune diseases [13]. The mechanism of disulfidptosis is incompletely understood, and further research on its regulatory mechanisms and applications could lead to the identification of new approaches for treating diabetes and autoimmune diseases.
The prospects and outlook of targeting disulfidptosis
The role and significance of disulfidptosis, which is a novel form of programmed cell death, in various diseases has been the subject of intensive study and exploration. There are several future research directions and application prospects for targeting disulfidptosis.
In-depth study of the mechanism and regulatory network of disulfidptosis
Under conditions of limited NADPH availability due to glucose deprivation, cells with elevated SLC7A11 expression face a serious challenge. Their heightened cystine uptake leads to dual consequences: the depletion of intracellular NADPH reserves and an excessive accumulation of cystine alongside other disulfide-containing molecules. This surplus disrupts normalcy by instigating aberrant disulfide bond formation within actin cytoskeleton proteins, ultimately causing the collapse of the actin network and a state referred to as disulfidptosis [2,3,4].
The process is also tightly controlled by molecules like GAPDH, Trx, and Prx [108,109,110], and pathways such as NF-κB [111, 112] and JNK [113], which regulate redox levels, protein synthesis, and function. These interactions reveal a complex regulatory network underlying disulfidptosis, with profound impacts on cellular health and disease susceptibility.
Presently, many factors that are involved in the mechanism of disulfidptosis have yet to be identified, requiring further research to elucidate the specific molecular mechanism and regulatory network.
Comparison of disulfidptosis with other forms of cell death
Disulfidptosis, a unique form of cell stress and death characterized by the abnormal accumulation of disulfide-bonded proteins and actin cytoskeleton collapse, stands apart from more conventionally recognized modes of cell death such as apoptosis, necrosis, autophagy, and ferroptosis. Here's a comparison highlighting the distinctive features of disulfidptosis against these other cell death processes:
Apoptosis
Often described as programmed cell death, apoptosis is a regulated process marked by cell shrinkage, membrane blebbing, chromatin condensation, and DNA fragmentation [136]. It plays a crucial role in development, tissue homeostasis, and immune function. Unlike disulfidptosis, apoptosis does not typically involve oxidative stress-induced protein aggregation or actin network dysfunction as central features.
Necrosis
Necrosis is an uncontrolled and accidental cell death usually triggered by severe physical or chemical insults. It is characterized by swelling of organelles, plasma membrane rupture, and inflammation due to cellular content release [137]. While both necrosis and disulfidptosis can be induced by oxidative stress, necrosis lacks the specific disulfide bond abnormalities and actin cytoskeleton collapse seen in disulfidptosis.
Autophagy
Autophagy is a lysosome-dependent degradation process that cells use to recycle damaged organelles and long-lived proteins [138]. It serves as a survival mechanism during starvation but can also contribute to cell death when overly activated or dysregulated. Unlike disulfidptosis, autophagy involves vesicular sequestration of cytoplasmic components rather than direct protein misfolding and aggregation due to disulfide bond anomalies.
Ferroptosis
Ferroptosis is a form of regulated cell death driven by iron-dependent lipid peroxidation. It is characterized by the accumulation of toxic lipid ROS (reactive oxygen species) and membrane damage [139]. Although both ferroptosis and disulfidptosis involve oxidative stress, they differ in that ferroptosis specifically targets lipids, whereas disulfidptosis centers around protein misfolding and aggregation due to disulfide bond imbalances.
In summary, while all these forms of cell death share some common elements like oxidative stress responses, each has distinct mechanisms and hallmarks. Disulfidptosis, with its focus on abnormal disulfide bond formation and cytoskeletal collapse, offers a unique perspective on how disruptions in protein homeostasis and redox balance can lead to cell demise, diverging from the pathways of more classical cell death modalities. Understanding these differences is crucial for developing targeted therapeutic interventions for various diseases where dysregulated cell death plays a significant role.
Prospects for targeting disulfidptosis in disease treatment
Targeting disulfidptosis for disease treatment holds significant promise given its involvement in a range of pathologies, including neurodegenerative disorders, cancer, and aging. Here are some prospective strategies and areas of focus for therapeutic intervention:
Modulating SLC7A11 expression or activity
Since high SLC7A11 expression contributes to cystine uptake and subsequent NADPH depletion, therapeutics that downregulate SLC7A11 could help mitigate disulfidptosis. Small molecule inhibitors or RNA-based therapies could be explored to suppress SLC7A11 function.
Enhancing intracellular redox balance
Strategies to increase NADPH levels or improve the efficiency of antioxidant systems, such as boosting the activity of enzymes like glutathione reductase or NADPH-producing enzymes, could counteract oxidative stress and disulfide bond imbalances.
Disulfide bond modifiers
Developing compounds that selectively break abnormal disulfide bonds or enhance the activity of enzymes involved in disulfide bond formation and reduction (thioredoxin, glutaredoxins) could help restore protein homeostasis and prevent cytoskeletal collapse.
Actin cytoskeleton stabilizers
Agents that stabilize the actin cytoskeleton could potentially counteract the effects of disulfidptosis on actin dynamics, preserving cellular integrity and function.
Targeting signaling pathways
Modulating signaling pathways like NF-κB and JNK, which influence intracellular redox status and protein homeostasis, could offer a systemic approach to managing disulfidptosis. Small molecule inhibitors or activators of these pathways could be developed for therapeutic use.
Autophagy inducers
Given the role of autophagy in protein quality control, inducing autophagic flux could help clear abnormal disulfide-bonded proteins and mitigate the downstream effects of disulfidptosis.
Research into these areas is still nascent, but the growing recognition of disulfidptosis's importance in disease etiology underscores the potential for innovative therapeutic strategies. By studying and exploring the mechanism of disulfidptosis, a deeper understanding of its mechanism and influencing factors can be achieved, thus yielding insights for new treatment methodologies.
Prospects for targeting disulfidptosis in drug development
Targeting disulfidptosis in drug development is an emerging area of research with promising implications for several diseases where disruptions in protein disulfide homeostasis play a key role. Here are some prospects and challenges for incorporating disulfidptosis targets into the drug development pipeline:
Discovery of novel targets
Identifying specific enzymes, transporters, or signaling molecules involved in disulfide bond formation, reduction, or regulation can lead to new drug targets. For example, inhibitors of cystine-glutamate antiporter (xCT/SLC7A11) have gained attention for their potential in modulating oxidative stress in cancer and neurodegeneration.
Small molecule therapeutics
Developing small molecules capable of modulating the activity of target proteins related to disulfide metabolism, such as glutathione peroxidases, thioredoxins, or protein disulfide isomerases (PDIs), could correct imbalances in redox state.
Biologicals and protein therapeutics
Monoclonal antibodies or recombinant proteins designed to bind and modulate the activity of specific proteins in the disulfide metabolism pathway may offer targeted interventions with fewer off-target effects.
Repurposing existing drugs
Investigating whether drugs already approved for other indications can also modulate disulfide homeostasis could accelerate the development process. For instance, some chemotherapeutic agents and antioxidants may have unexplored effects on redox balance.
In-depth investigations of the molecular mechanisms and regulatory network involved in disulfidptosis may lead to the discovery of new targets and drugs, offering novel ideas and directions for drug development to treat diseases.
Conclusion
In conclusion, disulfidptosis is a crucial mode of cell death and is highly important for understanding biology. Through in-depth study of the molecular mechanism and regulatory network of disulfidptosis, we can reveal the mechanisms of various physiological and pathological processes in the body, provide new ideas and methods for disease prevention and treatment, offer a new theoretical basis for the development of life sciences, and make greater contributions to human health. Moreover, the study of disulfidptosis is highly important for the development of cell biology. This research can not only propel the field forward but also offer fresh perspectives and directions for the study of other forms of cell death.
Data availability
No datasets were generated or analysed during the current study.
References
Bertheloot D, Latz E, Franklin BS (2021) Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol 18:1106–1121. https://doi.org/10.1038/s41423-020-00630-3
Liu XG, Nie LT, Zhang YL et al (2023) Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol 25:404–414. https://doi.org/10.1038/s41556-023-01091-2
Machesky LM (2023) Deadly actin collapse by disulfidptosis. Nat Cell Biol 25:375–376. https://doi.org/10.1038/s41556-023-01100-4
Liu XG, Zhuang L, Gan BY (2023) Disulfidptosis: disulfide stress-induced cell death. Trends Cell Biol S0962–8924(23):00141–00151. https://doi.org/10.1016/j.tcb.2023.07.009
Zheng TJ, Liu QB, Xing FY et al (2023) Disulfidptosis: a new form of programmed cell death. J Exp Clin Cancer Res 42:137. https://doi.org/10.1186/s13046-023-02712-2
Zheng PJ, Zhou CT, Ding YM et al (2023) Disulfidptosis: a new target for metabolic cancer therapy. J Exp Clin Cancer Res 42:103. https://doi.org/10.1186/s13046-023-02675-4
Zhu YD, Kong LY, Han TX et al (2023) Machine learning identification and immune infiltration of disulfidptosis-related Alzheimer’s disease molecular subtypes. Immun Inflamm Dis 11:e1037. https://doi.org/10.1002/iid3.1037
Ma SJ, Wang D, Xie DJ (2023) Identification of disulfidptosis-related genes and subgroups in Alzheimer’s disease. Front Aging Neurosci 15:1236490. https://doi.org/10.3389/fnagi.2023.1236490
Wang XX, Lin JY, Li Z et al (2023) In what area of biology has a “new” type of cell death been discovered? Biochim Biophys Acta Rev Cancer 1878:188955. https://doi.org/10.1016/j.bbcan.188955
Chen YX, Xue WY, Zhang YT et al (2023) A novel disulfidptosis related immune checkpoint genes signature: forecasting the prognosis of hepatocellular carcinoma. J Cancer Res Clin Oncol 149:12843–12854. https://doi.org/10.1007/s00432-023-05076-4
Yu XX, Guo ZH, Fang ZH et al (2023) Identification and validation of disulfidptosis-associated molecular clusters in non-alcoholic fatty liver disease. Front Genet 14:1251999. https://doi.org/10.3389/fgene.2023.1251999
Chen XN, Wang ZJ, Wu YL et al (2023) Typing and modeling of hepatocellular carcinoma based on disulfidptosis-related amino acid metabolism genes for predicting prognosis and guiding individualized treatment. Front Oncol 13:1204335. https://doi.org/10.3389/fonc.2023.1204335
Zhong ZY, Zhang CJ, Ni S et al (2023) NFATc1-mediated expression of SLC7A11 drives sensitivity to TXNRD1 inhibitors in osteoclast precursors. Redox Biol 63:102711. https://doi.org/10.1016/j.redox.2023.102711
Chen HL, Yang WJ, Li YJ et al (2023) Leveraging a disulfidptosis based signature to improve the survival and drug sensitivity of bladder cancer patients. Front Immunol 14:1198878. https://doi.org/10.3389/fimmu.2023.1198878
Qi C, Ma JM, Sun JJ et al (2023) The role of molecular subtypes and immune infiltration characteristics based on disulfidptosis-associated genes in lung adenocarcinoma. Aging (Albany NY) 15:5075–5095. https://doi.org/10.18632/aging.204782
Liu XG, Olszewski K, Zhang YL et al (2020) Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat Cell Biol 22:476–486. https://doi.org/10.1038/s41556-020-0496-x
Green DR, Llambi F (2015) Cell Death Signaling. Cold Spring Harb Perspect Biol 7:a006080. https://doi.org/10.1101/cshperspect.a006080
Boatright KM, Salvesen GS (2003) Mechanisms of caspase activation. Curr Opin Cell Biol 15:725–731. https://doi.org/10.1016/j.ceb.2003.10.009
Li J, Yuan J (2008) Caspases in apoptosis and beyond. Oncogene 27:6194–6206. https://doi.org/10.1038/onc.2008.297
Herrmann BI, Grayczyk JP, Brodsky IE (2023) Collab or Cancel? Bacterial Influencers of Inflammasome Signaling. Annu Rev Microbiol 77:451–477. https://doi.org/10.1146/annurev-micro-032521-024017
Wu TT, Li ZG, Wei YJ (2023) Advances in understanding mechanisms underlying mitochondrial structure and function damage by ozone. Sci Total Environ 861:160589. https://doi.org/10.1016/j.scitotenv.2022.160589
Wang YP, Gao WQ, Shi XY et al (2017) Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547:99–103. https://doi.org/10.1038/nature22393
Fritsch M, Günther SD, Schwarzer R et al (2019) Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 575:683–687. https://doi.org/10.1038/s41586-019-1770-6
Grunwald DS, Otto NM, Park JM et al (2020) GABARAPs and LC3s have opposite roles in regulating ULK1 for autophagy induction. Autophagy 16:600–614. https://doi.org/10.1080/15548627.2019.1632620
Shim MS, Nettesheim A, Hirt J et al (2020) The autophagic protein LC3 translocates to the nucleus and localizes in the nucleolus associated to NUFIP1 in response to cyclic mechanical stress. Autophagy 16:1248–1261. https://doi.org/10.1080/15548627.2019.1662584
Jiang JW, Zhang L, Chen HN et al (2020) Regorafenib induces lethal autophagy arrest by stabilizing PSAT1 in glioblastoma. Autophagy 16:106–122. https://doi.org/10.1080/15548627.2019.1598752
Wang XC, Kong XH, Feng X et al (2023) Effects of DNA, RNA, and Protein Methylation on the Regulation of Ferroptosis. Int J Biol Sci 19:3558–3575. https://doi.org/10.7150/ijbs.85454
Liu XG, Zhang YL, Zhuang L et al (2020) NADPH debt drives redox bankruptcy: SLC7A11/xCT-mediated cystine uptake as a double-edged sword in cellular redox regulation. Genes Dis 8:731–745. https://doi.org/10.1016/j.gendis.2020.11.010
Koppula P, Zhuang L, Gan BY (2021) Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 12:599–620. https://doi.org/10.1007/s13238-020-00789-5
Liu XG, Gan BY (2021) Glucose starvation induces NADPH collapse and disulfide stress in SLC7A11high cancer cells. Oncotarget 12:1629–1630. https://doi.org/10.18632/oncotarget.27993
Hadian K, Stockwell BR (2023) The therapeutic potential of targeting regulated non-apoptotic cell death. Nat Rev Drug Discov 22:723–742. https://doi.org/10.1038/s41573-023-00749-8
Wang LY, Liu XJ, Li QQ et al (2023) The romantic history of signaling pathway discovery in cell death: an updated review. Mol Cell Biochem Oct 18. https://doi.org/10.1007/s11010-023-04873-2
Chen FQ, Kang R, Liu J et al (2023) Mechanisms of alkaliptosis. Front Cell Dev Biol 11:1213995. https://doi.org/10.3389/fcell.2023.1213995
Liu J, Kuang FM, Kang R et al (2020) Alkaliptosis: a new weapon for cancer therapy. Cancer Gene Ther 27:267–269. https://doi.org/10.1038/s41417-019-0134-6
Scaturro P, Pichlmair A (2019) Oxeiptosis: a discreet way to respond to radicals. Curr Opin Immunol 56:37–43. https://doi.org/10.1016/j.coi.2018.10.006
Holze C, Michaudel C, Mackowiak C et al (2018) Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat Immunol 19:130–140. https://doi.org/10.1038/s41590-017-0013-y
Denton D, Kumar S (2019) Autophagy-dependent cell death. Cell Death Differ 26(4):605–616. https://doi.org/10.1038/s41418-018-0252-y
Chung C, Seo W, Silwal P et al (2020) Crosstalks between inflammasome and autophagy in cancer. J Hematol Oncol 13:100. https://doi.org/10.1186/s13045-020-00936-9
Fleming A, Bourdenx M, Fujimaki M et al (2022) The different autophagy degradation pathways and neurodegeneration. Neuron 110:935–966. https://doi.org/10.1016/j.neuron.2022.01.017
Galluzzi L, Vitale I, Aaronson SA et al (2018) Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25:486–541. https://doi.org/10.1038/s41418-017-0012-4
Gao WT, Wang XY, Zhou Y et al (2022) Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct Target Ther 7:196. https://doi.org/10.1038/s41392-022-01046-3
Wen SM, Niu YJ, Lee SO et al (2014) Androgen receptor (AR) positive vs negative roles in prostate cancer cell deaths including apoptosis, anoikis, entosis, necrosis and autophagic cell death. Cancer Treat Rev 40:31–40. https://doi.org/10.1016/j.ctrv.2013.07.008
Mou YH, Wang J, Wu JC et al (2019) Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol 12:34. https://doi.org/10.1186/s13045-019-0720-y
Jiang XJ, Stockwell BR, Conrad M (2021) Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 22:266–282. https://doi.org/10.1038/s41580-020-00324-8
Fricker M, Tolkovsky AM, Borutaite V et al (2018) Neuronal Cell Death. Physiol Rev 98:813–880. https://doi.org/10.1152/physrev.00011.2017
Zheng DD, Liu J, Piao HL et al (2022) ROS-triggered endothelial cell death mechanisms: Focus on pyroptosis, parthanatos, and ferroptosis. Front Immunol 13:1039241. https://doi.org/10.3389/fimmu.2022.1039241
Zhang T, Luu MDA, Dolga AM et al (2023) The old second messenger cAMP teams up with novel cell death mechanisms: potential translational therapeutical benefit for Alzheimer’s disease and Parkinson’s disease. Front Physiol 14:1207280. https://doi.org/10.3389/fphys.2023.1207280
Liu SQ, Luo WB, Wang YF (2022) Emerging role of PARP-1 and PARthanatos in ischemic stroke. J Neurochem 160:74–87. https://doi.org/10.1111/jnc.15464
Fatokun AA, Dawson VL, Dawson TM (2014) Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol 171:2000–2016. https://doi.org/10.1111/bph.12416
Koehler RC, Dawson VL, Dawson TM (2021) Targeting Parthanatos in Ischemic Stroke. Front Neurol 12:662034. https://doi.org/10.3389/fneur.2021.662034
Robinson N, Ganesan R, Hegedűs C et al (2019) Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol 26:101239. https://doi.org/10.1016/j.redox.2019.101239
Florey O, Kim SE (2015) Overholtzer M (2015) Entosis: Cell-in-Cell Formation that Kills Through Entotic Cell Death. Curr Mol Med 15:861–866. https://doi.org/10.2174/1566524015666151026100042
Sarcognato S, Jong IEM, Fabris L et al (2020) Necroptosis in Cholangiocarcinoma. Cells 9:982. https://doi.org/10.3390/cells9040982
Inoue M, Enomoto M, Yoshimura M et al (2021) Pharmacological inhibition of sodiumcal-cium exchange activates NADPH oxidase and induces infection-independent NETotic cell death. Redox Biol 43:101983. https://doi.org/10.1016/j.redox.2021.101983
Kovacs SB, Miao EA (2017) Gasdermins: Effectors of Pyroptosis. Trends Cell Biol 27:673–684. https://doi.org/10.1016/j.tcb.2017.05.005
Fang Y, Tian SW, Pan YT et al (2020) Pyroptosis: A new frontier in cancer. Biomed Pharmacother 121:109595. https://doi.org/10.1016/j.biopha.2019.109595
Loveless R, Bloomquist R, Teng Y (2021) Pyroptosis at the forefront of anticancer immunity. J Exp Clin Cancer Res 40:264. https://doi.org/10.1186/s13046-021-02065-8
Du TT, Gao J, Li PL et al (2021) Pyroptosis, metabolism, and tumor immune microenvironment. Clin Transl Med 11:e492. https://doi.org/10.1002/ctm2.492
Zeng ZL, Li GH, Wu SY et al (2019) Role of pyroptosis in cardiovascular disease. Cell Prolif 52:e12563. https://doi.org/10.1111/cpr.12563
Milani M, Pihán P, Hetz C (2023) Calcium signaling in lysosome-dependent cell death. Cell Calcium 113:102751. https://doi.org/10.1016/j.ceca.2023.102751
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35(4):495–516. https://doi.org/10.1080/01926230701320337
Takahashi JDB, Takahashi LS, Mingatto FE et al (2015) The immune system is limited by oxidative stress: Dietary selenium promotes optimal antioxidative status and greatest immune defense in pacu Piaractus mesopotamicus. Fish Shellfish Immunol 47:360–367. https://doi.org/10.1016/j.fsi.2015.09.022
Wu GY, Fang YZ, Yang S et al (2004) (2004) Glutathione metabolism and its implications for health. J Nutr 134:489–492. https://doi.org/10.1093/jn/134.3.489
Badgley MA, Kremer DM, Maurer HC et al (2020) Cysteine depletion induces pancreatic tumorferroptosis in mice. Science 368:85–89. https://doi.org/10.1126/science.aaw9872
Costa I, Barbosa DJ, Benfeito S et al (2023) Molecular mechanisms of ferroptosis and their involvement in brain diseases. Pharmacol Ther 244:108373. https://doi.org/10.1016/j.pharmthera.2023.108373
Collet JF, Cho SH, Iorga BI et al (2020) How the assembly and protection of the bacterial cell envelope depend on cysteine residues. J Biol Chem 295:11984–11994. https://doi.org/10.1074/jbc.REV120.011201
Galván I (2018) Predation risk determines pigmentation phenotype in nuthatches by Melanin-related gene expression effects. J Evol Biol 31:1760–1771. https://doi.org/10.1111/jeb.13379
Yang Y, Li L, Hang QY et al (2019) γ-glutamylcysteine exhibits anti-inflammatory effects by increasing cellular glutathione level. Redox Biol 20:157–166. https://doi.org/10.1016/j.redox.2018.09.019
Quintana-Cabrera R, Fernandez-Fernandez S, Bobo-Jimenez V et al (2012) γ-Glutamylcysteine detoxifies reactive oxygen species by acting as glutathione peroxidase-1 cofactor. Nat Commun 3:718. https://doi.org/10.1038/ncomms1722
Ren Y, Chen J, Chen P et al (2021) Oxidative stress-mediated AMPK inactivation determines the high susceptibility of LKB1-mutant NSCLC cells to glucose starvation. Free Radic Biol Med 166:128–139. https://doi.org/10.1016/j.freeradbiomed.2021.02.018
Ying M, You D, Zhu X et al (2021) (2021) Lactate and glutamine support NADPH generation in cancer cells under glucose deprived conditions. Redox Biol 46:102065. https://doi.org/10.1016/j.redox.2021.102065
Soeters MR, Soeters PB (2012) The evolutionary benefit of insulin resistance. Clin Nutr 31(6):1002–1007. https://doi.org/10.1016/j.clnu.2012.05.011
Njeim R, Alkhansa S, Fornoni A (2023) Unraveling the Crosstalk between Lipids and NADPH Oxidases in Diabetic Kidney Disease. Pharmaceutics 15(5):1360. https://doi.org/10.3390/pharmaceutics15051360
Yao XM, Li W, Fang D et al (2021) Emerging Roles of Energy Metabolism in Ferroptosis Regulation of Tumor Cells. Adv Sci (Weinh) 8:e2100997. https://doi.org/10.1002/advs.202100997
Patra KC, Hay N (2014) The pentose phosphate pathway and cancer. Trends Biochem Sci 39:347–354. https://doi.org/10.1016/j.tibs.2014.06.005
Ying MF, You D, Zhu XB et al (2021) Lactate and glutamine support NADPH generation in cancer cells under glucose deprived conditions. Redox Biol 102065. https://doi.org/10.1016/j.redox.2021.102065
Wang YT, Trzeciak AJ, Rojas WS et al (2023) Metabolic adaptation supports enhanced macrophage efferocytosis in limited-oxygen environments. Cell Metab 35:316-331.e6. https://doi.org/10.1016/j.cmet.2022.12.005
Rahman I, Kode A, Biswas SK (2006) Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc 1(6):3159–3165. https://doi.org/10.1038/nprot.2006.378
Lood C, Blanco LP, Purmalek MM et al (2016) Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 22(2):146–153. https://doi.org/10.1038/nm.4027
Liu N, Lin MM, Huang SS et al (2021) NADPH and Mito-Apocynin Treatment Protects Against KA-Induced Excitotoxic Injury Through Autophagy Pathway. Front Cell Dev Biol 9:612554. https://doi.org/10.3389/fcell.2021.612554.eCollection2021
Ortiz-Rodríguez JM, Martín-Cano FE, Gaitskell-Phillips G et al (2020) The SLC7A11: sperm mitochondrial function and non-canonical glutamate metabolism. Reproduction 160:803–818. https://doi.org/10.1530/REP-20-0181
Chen QP, Zheng W, Guan J et al (2023) SOCS2-enhanced ubiquitination of SLC7A11 promotes ferroptosis and radiosensitization in hepatocellular carcinoma. Cell Death Differ 30:137–151. https://doi.org/10.1038/s41418-022-01051-7
Potter PGW, Ellacott KLJ, Randall AD et al (2022) Glutamate Prevents Altered Mitochondrial Function Following Recurrent Low Glucose in Hypothalamic but Not Cortical Primary Rat Astrocytes. Cells 11:3422. https://doi.org/10.3390/cells11213422
Stockwell BR, Angeli JPF, Bayir H et al (2017) Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171:273–285. https://doi.org/10.1016/j.cell.2017.09.021
Li J, Cao F, Yin HL et al (2020) Ferroptosis: past, present and future. Cell Death Dis 11:88. https://doi.org/10.1038/s41419-020-2298-2
Chen H, Cao LX, Han K et al (2022) Patulin disrupts SLC7A11-cystine-cysteine-GSH antioxidant system and promotes renal cell ferroptosis both in vitro and in vivo. Food Chem Toxicol 166:113255. https://doi.org/10.1016/j.fct.2022.113255
Yuan Y, Zhai YY, Chen JJ et al (2021) Kaempferol Ameliorates Oxygen-Glucose Deprivation/Reoxygenation-Induced Neuronal Ferroptosis by Activating Nrf2/SLC7A11/GPX4 Axis. Biomolecules 11:923. https://doi.org/10.3390/biom11070923
Fu C, Wu YF, Liu SJ et al (2022) Rehmannioside A improves cognitive impairment and alleviates ferroptosis via activating PI3K/AKT/Nrf2 and SLC7A11/GPX4 signaling pathway after ischemia. J Ethnopharmacol 289:115021. https://doi.org/10.1016/j.jep.2022.115021
Ye YZ, Chen A, Li L et al (2022) Repression of the antiporter SLC7A11/glutathione/glutathione peroxidase 4 axis drives ferroptosis of vascular smooth muscle cells to facilitate vascular calcification. Kidney Int 102:1259–1275. https://doi.org/10.1016/j.kint.2022.07.034
Wang XM, Wang YL, Huang DM et al (2022) Astragaloside IV regulates the ferroptosis signaling pathway via the Nrf2/SLC7A11/GPX4 axis to inhibit PM2.5-mediated lung injury in mice. Int Immunopharmacol 112:109186. https://doi.org/10.1016/j.intimp.2022.109186
Yuan SY, Wei C, Liu GF et al (2022) Sorafenib attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis via HIF-1α/SLC7A11 pathway. Cell Prolif 55:e13158. https://doi.org/10.1111/cpr.13158
Ayala A, Muñoz MF, Argüelles S (2014) Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev 2014:360438. https://doi.org/10.1155/2014/360438
Kajarabille N, Latunde-Dada GO (2019) Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int J Mol Sci 20(19):4968. https://doi.org/10.3390/ijms20194968
Li Y, Liu Y, Wu P et al (2021) Inhibition of Ferroptosis Alleviates Early Brain Injury After Subarachnoid Hemorrhage In Vitro and In Vivo via Reduction of Lipid Peroxidation. Cell Mol Neurobiol 41(2):263–278. https://doi.org/10.1007/s10571-020-00850-1
Davaanyam D, Lee H, Seol SI et al (2023) HMGB1 induces hepcidin upregulation in astrocytes and causes an acute iron surge and subsequent ferroptosis in the postischemic brain. Exp Mol Med 55(11):2402–2416. https://doi.org/10.1038/s12276-023-01111-z
Sun Y, Ren Y, Song LY et al (2024) Targeting iron-metabolism: a potential therapeutic strategy for pulmonary fibrosis. Biomed Pharmacother 172:116270. https://doi.org/10.1016/j.biopha.2024.116270
Chen Z, Li J, Peng H et al (2023) Meteorin-like/Meteorin-β protects LPS-induced acute lung injury by activating SIRT1-P53-SLC7A11 mediated ferroptosis pathway. Mol Med 29(1):144. https://doi.org/10.1186/s10020-023-00714-6
Chen X, Wang ZC, Li C et al (2024) SIRT1 activated by AROS sensitizes glioma cells to ferroptosis via induction of NAD+ depletion-dependent activation of ATF3. Redox Biol 69:103030. https://doi.org/10.1016/j.redox.2024.103030
Liu L, He JT, Sun GF et al (2022) The N6-methyladenosine Modification enhances ferroptosis resistance through inhibiting SLC7A11 mRNA deadenylation in hepatoblastoma. Clin Transl Med 12:e778. https://doi.org/10.1002/ctm2.778
Soria FN, Zabala A, Pampliega O et al (2016) Cystine/glutamate antiporter blockage induces myelin degeneration. Glia 64:1381–1395. https://doi.org/10.1002/glia.23011
Li R, Li SL, Shen L et al (2024) SNHG1, interacting with SND1, contributes to sorafenib resistance of liver cancer cells by increasing m6A-mediated SLC7A11 expression and promoting aerobic glycolysis. Environ Toxicol 39:1269–1282. https://doi.org/10.1002/tox.24014
Robinson PJ, Bulleid NJ (2020) Mechanisms of Disulfide Bond Formation in Nascent Polypeptides Entering the Secretory Pathway. Cells 9:1994. https://doi.org/10.3390/cells9091994
Jeong HR (2015) An SSA (2015) Causative factors for formation of toxic islet amyloid Polypeptide oligomer in type 2 diabetes mellitus. Clin Interv Aging 10:1873–1879. https://doi.org/10.2147/CIA.S95297
Chen XB, Roeters SJ, Cavanna F et al (2023) Crowding alters F-actin secondary structure and hydration. Commun Biol 6:900. https://doi.org/10.1038/s42003-023-05274-3
Zhu AK, Zong Y, Wei S et al (2023) Pan-cancer Analysis of the Disulfidptosis-related Gene NCKAP1 and Its Prognostic Value for Lung Adenocarcinoma. J Cancer 14:3351–3367. https://doi.org/10.7150/jca.88650
Chen BY, Chou HT, Brautigam CA et al (2017) Rac1 GTPase activates the WAVE regulatory complex through two distinct binding sites. Elife 6:e29795. https://doi.org/10.7554/eLife.29795
Jiang SW, Carroll L, Rasmussen LM et al (2021) Oxidation of protein disulfide bonds by singlet oxygen gives rise to glutathionylated proteins. Redox Biol 38:101822. https://doi.org/10.1016/j.redox.2020.101822
Meyer Y, Belin C, Hinoux VD et al (2012) Thioredoxin and glutaredoxin systems in plants: molecular mechanisms, crosstalks, and functional significance. Antioxid Redox Signal 17:1124–1160. https://doi.org/10.1089/ars.2011.4327
Yafee YAA, Ayadhi LYA, Haq SH et al (2011) Novel metabolic biomarkers related to sulfur-dependent detoxification pathways in autistic patients of Saudi Arabia. BMC Neurol 11:139. https://doi.org/10.1186/1471-2377-11-139
Dokainish HM, Simard DJ, Gauld JW (2017) A Pseudohypervalent Sulfur Intermediate as an Oxidative Protective Mechanism in the Archaea Peroxiredoxin Enzyme ApTPx. J Phys Chem B 121:6570–6579. https://doi.org/10.1021/acs.jpcb.7b04671
Ji PY, Li ZY, Wang H et al (2019) Arsenic and sulfur dioxide coexposure induce renal injury via activation of the NF-κB and caspase signaling pathway. Chemosphere 224:280–288. https://doi.org/10.1016/j.chemosphere.2019.02.111
Ban JO, Oh JH, Kim TM et al (2009) Antiinflamma-tory and arthritic effects of thiacremonone, a novel sulfur compound isolated from garlic via inhibition of NF-kappaB. Arthritis Res Ther 11:R145. https://doi.org/10.1186/ar2819
Brancaccio M, Milito A, Viegas CA et al (2022) First evidence of dermo-protective activity of marine sulfur containing histidine compounds. Free Radic Biol Med 192:224–234. https://doi.org/10.1016/j.freeradbiomed.2022.09.017
Zhang WJ, Sun Y, Bai L et al (2021) RBMS1 regulates lung cancer ferroptosis through translational control of SLC7A11. J Clin Invest 131:e152067. https://doi.org/10.1172/JCI152067
Wang XB, Chen YQ, Wang XD et al (2021) Stem Cell Factor SOX2 Confers Ferroptosis Resistance in Lung Cancer via Upregulation of SLC7A11. Cancer Res 81:5217–5229. https://doi.org/10.1158/0008-5472.CAN-21-0567
Ishii T, Mimura I, Nagaoka K et al (2022) Effect of M2-like macrophages of the injured kidney cortex on kidney cancer progression. Cell Death Discov 8:480. https://doi.org/10.1038/s41420-022-01255-3
Ren CZ, Wang QH, Xu ZN et al (2023) Development and validation of a disulfidptosis and M2 TAM-related classifier for bladder cancer to explore tumor subtypes, immune landscape and drug treatment. J Cancer Res Clin Oncol 149:15805–15818. https://doi.org/10.1007/s00432-023-05352-3
Liu L, Liu J, Lyu QB et al (2023) Disulfidptosis-associated LncRNAs index predicts prognosis and chemotherapy drugs sensitivity in cervical cancer. Sci Rep 13:12470. https://doi.org/10.1038/s41598-023-39669-3
Jiang CH, Xiao YG, Xu DP et al (2023) Prognosis Prediction of Disulfidptosis-Related Genes in Bladder Cancer and a Comprehensive Analysis of Immunotherapy. Crit Rev Eukaryot Gene Expr 33(6):73–86. https://doi.org/10.1615/CritRevEukaryotGeneExpr.2023048536
Wu YH, Shang J, Ruan Q et al (2023) Integrated single-cell and bulk RNA sequencing in pancreatic cancer identifies disulfidptosis-associated molecular subtypes and prognostic signature. Sci Rep 13:17577. https://doi.org/10.1038/s41598-023-43036-7
Zhao DZ, Meng Y, Dian YT et al (2023) Molecular landmarks of tumor disulfidptosis across cancer types to promote disulfidptosis-target therapy. Redox Biol 68:102966. https://doi.org/10.1016/j.redox.2023.102966
Fan DQ, Zhu YH (2022) Circ_0120175 promotes laryngeal squamous cell carcinoma development through up-regulating SLC7A11 by sponging miR-330-3p. J Mol Histol 53:159–171. https://doi.org/10.1007/s10735-022-10061-1
Ke Y, Chen XY, Su YT et al (2021) Low Expression of SLC7A11 Confers Drug Resistance and Worse Survival in Ovarian Cancer via Inhibition of Cell Autophagy as a Competing Endogenous RNA. Front Oncol 11:744940. https://doi.org/10.3389/fonc.2021.744940
Drayton RM, Dudziec E, Peter S et al (2014) Reduced expression of miRNA-27a modulates cisplatin resistance in bladder cancer by targeting the cystine/glutamate exchanger SLC7A11. Clin Cancer Res 20(7):1990–2000. https://doi.org/10.1158/1078-0432.CCR-13-2805
Wu XD, Shen SZ, Qin JL et al (2022) High co-expression of SLC7A11 and GPX4 as a predictor of platinum resistance and poor prognosis in patients with epithelial ovarian cancer. BJOG 129(Suppl 2):40–49. https://doi.org/10.1111/1471-0528.17327
Xiao LJ, Yin W, Chen XQ et al (2023) A disulfidptosis-related lncRNA index predicting prognosis and the tumor microenvironment in colorectal cancer. Sci Rep 13:20135. https://doi.org/10.1038/s41598-023-47472-3
Liu TH, Ren YL, Wang QX et al (2023) Exploring the role of the disulfidptosis-related gene SLC7A11 in adrenocortical carcinoma: implications for prognosis, immune infiltration, and therapeutic strategies. Cancer Cell Int 23:259. https://doi.org/10.1186/s12935-023-03091-6
Dai Y, Hu L (2022) HSPB1 overexpression improves hypoxic-ischemic brain damage by attenuating ferroptosis in rats through promoting G6PD expression. J Neurophysiol 128:1507–1517. https://doi.org/10.1152/jn.00306.2022
Zhu KY, Zhu X, Sun SH et al (2021) Inhibition of TLR4 prevents hippocampal hypoxic-ischemic injury by regulating ferroptosis in neonatal rats. Exp Neurol 345:113828. https://doi.org/10.1016/j.expneurol.2021.113828
Ye J, Lyu TJ, Li LY et al (2023) Ginsenoside Re attenuates myocardial ischemia/reperfusion induced ferroptosis via miR-144-3p/SLC7A11. Phytomedicine 113:154681. https://doi.org/10.1016/j.phymed.2023.154681
Pang P, Si W, Wu H et al (2023) YTHDF2 Promotes Cardiac Ferroptosis via Degradation of SLC7A11 in Cardiac Ischemia-reperfusion Injury. Antioxid Redox Signal 10. https://doi.org/10.1089/ars.2023.0291.
Li HL, Ding JX, Liu W et al (2023) Plasma exosomes from patients with acute myocardial infarction alleviate myocardial injury by inhibiting ferroptosis through miR-26b-5p/SLC7A11 axis. Life Sci 322:121649. https://doi.org/10.1016/j.lfs.2023.121649
Wang ZR, Yao MR, Jiang LY et al (2022) Dexmedetomidine attenuates myocardial ischemia/reperfusion-induced ferroptosis via AMPK/GSK-3β/Nrf2 axis. Biomed Pharmacother 154:113572. https://doi.org/10.1016/j.biopha.2022.113572
Zhang XY, Zheng CT, Gao ZQ et al (2022) SLC7A11/xCT Prevents Cardiac Hypertrophy by Inhibiting Ferroptosis. Cardiovasc Drugs Ther 36:437–447. https://doi.org/10.1007/s10557-021-07220-z
Xu DP, Jiang CH, Xiao YG et al (2023) Identification and validation of disulfidptosis-related gene signatures and their subtype in diabetic nephropathy. Front Genet 14:1287613. https://doi.org/10.3389/fgene.2023.1287613
Khanna N, Reddy VG, Tuteja N et al (2000) Differential gene expression in apoptosis: identification of ribosomal protein S29 as an apoptotic inducer. Biochem Biophys Res Commun 277(2):476–486. https://doi.org/10.1006/bbrc.2000.3688
Martinet W, Schrijvers DM, Meyer GRYD (2011) Necrotic cell death in atherosclerosis. Basic Res Cardiol 106(5):749–760. https://doi.org/10.1007/s00395-011-0192-x
Li RY, Gu ZH, Zhang X et al (2020) RNF115 deletion inhibits autophagosome maturation and growth of gastric cancer. Cell Death Dis 11(9):810. https://doi.org/10.1038/s41419-020-03011-w
Rochette L, Dogon G, Rigal E et al (2022) Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis. Int J Mol Sci 24(1):449. https://doi.org/10.3390/ijms24010449
Acknowledgements
We sincerely acknowledge the professor of Zhengyuan Xia from Guangdong Medical University for providing helpful comments on revision of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82160157 and No. 81970290) , the Joint Funds of the National Natural Science Foundation of China (No. U20A2018) , and the Natural Science Foundation of Beijing (No. 7242046 and No. 7222044).
Author information
Authors and Affiliations
Contributions
Fei Xiao, Hui-Li Li, and Bei Yang helped search the literature and prepare the manuscript; Hao Che, Fei Xu, and Gang Li prepared figures; Cheng-Hui Zhou and Sheng Wang helped with the article modification. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Consent for publication
All authors consent this manuscript for publication.
Ethical approval and Consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiao, F., Li, HL., Yang, B. et al. Disulfidptosis: A new type of cell death. Apoptosis (2024). https://doi.org/10.1007/s10495-024-01989-8
Accepted:
Published:
DOI: https://doi.org/10.1007/s10495-024-01989-8