
Abstract
Immune checkpoint factors, such as programmed cell death protein-1/2 (PD-1, PD-2) or cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) receptors, are targets for monoclonal antibodies (MAbs) developed for cancer immunotherapy. Indeed, modulating immune inhibitory pathways has been considered an important breakthrough in cancer treatment. Although immune checkpoint blockade therapy used to treat malignant diseases has provided promising results, both solid and haematological malignancies develop mechanisms that enable themselves to evade the host immune system. To overcome some major limitations and ensure safety in patients, recent strategies have shown that combining epigenetic modulators, such as inhibitors of histone deacetylases (HDACi) or DNA methyltransferases (DNMTi), with immunotherapeutics can be useful. Preclinical data generated using mouse models strongly support the feasibility and effectiveness of the proposed approaches. Indeed, co-treatment with pan- or class I-selective HDACi or DNMTi improved beneficial outcomes in both in vitro and in vivo studies. Based on the evidence of a pivotal role for HDACi and DNMTi in modulating various components belonging to the immune system, recent clinical trials have shown that both HDACi and DNMTi strongly augmented response to anti-PD-1 immunotherapy in different tumour types. This review describes the current strategies to increase immunotherapy responses, the effects of HDACi and DNMTi on immune modulation, and the advantages of combinatorial therapy over single-drug treatment.
Similar content being viewed by others


Background
Accumulation of genetic alterations might be caused by abnormal expression of genes that play a key role in regulation of cell survival, proliferation, and death. In addition, many studies have evaluated the capability of epigenetic regulators to modulate gene expression in cancer cells through covalent modification of DNA as well as histone and non-histone proteins [1]. The most important epigenetic processes reported in the clinical settings involve DNA methylation and histone modifications without altering the DNA sequence of bases. DNA methyltransferases (DNMTs) catalyse the methylation at cytosine-C5 mainly in a CpG dinucleotide context at the promoters of selected genes [2]. Although DNA methylation is essential for fundamental processes like embryonic development or differentiation, aberrant expression and/or activities of DNMTs are involved in several pathologies, from neurodegeneration to cancer [3,4,5,6]. DNMT enzymes are classified into three distinct families: DNMT1; DNMT2, also known as TRDMT1 (t-RNA cytosine-5-methyltransferase); and DNMT3 (consisting of DNMT3a, DNMT3b, and DNMT3L) [2, 7]. Currently, two DNMT inhibitors (DNMTi), the nucleoside analogues azacitydine (5-AZA) and decitabine (5-AZA-CdR), have been approved by FDA and the European Medicines Agency (EMA) against myelodysplastic syndromes (MDS), acute myeloid leukaemia (AML), and chronic myelomonocytic leukaemia (CMML). DNMT overexpression is described in numerous cancer types. DNMTi can arrest tumour growth and cell invasiveness and can induce cell differentiation [8]. Among histone-modifying enzymes, histone acetyltransferases (HATs) and histone deacetylases (HDACs) are among the most studied targets for chromatin remodelling, control of gene expression, and anticancer therapy. HDACs are divided into four groups: “classical HDACs” are expressed in the nucleus and/or cytoplasm, share a Zn2+-dependent catalytic activity, and include class I (HDAC1, 2, 3, 8), IIa (HDAC4, 5, 7, 9), IIb (HDAC6 and 10), and IV (HDAC11) enzymes. Class III HDACs, known as sirtuins, possess NAD+-dependent deacetylase activity and share no sequence similarity with the classical deacetylases [9]. HDAC inhibitors (HDACi) can induce, among others, tumour cell apoptosis, growth arrest, differentiation, inhibition of angiogenesis, and immunogenicity [10]. Among them, vorinostat and romidepsin have been approved for treatment of refractory cutaneous T cell lymphoma (CTCL), belinostat and chidamide (the latter approved only in China) for peripheral T cell lymphoma (PTCL), and panobinostat for multiple myeloma (MM), all from 2006 to 2015. Interestingly, most HDACi and DNMTi have shown a potent immunomodulatory activity, thus justifying their application in cancer immunotherapies. In fact, there is a growing interest in understanding how these potential therapies can modulate the host immune system in order to achieve beneficial antitumour effects [11]. The cancer immunotherapy field is under intense investigation to ameliorate cancer cell recognition by immune cells and to make them more sensitive to cytotoxic antitumour treatment. Cancer immunotherapy refers to a diverse range of therapeutic approaches to improve the capability of T cells and other immune effector cells in recognition and elimination of cancer cells through overcoming of cancer cell resistance in different tumour types [12]. Moreover, cancer cell immune recognition provides the tools to modulate immune signalling pathways that drive tumour growth and progression, suggesting rational combinatorial approaches [13]. This review will also focus on current immunomodulatory combinatorial treatment strategies aiming to improve the effectiveness of cancer immunotherapy.
Cancer cell escape mechanisms and the role of the immune system
Actually, cancer immunotherapy strategies designed to break the immune tolerance can be broadly classified on the basis of the mechanisms involved in resistance processes. Such strategies include several factors: (i) adoptive transfer of immune effectors, (ii) vaccination, and (iii) immunomodulatory therapy. In particular, effector cells of innate immunity, such as natural killer (NK) cells and macrophages, and adaptive immunity (such as T and B cells) can eliminate immunogenic malignant cells [14]. Nevertheless, the main escape pathways, including anti-apoptotic signalling, mitogen-activated protein kinase (MAPK) pathway, microphthalmia-associated transcription factor (MITF), cyclic adenosyl monophosphate (cAMP), and nuclear factor kappa light chain enhancer of activated B cells (NF-κB)-related mechanisms, negatively influence the therapeutic success. Part of this failure is due to immune suppression by the tumour microenvironment (TME). So far, defective antigen presentation, tumour-induced inhibitory checkpoint pathways against effector T cell activity, infiltrating regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and secretion of immunosuppressive cytokines, such as transforming growth factor β (TGF-β), interleukin-6 (IL-6), and vascular endothelial growth factor (VEGF), represent the major mechanisms for escaping [15]. It is well known that the immune system protects the host against tumour development on one side and promotes tumour growth by selecting tumours of lower immunogenicity on the other side. These two effects create a dynamic process also called “cancer immunoediting” that includes three phases: elimination, equilibrium, and escape [16]. However, due to their heterogeneity, tumour cells with a less immunogenic phenotype are able to escape this elimination phase also called immunosurveillance and to expand during the equilibrium phase. These considerations have encouraged many researchers to develop new therapeutic strategies to fight different cancer types with immunotherapy.
Rationale for the development of cancer immunotherapy strategies
Active research in tumour immunology includes studies on adoptive T cell therapy and cancer vaccination, as well as clinical investigation regarding immune checkpoint blockade in combination therapy.
The immune system plays a key role in maintaining self-tolerance and regulating T cell responses. For this reason, it is very important to understand the complex and dynamic nature of host immune responses and the regulation of additional molecules in the TME in order to develop strategies to improve clinical efficacy. Activation of antigen-specific T cells is a key step in immune responses, and it is provided by the interaction between the peptide-major histocompatibility complex (MHC) complex and the T cell receptor (TCR) in the presence of other co-stimulatory molecules. Among these molecules, cluster of differentiation 28 (CD28), expressed on the surface of naive CD4+ and CD8+ cells, is one of the most important proteins involved in the initial activation of the immune system response.
Conversely, the interaction between molecule and antigenic peptide in the absence of co-stimulation results in T cell “anergy” instead of activation [17]. Immune system homeostasis includes the presence of both stimulatory and inhibitory signals such as cytotoxic T lymphocyte-associated antigen-4 (CTLA-4, a CD28 homolog), which acts by a competitive mechanism with CD28 for binding to its cognate ligands such as CD80/86 expressed on the surface of antigen-presenting cells (APCs) [18]. Another important immune checkpoint is mediated by programmed cell death protein-1 (PD-1). In comparison with CTLA-4, PD-1 regulates immune activity when effector T cell tissue infiltration occurs. Besides activated T cells, PD-1 is mainly expressed on the surface of activated B cells, NK cells, dendritic cells (DCs), and Tregs [13]. Engagement of PD-1 by its ligands, either PD-L1 or PD-L2, induces a negative control signal resulting in the inhibition of T cell proliferation, cytokine production, and cytotoxic activity [19]. Moreover, upregulation of PD-L1 on different tumour types and production of cytokines as a consequence of inflammatory signals induces an innate (tumour cell intrinsic) and an adaptive resistance, respectively. Preclinical studies have demonstrated that blocking the PD-L1/PD-1 interaction augments antitumour T cell responses [20]. About 20% of advanced non-small cell lung cancer (NSCLC) patients and 30–40% of advanced melanoma patients have provided tumour responses to PD-1 blockade monotherapy [21]. Therefore, modulation of immune inhibitory pathways is considered as an important breakthrough in cancer treatment. In particular, since 2011 with the approval by FDA of the monoclonal antibody (MAb) ipilimumab for advanced melanoma, and 3 years later of pembrolizumab and nivolumab as well, there has been an increasing interest in this field. Notably, ipilimumab, targeting CTLA-4 on T cells, allows T cell activation for immune responses in several cancers as well as inhibition of Treg function [22]. Early clinical trials evaluated ipilimumab in patients with a variety of malignancies, including melanoma, prostate cancer, renal cell carcinoma, and non-Hodgkin lymphoma [23, 24]. Similarly, an intense investigation has been conducted for nivolumab and pembrolizumab, fully human and humanized IgG4 anti-PD-1 MAbs, respectively [25, 26]. In general, different tumour type responses to checkpoint blockade are more closely associated with inherent immunogenicity (mutational burden or dominant neoantigens) than with the tumour tissue origin [27]. In preclinical models, combined blockade of PD-1 and CTLA-4 achieved more pronounced antitumour activity than blockade of either pathway alone [28,29,30,31]. Indeed, the first reported ipilimumab and nivolumab combination with response in melanoma has provided a rationale for the development of immune checkpoint combination strategies (NCT01024231) [32]. Additionally, recent studies have shown a synergistic antitumour activity in mouse MC38 and CT26 colorectal tumour models with concurrent, but not sequential, CTLA-4 and PD-1 blockade (ipilimumab and nivolumab) [33]. Updated reviews about the current status of immunotherapy and clinical developments of immune checkpoint inhibitors have been recently reported [34,35,36].
Epigenetic regulation of the immune system
Immune checkpoint regulation mechanisms include covalent modifications, microRNAs (miRNAs), long noncoding RNAs (lncRNAs), and histone modifications [37]. Epigenetic modifiers can either turn on or turn off immune responses, resulting in immune evasion [38]. Since some epigenetic regulators have shown a potent immunomodulatory activity, their combination with immune checkpoint inhibitors could represent a promising therapeutic strategy. Currently, many researchers are investigating the link between epigenetic modulation of the immune system and cancer development. Among the epigenetic processes implicated in immune regulation, DNA methylation and histone acetylation are likely the most important modifications in controlling development, differentiation, and functions of T cells [39]. During immune responses, naive CD4+ T cells differentiate into several T helper (Th) cell subsets, including Th1, Th2, Th17, and induced regulatory T (iTreg) cells, as defined by their pattern of cytokine production [40]. Moreover, CD4+ Th subsets are distinguished by their phenotype as well as by the transcription factors that control their differentiation, including T-bet in Th1, GATA-3 in Th2, RAR-related orphan receptor γ (RORγT) in Th17, and forkhead box protein 3 (Foxp3) in Treg [41]. The first studies in humans showed that Th1 and Th2 cells are true lineages regulated by epigenetic modifications occurring on interferon-γ (IFN-γ), IL-4, and IL-13 genes. The IFN-γ promoter is hypermethylated in human naive T cells and is demethylated during the differentiation to Th1 cells [42]. Conversely, Th2 cell differentiation results in the selective demethylation of several specific CpG dinucleotides in the IL-4 and IL-13 genes, which are expressed in activated Th2 but not Th1 cells [43]. Moreover, epigenetic histone marks are also essential for the Th1/Th2 cell fate decisions. Signal transducer and activator of transcription 4 (STAT4) and T-bet or STAT6 and GATA-3 are key transcription factors for the Th1 and Th2 lineages, respectively [44]. The histone methyltransferase (HMT) SUV39H1, which is involved in H3K9 trimethylation (H3K9me3), has recently been implicated in the silencing of the Th1 locus and the subsequent promotion of stability of Th2 cells [45]. Chang et al. explored the mechanisms establishing long-range H4 acetylation marks at the IFN-γ locus, during Th1 lineage commitment. T-bet displaced the Sin3 transcription regulator family member A (Sin3A)-histone deacetylase (HDAC1, HDAC2) complexes, to facilitate the differentiation of Th1 cells [46]. In response to IL-12 signals, the activation of STAT4 required for the development of Th1 cells facilitates chromatin remodelling at the enhancer regions of Th1 genes. Similarly, Th2 commitment requires STAT6 and GATA-3 activities in response to IL-4 stimulation [47]. Therefore, transcription factors not only promote T cell differentiation but also influence epigenetic states and gene expression programs that define a particular lineage. Furthermore, epigenetic histone modifications by enhancer of zeste homolog 2 (EZH2), a member of polycomb repressive complex 2 (PRC2), regulate differentiation and plasticity of CD4+ T cells. Notably, EZH2 directly binds and facilitates correct expression of T-box transcription factor 21 (Tbx21) and GATA-3 for differentiating Th1 and Th2 cells, accompanied by increased H3K27 trimethylation (H3K27me3) [48]. Finally, in Tregs, Foxp3 is acting predominantly as a transcriptional repressor and is required for establishment of the chromatin repressive mark H3K27me3 in activated Tregs. Indeed, Foxp3 has been found to interact with EZH2 exclusively in activated Tregs, suggesting that Foxp3 recruits the PRC2 complex to target genes and forms repressive chromatin under inflammatory conditions [49]. Morinobu et al. analysed the histone acetylation levels of Th1 genes, IFN-γ, T-bet, and IL18RAP in response to different cytokines [50]. Multiple levels of regulation of IFN-γ histone acetylation may reflect critical checkpoints for Th1 differentiation. In addition, basic leucine zipper transcription factor (BATF) regulates Th1 gene expression via acetylation of T-bet and IFN-γ, considered as an important checkpoint in T cell differentiation [51]. Several other findings suggest that miRNA epigenetic modifications in cancer can promote an immune evasion [52]. More recently, Cortez et al. have identified a novel mechanism of PD-L1 epigenetic regulation by which tumour immune evasion is regulated by the p53/miR-34/PD-L1 axis [53]. Indeed, p53 influences immune response by monitoring T cell activation and inflammatory cytokines and enhancing tumour cell recognition by NK cells [54, 55]. Furthermore, the overexpression of T cell immunoglobulin and mucin domain 3 (Tim-3) on T cells negatively controls the antitumour T cell responses, with important implications for anti-PD1 immunotherapy [56]. Another important immune checkpoint is lymphocyte-activation gene 3 (LAG-3), highly expressed on activated T cells in many cancer types, that can be used as an immunotherapy target [57]. miR-138 has been reported with a multifaceted role in carcinomas, although its ability to interact with the immune system is unknown. Wei et al. have demonstrated that the combination of miR-138 with a MAb therapy against CTLA-4 provided a strong therapeutic synergism. Transfection of human CD4+ T cells with miR-138 suppressed expression of CTLA-4, PD-1, and Foxp3 in glioma preclinical models [58]. Moreover, previous studies described a novel biological role of other miRNAs in regulating the expression of immune checkpoints [59, 60]. Hence, targeting these miRNAs in combination with traditional immune checkpoint inhibitors is certainly a potent immunotherapeutic strategy. At last, lncRNAs are also critical mediators in various tumours associated with cancer progression [61, 62]. Notably, Zeng et al. have found that the nuclear paraspeckle assembly transcript 1 (NEAT1) expression was repressed by PML-RARα, a leukemic-specific antigen and part of the PD-1 pathway. Moreover, reduced NEAT1 expression may play a role in the myeloid differentiation of acute promyelocytic leukaemia (APL) cells [63]. Many lncRNAs are bound and regulated by the key T cell transcription factors T-bet, GATA-3, STAT4, and STAT6. Hu et al. have found that LincR-Ccr2-5′AS, together with GATA-3, is essential for regulation of several chemokine receptor genes and for Th2 cell migration, but the exact mechanism of action of LincR-Ccr2-5′AS is currently unknown [64].
The potential role of epi-drugs as “immune-regulators”
HDACi are being used as a novel, therapeutic approach for treatment of leukaemia and other haematological malignancies [2, 65]. However, their effect on immune cells remains ill-defined, as HDACi may impair immune surveillance. Cancer arises as a result of accumulation of genetic mutations and epigenetic aberrations regulated by many players including HDACs. Abnormal expression of HDACs has been reported in tumours, whereas knockdown of HDACs inhibits tumour growth [66]. Tumour cell-intrinsic responses to HDACi treatment involving cell death, arrest of proliferation, and modulation of tumour immunogenicity have already been well described and reviewed [2, 67]. In particular, cell death is one of the deepest studied antitumour activity of HDACi, which are able to induce apoptosis by various pathways and processes, including activation of both intrinsic and extrinsic apoptosis pathways by modulating expression of pro- and anti-apoptotic genes, and by activating and/or inducing transcription factors such as E2F1, forkhead box protein O1 (FOXO1), p53, and specificity protein 1 (Sp1) [68, 69]. Another important mechanism by which HDACi can induce tumour cell death is the generation of reactive oxygen species (ROS) that decrease the expression of free radical scavengers. It has been reported that vorinostat and entinostat treatment can induce selective accumulation of ROS and caspase activation only in transformed cells [70]. Hui et al. have demonstrated that synergistic killing of gastric carcinoma (GC) cells by bortezomib/romidepsin combination was dependent on ROS generation and caspase activation. Collectively, this combinatorial effect could also induce autophagy by the activation of MAPK family members (ERK1/2 and JNK) [71]. Furthermore, a synergistic antiproliferative effect has been observed by combination treatment with vorinostat and gefitinib or erlotinib, two epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs), through reduction of cell migration in NSCLC cells. However, the key finding of this study is that the upregulation of the major mitochondrial porin, the voltage-dependent anion-selective channel protein 1 (VDAC1), by vorinostat and TKIs could be involved in oxidative stress-dependent apoptosis. In addition, the usage of vorinostat alone or in combination modulated the c-Myc-NRF2-KEAP1 pathway, crucial for the redox stress response [72]. Further important biological responses to HDACi include cell cycle arrest at the G1/S and G2/M checkpoints, cellular senescence, and autophagy. A recent study has shown that activation of FOXO1 transcription factor by HDACi is an important mediator of autophagic response [73]. HDACi have been recently tested in combination with immunotherapeutic approaches. In addition to their direct antitumour effects, these agents could facilitate recognition and sensitivity to effector functions by cytotoxic T lymphocytes (CTLs) and NK cells, thereby sensitizing cancer cells to immunotherapy. Conversely, in cancer patients, immunological side effects of HDACi such as lymphopenia, leukopenia, neutropenia, and thrombocytopenia might be contradictory for their application in cancer immunotherapy. On the other hand, there is an increasing number of studies showing beneficial effects and immunomodulatory properties of these agents. To date, a number of studies referring to the ability of HDACi in upregulating MHC, co-stimulatory molecule expression, components involved in tumour necrosis factor (TNF) superfamily signalling have been performed [74]. Nevertheless, the molecular mechanisms underlying the involvement of HDACi-regulated genes in immune recognition are not fully understood. Trichostatin A (TSA), a pan-HDACi, in combination with valproic acid (VPA), a class I/IIa HDACi, has been reported to enhance cell surface expression of class I MHC and co-stimulatory molecules CD40 and CD86 in melanoma cells [75]. In the same way, sodium butyrate, a class I/IIa HDACi, and TSA activated expression of class I and II MHC and CD40 in multiple human neuroblastoma (NB) or mouse plasmacytoma J558 tumour cell lines [76]. Furthermore, romidepsin promotes tumour-specific T cell-mediated killing of B16/F10 murine melanoma cells and enhances the expression of class II MHC, CD40, and B7-1/2 [77]. Many studies reported that HDACi sensitize tumour cells to NK cell lysis by promoting expression of NK cell ligands [78,79,80]. Moreover, low cell cytotoxicity by reducing the NK cell activation receptors has been documented using therapeutic concentration of vorinostat and VPA. In a further study, Rossi et al. have demonstrated the reduction of NK cell production by IFN-γ after TSA, VPA, and sodium butyrate treatment [81]. HDACi are also important for macrophage differentiation, polarization, and innate defence function [82]. Multiple studies showed a suppressive role of HDAC inhibition during macrophage activation status. Roger et al. have described that the blockage of class I and II HDACs enhances the recruitment of the repressive complex Mi-2b to the promoters of M1 activation state genes, such as II6 [83]. Cabanel et al. have highlighted the role of TSA as a macrophage differentiation and elongation regulator. They assessed, for the first time, that macrophage plasticity is kept by HDAC inhibition. Furthermore, simultaneous inhibition of class I and II HDACs in several macrophage populations results in reduced levels of recognition receptors, activation markers, cytokines, and chemokines [84]. Moreover, HDAC inhibition can functionally target Tregs and helps to break the immune tolerance. Low levels of Tregs exist under normal physiological conditions, where they mediate the suppression of sustained inflammation, prevent autoimmune responses, and keep homeostasis of immune response. In cancer patients, Tregs are induced by tumour or stroma-secreted factors and also regulated by effector B, T cells, and OX40/OX40L expressed on activated CD4+ and CD8+ T cells, members of the TNFR/TNF superfamily [85, 86]. Tregs are capable of inhibiting NK and T cell function in TME, thus impairing both innate and tumour antigen-specific antitumour immune responses. Nowadays, it is well established that Foxp3 is the major key regulator of Treg development and function. Among the epigenetic modifications, acetylation, together with methylation, regulates the stability and activity of Foxp3 [87]. Furthermore, recent reports have described opposite mechanisms by which different HDAC isoforms modulate Treg and Treg-Foxp3 expression. For instance, by enhancing Foxp3 acetylation, entinostat has been found to increase Treg suppression function. The mechanism of Foxp3 expression regulation by entinostat may involve acetylation of STAT3 protein, which is a substrate of HDAC3 [88]. Conversely, other authors have shown Treg and Foxp3 downregulation following entinostat treatment [89]. Beier et al. suggested that Sirt1, HDAC6, or HDAC9 have different effects on Treg biology. Although HDAC inhibition increased the expression of the Foxp3-encoding gene, the transcription factors involved are different. In particular, loss of HDAC9 stabilizes STAT5 acetylation (K694, K701, and K359) and phosphorylation (Y694) and increases Treg function [90]. On the contrary, HDAC5 decreased Treg suppressive function and impairs iTreg formation as well as IFN-γ production [91]. Other researchers have investigated the effect of HDACi on suppressive myeloid cells, including myeloid-derived suppressor cells (MDSCs) that are comprised of monocytic (M-MDSC) and polymorphonuclear (PMN-MDSC) cells. Suppressive myeloid cells, also including tumour-associated macrophages (TAMs), are induced by tumour growth and accumulated in TME. These cells impair host immunity against tumour cells and facilitate tumour progression and metastasis. Youn et al. have reported that HDAC2 inhibitors can directly interact with the retinoblastoma 1 (Rb1) promoter and participate in silencing Rb1 expression in tumour-bearing hosts. This evidence suggested that HDAC2 inhibitors may regulate pathologic differentiation of myeloid cells in cancer [92]. It has been shown that treatment with TSA and vorinostat led to the expansion of MDSCs in bone marrow cells in vitro, and this effect has been confirmed also in vivo by TSA treatment [93]. Recently, to deeply explore the HDACi impact on antigen presentation, Tiper and Webb have provided evidence on combination of HDACi and NK T cell-based immunotherapy. Importantly, HDACi treatment not only enhances both CD1d- and class II MHC-mediated antigen presentation but also inhibits inflammatory cytokine secretion, which may contribute to the suppression of antitumour NK T cell responses. Moreover, the same authors demonstrated the efficacy of HDACi in restoring antitumour responses to mantle cell lymphoma (MCL) through both cell-intrinsic and cell-extrinsic mechanisms [94].
Also, DNMTi seem to be implicated in the modulation of various immune system components, including tumour-associated antigen (TAA) and antigen presentation machinery (APM). Cancer testis antigens (CTAs) are a large family of tumour-associated antigens expressed in histologically different human tumours, but not in normal tissues except for the testis and placenta. CTAs include the melanoma-associated antigen (MAGE), NY-ESO-1, and SSX gene families and the GAGE/PAGE/XAGE superfamilies, and they are expressed by neoplastic cells and recognized by CTLs. These tumour-restricted expression patterns, together with their strong in vivo immunogenicity, identified CTAs as ideal targets for tumour-specific immunotherapeutic approach, and several clinical trials for a CTA-based vaccine therapy have been developed after these findings [95,96,97]. DNA methylation can lead to induction or upregulation of CTA expression in histologically different solid tumour cells, as well as in stem cells [98]. CTAs, such as preferentially expressed antigen in melanoma (PRAME), have been induced by pharmacological inhibition (5-AZA-CdR) or genetic knockdown of DNMTs, in epithelial ovarian cancer (EOC) or in HGSC [99]. DNMTi are capable of regulating APM on tumour cells through different mechanisms. APM plays an important role during the recognition phase and lysis of neoplastic cells by antigen-specific CTLs and represents a good candidate for immunotherapy likewise CTAs. In addition, to boost immune response, DNMTi can decrease immunosuppression by reducing Treg function [100]. Interestingly, DNMTi and HDACi were strongly effective in inducing upregulation of APM component expression in a broad spectrum of tumour types, suggesting a contribution by indirect epigenetic mechanisms not yet identified [101]. Recently, the role of histone methyltransferases (HMTs) in tumour immunity has been investigated. For instance, combination of EZH2 inhibitors such as deazaneplanocin A (DZNep) or tazemetostat (EPZ6438) with 5-AZA displayed improved therapeutic efficacy of anti-PD-L1 treatment by increasing Teff tumour infiltration and decreasing tumour progression [102]. EZH2 and other PRC2 components have been found to repress the expression of CXCL9 and CXCL10 (Th1-type chemokines) even in colon cancer [103]. However, initial support for the immunological efficacy of DNMTi, alone or combined with HDACi, came out from studies in haematological malignancies, such as AML and MDS, for which 5-AZA and 5-AZA-CdR have been approved by FDA, as already mentioned. In the last decade, a clinical study has reported that the administration of 5-AZA-CdR with VPA induces anti-MAGE CD8+ response in 50% of patients with AML (Fig. 1) [104].

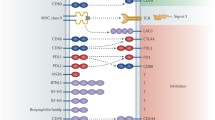
Interaction between tumour and immune cells. T cell stimulation is driven by antigens and requires a coordinated participation of several other receptors and molecules expressed on the T cell surface and antigen-presenting cells (APCs) or tumour cells. HDACi and/or DNMTi can inhibit different signalling pathways involved in adaptive immune responses, enhancing antitumour effects by combination with immune checkpoint inhibitors
The structures of the epi-drugs discussed in the above section are shown in Fig. 2.

Structures of epi-drugs discussed in this review
Preclinical studies of HDACi and DNMTi in combination with immunotherapies
In this section, main preclinical studies are described involving immunotherapy strategies in combination with HDACi or DNMTi (Fig. 2). As aforementioned, HDACi can enhance T cell survival and function and induce expression of multiple chemokines in tumour cells, tumour-infiltrating macrophages, and T cells, thus augmenting the response to anti-PD-1 immunotherapy in lung adenocarcinoma [105]. Over the last decade, extensive studies have been carried out to evaluate the efficacy of combining HDACi with various immunotherapy treatments to overcome cancer cell resistance and strongly improve clinical responses.
Recently, Kim et al. have reported that treatment with both anti-PD-1 and anti-CTLA-4 antibodies was unable to eradicate two types of immunogenic tumours, namely CT26 and 4T1. Nevertheless, co-treatment with epigenetic modulating drugs and checkpoint inhibitors improved treatment outcomes, curing more than 80% of the tumour-bearing mice. In this study, animals bearing large CT26 tumours (>600 mm3) were treated with anti-PD-1 and anti-CTLA-4 antibodies, in combination with 5-AZA or entinostat. The resulting data showed the eradication of primary tumours in 10 out of 11 mice. Interestingly, the primary tumours and metastases were not found in any of the mice treated with both antibodies plus entinostat, whereas only the primary tumour was detected in the mice treated with both antibodies plus 5-AZA [106]. Preclinical studies suggest that HDAC inhibition induces upregulation of PD-L1, and to a lesser extent PD-L2, in a dose-dependent manner. These results have been obtained in vitro and further confirmed in vivo using a murine B16F10 cell melanoma model. Among the inhibitors evaluated, panobinostat displayed the greatest ability to enhance PD-L1 expression, providing a rationale for panobinostat/anti-PD1 combinatorial treatment [107]. Currently, a better understanding of the molecular mechanisms by which HDACi elicit immunostimulatory effects would contribute to their clinical development as anticancer agents. More recently, in order to test whether HDACi could have a synergistic effect with immunotherapy, panobinostat has been administered in an in vivo B16 melanoma model in combination with T cell transfer therapy [108]. In this study, panobinostat improved the effectiveness of gp100-specific T cell immunotherapy and maintained systemic pro-inflammatory levels. Moreover, it enhanced proliferation, retention, and polyfunctional status of tumour-specific T cells, leading to decreased tumour burden and highly decreased Treg populations. Kroesen et al. have shown that combination of anti-GD2 plus vorinostat reduces NB tumour growth [109]. Further studies have also shown that HDACi upregulate the expression of various components of the immune system, in particular molecules involved in APM as well as those involved in immune co-stimulation. Horing et al. described how TSA, in addition to induction of apoptosis in tumour cells, can augment anti-glioblastoma multiforme (GBM) innate immune responses. Systemic treatment with TSA delayed GBM xenograft growth by enhancing tumour recognition by NK cells [110]. As already aforementioned, HDACi may regulate APM through different ways including activation of class II transactivator (CIITA), a master regulator of MHC II [111]. Additionally, it has been demonstrated that entinostat increased the level of MHC II by transcriptional activation of CIITA in diffuse large B cell lymphoma (DLBCL) [112]. Furthermore, NB and hepatoma cells treated with VPA have shown increased sensitivity to NK cell killing through transcription of MHC class I-related chain A and B (MICA and MICB). In addition, VPA at non-toxic pharmacological concentration arrested growth, induced differentiation, and increased immunogenicity of NB cells through non-toxic mechanisms [113]. Further experiments have been carried out to evaluate the effect of vorinostat on regulation of MICA/B expression. In this study, Yang et al. have reported that vorinostat upregulates the transcription of MICA/B by promoting MICA-associated histone acetylation and by suppressing the MICA/B-targeting miRNAs, such as miR-20a, miR-93, and miR-106b. Vorinostat can regulate miR-17-92 cluster and MCM7 to upregulate MICA expression in hepatoma [114].
Romidepsin displayed an antiproliferative effect on T cells by inhibition of the production of IL-2 and downregulation of CD25 (part of the IL-2 receptor) [115]. Although poor efficacy was observed in the antitumour immune response in vivo, Cao et al. have demonstrated that in vivo treatment with TSA induced suppression of nuclear factor of activated T cells 1 (NFAT1)-regulated FasL expression on activated CD4+ T cells. Importantly, they also found that the co-administration of HDACi and anti-CTLA-4 could further enhance the infiltration of CD4+ T cells and achieve a synergistic anticancer effect. In fact, within this study, modulation of activation-induced cell death (AICD) of tumour-infiltrating CD4+ T cells by TSA enhanced antitumour immune responses, uncovering a novel mechanism underlying the antitumour effect of HDACi [116]. Previous studies have evaluated the capability of rocilinostat (ACY-1215), a HDAC6-specific inhibitor, to prevent skin inflammation through blocking the effector CD8+ T cells and impairing the MAPK pathway [117]. Furthermore, since tumour growth induces accumulation of immunosuppressive cells including Tregs, a huge number of studies addressing the effect of HDACi on Tregs and other immunosuppressive cells have been performed. Entinostat inhibited Foxp3 expression and Treg suppressive function in a dose-dependent manner and, at lower doses, enhanced cytokine and vaccine therapies in murine renal cell carcinoma and prostate cancer models, respectively [88]. In contrast, in murine colitis models of inflammation and autoimmunity, the HDAC6-selective inhibitors tubacin and tubastatin A enhanced Treg suppressive function [118]. More studies have revealed that co-treatment with HDACi or DNMTi and checkpoint inhibitors were capable of suppressing MDSCs and eradicate metastatic mouse cancer resistant to immune checkpoint blockade [106]. A very recent study has demonstrated that prostate (LNCAP) and breast (MDA-MB-231) carcinoma cells are more sensitive to T cell-mediated lysis in vitro after clinically relevant exposure to epigenetic therapy with either vorinostat or entinostat and that genetic or pharmacological inhibition studies identified HDAC1 as a key determinant to reverse carcinoma immune escape [119]. Moreover, in two non-epithelial cancers (glioma and mesothelioma), it was found that the epigenetic regulation of the NY-ESO1 gene requires the sequential recruitment of the HDAC1-mSin3a-NCOR and DNMT1-PCNA-UHRF1-G9a complexes [120].
Treatment with DNMTi allows immunological recognition and cytolysis of cancer cells overcoming the resistance to CTA-specific CTLs. 5-AZA-CdR has been reported to modulate the expression of both CTA and class I human leucocyte antigen (HLA) and the resulting modification in neoplastic cell immunogenicity [121]. 5-AZA has been shown to upregulate PD-L1 in EOC and NSCLC cell lines, eliciting the activation of the viral/IFN response [122]. Nevertheless, another recent report has shown that PD-1 promoter demethylation was associated with PD-1 mRNA upregulation and worse overall response in MDS patients [123]. Notably, patients with MDS resistant to DNMTi showed elevated levels of PD-L1, PD-L2, and CTLA-4, suggesting a putative involvement of PD-1 signalling in resistance mechanisms to hypomethylating agents [124]. Importantly, Odunsi et al. performed a phase I dose escalation of 5-AZA-CdR, in addition to NY-ESO-1 vaccine and doxorubicin liposome chemotherapy, in 12 patients with relapsed EOC. Increased NY-ESO-1 serum antibodies and T cell responses were observed in most patients, encouraging further evaluation in other tumour types [125].
Based on the evidence above highlighted, the cooperation between DNA methylation and histone acetylation in controlling gene transcription prompted some researchers to explore new combined therapies using both HDACi and DNMTi. A synergistic upregulation of MAGE-A genes in selected cancer cell lines by 5-AZA-CdR/TSA combination has been reported [126]. Despite these results, a non-durable synergistic effect was observed for such a combination, with DNMTi remaining the most effective epigenetic drugs in modulating CTA expression in cancer cells. Additional preclinical data confirmed the upregulation of cellular CTA expression by systemic administration of 5-AZA-CdR and modification of class I HLA antigen expression [127]. These in vivo modulations, including NY-ESO expression, were still detectable on melanoma xenografts 30 days after the end of 5-AZA-CdR administration, and injection of BALB/c mice generated high-titre anti-NY-ESO-1 antibodies [127]. Furthermore, 5-AZA-CdR induced demethylation of the Toll-like receptor 4 (TLR4) promoter, an important modulator of the immune response in various cancers, and increased H3K4 trimethylation and Sp1 binding to reactivate silenced TLR4.
In addition, it was demonstrated that the recruitment of the MeCP2/HDAC1 repressor complex increased the low levels of TLR4 expression through epigenetic modification of DNA and histones on the TLR4 promoter in gastric cancer cells [128]. A phase I trial showed that 5-AZA-CdR may be a potential modulator of the immune-activating properties of high-dose IL-2 in melanoma and renal cell carcinoma patients. While upregulation of chemokines and genes involved in IL-1, IL-17, IL-22, and IFN signalling might favour the activity of administered IL-2, downregulation of IL-2Ra, CD3-ε, CD2, and genes involved in IL-2 signalling can be expected to impair IL-2 activity [129].
Clinical investigation of HDACi or DNMTi treatment in combination with immunotherapies
In the light of the above considerations, researchers have proposed the clinical use of some epigenetic drugs in order to overcome some major limitations of current therapeutic strategies to fight cancer and to evaluate their efficacy and clinical tolerability. Many preclinical studies have been conducted using different classes of HDACi, also corroborated by an increasing number of clinical investigations started by combining HDACi with immunotherapeutics. Immune evasion is the major obstacle to the efficacy of cancer immunotherapies, by preventing long-lasting tumour control. Hence, there is a strong need to restore tumour immune recognition of malignant tumours in order to increase the clinical benefit for patients. HDACi appear to be able to improve the in vivo therapy efficacy, and, although additional preclinical data are needed to assess the efficacy and toxicity of these drugs alone or in combination with other chemotherapeutics and immunotherapy strategies, several clinical studies are being investigated (Table 1). Among current clinical trials, in patients with advanced renal or urothelial cell carcinoma, pembrolizumab and vorinostat will be administered to evaluate the antitumour activity by estimation of serious adverse events (AEs), maximum tolerated dose (MTD), and progression-free survival (PFS). These clinical studies have a run-in phase with sequential single agents and then a combination phase. Thirty patients will be enrolled in two expansion cohorts: 15 anti-PD1-naive patients and 15 anti-PD1-resistant patients (NCT02619253, Table 1) [130]. More interesting evaluation of the potential combined therapy targeting cancer cells will be represented by the study that keeps in consideration the possibility to treat metastatic eye melanoma (PEMDAC) with pembrolizumab and entinostat. Their co-administration will be, respectively, intravenously (IV) for pembrolizumab at 200 mg and orally for entinostat at 5 mg for a period of 24 months. PFS and clinical beneficial rate (CBR) are some of the parameters that should be characterized to evaluate patient responses (NCT02697630, Table 1) [130]. Focusing on DNMTi, 5-AZA or entinostat will be orally administered to metastatic NSCLC patients together with the monoclonal anti-PD1 antibody nivolumab (NCT01928576, Table 1) [130]. In a phase I study, the safety of a combination between orally administered pembrolizumab and 5-AZA will be evaluated (NTC02546986, Table 1) [130]. Likewise, in a phase II study, 60 patients with NSCLC will be enrolled to evaluate the efficacy of 5-AZA-CdR plus nivolumab treatment vs nivolumab alone (NCT02664181, Table 1) [130]. An overview of all other combinations is shown in Table 1.
Analysing the recent clinical trials, vorinostat and 5-AZA are the drugs most frequently used, likely due to their intense preclinical and clinical investigations.
Conclusions
Robust data support the role of epigenetic drugs in facilitating immunological targeting of cancer cells by their ability to modulate different mediator factors and pathways involved in the interaction between tumour cells and the immune system. Following this observation, HDACi or DNMTi have been combined with immune checkpoint therapies to provide more significant benefit for cancer patients than monotherapy. In this review, we have summarized preclinical and clinical results combining HDACi or DNMTi with immune checkpoint inhibitors and their direct effects on various components of the immune system. Although more and more preclinical trials are being conducted to enhance safety and efficacy, especially for DNMTi, these findings will help along the road for the discovery and the development of novel therapeutic approaches in cancer immunotherapy. Focused on the results from preclinical studies of HDACi on Tregs, either class I or class II HDAC inhibition may have opposite effects on Treg function as inhibition or promotion, respectively. Thus, it is currently believed that cancer treatments with class I-specific HDACi could provide future interesting outcomes in patients. Indeed, different studies have shown that immunomodulatory effects of HDAC inhibition with high specificity may lead to a selective immune regulation, when compared with pan-HDACi treatment. Even if selective HDACi may provide greater efficacy, the identification of the proper dose could reduce the adverse effects associated with HDAC inhibition. Regardless of which epigenetic modulator is used in preclinical or clinical studies, the toxicity on different tumours types remains a great challenge. A better understanding of the molecular mechanisms by which HDACi and DNMTi elicit immunomodulatory effects could help to ameliorate their clinical development. Nevertheless, to achieve beneficial responses in patients, a deep investigation on the main molecular processes on which the immune system relies remains of high interest. Future inquiry on immuneregulatory mechanisms could provide more interesting targets for epigenetic drugs in order to improve cancer cell recognition by T cells and overcome cancer therapy failure. In addition, it could be interesting to evaluate the immunomodulatory activity of other epigenetic modulators (i.e. HMTs and demethylase inhibitors), even highlighting their clinical effects by combination with the already described HDACi or DNMTi.
Abbreviations
- 5-AZA:
-
Azacitydine
- 5-AZA-CdR:
-
Decitabine
- AEs:
-
Adverse events
- AICD:
-
Activation-induced cell death
- AML:
-
Acute myeloid leukaemia
- APC:
-
Antigen-presenting cell
- APL:
-
Acute promyelocytic leukaemia
- APM:
-
Antigen presentation machinery
- BATF:
-
Basic leucine zipper transcription factor
- cAMP:
-
Cyclic adenosyl monophosphate
- CBR:
-
Clinical beneficial rate
- CD28:
-
Cluster of differentiation 28
- CIITA:
-
Class II transactivator
- CMML:
-
Chronic myelomonocytic leukaemia
- CTA:
-
Cancer testis antigen
- CTCL:
-
Cutaneous T cell lymphoma
- CTL:
-
Cytotoxic T lymphocytes
- CTLA-4:
-
Cytotoxic T lymphocyte-associated antigen-4
- DC:
-
Dendritic cell
- DLBCL:
-
Diffuse large B cell lymphoma
- DNMT:
-
DNA methyltransferase
- DZNeP:
-
Deazaneplanocin A
- EGFR:
-
Epidermal growth factor receptor
- EMA:
-
European Medicines Agency
- EOC:
-
Epithelial ovarian cancer
- EZH2:
-
Enhancer of zeste homolog 2
- FOXO1:
-
Forkhead box protein O1
- Foxp3:
-
Forkhead box protein 3
- GC:
-
Gastric carcinoma
- HATs:
-
Histone acetyltransferases
- HDAC:
-
Histone deacetylase
- HLA:
-
Human leucocyte antigen
- HMTs:
-
Histone methyltransferases
- IFN-γ:
-
Interferon-γ
- IL-6:
-
Interleukin-6
- iTreg:
-
Induced regulatory T
- lncRNAs:
-
Long noncoding RNAs
- MAbs:
-
Monoclonal antibodies
- MAGE-A:
-
Melanoma-associated antigen
- MAPK:
-
Mitogen-activated protein kinases
- MCL:
-
Mantle cell lymphoma
- MDS:
-
Myelodysplastic syndromes
- MDSCs:
-
Myeloid-derived suppressor cells
- MHC:
-
Major histocompatibility complex
- MICA:
-
MHC class I-related chain A
- MICB:
-
MHC class I-related chain B
- miRNAs:
-
MicroRNAs
- MITF:
-
Microphthalmia-associated transcription factor
- MM:
-
Multiple myeloma
- MTD:
-
Maximum tolerated dose
- NB:
-
Neuroblastoma
- NEAT1:
-
Nuclear paraspeckle assembly transcript 1
- NFAT1:
-
Nuclear factor of activated T cells 1
- NF-κB:
-
Nuclear factor kappa light chain enhancer of activated B cells
- NK:
-
Natural killer
- NSCLC:
-
Non-small cell lung cancer
- PD:
-
Programmed cell death protein
- PFS:
-
Progression-free survival
- PRAME:
-
Preferentially expressed antigen in melanoma
- PRC2:
-
Polycomb repressive complex 2
- PTCL:
-
Peripheral T cell lymphoma
- Rb1:
-
Retinoblastoma 1
- RORγT:
-
RAR-related orphan receptor
- ROS:
-
Reactive oxygen species
- Sin3A:
-
Transcription regulator family member A
- Sp1:
-
Specificity protein 1
- STAT4:
-
Signal transducer and activator of transcription 4
- TAA:
-
Tumour-associated antigen
- TAM:
-
Tumour-associated macrophage
- Tbx21:
-
T-box transcription factor 21
- TCR:
-
T cell receptor
- TGF-β:
-
Transforming growth factor β
- Th:
-
T helper
- Tim-3:
-
T cell immunoglobulin and mucin domain 3
- TKIs:
-
Tyrosine kinase inhibitors
- TLR4:
-
Toll-like receptor 4
- TME:
-
Tumour microenvironment
- TNF:
-
Tumour necrosis factor
- Treg:
-
T regulatory cell
- TSA:
-
Trichostatin A
- VDAC1:
-
Voltage-dependent anion- selective channel protein 1
- VEGF:
-
Vascular endothelial growth factor
- VPA:
-
Valproic acid
References
Thurn KT, Thomas S, Moore A, Munster PN. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol. 2011;7:263–83.
Zwergel C, Valente S, Mai A. DNA methyltransferases inhibitors from natural sources. Curr Top Med Chem. 2016;16:680–96.
Qureshi IA, Mehler MF. Advances in epigenetics and epigenomics for neurodegenerative diseases. Curr Neurol Neurosci Rep. 2011;11:464–73.
Feinberg AP. Epigenetics at the epicenter of modern medicine. JAMA. 2008;299:1345–50.
Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92.
Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–54.
Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514.
Robertson KD. DNA methylation, methyltransferases, and cancer. Oncogene. 2001;20:3139–55.
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705.
Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84.
Shen L, Orillion A, Pili R. Histone deacetylase inhibitors as immunomodulators in cancer therapeutics. Epigenomics. 2016;8:415–28.
Topalian SL, Weiner GJ, Pardoll DM. Cancer immunotherapy comes of age. J Clin Oncol. 2011;29:4828–36.
Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–14.
Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–71.
Stewart TJ, Abrams SI. How tumours escape mass destruction. Oncogene. 2008;27:5894–903.
Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–60.
Suh WK, Tafuri A, Berg-Brown NN, Shahinian A, Plyte S, Duncan GS, Okada H, Wakeham A, Odermatt B, Ohashi PS, Mak TW. The inducible costimulator plays the major costimulatory role in humoral immune responses in the absence of CD28. J Immunol. 2004;172:5917–23.
Gardner D, Jeffery LE, Sansom DM. Understanding the CD28/CTLA-4 (CD152) pathway and its implications for costimulatory blockade. Am J Transplant. 2014;14:1985–91.
Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704.
Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. 2014;2:3.
Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, Vilgelm AE, Feld E, Johnson AS, Greenplate AR, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun. 2016;7:10582.
El-Osta H, Shahid K, Mills GM, Peddi P. Immune checkpoint inhibitors: the new frontier in non-small-cell lung cancer treatment. Onco Targets Ther. 2016;9:5101–16.
Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, Restifo NP, Haworth LR, Seipp CA, Freezer LJ, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A. 2003;100:8372–7.
Maker AV, Phan GQ, Attia P, Yang JC, Sherry RM, Topalian SL, Kammula US, Royal RE, Haworth LR, Levy C, et al. Tumor regression and autoimmunity in patients treated with cytotoxic T lymphocyte-associated antigen 4 blockade and interleukin 2: a phase I/II study. Ann Surg Oncol. 2005;12:1005–16.
Krasner C, Joyce RM. Zevalin: 90yttrium labeled anti-CD20 (ibritumomab tiuxetan), a new treatment for non-Hodgkin’s lymphoma. Curr Pharm Biotechnol. 2001;2:341–9.
Menon S, Shin S, Dy G. Advances in cancer immunotherapy in solid tumors. Cancers (Basel). 2016;8.
Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–81.
Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275–80.
Grosso JF, Jure-Kunkel MN. CTLA-4 blockade in tumor models: an overview of preclinical and translational research. Cancer Immun. 2013;13:5.
Larkin J, Hodi FS, Wolchok JD. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:1270–1.
Carosella ED, Ploussard G, LeMaoult J, Desgrandchamps F. A systematic review of immunotherapy in urologic cancer: evolving roles for targeting of CTLA-4, PD-1/PD-L1, and HLA-G. Eur Urol. 2015;68:267–79.
Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33.
Selby MJ, Engelhardt JJ, Johnston RJ, Lu LS, Han M, Thudium K, Yao D, Quigley M, Valle J, Wang C, et al. Preclinical development of ipilimumab and nivolumab combination immunotherapy: mouse tumor models, in vitro functional studies, and cynomolgus macaque toxicology. PLoS One. 2016;11:e0161779.
Ito A, Kondo S, Tada K, Kitano S. Clinical development of immune checkpoint inhibitors. Biomed Res Int. 2015;2015:605478.
Nicodemus CF. Antibody-based immunotherapy of solid cancers: progress and possibilities. Immunotherapy. 2015;7:923–39.
Suzuki S, Ishida T, Yoshikawa K, Ueda R. Current status of immunotherapy. Jpn J Clin Oncol. 2016;46:191–203.
Heninger E, Krueger TE, Lang JM. Augmenting antitumor immune responses with epigenetic modifying agents. Front Immunol. 2015;6:29.
Nelson HH, Kelsey KT. Epigenetic epidemiology as a tool to understand the role of immunity in chronic disease. Epigenomics. 2016;8:1007–9.
Reiner SL. Inducing the T cell fates required for immunity. Immunol Res. 2008;42:160–5.
Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. 2010;28:445–89.
Kanno Y, Vahedi G, Hirahara K, Singleton K, O'Shea JJ. Transcriptional and epigenetic control of T helper cell specification: molecular mechanisms underlying commitment and plasticity. Annu Rev Immunol. 2012;30:707–31.
Schoenborn JR, Dorschner MO, Sekimata M, Santer DM, Shnyreva M, Fitzpatrick DR, Stamatoyannopoulos JA, Wilson CB. Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nat Immunol. 2007;8:732–42.
Santangelo S, Cousins DJ, Winkelmann N, Triantaphyllopoulos K, Staynov DZ. Chromatin structure and DNA methylation of the IL-4 gene in human T(H)2 cells. Chromosome Res. 2009;17:485–96.
Aune TM, Collins PL, Chang S. Epigenetics and T helper 1 differentiation. Immunology. 2009;126:299–305.
Allan RS, Zueva E, Cammas F, Schreiber HA, Masson V, Belz GT, Roche D, Maison C, Quivy JP, Almouzni G, Amigorena S. An epigenetic silencing pathway controlling T helper 2 cell lineage commitment. Nature. 2012;487:249–53.
Chang S, Collins PL, Aune TM. T-bet dependent removal of Sin3A-histone deacetylase complexes at the Ifng locus drives Th1 differentiation. J Immunol. 2008;181:8372–81.
Vahedi G, Takahashi H, Nakayamada S, Sun HW, Sartorelli V, Kanno Y, O'Shea JJ. STATs shape the active enhancer landscape of T cell populations. Cell. 2012;151:981–93.
Tumes DJ, Onodera A, Suzuki A, Shinoda K, Endo Y, Iwamura C, Hosokawa H, Koseki H, Tokoyoda K, Suzuki Y, et al. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity. 2013;39:819–32.
Arvey A, van der Veeken J, Samstein RM, Feng Y, Stamatoyannopoulos JA, Rudensky AY. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat Immunol. 2014;15:580–7.
Morinobu A, Kanno Y, O'Shea JJ. Discrete roles for histone acetylation in human T helper 1 cell-specific gene expression. J Biol Chem. 2004;279:40640–6.
Kurachi M, Barnitz RA, Yosef N, Odorizzi PM, DiIorio MA, Lemieux ME, Yates K, Godec J, Klatt MG, Regev A, et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat Immunol. 2014;15:373–83.
Weber B, Stresemann C, Brueckner B, Lyko F. Methylation of human microRNA genes in normal and neoplastic cells. Cell Cycle. 2007;6:1001–5.
Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, Araujo L, Carbone DP, Shilo K, Giri DK, et al. PDL1 regulation by p53 via miR-34. J Natl Cancer Inst. 2016;108.
Shatz M, Menendez D, Resnick MA. The human TLR innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells. Cancer Res. 2012;72:3948–57.
Textor S, Fiegler N, Arnold A, Porgador A, Hofmann TG, Cerwenka A. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res. 2011;71:5998–6009.
Li J, Shayan G, Avery L, Jie HB, Gildener-Leapman N, Schmitt N, Lu BF, Kane LP, Ferris RL. Tumor-infiltrating Tim-3+ T cells proliferate avidly except when PD-1 is co-expressed: evidence for intracellular cross talk. Oncoimmunology. 2016;5:e1200778.
Thommen DS, Schreiner J, Muller P, Herzig P, Roller A, Belousov A, Umana P, Pisa P, Klein C, Bacac M, et al. Progression of lung cancer is associated with increased dysfunction of T cells defined by coexpression of multiple inhibitory receptors. Cancer Immunol Res. 2015;3:1344–55.
Wei J, Nduom EK, Kong LY, Hashimoto Y, Xu S, Gabrusiewicz K, Ling X, Huang N, Qiao W, Zhou S, et al. MiR-138 exerts anti-glioma efficacy by targeting immune checkpoints. Neuro Oncol. 2016;18:639–48.
Cheng Y, Li Y, Nian Y, Liu D, Dai F, Zhang J. STAT3 is involved in miR-124-mediated suppressive effects on esophageal cancer cells. BMC Cancer. 2015;15:306.
Xu S, Tao Z, Hai B, Liang H, Shi Y, Wang T, Song W, Chen Y, OuYang J, Chen J. miR-424(322) reverses chemoresistance via T-cell immune response activation by blocking the PD-L1 immune checkpoint. Nat Commun. 2016;7:11406.
Li H, Liu C, Lu Z, Chen L, Wang J, Li Y, Ma H. Upregulation of the long non-coding RNA SPRY4-IT1 indicates a poor prognosis and promotes tumorigenesis in ovarian cancer. Biomed Pharmacother. 2017;88:529–34.
Spizzo R, Almeida MI, Colombatti A, Calin GA. Long non-coding RNAs and cancer: a new frontier of translational research? Oncogene. 2012;31:4577–87.
Zeng C, Xu Y, Xu L, Yu X, Cheng J, Yang L, Chen S, Li Y. Inhibition of long non-coding RNA NEAT1 impairs myeloid differentiation in acute promyelocytic leukemia cells. BMC Cancer. 2014;14:693.
Hu G, Tang Q, Sharma S, Yu F, Escobar TM, Muljo SA, Zhu J, Zhao K. Expression and regulation of intergenic long noncoding RNAs during T cell development and differentiation. Nat Immunol. 2013;14:1190–8.
Chun P. Histone deacetylase inhibitors in hematological malignancies and solid tumors. Arch Pharm Res. 2015;38:933–49.
Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015;20:3898–941.
Newbold A, Falkenberg KJ, Prince MH, Johnstone RW. How do tumor cells respond to HDAC inhibition? FEBS J. 2016;283:4032–46.
Waby JS, Chirakkal H, Yu C, Griffiths GJ, Benson RS, Bingle CD, Corfe BM. Sp1 acetylation is associated with loss of DNA binding at promoters associated with cell cycle arrest and cell death in a colon cell line. Mol Cancer. 2010;9:275.
Yang Y, Zhao Y, Liao W, Yang J, Wu L, Zheng Z, Yu Y, Zhou W, Li L, Feng J, et al. Acetylation of FoxO1 activates Bim expression to induce apoptosis in response to histone deacetylase inhibitor depsipeptide treatment. Neoplasia. 2009;11:313–IN311.
Ungerstedt JS, Sowa Y, Xu WS, Shao Y, Dokmanovic M, Perez G, Ngo L, Holmgren A, Jiang X, Marks PA. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102:673–8.
Hui KF, Yeung PL, Chiang AK. Induction of MAPK- and ROS-dependent autophagy and apoptosis in gastric carcinoma by combination of romidepsin and bortezomib. Oncotarget. 2016;7:4454–67.
Leone A, Roca MS, Ciardiello C, Terranova-Barberio M, Vitagliano C, Ciliberto G, Mancini R, Di Gennaro E, Bruzzese F, Budillon A. Vorinostat synergizes with EGFR inhibitors in NSCLC cells by increasing ROS via up-regulation of the major mitochondrial porin VDAC1 and modulation of the c-Myc-NRF2-KEAP1 pathway. Free Radic Biol Med. 2015;89:287–99.
Zhang J, Ng S, Wang J, Zhou J, Tan SH, Yang N, Lin Q, Xia D, Shen HM. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy. 2015;11:629–42.
Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–42.
Khan AN, Gregorie CJ, Tomasi TB. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol Immunother. 2008;57:647–54.
Magner WJ, Kazim AL, Stewart C, Romano MA, Catalano G, Grande C, Keiser N, Santaniello F, Tomasi TB. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol. 2000;165:7017–24.
Murakami T, Sato A, Chun NA, Hara M, Naito Y, Kobayashi Y, Kano Y, Ohtsuki M, Furukawa Y, Kobayashi E. Transcriptional modulation using HDACi depsipeptide promotes immune cell-mediated tumor destruction of murine B16 melanoma. J Invest Dermatol. 2008;128:1506–16.
Pfeiffer MM, Burow H, Schleicher S, Handgretinger R, Lang P. Influence of histone deacetylase inhibitors and DNA-methyltransferase inhibitors on the NK cell-mediated lysis of pediatric B-lineage leukemia. Front Oncol. 2013;3:99.
Morgado S, Sanchez-Correa B, Casado JG, Duran E, Gayoso I, Labella F, Solana R, Tarazona R. NK cell recognition and killing of melanoma cells is controlled by multiple activating receptor-ligand interactions. J Innate Immun. 2011;3:365–73.
Son CH, Keum JH, Yang K, Nam J, Kim MJ, Kim SH, Kang CD, Oh SO, Kim CD, Park YS, Bae J. Synergistic enhancement of NK cell-mediated cytotoxicity by combination of histone deacetylase inhibitor and ionizing radiation. Radiat Oncol. 2014;9:49.
Rossi LE, Avila DE, Spallanzani RG, Ziblat A, Fuertes MB, Lapyckyj L, Croci DO, Rabinovich GA, Domaica CI, Zwirner NW. Histone deacetylase inhibitors impair NK cell viability and effector functions through inhibition of activation and receptor expression. J Leukoc Biol. 2012;91:321–31.
Han SB, Lee JK. Anti-inflammatory effect of Trichostatin-A on murine bone marrow-derived macrophages. Arch Pharm Res. 2009;32:613–24.
Roger T, Lugrin J, Le Roy D, Goy G, Mombelli M, Koessler T, Ding XC, Chanson AL, Reymond MK, Miconnet I, et al. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood. 2011;117:1205–17.
Cabanel M, Brand C, Oliveira-Nunes MC, Cabral-Piccin MP, Lopes MF, Brito JM, de Oliveira FL, El-Cheikh MC, Carneiro K. Epigenetic control of macrophage shape transition towards an atypical elongated phenotype by histone deacetylase activity. PLoS One. 2015;10:e0132984.
Burocchi A, Pittoni P, Gorzanelli A, Colombo MP, Piconese S. Intratumor OX40 stimulation inhibits IRF1 expression and IL-10 production by Treg cells while enhancing CD40L expression by effector memory T cells. Eur J Immunol. 2011;41:3615–26.
Sojka DK, Huang YH, Fowell DJ. Mechanisms of regulatory T-cell suppression—a diverse arsenal for a moving target. Immunology. 2008;124:13–22.
Pan F, Fan H, Liu Z, Jiang S. T cell signaling targets for enhancing regulatory or effector function. Sci Signal. 2012;5:pe32.
Shen L, Ciesielski M, Ramakrishnan S, Miles KM, Ellis L, Sotomayor P, Shrikant P, Fenstermaker R, Pili R. Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. PLoS One. 2012;7:e30815.
Bridle BW, Chen L, Lemay CG, Diallo JS, Pol J, Nguyen A, Capretta A, He R, Bramson JL, Bell JC, et al. HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy. Mol Ther. 2013;21:887–94.
Beier UH, Wang L, Han R, Akimova T, Liu Y, Hancock WW. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci Signal. 2012;5:ra45.
Xiao H, Jiao J, Wang L, O'Brien S, Newick K, Wang LC, Falkensammer E, Liu Y, Han R, Kapoor V, et al. HDAC5 controls the functions of Foxp3(+) T-regulatory and CD8(+) T cells. Int J Cancer. 2016;138:2477–86.
Youn JI, Kumar V, Collazo M, Nefedova Y, Condamine T, Cheng P, Villagra A, Antonia S, McCaffrey JC, Fishman M, et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol. 2013;14:211–20.
Rosborough BR, Castellaneta A, Natarajan S, Thomson AW, Turnquist HR. Histone deacetylase inhibition facilitates GM-CSF-mediated expansion of myeloid-derived suppressor cells in vitro and in vivo. J Leukoc Biol. 2012;91:701–9.
Tiper IV, Webb TJ. Histone deacetylase inhibitors enhance CD1d-dependent NKT cell responses to lymphoma. Cancer Immunol Immunother. 2016;65:1411–21.
Valmori D, Ayyoub M. Prospective strategies to combine conventional, targeted and immunotherapies in non-small cell lung cancer. Oncoimmunology. 2016;5:e947175.
Geldmacher A, Freier A, Losch FO, Walden P. Therapeutic vaccination for cancer immunotherapy: antigen selection and clinical responses. Hum Vaccin. 2011;7(Suppl):115–9.
Wang M, Li J, Wang L, Chen X, Zhang Z, Yue D, Ping Y, Shi X, Huang L, Zhang T, et al. Combined cancer testis antigens enhanced prediction accuracy for prognosis of patients with hepatocellular carcinoma. Int J Clin Exp Pathol. 2015;8:3513–28.
Fratta E, Coral S, Covre A, Parisi G, Colizzi F, Danielli R, Nicolay HJ, Sigalotti L, Maio M. The biology of cancer testis antigens: putative function, regulation and therapeutic potential. Mol Oncol. 2011;5:164–82.
Zhang W, Barger CJ, Eng KH, Klinkebiel D, Link PA, Omilian A, Bshara W, Odunsi K, Karpf AR. PRAME expression and promoter hypomethylation in epithelial ovarian cancer. Oncotarget. 2016;7:45352–69.
Costantini B, Kordasti SY, Kulasekararaj AG, Jiang J, Seidl T, Abellan PP, Mohamedali A, Thomas NS, Farzaneh F, Mufti GJ. The effects of 5-azacytidine on the function and number of regulatory T cells and T-effectors in myelodysplastic syndrome. Haematologica. 2013;98:1196–205.
Seliger B. Molecular mechanisms of MHC class I abnormalities and APM components in human tumors. Cancer Immunol Immunother. 2008;57:1719–26.
Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249–53.
Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, Zhao L, Wei S, Frankel T, Vatan L, et al. PRC2 epigenetically silences Th1-type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer Res. 2016;76:275–82.
Goodyear O, Agathanggelou A, Novitzky-Basso I, Siddique S, McSkeane T, Ryan G, Vyas P, Cavenagh J, Stankovic T, Moss P, Craddock C. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood. 2010;116:1908–18.
Zheng H, Zhao W, Yan C, Watson CC, Massengill M, Xie M, Massengill C, Noyes DR, Martinez GV, Afzal R, et al. HDAC inhibitors enhance T-cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin Cancer Res. 2016;22:4119–32.
Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, Diaz Jr LA, Papadopoulos N, Kinzler KW, Vogelstein B, Zhou S. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. 2014;111:11774–9.
Woods DM, Sodre AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res. 2015;3:1375–85.
Lisiero DN, Soto H, Everson RG, Liau LM, Prins RM. The histone deacetylase inhibitor, LBH589, promotes the systemic cytokine and effector responses of adoptively transferred CD8+ T cells. J Immunother Cancer. 2014;2:8.
Kroesen M, Bull C, Gielen PR, Brok IC, Armandari I, Wassink M, Looman MW, Boon L, den Brok MH, Hoogerbrugge PM, Adema GJ. Anti-GD2 mAb and vorinostat synergize in the treatment of neuroblastoma. Oncoimmunology. 2016;5:e1164919.
Horing E, Podlech O, Silkenstedt B, Rota IA, Adamopoulou E, Naumann U. The histone deacetylase inhibitor trichostatin a promotes apoptosis and antitumor immunity in glioblastoma cells. Anticancer Res. 2013;33:1351–60.
Woan KV, Sahakian E, Sotomayor EM, Seto E, Villagra A. Modulation of antigen-presenting cells by HDAC inhibitors: implications in autoimmunity and cancer. Immunol Cell Biol. 2012;90:55–65.
Cycon KA, Mulvaney K, Rimsza LM, Persky D, Murphy SP. Histone deacetylase inhibitors activate CIITA and MHC class II antigen expression in diffuse large B-cell lymphoma. Immunology. 2013;140:259–72.
Cinatl Jr J, Cinatl J, Scholz M, Driever PH, Henrich D, Kabickova H, Vogel JU, Doerr HW, Kornhuber B. Antitumor activity of sodium valproate in cultures of human neuroblastoma cells. Anticancer Drugs. 1996;7:766–73.
Yang H, Lan P, Hou Z, Guan Y, Zhang J, Xu W, Tian Z, Zhang C. Histone deacetylase inhibitor SAHA epigenetically regulates miR-17-92 cluster and MCM7 to upregulate MICA expression in hepatoma. Br J Cancer. 2015;112:112–21.
Kelly-Sell MJ, Kim YH, Straus S, Benoit B, Harrison C, Sutherland K, Armstrong R, Weng WK, Showe LC, Wysocka M, Rook AH. The histone deacetylase inhibitor, romidepsin, suppresses cellular immune functions of cutaneous T-cell lymphoma patients. Am J Hematol. 2012;87:354–60.
Cao K, Wang G, Li W, Zhang L, Wang R, Huang Y, Du L, Jiang J, Wu C, He X, et al. Histone deacetylase inhibitors prevent activation-induced cell death and promote anti-tumor immunity. Oncogene. 2015;34:5960–70.
Glauben R, Sonnenberg E, Wetzel M, Mascagni P, Siegmund B. Histone deacetylase inhibitors modulate interleukin 6-dependent CD4+ T cell polarization in vitro and in vivo. J Biol Chem. 2014;289:6142–51.
de Zoeten EF, Wang L, Butler K, Beier UH, Akimova T, Sai H, Bradner JE, Mazitschek R, Kozikowski AP, Matthias P, Hancock WW. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells. Mol Cell Biol. 2011;31:2066–78.
Gameiro SR, Malamas AS, Tsang KY, Ferrone S, Hodge JW. Inhibitors of histone deacetylase 1 reverse the immune evasion phenotype to enhance T-cell mediated lysis of prostate and breast carcinoma cells. Oncotarget. 2016;7:7390–402.
Cartron PF, Blanquart C, Hervouet E, Gregoire M, Vallette FM. HDAC1-mSin3a-NCOR1, Dnmt3b-HDAC1-Egr1 and Dnmt1-PCNA-UHRF1-G9a regulate the NY-ESO1 gene expression. Mol Oncol. 2013;7:452–63.
Coral S, Sigalotti L, Altomonte M, Engelsberg A, Colizzi F, Cattarossi I, Maraskovsky E, Jager E, Seliger B, Maio M. 5-aza-2'-deoxycytidine-induced expression of functional cancer testis antigens in human renal cell carcinoma: immunotherapeutic implications. Clin Cancer Res. 2002;8:2690–5.
Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R, Topper MJ, Luo J, Connolly RM, Azad NS, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014;5:587–98.
Orskov AD, Treppendahl MB, Skovbo A, Holm MS, Friis LS, Hokland M, Gronbaek K. Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS/AML patients: a rationale for combined targeting of PD-1 and DNA methylation. Oncotarget. 2015;6:9612–26.
Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, Fang Z, Nguyen M, Pierce S, Wei Y, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014;28:1280–8.
Odunsi K, Matsuzaki J, James SR, Mhawech-Fauceglia P, Tsuji T, Miller A, Zhang W, Akers SN, Griffiths EA, Miliotto A, et al. Epigenetic potentiation of NY-ESO-1 vaccine therapy in human ovarian cancer. Cancer Immunol Res. 2014;2:37–49.
Sigalotti L, Fratta E, Coral S, Cortini E, Covre A, Nicolay HJ, Anzalone L, Pezzani L, Di Giacomo AM, Fonsatti E, et al. Epigenetic drugs as pleiotropic agents in cancer treatment: biomolecular aspects and clinical applications. J Cell Physiol. 2007;212:330–44.
Coral S, Sigalotti L, Colizzi F, Spessotto A, Nardi G, Cortini E, Pezzani L, Fratta E, Fonsatti E, Di Giacomo AM, et al. Phenotypic and functional changes of human melanoma xenografts induced by DNA hypomethylation: immunotherapeutic implications. J Cell Physiol. 2006;207:58–66.
Kim TW, Lee SJ, Oh BM, Lee H, Uhm TG, Min JK, Park YJ, Yoon SR, Kim BY, Kim JW, et al. Epigenetic modification of TLR4 promotes activation of NF-kappaB by regulating methyl-CpG-binding domain protein 2 and Sp1 in gastric cancer. Oncotarget. 2016;7:4195–209.
Gollob JA, Sciambi CJ, Peterson BL, Richmond T, Thoreson M, Moran K, Dressman HK, Jelinek J, Issa JP. Phase I trial of sequential low-dose 5-aza-2'-deoxycytidine plus high-dose intravenous bolus interleukin-2 in patients with melanoma or renal cell carcinoma. Clin Cancer Res. 2006;12:4619–27.
ClinicalTrials.gov 2016, http://clinicaltrials.gov, Accessed: 31 Jan 2017
Acknowledgements
Not applicable.
Funding
This work was supported by the IIT-Sapienza Project, by the COST Action CM1406 Epigenetic Chemical Biology, and by PRIN 2016 (prot. 20152TE5PK).
Availability of data and materials
Not applicable.
Authors’ contributions
RM and CZ reviewed the main literature for biology of epigenetic compounds and immunotherapy. RM, CZ, SV, and AM wrote the manuscript and the general concept of this review. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mazzone, R., Zwergel, C., Mai, A. et al. Epi-drugs in combination with immunotherapy: a new avenue to improve anticancer efficacy. Clin Epigenet 9, 59 (2017). https://doi.org/10.1186/s13148-017-0358-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-017-0358-y